

Abstract
Psymberin is the only member of the pederin natural product family that contains a dihydroisocoumarin side chain. Structural modifications of psymberin uncoupled inhibition of protein translation from cytotoxicity, suggesting that psymberin has more than one bioactivity. A forward genetic screen in Caenorhabditis elegans was conducted to identify the molecular target(s) of psymberin. Multiple independent psymberin-resistant mutants were isolated, each containing the same point mutation in a gene encoding a ribosomal protein. However, a psymberin-resistant mutant strain bearing this mutation was not cross-resistant to the pederin family member mycalamide A, which binds to the archaeal form of the same protein. Thus, two pederin family members likely differ in how they bind the same molecular target. The accumulation of psymberin in cells was sensitive to the stereochemistry of the amide side chain at C4 or C8 and the presence of the dihydroisocoumarin side chain. The observation that psymberin diastereomers or dihydroisocoumarin-truncated analogs lose all cytotoxic activity while retaining the ability to inhibit protein translation in a cell-free in vitro assay can be explained in the context of these differential cell uptake issues. Finally, we also demonstrate that the blistering activity associated with pederin and other members of the family is not due to their protein synthesis inhibiting activity. Unlike pederin and mycalamide, psymberin does not display irritant or blistering activity.
INTRODUCTION
In a preceding article,1 we described the full structural elucidation, two alternative total syntheses, and analog generation of psymberin (1a), the latest member and a structural outlier of the pederin family of natural products.2 Psymberin shares a common N-acyl aminal-substituted tetrahydropyranyl core with all pederin family members, several of which exhibit dermatotoxicity (blistering activity) and pharmacologically act as protein synthesis inhibitors.3 However, psymberin differs structurally in two significant ways from the rest of the pederin family. Without exception, all 36 members of the pederin family,4 isolated from various sources around the globe, display an identical cyclic pederate side chain – a structural (and evolutionary) conservation which we postulate to be functionally relevant. In contrast, psymberin uniquely possesses a less complex acyclic side chain (psymberate). Second, the dihydrosiocoumarin bottom fragment is unique to psymberin, indicating divergent biosynthetic machinery not found in any of the other pederin-type producing organisms.5 Finally, psymberin was described to exhibit an unprecedented differential cytotoxicity profile,2 whereas pederin-like compounds display much more uniform cytotoxicity profiles.6 In light of these observations, we hypothesized that psymberin could be endowed with unique biological activities. In this paper, we describe detailed mode-of-action studies through a combination of structure-activity relationships, biochemical studies, and a forward genetic screen in the nematode worm Caenorhabditis elegans.
RESULTS AND DISCUSSION
To study meaningful structure-activity relationships of psymberin, several analogs were synthesized1 to probe the influence of the individual psymberate and dihydroisocoumarin side chains on biological output as compared to pederin/mycalamide structures containing a cyclic pederate side-chain. A selection of analogs relevant to this study is depicted in Figure 1 and includes: psymberin (1a) and its 4-epi and 8-epi diastereomers 3 and 4; a psymberate-truncated analog 7; a dihydroisocoumarin-truncated psymberin analog which also represents a pederin analog in which the cyclic pederate side chain is substituted with psymberin’s acyclic psymberate side chain and termed psympederin 5 (basically a hybrid between pederin and psymberin), and the corresponding 8-epi psympederin diastereomer 6; pederin (2a) and the corresponding desmethylene pederin analog 2b;7 and mycalamide A (8) which serves as a representative of the pederin family.8
Figure 1.

