

Summary
Antisense noncoding transcripts, genes-within-genes, and convergent gene pairs are prevalent among eukaryotes. The existence of such transcription units raises the question of what happens when RNA polymerase II (RNAPII) molecules collide head-to-head. Here we use a combination of biochemical and genetic approaches in yeast to show that polymerases transcribing opposite DNA strands cannot bypass each other. RNAPII stops but does not dissociate upon head-to-head collision in vitro, suggesting that opposing polymerases represent insurmountable obstacles for each other. Head-to-head collision in vivo also results in RNAPII stopping, and removal of collided RNAPII from the DNA template can be achieved via ubiquitylation-directed proteolysis. Indeed, in cells lacking efficient RNAPII polyubiquitylation, the half-life of collided polymerases increases, so that they can be detected between convergent genes. These results provide insight into fundamental mechanisms of gene traffic control and point to an unexplored effect of antisense transcription on gene regulation via polymerase collision.
Graphical Abstract

Highlights
► Convergently transcribing RNAPIIs cannot transcribe past one another in vivo ► In vitro, RNAPII stops when the front edges of the colliding proteins touch ► Collided polymerases remain stably associated with the template ► Collided RNAPII accumulates between convergent genes in elc1Δ strains
Introduction
Recent advances in genomics have provided evidence for a highly complex and dynamic transcription landscape in eukaryotes. It is now clear that transcription is surprisingly pervasive, giving rise to both stable mRNAs and a large variety of noncoding RNAs (ncRNAs) (Berretta and Morillon, 2009; Jacquier, 2009; Carninci, 2010). In budding yeast, approximately 55% of stable, uncharacterized transcripts (SUTs) are produced in the antisense direction relative to an annotated open reading frame (ORF), with SUTs often being initiated from the 3′ end of active genes (Xu et al., 2009, 2011). Moreover, ∼1,500 gene pairs are convergent in the compact budding yeast genome, and these are sometimes overlapping or without a terminator sequence between them. In mammalian cells, numerous genes are embedded in, and transcribed in the opposite direction of, another gene (Yu et al., 2005; Mourier and Willerslev, 2008), and antisense ncRNAs are also produced genome wide, with a substantial fraction of human genes being associated with an antisense transcript (Chen et al., 2004; Vallon-Christersson et al., 2007). The possible functions of antisense transcripts in the regulation of transcription are a subject of obvious interest, but their existence also raises a more fundamental question: what happens when convergently transcribing RNA polymerase II (RNAPII) elongation complexes (ECs) collide with each other on DNA?
We have previously studied collision between RNAPII ECs transcribing the same DNA strand (head-to-tail collision). This study indicated that dynamic interactions between conformationally elastic ECs make significant and fundamental contributions to transcript elongation (Saeki and Svejstrup, 2009). The situation is different when RNAPII molecules transcribe opposite DNA strands; here, approaching transcription “bubbles” should in theory be able to pass each other (Figure 1A, left). Indeed, crystallographic data suggest that the nontranscribed strand may be held fairly loosely in the RNAPII groove accommodating the DNA template (Kornberg, 2007), and a study using T3 and T4 bacteriophage RNAP showed that these single-subunit polymerases can transcribe past one another in vitro (Ma and McAllister, 2009). On the other hand, the large size and extraordinary stability of the eukaryotic EC (Kornberg, 2007) might make bypass difficult or impossible (Figure 1A, right).
Figure 1.

RNAPII Collision Is a Block to Transcript Elongation In Vivo
(A) Left: Nucleic acid-centric view of convergent transcription. Right: Protein-centric view of elongation, showing a crystallographic model of convergently transcribing RNAPII elongation complexes (Kornberg, 2007) (DNA in green/blue; RNA in red).
(B) GAL10-GAL7 and GAL10-GAL7ΔTATA constructs, with short and long G-less cassettes indicated.
(C) Autoradiograph of RNA isolated from GAL10-GAL7ΔTATA (“−,” convergent transcription, lanes 1 and 3) or GAL10-GAL7 (“+,” 2 and 4) incubated for 75 min in the presence of glucose (Glc, lanes 1 and 2) or galactose (Gal, lanes 3 and 4). Mean percent distal cassette transcribed is indicated below, after normalizing to proximal G-less cassette levels (distal/proximal set to 100 in GAL10-GAL7ΔTATA). The standard error calculated from two biological replicates is presented below.
(D) Schematic showing experimental layout and results.
While bypass is a possibility, collision-induced RNAPII stopping, or pausing, seems highly likely. Such pausing could result in back-tracking and transcriptional arrest or, if bypass is impossible, lead to gene blockage. This would be highly problematic for cells, as even a single persistently arrested RNAPII molecule in an essential gene is potentially lethal (Svejstrup, 2007). Indeed, transcriptional arrest resulting from DNA damage or backtracking triggers a “last resort” mechanism, namely polyubiquitylation and degradation of RNAPII (Woudstra et al., 2002; Somesh et al., 2005; Sigurdsson et al., 2010). This pathway works through a two-step mechanism: Rsp5 (NEDD4) monoubiquitylates RNAPII, followed by Elongin-Cullin complex-mediated polyubiquitylation and proteasomal degradation (Harreman et al., 2009).
In this study we use an in vitro transcription system to show that RNAPII molecules cannot transcribe past one another and remain bound to DNA following collision. We also show that collision is problematic in vivo as well, and that it can trigger removal of collided RNAPII via the ubiquitin-proteasome system.
Results
Elongation Complex Collision between Active Genes In Vivo
RNAPII head-to-head collisions are potentially rare and stochastically distributed across large areas of genomic DNA, which might lead to detection difficulties under normal circumstances. To investigate the consequences of RNAPII collision in vivo, we therefore first generated a transcription template to study the process in budding yeast (Figure 1B). Similar templates were used by others previously (Prescott and Proudfoot, 2002; García-Rubio and Aguilera, 2012), but those were not designed to specifically study elongation and therefore could not distinguish between the effects of promoter occlusion and polymerase collision. In our system, head-to-head transcription is directed by two inducible promoters (GAL10 and GAL7, respectively) separated by a coding region lacking transcriptional terminators (Figure 1B, GAL10-GAL7). As a negative control, the TATA box of the GAL7 promoter was mutated to eliminate transcription from this promoter (Prescott and Proudfoot, 2002) (Figure 1B, GAL10-GAL7ΔTATA). Importantly, we also inserted two cassettes downstream of the GAL10 promoter producing tracts of guanine-free (G-less) mRNA: a short cassette (“proximal”) was inserted immediately after the promoter while a longer (“distal”) cassette was placed as an extension of the GAL10 ORF, approximately 2 kb further downstream. Progression of the EC driven by the GAL10 promoter was then assessed following digestion of the isolated RNA with RNase T1, which cleaves RNA at guanines and thus removes all RNA except for the two G-less cassettes. The proximal G-less cassette served as an internal control: any decrease in transcription of the distal cassette relative to the proximal cassette would thus be due to collision between elongating RNAPII molecules in the ∼2 kb region between the two cassettes.
As expected, no transcripts were detected in repressive conditions (Figure 1C, lanes 1 and 2). However, signals from both G-less cassettes were observed when the GAL10 promoter was active (lanes 3 and 4). Importantly, when both promoters (GAL10-GAL7 construct) were active, transcription of the distal cassette was only ∼21% relative to the situation in the control construct (GAL10-GAL7ΔTATA) (Figure 1C, compare lanes 3 and 4). This indicates that RNAPII head-to-head collision is a significant block to transcript elongation in vivo (Figure 1D). These observations confirm and extend conclusions from run-on assays performed with a GAL10-GAL7 construct in the Proudfoot laboratory, which showed the generation of truncated transcripts originating from both promoters (Prescott and Proudfoot, 2002).
Lack of Bypass upon Collision In Vitro
We next studied the mechanistic basis of collision using a previously described biochemical system that faithfully recapitulates RNAPII transcript elongation in vitro (Kireeva et al., 2000; Saeki and Svejstrup, 2009). With this system, highly purified yeast RNAPII, DNA-, and RNA-oligonucleotides were used to generate ECs facing one another (Figure 2A, Di-EC, upper). Mono-ECs were assembled as a control (Figure 2A, Mono-EC, upper). One of the ECs was formed with a radioactively labeled RNA oligonucleotide so that transcription in the presence of all NTPs resulted in a 119 nucleotide (nt) product when the polymerase reached the end of the template (Figure 2A, Mono-EC, middle). Two different approaches were used to purify and characterize transcription templates harboring the desired number of ECs. In the first approach, di-ECs were formed using two different versions of RNAPII (FLAG- and HA-tagged, respectively; Figure 2B, upper left), while mono-ECs were formed using only FLAG-RNAPII. Mono-ECs were then purified by FLAG affinity chromatography, while di-ECs were isolated via tandem FLAG- and HA-affinity chromatography (Figure 2B, left box). Upon the addition of all NTPs, mono-ECs produced RNA of 119 nt (Figure 2B, lane 1), as expected, whereas di-ECs did not (lane 2). Instead, head-to-head transcription with di-ECs produced a series of RNA products ranging from ∼26 to 55 nt. It seems reasonable to presume that these products result from RNAPII molecules colliding at different points on the DNA template, unable to pass each other. We note that stops were especially prevalent at pause sites that were also observed with mono-ECs, presumably because the longer residence time at pause sites increased the likelihood of an encounter with the opposing polymerase. A lack of noteworthy bypass was also observed when transcription was allowed to proceed for extended periods of time (Figure 2C), indicating that ECs are stable upon collision.
Figure 2.