Structures of psymberin, pederin, mycalamide, and analogs
Cytotoxicity of psymberin and related compounds
As noted in the introduction, psymberin was reported to exhibit a highly differential cytotoxicity profile (>10,000-fold differences among cancer cell lines in the NCI 60 cell line panel). We were therefore surprised to see that in our hands, synthetic psymberin was highly cytotoxic to every human cancer cell line tested. For example, synthetic psymberin inhibited the PC3 prostate and SK-MEL-5 melanoma cancer cell lines with an IC50 of 0.98 and 2.29 nM respectively,9 whereas the same two cancer cell lines were reported to respond highly differentially to natural psymberin (>25 μM for PC3 and <2.5 nM for SK-MEL-5).2 The cytotoxicity of synthetic psymberin against HeLa and SK-MEL-5 human tumor cell lines is listed in Table 1 (selected dose-response curves are shown in Figure 2).10 Compared to psymberin (1a), mycalamide A (8) was nearly equipotent against the same cell lines. Because mycalamide is a known eukaryotic protein translation inhibitor,6b,11 we also included cycloheximide, a compound with a similar mode-of-action – namely inhibition of elongation-translocation via binding to the large 60S ribosomal subunit.12 In the HeLa cell line, cycloheximide was over a 1,000-fold less potent as a cytotoxic agent (Table 1 and Fig. 2B). Stereochemical permutations, as for example in psymberin diastereomers 3 and 4, resulted in dramatically reduced antiproliferative activity.13 Similar structural changes (methoxyaminal epimer) in the pederin/mycalamide family were reported to result in similar dramatic reductions in cytotoxic activity.6c,14 Removal of the psymberate side chain as in compound 7 also completely abolished cytotoxic activity. Moving to the dihydroisocoumarin side chain, acetylation of one of the phenols as in compound 1b resulted in similar cytotoxicity as psymberin (Table 1). The corresponding phenolic methylether (Fig. 1, 1c) neither reduced nor increased activity compared to psymberin. The antiproliferative activity of the psymberin-pederin hybrid (psympederin 5) provided the most informative piece of structure-function information: this compound, and its diastereomer 6, was devoid of activity up to 1 μM concentrations, a loss of >1,000-fold compared to psymberin or mycalamide. If pederin/mycalamide and psymberin share the same mode-of-action, then the dihydroisocoumarin side chain in psymberin should not be critically important given the absence of this fragment in the potent cytotoxin mycalamide. Moreover, we also know that a cyclic pederate side chain is not critical for activity given the potent cytotoxicity associated with psymberin. Thus the inactivity of psympederin 5 strongly suggests that the dihydroisocoumarin fragment is vitally important for psymberin cytotoxicity, and at the same time reveals that the cyclic pederate side chain is critically important for the cytotoxic phenotype of pederin/mycalamide family members.15
Table 1.
Cytotoxicitya and translation inhibition of psymberin, mycalamide and analogs against human tumor cell lines.b
| Compound | Cytotoxicity (IC50, nM) | Translation Inhibition (EC50, nM)***
|
|||
|---|---|---|---|---|---|
| in vitro assay | cell-based assay | ||||
|
| |||||
| Hela* | SK-MEL-5** | HeLa | SK-MEL-5 | ||
| Cycloheximide | 2242±1515 | 3116±754 | 3150±2152 | 3325±834 | 2670 |
| 1a | 0.64±0.14 | 0.27±0.04 | 28±7 | 2.2±1.4 | 11±10 |
| 1b | 0.54±0.01 | 0.35±0.07 | 142±21 | 5.8±1.7 | 4.2±3.2 |
| 1c | 2.34±0.53 | 1.58±0.42 | 120±47 | 9.6±8.9 | 9.3±8.5 |
| 8 | 2.52±1.39 | 3.79±0.04 | 238±44 | 59±32 | 64 |
| 3 | 618.6±267.0 | 352.0±12.1 | 346±64 | 4950±4870 | 496 |
| 4 | >1000 | 762.8±70.0 | 318±182 | 2200±1410 | 843 |
| 5 | >1000 | >1000 | 641±262 | 1650±1060 | 578 |
| 6 | >1000 | >1000 | >10000 | >10000 | >10000 |
| 7 | >1000 | >1000 | >10000 | >10000 | >10000 |
A CellTiter-Glo® luminescent assay, which measures cellular ATP concentrations, was used to measure cell viability with and without compound treatment. IC50 values were calculated by fitting the luminescence data to an equation representing the dose-response of inhibiting luminescence.
Data are means ± standard deviation from at least two independent experiments conducted in triplicate.
R2 values range from 0.931 to 0.994.
R2 values range from 0.865 to 0.997.
R2 ranges from 0.90 to 0.995.
Figure 2.