Reconstitution of Convergent Transcription In Vitro
(A) Mono-EC (left) and di-EC (right) schematics showing EC constituents and predicted transcript lengths. 32P end-labeled transcript is indicated by black sphere.
(B) Upper left: Schematic showing the position of epitope-tagged RNAPII molecules in di-ECs. Lower left: experimental strategy. Right: Autoradiograph of transcripts from mono- (M) and di-ECs (D) after adding all four NTPs (lanes 1 and 2), or leaving out GTP (lanes 3 and 4), respectively. Transcript length indicated on right.
(C) A time course of collision. Di-ECs were incubated with all NTPs for the times indicted.
(D) Schematic showing ECs before (upper) and after (lower) transcription. RNAPII “footprint” is based on crystal structures (Gnatt et al., 2001; Kettenberger et al., 2004; Wang et al., 2006).
To further characterize polymerase collision, we took advantage of transcription stops (C-stops) engineered into the DNA template. By adding only a subset of NTPs (ATP, UTP, and CTP), the EC containing the unlabelled RNA could only transcribe one base pair before requiring GTP for further translocation, while the labeled EC could, potentially, transcribe nearly the whole template (117 nt, see Figure 2A, lower, and Figure 2D, upper). This method allowed us to map the point of interaction between ECs at single-nucleotide resolution. In the absence of GTP, mono-ECs produced the expected 117 nt RNA product (Figure 2B, lane 3), whereas di-ECs produced RNA products between 51 and 55 nt (Figure 2B, lane 4). As expected, similar RNA products were also observed when assaying transcription with RNAPII from the opposite direction (Figure S1). Upon transcription in the absence of GTP, the shorter RNA products observed in the presence of all NTPs were not observed (Figure 2B, compare lanes 2 and 4), indicating that these were caused by collision between two actively transcribing ECs. The fact that RNA products between 51 and 55 nt were predominant also under conditions where both ECs could transcribe (Figure 2B, lane 2) indicates that a substantial fraction of HA-tagged EC failed to move forward before being hit by the FLAG-tagged EC.
The precise point at which transcription stopped upon head-to-head collision could be inferred from the length of the RNA synthesized in the absence of GTP and the known length of DNA protected by yeast RNAPII (Gnatt et al., 2001; Kettenberger et al., 2004; Wang et al., 2006). This analysis showed that the transcribing polymerases stop as soon as the proteins touch each other (see schematic in Figure 2D, lower). Interestingly, this is in sharp contrast to head-to-tail collision, where temporal but extensive structural “intermingling” between colliding polymerases was shown to occur (Saeki and Svejstrup, 2009).
Collided Elongation Complexes Are Stable
We used a second approach to more directly examine the stability of the collided ECs. Here, mono- and di-ECs were reconstituted (Figure 3A, box) and subjected to native agarose electrophoresis, either before (Figure 3A, lanes 1 and 2) or after transcription (lanes 3 and 4). There was a significant difference in the electrophoretic mobility of mono- and di-ECs, as previously observed (Saeki and Svejstrup, 2009). Interestingly, however, there was no marked dissociation of di-ECs following transcriptional collision (compare band labeled “Di” in Figure 3A, lane 2, to lane 4), showing that RNAPII remains in the EC after collision. Indeed, RNA isolated from native agarose gel slices representing the various ECs (band labeled “Mono” in 3A, lane 3, and band labeled “Di” in 3A, lane 4, respectively) showed that transcribed di-ECs contained the 51–55 nt collision products, as expected (Figure 3B, lane 2). Similarly, nascent RNA could be isolated from the di-ECs following transcription with all NTPs, indicating that collided complexes are stable also when both polymerases are transcribing (Figure S2).
Figure 3.

Elongation Complexes Remain on DNA Following Collision
(A) Experimental schematic (left) and autoradiograph showing native agarose electrophoresis of mono- (M) and di-ECs (D), before (lanes 1 and 2) or after (lanes 3 and 4) transcription.
(B) Autoradiograph of transcripts isolated from gel slices containing mono- (lane 3 in 3A) or di- (lane 4 in 3A) ECs, respectively, resolved by denaturing PAGE.
Ubiquitylation and Degradation of Collided Polymerases In Vivo
Together, the data above indicate that RNAPII collision may be a potent block to transcription both in vivo and in vitro. Unless removed, persistently stalled ECs are likely to pose severe problems for normal gene expression and for other DNA-related events. We thus consider it possible, even likely, that factors allowing some polymerase bypass in vivo have evolved, although the identity of such factors, should they exist, remains unknown (see Discussion). We have previously shown that abrogating elongation by inducing DNA damage or otherwise impeding transcript elongation can trigger RNAPII polyubiquitylation and degradation, presumably as a last resort when an irreversibly stalled polymerase cannot be dealt with effectively by other means (Woudstra et al., 2002; Somesh et al., 2005; Svejstrup, 2007; Harreman et al., 2009; Sigurdsson et al., 2010). To determine if ECs stalled as a result of transcriptional collision might also be dealt with, at least partly, by ubiquitylation and proteolysis, we used the GAL10-GAL7 collision templates for RNAPII chromatin immunoprecipitation (ChIP) experiments in a strain defective for RNAPII polyubiquitylation (elc1Δ) (Ribar et al., 2006; Harreman et al., 2009). Interestingly, even though two promoters (rather than one) are active in the GAL10-GAL7 construct (Figure 4, schematic), the density of polymerases in the ORF between the promoters did not increase compared to the GAL10-GAL7ΔTATA construct in wild-type cells. Instead, it decreased (Figure 4, WT, compare white and black bars), suggesting active removal of collided ECs. In agreement with this model, there was an increase in RNAPII density after collision in the elc1Δ mutant (Figure 4, elc1Δ), which cannot efficiently remove stalled RNAPII via ubiquitylation (Ribar et al., 2006; Harreman et al., 2009). Similar results were obtained using another mutant (def1Δ) that also affects RNAPII polyubiquitylation and degradation (Woudstra et al., 2002) (Figure 4, def1Δ). This suggests that polyubiquitylation and proteolysis of RNAPII plays a role by removing collided ECs that cannot otherwise be resolved.
Figure 4.
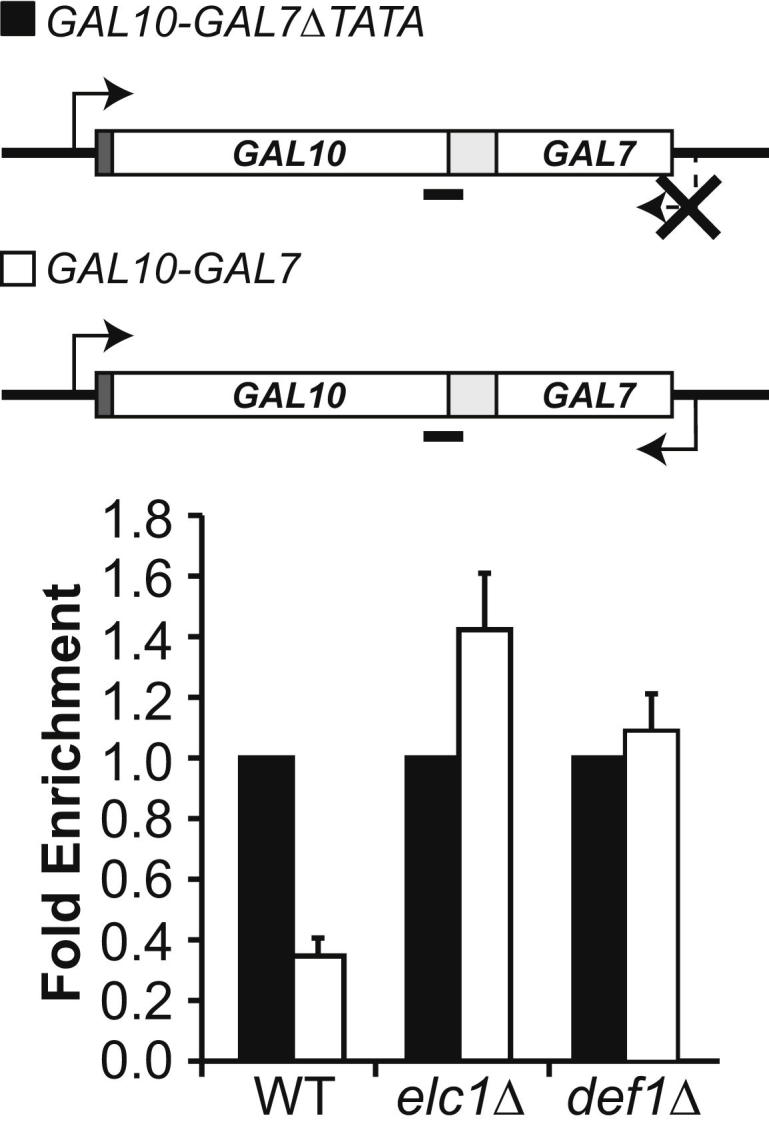
RNAPII Is Rapidly Removed from Convergent Genes in a Ubiquitylation-Dependent Manner
Upper: Collision gene constructs with position of qPCR product indicated below (black line). Lower: RNAPII ChIP in WT and elc1Δ, in the absence (black) and presence of collision (white). ChIP values were divided by the input, telomere, and IgG control values. Density in the absence of collision was set to one, and the value in the presence of collision was expressed relative to that. Error bars show standard error (from three biological replicates).