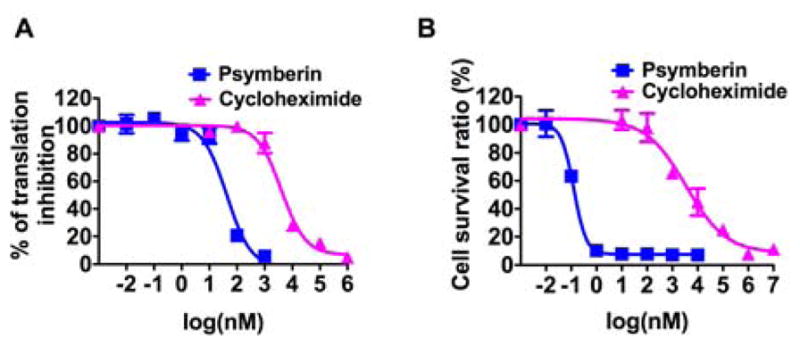
A. Representative translation-inhibition curves from a cell-based assay. B. Representative cytotoxicity curves from the cell-based CellTiter-Glo® assay. Means with standard deviations are plotted for each treatment condition.
Inhibition of protein translation in cell-based and in vitro assays
Given the structural relationship between psymberin and pederin, we tested the ability of psymberin and analogs to inhibit protein synthesis. Two assays were used to measure the inhibition of translation induced by psymberin and analogs. The first assay measured total protein translation in HeLa or SK-MEL-5 cells through incorporation of radioactive 35S-methionine into TCA-precipitable counts in the presence or absence of the compounds. As shown in Table 1, psymberin (1a) and mycalamide (8) potently inhibited translation in HeLa (EC50 = 2.2 and 60 nM) and SK-MEL-5 cells (EC50 = 11 and 64 nM) as compared to the positive control cycloheximide. In HeLa cells, the modification on dihydroisocoumarin slightly reduced the potency in translation, whereas the EC50 concentrations of psymberin epimers 3 and 4, and psympederin (5) increased 200 to a 400 fold compared to psymberin. Psympederin diastereomer 6 and psymberate-truncated analog 7 lost all activity. Thus it appears that the SAR data obtained in the cell-based protein translation inhibition assay mirrored those observed in the cytotoxicity assay (Table 1).
Compounds were also evaluated in a cell-free in vitro translation assay using rabbit reticulocyte extracts, where translation of firefly luciferase was measured as a function of compound concentration. Surprisingly, in this assay the psymberin analogs 3–5 were only about 10 fold less potent than psymberin (1a).
We also observed that the EC50 values for the natural products psymberin and mycalamide were higher in the in vitro assay than the cell-based translation inhibition assay, unlike the analogs 3–5 which were more potent in the in vitro assay. This data suggest that stereochemical changes (3 and 4) or the elimination of the dihydroisocoumarin side chain (psympederin 5) must affect processes in the cell-based assays other than those occurring on the ribosome. Indeed, psympederin 5 gave no more than a 30-fold change in inhibition of protein synthesis in vitro compared to psymberin, but more than a 1,000-fold change in cell-based cytotoxicity assay. Thus, the dihydroisocoumarin side chain of psymberin appears to be important for inducing cytotoxicity but not for inhibiting protein translation. This significant result was surprising given the generally accepted notion that protein synthesis inhibition fully accounts for the cytotoxic effects of the pederin/mycalamide family of natural products. However, we note that the majority of pederin/mycalamide analogs have been tested for cytotoxic effects only, and not for protein synthesis inhibition. The similar IC50 and EC50 values of cycloheximide suggest that its cytotoxicity may originate entirely from inhibiting translation.
Differential accumulation of analogs in cells
Based on our observation that cytotoxic effects and protein synthesis inhibition did not coincide for some analogs tested (most notably psympederin 5), we argued that the dihydroisocoumarin side chain was key to psymberin’s cytotoxic activity, but not for translation inhibition. However, the genetics (described in the following section) does indicate that the ribsome is the main target for inducing toxicity, at least in worms. Aside from the vesicant activity, psymberin and mycalamide show very similar cellular phenotypes, inhibit translation, induce ribotoxic stress and bind the same site on the ribosome. To explain the discrepancies between the in vitro and cell-based potency of compounds 3–5 (Table 1), we measured their intracellular concentration. As shown in Table 2, after incubating HeLa cells for two hours with 100 nM of each compound, the intracellular concentration of the two psymberin epimers 3 and 4 was about 20-fold less than psymberin. The intracellular concentration of psympederin 5 was below the limit of detection. This difference could be due to a change in cellular uptake of compounds, a change in efflux, or a change in metabolism of the compounds and indicates that structural features of psymberin important for its intracellular concentration control biological activities.
Table 2.
The intracellular concentration of different psymberin analogs in HeLa cells.a
| Compound | Intracellular Concentration (μM) |
|---|---|
| 1a | 7.14±2.93 |
| 3 | 0.21±0.08 |
| 4 | 0.31±0.11 |
| 5 | < LD |
Data shown are means ± standard deviation from at least two independent experiments conducted in duplicate. LD, limit of detection. See Supporting Information for experimental details.
Forward Genetic Screen in C. elegans
Parallel to the SAR study, we also searched for targets of psymberin responsible for toxicity in an unbiased fashion. We used a forward genetic screen in the model organism C. elegans.16 We determined the concentrations of psymberin that caused wild-type Bristol strain (N2) C. elegans to fail to reproduce.17 Psymberin killed more than 95% of N2 worms above 2 μM concentrations, and no L1 larva could grow or survive at 5 μM. Thus, 4.5 and 5 μM of psymberin were used in the forward genetic screen. N2 worms at the L4/young adult stage were treated with ethyl methanesulfonate (EMS) to generate random mutations. Psymberin-resistant F2 (second generation) progeny were identified as the few viable adult worms with eggs that grew in the presence of the compound. From two separate genetic screens, we isolated 7 toxin-resistant worms under conditions that precluded isolating sibling worms. The drug-resistant mutation in one of these worms, strain DA2312, was identified by a combination of standard genetic techniques and DNA sequencing, and confirmed by micro-injecting cDNA containing the dominant mutant gene into wild-type worms, which conferred drug resistance to their progeny. DA2312 worms were 20 fold more resistance to psymberin compared to wild type worms (Fig. 3A). The drug-resistant mutation in DA2312 causes a proline to leucine transition in the ribosomal large subunit protein, RPL41, which is the ortholog of human RPL36a and RPL36al and of archaeal protein L44e.18 The amino acid sequence surrounding this proline is highly conserved in eukaryotic organisms and forms a loop of protein between the P site and E site on the ribosome.19 During the course of our studies, Steitz and coworkers published the crystal structure of mycalamide A bound to an archaeal ribosome.20,21 Their data suggest that there is interaction between mycalamide A and the two conserved lysines in the ribosomal protein L44e (the homolog of RPL41). To determine if the mutation in rpl-41 would confer cross-resistance to mycalamide, wild-type N2 and psymberin-resistant DA2312 worms were treated with various concentrations of mycalamide A (8). Interestingly, DA2312 showed the same sensitivity to mycalamide A as the wild-type N2 worms (Fig. 3B). Thus, the mutation in rpl-41 is psymberin-specific.
Figure 3.

The psymberin-resistant mutation does not confer resistance to mycalamide A. The toxicity curves for psymberin on DA2312 and N2 worms (A), and the toxicity curves for mycalamide A on DA2312 and N2 worms (B) are presented. Blue squares indicate data from N2 samples, pink solid circle the data from DA2312. The concentration of compound is indicated on the x-axis. The y-axis is the ratio of survived worms. Means with standard deviations are plotted for each treatment condition.
When the gene encoding RPL41 was sequenced in the parental wild type worm, it was found to conform to the published sequence and encode proline. However, the genes for RPL41 in each of the other mutant worms contained the identical mutation as DA2312. We had expected to find multiple targets of psymberin, yet in C. elegans, we repeatedly found a single point mutation, suggesting that relatively few changes on the ribosome allow it to remain active in the presence of psymberin. The change from a rigid proline to a relatively flexible leucine may cause structural changes in the pocket where psymberin binds to the ribosome. If so, these changes do not affect the binding of the structurally similar toxin, mycalamide A, and this suggests that the changes induced by the P65L mutation must be rather local. A comparison of the binding pocket between archeal20 and mammalian ribosomes22 reveals that the conformations of the rRNA surrounding the binding pocket are totally different in the two ribosomes. The proline that was mutated to produce resistance to psymberin is in a small loop of sequence absent from the archaeal ribosome. Thus, the binding conformation of mycalamide A on the archaeal ribosome may suggest one of several types of interactions that can stabilize binding, but not necessarily indicate the interactions on the eukaryotic ribosome. The answers to these arguments will have to come from higher resolution structures of mammalian ribosomes with these inhibitors bound.