Slow, Ubiquitylation-Dependent Clearance of Transcription Traffic Jams
To explore the mechanism of polymerase removal upon collision further, we next investigated the rate of RNAPII removal from the GAL10-GAL7 collision gene after repression of new transcription by addition of glucose (Figure 5). We surmised that collided polymerases on this gene might have a longer half-life than polymerases on the endogenous GAL genes (from which it disappears within a few minutes after promoter repression [Mason and Struhl, 2005]), so that if the clearance of a significant proportion of collided polymerases requires ubiquitylation/degradation, then the rate of RNAPII disappearance should be affected in elc1Δ. As expected (Mason and Struhl, 2005), RNAPII disappeared from the endogenous GAL1 gene (which is coregulated with endogenous GAL10, with which it shares a promoter) within 6 min upon repression (∼20% RNAPII remaining) (Figure 5A), with slightly slower initial kinetics in elc1Δ, possibly owing to the presence of CUT445 transcribed in the antisense orientation across GAL1. In contrast, removal of the polymerase from the GAL10-GAL7 collision construct in the same cells was much less pronounced, and importantly, removal of the collided RNAPIIs was markedly reduced in the elc1Δ cells relative to wild-type, with almost 90% RNAPII remaining on the gene after 10 min in these cells (Figure 5B). This further supports the idea that ubiquitylation/degradation contributes to the clearance of collided RNAPII molecules.
Figure 5.
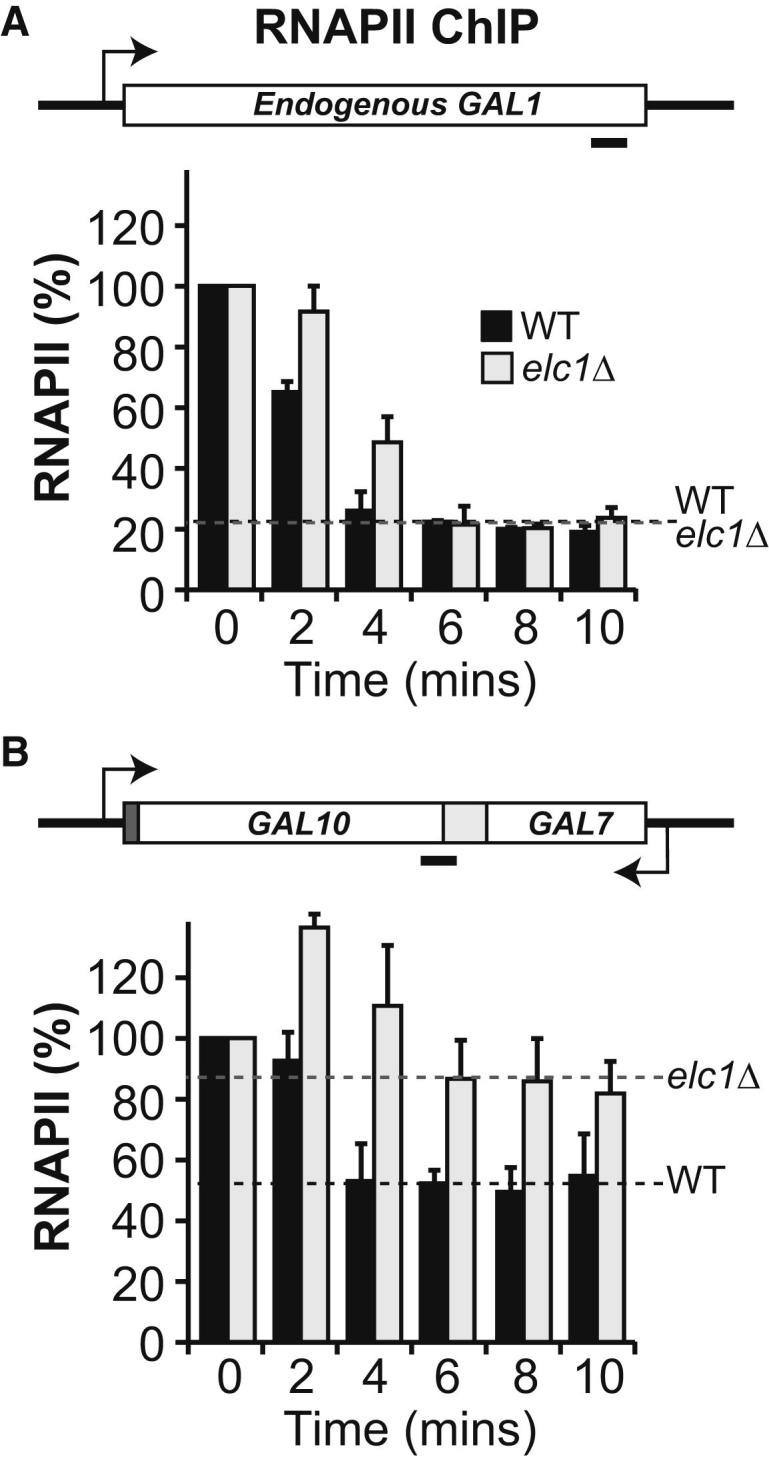
Slow Clearance of RNAPII from the Deactivated Gene Due to Collision
(A) RNAPII density (ChIP) in the endogenous GAL1 gene in WT and elc1Δ cells as genes are switched off (x axis shows time in minutes after glucose addition). Error bars show standard error (from three biological replicates). ChIP values were divided by the input and telomere signal. Values obtained at time = 0 in each case were set to 100, and other values are shown relative to that.
(B) As in (A), but with the GAL10-GAL7 collision construct.
Evidence for RNAPII Collision at Natural Genes
Because collision is likely to be stochastically distributed under normal circumstances, it is difficult to ascertain the possible physiological relevance of RNAPII collision at natural genes. To try and overcome this challenge, we took advantage of the finding that the half-life of collided ECs is extended significantly in elc1Δ cells. Thus, if collision does indeed occur between polymerases transcribing natural genes, one might expect to find elevated levels of RNAPII in the terminator region of convergent genes in elc1Δ cells. RNAPII ChIPs from wild-type and elc1Δ cells were subjected to deep sequencing (ChIP-Seq) and used to compare the average intergenic density of RNAPII across all pairs of convergent genes, where collision might be expected, and divergent genes, where little or no collision would be expected (Figure 6, upper panel schematics). The region between divergent rather than tandem genes was chosen as the control because substantial antisense, noncoding transcription is observed from both terminators and promoters, which might be detected as collision events between tandem genes as well. Remarkably, although there was no significant difference in RNAPII density inside the coding regions of genes, a clear increase was indeed observed in the region between convergent genes, but not between divergent genes in elc1Δ cells (Figure 6A). Deep sequencing of total RNA (treated with Ribo-Zero to remove rRNA) revealed no evidence for an accompanying change in the amounts or length of the transcripts (Figure 6B), suggesting that these changes were not due to terminator read-through in elc1Δ strains, but rather due to polymerases with increased residence time in areas of potential collision. Taken together with the previous results, these data support the idea that RNAPII head-to-head collision occurs naturally throughout the genome.
Figure 6.

Evidence for Collision between Natural Convergent Genes
(A) Average RNAPII density between the 1,478 convergent (left) and 1,492 divergent (right) genes in WT (blue lines) and elc1Δ (red lines), as measured by ChIP-Seq. Log2 ChIP signal divided by IgG control signal is shown relative to the midpoint (0) between the genes. Colored lines represent the average of two independent experiments. A schematic is shown above graphs, with arrows indicating the direction of transcription. Note that the distance from the end (convergent), or beginning (divergent), of an ORF to the midpoint obviously differs from gene to gene (see Figure S4). The compared gene pairs are listed in Table S1.
(B) RNA levels, measured by nascent RNA-Seq, downstream of convergent genes. 0 indicates end of ORF. Lines represent the average of three independent experiments.
Discussion
The emerging view from numerous genome-wide studies is that RNA polymerases constantly and stochastically initiate transcription across the genome, often from introns or the 3′ end of genes, and in the antisense direction relative to the protein-encoding gene (Berretta and Morillon, 2009; Jacquier, 2009; Carninci, 2010). The consequences of the ensuing transcription “traffic” therefore need to be understood.
At first glance, it might be expected that collided polymerases resulting from convergent transcription would simply dissociate from the template. However, RNAPII ECs are remarkably stable, so collision is likely to pose a considerable problem to the polymerases involved and thereby to gene transcription in general. In many ways, the problem with collision is similar to that encountered during DNA damage. Like polymerase-polymerase collisions, transcription-impeding DNA lesions are rare and stochastically distributed, yet the resolution of damage-stalled RNAPII ECs is one of the most crucial aspects of the DNA damage response (Svejstrup, 2002; Hanawalt and Spivak, 2008). Indeed, at least one of two processes is required for an efficient response to bulky DNA damage: cells need to either remove the DNA lesion through efficient repair mechanisms such as transcription-coupled nucleotide excision repair (TC-NER), or remove the polymerase via ubiquitylation-mediated proteolysis of RNAPII so that the lesion can be dealt with by other means, allowing transcription to resume. Otherwise, even a few DNA lesions are cell lethal (Woudstra et al., 2002; Svejstrup, 2007). Likewise, even if it were a rare occurrence, polymerase-polymerase collisions need to be resolved as well. We note that it is difficult to estimate how often RNAPII collision occurs in normal cells. In all likelihood, it is very rare at any individual site, but very frequent across the genome. Indeed, evidence for collision between natural genes is clear from averaging across the ∼1,500 convergent gene pairs (Figure 6A), but is difficult to spot at any individual pair.