Vesicant activity
One of the earliest described effects of pederin is a vesicant activity causing severe dermatitis.3 Many related compounds, such as mycalamide and the onnamide family, are also known to share this activity. The structural features of the pederin family of compounds responsible for this activity are unknown. To determine if psymberin is also a vesicant, a mouse ear-swelling test (MEST)23,24 was established and psymberin vesicant activity evaluated in comparison to that shown by mycalamide, pederin, and a synthetic variant of pederin, desmethylene pederin 2b.7 The left and right ears of C57BL/6 mice were painted with either vehicle or compound and ear thickness was monitored daily with a modified Mitutoyo micrometer by an investigator blinded to the treatment. Both acute vesicant and delayed contact hypersensitivity were monitored by pre-treatment of the mice on their abdomen with vehicle or compound. As shown in Figure 4a, mycalamide (8), but not psymberin (1a), clearly induces swelling of the ear treated with compound relative to the vehicle treated ear. Pre-treatment of the abdomen of mice with mycalamide but not psymberin resulted in a blistering effect on the abdominal skin but did not consistently or significantly increased swelling of subsequently treated ears (data not shown), suggesting the effect was acute and not due to an immune mediated delayed type hypersensitivity reaction. As expected, pederin (2a) also showed significant vesicant activity as measured in the MEST (Fig. 4b). Significantly, a synthetic variant of pederin in which a methylene group has been removed (desmethylene pederin 2b) showed no activity in this assay. A hydrogenated derivative of pederin (dihydropederin) was reported to have lost its vesicant activity though it remained a potent inhibitor of protein synthesis.3 This uncoupling of vesicant activity and protein translation inhibition in desmethylene pederin (2b), psymberin (1a), and dihydropederin3 suggests that the homoallylic acetal in pederin (2a), mycalamide A (8) and related natural products is the chemical pharmacophore responsible for the blistering/vesicant activity. The absence of an acetal in psymberin and removal of the exo-methylene double bond in pederin/mycalamide relatives is sufficient to eliminate the blistering/vesicant activity of these compounds.
Figure 4.
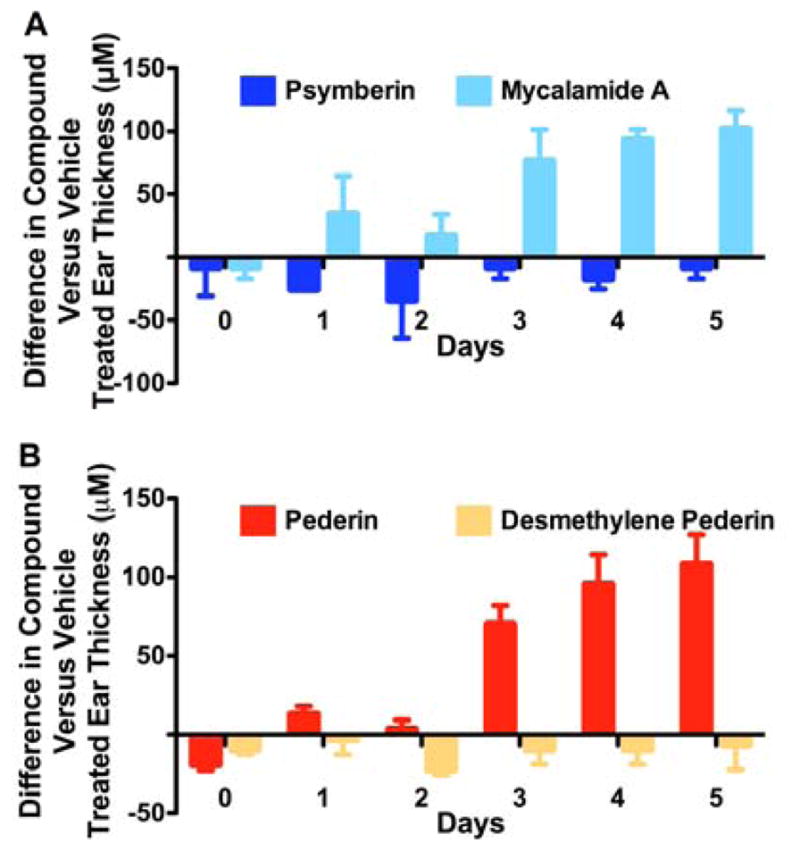
The vesicant activity of pederin, mycalamide, and psymberin differ. The abdomen of C57BL/6 mice was shaved using an electrical razor and 100 ml of vehicle solution (5% ethanol in 3:1 acetone:olive oil) was applied. Seven days later, 25 ml of a 0.00125% w/v solution of psymberin or mycalamide A. (A) or pederin or its synthetic analog, demethylene pederin (B) prepared in the same vehicle were applied to either the right or left ear of the treated mice. The opposing ear was treated with vehicle only. Prior to the ear application and then each day thereafter for 5 days, the thickness of both ears was measured by an investigator blinded to the treatment using a modified Mitutoyo micrometer. The differences in ear thickness between compound and vehicle treated ears in mm are plotted.