The failure of RNAPII molecules to bypass each other in vitro in a highly defined system raises the question of whether bypass is also impossible in vivo. It is presently our hypothesis that a factor(s) facilitating bypass does exist. Future efforts will be focused on trying to identify it. We have tested the ability of crude yeast extracts to promote bypass. Although weak bypass in the presence of extract was observed, we have so far been unable to enrich for the putative, responsible activity by fractionation. We also tested the effect of RNAPII monoubiquitylation. Rsp5-mediated RNAPII monoubiquitylaton is required for subsequent polyubiquitylation (by Elc1-Cul3 complex [Harreman et al., 2009]) and degradation, but in itself, RNAPII monoubiquitylation might affect fundamental aspects of transcription, such as transcriptional bypass. However, we failed to observe a significant effect of this modification on bypass in the highly defined transcription system (Figure S3).
If bypass factors for RNAPII collision are regarded as the equivalent of TC-NER factors for removing transcription-impeding DNA lesions, it follows that RNAPII ubiquitylation/degradation might also function as a last resort mechanism if bypass cannot occur—as it does in the DNA damage response. Indeed, we found evidence that ubiquitylation/degradation is involved in clearing polymerases from the region between convergently transcribing GAL genes. We also observed a peak of RNAPII density in the region between convergent genes across the genome in cells lacking ELC1, suggesting that collisions also occur in these natural intergenic regions. Interestingly, the fact that genes required for RNAPII ubiquitylation/degradation (such as ELC1 and DEF1) are not required for cell viability represents an argument for the existence of alternative mechanisms for dealing with collided polymerases, such as bypass. Indeed, deletion of ELC1 had surprisingly little effect on the stable transcriptome (Figure 6B, and data not shown).
The possible function of antisense transcription in the regulation of sense transcription is a subject of obvious interest. Recent data indicate that antisense expression increases gene expression variability, characterized by more “switching-off” at low levels of expression for genes with antisense transcription than for genes without (Xu et al., 2011). One intriguing possibility is that collision between polymerases generating sense-antisense transcripts plays a role in the fine-tuning of transcription regulation. It is also conceivable that antisense transcription, and the ensuing collision with RNAPII initiating unscheduled transcription from a protein-coding gene, might be used to keep uninduced genes silent. It thus seems appropriate that not only the role of the RNA transcripts generated by noncoding transcription (siRNAs, miRNAs, etc.), but also the impact of the polymerases that generate them, should be considered in the regulation of gene expression.
Experimental Procedures
Yeast Strains, Plasmids, and Proteins
All in vivo experiments were performed with S. cerevisiae strains congenic to W303 (Thomas and Rothstein, 1989) and include elc1Δ::URA3 (Harreman et al., 2009). Untransformed cells were grown in YPD. For activation of the GAL genes, cells transformed with pRS314-GAL10-GAL7 or pRS314-GAL10-GAL7ΔTATA were grown at 30°C overnight in synthetic complete media (lacking tryptophan, with raffinose as the only sugar source) to stationary phase before being diluted and grown overnight to mid-log phase. Galactose or glucose was added to cells (as indicated) at a final concentration of 2% for the indicated time.
The plasmid GAL10-GAL7 used throughout this study was constructed by obtaining a GAL10 PCR product using primers targeted 343 bp upstream of the ORF to include all promoter elements (5′AGAGAAAGCTCGAG CTTTATTGTTCGGAGCAGTGCGG3′) and 47 bp upstream of the stop codon, to ensure no terminator elements were present (5′AGAGAGAGATCGAT TCAAGGTTACACAATCTTTCCAGTTCTC3′). This product was cloned between the XhoI and ClaI sites of pRS314. A GAL7 PCR product (or GAL7ΔTATA PCR product for the control) was obtained with primers hybridizing 446 bp upstream of the ORF (5′AGAGAGAGGAGCTC ATATCACTCACAACTATTGCGAAGCG3′) and 38 bp upstream of the GAL7 stop codon (5′AGAGAGAGACTAG TTCTTAGTTTTTCAGCAGCTTGTTCCG3′). The fragment was cloned into the SacI and SpeI sites in a convergent orientation to the GAL10 gene. A 100 bp G-less cassette was PCR amplified from pGAL4CG (Lue et al., 1989) and cloned into an EcoRI site at the 5′ end of the GAL10 ORF to form the promoter proximal G-less cassette (forward 5′AGAGAGAGGAATTCACTCACCCAATACTCCCTACTC3′; reverse 5′AGAGAGAGGAATTCGGGAGTGGAATGAGAAATG3′). Finally, a 371 bp G-less cassette obtained from the 365 bp G-less cassette of pGAL4CG (forward 5′AGAGAGAGATCGATCCTCCATACCCTTCCTCC3′; reverse 5′AGAGAGAGACTAGTGGGAGTGGAATGAGAAATG3′) was cloned into the SpeI and ClaI sites between the 3′ ends of the GAL7 (or GAL7ΔTATA) and GAL10 ORFs. The plasmids pYC10-7Fus and pYC10-7Fus-Δ7 (Prescott and Proudfoot, 2002) used as templates for the PCR amplifications above were kindly provided by Nick Proudfoot.