CONCLUSION
With its dihydroisocoumarin and acyclic N-acyl side chains, psymberin represents a structural outlier of the pederin/mycalamide family of natural products. Founded on a comprehensive synthetic footing, we were able to study the chemical biology of this family in more detail. Initial observations that cytotoxic activity and protein synthesis inhibition did not track for all analogs suggested that psymberin might differ from other pederin family members in it cellular targets. However, Forward genetic studies in C. elegans indicated that the primary target of psymberin is the ribosome.25 Drug-resistant worms were not cross-resistant to mycalamide and this could indicate subtle binding differences of these two compounds for the same target. We also demonstrated that psymberin is not a blistering agent, and we determined the chemical features responsible for vesicant activity in other pederin family members. In the end, dramatic differences in intracellular concentrations explained discrepancies between pharmacological and cellular activity of structurally related analogs with similar physicochemical properties, a characteristic that is often not measured in SAR studies of natural products.
Supplementary Material
Acknowledgments
We thank Professor Leon Avery (Virginia Commonwealth University) and Iryna Zubovych for advice and technical support in the C. elegans genetic work. We thank Matthew Evans for experimental assistance. Financial support was provided by the National Institutes of Health (grant CA95471 supporting J.K.D.B., M.G.R., and N.W.); grant CA90349 to J.K.D.B.), the National Science Foundation (grant CHE-0848299 to P.E.F.) and the Robert A. Welch Foundation (grant I-1422 to J.K.D.B.).
ABBREVIATIONS
- rRNA
ribosomal RNA
- SAR
structure-activity-relationship
Footnotes
Supporting Information. Supporting figures, experimental methods for SAR assays, worm genetic and biochemical characterizations. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Feng Y, Jiang X, De Brabander JK. J Am Chem Soc. 2012;134:17083. doi: 10.1021/ja3057612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cichewicz RH, Valeriote FA, Crews P. Org Lett. 2004;6:1951. doi: 10.1021/ol049503q. [DOI] [PubMed] [Google Scholar]
- 3.For reviews, see: Narquizian R, Kocienski PJ. In The role of Natural Products in Drug Discovery. In: Mulzer J, Bohlmann R, editors. Ernst Schering Research Foundation Workshop. Vol. 32. Springer; New York: 2000. pp. 25–56.Mosey RA, Floreancig PE. Nat Prod Rep. 2012;29:980. doi: 10.1039/c2np20052j.
- 4.For a complete listing including references, see Fig. S1 in the Supporting Information, and the Supporting Information to ref. 2.
- 5.(a) Fisch KM, Gurgui C, Heycke N, van der Sar SA, Anderson SA, Webb VL, Taudien S, Platzer M, Rubio BK, Robinson SJ, Crews P, Piel J. Nat Chem Biol. 2009;5:494. doi: 10.1038/nchembio.176. [DOI] [PubMed] [Google Scholar]; (b) Piel J, Butzke D, Fusetani N, Hui D, Platzer M, Wen G, Matsunaga S. J Nat Prod. 2005;68:472. doi: 10.1021/np049612d. [DOI] [PubMed] [Google Scholar]
- 6.(a) Brega A, Falaschi A, De Carli L, Pavan M. J Cell Biol. 1986;36:485. doi: 10.1083/jcb.36.3.485. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Burres NS, Clement JJ. Canc Res. 1989;49:2935. [PubMed] [Google Scholar]; (c) Richter A, Kocienski P, Raubo P, Davies DA. Anti-Canc Drug Des. 1997;12:217. [PubMed] [Google Scholar]
- 7.Wan S, Wu F, Rech JC, Green ME, Balachandran R, Horne WS, Day BW, Floreancig PE. J Am Chem Soc. 2011;133:16668. doi: 10.1021/ja207331m. [DOI] [PubMed] [Google Scholar]
- 8.We gratefully acknowledge a gift of natural mycalamide A from Peter Northcote (Victoria University of Wellington). For a reference, see: West LM, Northcote PT, Hood KA, Miller JH, Page MJ. J Nat Prod. 2000;63:707. doi: 10.1021/np9904511.
- 9.Jiang X, Williams N, De Brabander JK. Org Lett. 2007;9:227. doi: 10.1021/ol062656o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.In addition to the cytotoxicity data reported in ref. 9, synthetic psymberin was tested against normal fibroblasts (BJ), telomerase immortalized fibroblasts (BJHtert, and a panel of ten human tumor cell lines. See Table S1 in the Supporting Information.
- 11.Dang Y, Schneider-Poetsch T, Eyler DE, Jewett JC, Bhat S, Rawal VH, Green R, Liu JO. RNA. 2011;17:1578. doi: 10.1261/rna.2624511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baliga BS, Pronczuk AW, Munro HN. J Biol Chem. 1969;244:4480. [PubMed] [Google Scholar]
- 13.For other psymberin SAR studies, see: ref. 7, 9, and Huang X, Shao N, Huryk R, Palani A, Aslanian R, Seidel-Dugan C. Org Lett. 2009;11:867. doi: 10.1021/ol802772s.Watanabe T, Imaizumi T, Chinen T, Nagumo Y, Shibuya M, Usui T, Kanoh N, Iwabuchi Y. Org Lett. 2010;12:1040. doi: 10.1021/ol1000389.
- 14.For extensive mycalamide SAR studies, see: Thompson AM, Blunt JW, Munro MHG, Perry NB. J Chem Soc Perkin Trans 1. 1992:1335.Thompson AM, Blunt JW, Munro MHG, Clark BM. J Chem Soc Perkin Trans 1. 1994:1025.Thompson AM, Blunt JW, Munro MHG, Perry NB. J Chem Soc Perkin Trans 1. 1995:1233.
- 15.Introducing the dihydroisocoumarin unit in pederin produced a compound termed pedestatin with a >100-fold increase of antiproliferative activity over pederin in the human cancer cell line HCT116, see ref. 7.
- 16.Kaletta T, Hengartner MO. Nat Rev Drug Discov. 2006;5:387. doi: 10.1038/nrd2031. [DOI] [PubMed] [Google Scholar]
- 17.See Fig. S3 in the Supporting Information.
- 18.See Fig. S4 in the Supporting Information.
- 19.Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. Science. 2011;334:1524. doi: 10.1126/science.1212642. [DOI] [PubMed] [Google Scholar]
- 20.Gurel G, Blaha G, Steitz TA, Moore PB. Antimicrob Agents Chemother. 2009;53:5010. doi: 10.1128/AAC.00817-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.One of us (P.E.F.) used this model to interpret the changes in toxicity observed in modified forms of pederin, see ref. 7.
- 22.Chandramouli P, Topf M, Menetret JF, Eswar N, Cannone JJ, Gutell RR, Sali A, Akey CW. Structure. 2008;16:535. doi: 10.1016/j.str.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gad SC. Toxicology. 1994;93:33. doi: 10.1016/0300-483x(94)90194-5. [DOI] [PubMed] [Google Scholar]
- 24.Stewart I, Seawright AA, Schluter PJ, Shaw GR. BMC Dermatol. 2006;6:5. doi: 10.1186/1471-5945-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.One of the reviewers suggested we perform a parallel DARTS assay to detect any protein binding partner(s) of psymberin in mammalian cells. We performed a DARTS assay of psymberin in HeLa cells twice without detecting any protein protection by psymberin as determined by silver staining. For a description of the DARTS assay, see: Lemonick, et al. Proc Natl Acad Sci USA. 2012;109:6811.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.