FLAG-tagged RNAPII and 3HA-tagged RNAPII were purified from 100 l of yeast culture using previously published techniques (heparin column, ammonium sulfate precipitation, 8WG16 affinity purification, and MonoQ anion-exchange chromatography) (Cramer et al., 2001), using strains with the endogenous Rpb3 or Rpb1 subunits (respectively) tagged at their genomic locus.
RNA Extraction and Northern Blotting
Cells were harvested following addition of 2% glucose or galactose for 75 min at 30°C, and RNA was extracted using the QIAGEN RNeasy kit and standard protocol.
Equal amounts of RNA were treated with 200 U of RNase T1 (Roche) for 1 hr at 37°C, prior to phenol-chloroform extraction. Formamide gel loading buffer (95% deionized formamide, 5 mM EDTA, 0.004% bromophenol blue, 0.004% xylene cyanol) was then added to the RNA, which was heated to 65°C for 10 min and separated by 7% denaturing (8.3 M urea) PAGE. RNA was transferred to Hybond-N+ nylon membrane (GE Healthcare) using semidry transfer blotting at 400 mA for 1 hr and UV crosslinked. Northern membranes were incubated with a random-primed 32P-labeled double-stranded DNA probe (corresponding to the long G-less cassette) for 1 hr at 65°C. This was followed by four washes with WB1 (2× SSC, 0.05% SDS) for 10 min at room temperature and two washes with WB2 (0.1× SSC, 0.1% SDS), each for 15 min at 50°C. Probed membranes were exposed to phosphor imager screens or Kodak BioMax MR film. “% Distal Cassette Transcribed” was calculated using data from the phosphor imager. The proximal cassette signals were equalized for “+/− Convergent Transcription.” The “+ Convergent Transcription” distal cassette signal was then calculated as a percentage of the “− Convergent Transcription” distal cassette signal (= 100%). The mean value and standard error were calculated from two biological replicates.
Reconstitution of Elongation Complexes
ECs were reconstituted using 150 nt DNA oligonucleotides from DNA technology, the sequences of which are as follows:
DNA Oligo1:
GCGTGGTATAGGAGTAGAGTATGGAGAGGTGTTGTTGTTGTGAGTTGGTTATGGTAGGTGAGTGTGTGATTGTGTGTTAGTAGTTGGTTAGTTGGGTGTATGTTTGATGTGTATCCTGGTAAGTGATGTGTGGTTGGAATGTGGTTGCGG
DNA Oligo2:
CCGCAACCACATTCCAACCACACATCACTTACCAGGATACACATCAAACATACACCCAACTAACCAACTACTAACACACAATCACACACTCACCTACCATAACCAACTCACAACAACAACACCTCTCCATACTCTACTCCTATACCACGC
The DNA oligonucleotides were received HPLC purified, but subjected to further purification by 5.2% denaturing PAGE (8.3 M urea).
The sequences of RNA oligonucleotides were as follows: RNA Oligo1 (CCAGGAUAC) and Oligo2 (AUGGAGAGG); these were purchased from Dharmacon and purified via 20% denaturing PAGE.
Mono-ECs were assembled in Reconstitution Buffer (20 mM Tris [pH 7.9], 40 mM KCl, 20 μM ZnCl2, 5 mM DTT, 0.2 mM MgCl2, 0.75 μg/μl BSA) by incubating 1 pmol of DNA Oligo1 with 2 pmol of RNA Oligo1 (including 400 counts per second [CPS] of 32P end-labeled RNA Oligo1) at 65°C for 5 min followed by step-wise cooling to 25°C over a 40 min period. Next, 3 pmol of FLAG-tagged RNAPII was added for 25 min at 25°C, followed by the addition of 6 pmol of DNA Oligo2 (which was placed at 65°C for 5 min then stored on ice before addition) at 37°C for 10 min.
Di-ECs were formed by incubation of 1 pmol of DNA Oligo1 with 2 pmol of RNA Oligo1 (including 400 CPS 32P end-labeled RNA1), while in a separate tube 6 pmol of DNA Oligo2 was incubated with 12 pmol of unlabelled RNA Oligo2. Next, 3 pmol of FLAG-tagged RNAPII was added to the Oligo1-RNA1 mixture, and 18 pmol of HA-tagged RNAPII was added to the DNA Oligo2-RNA Oligo2 mixture. Following incubation for 25 min at 25°C, the DNA-RNA-RNAPII mixtures from each tube were mixed and allowed to hybridize for 10 min at 37°C.
Purification of Elongation Complexes
Mono- and di-ECs were incubated with Anti-FLAG M2 Affinity Gel (Sigma) for 1 hr at 4°C with shaking, prior to washing with EC-WB (50 mM Tris [pH 7.5], 500 mM NaCl, 0.5 mM EDTA, 10 μM ZnCl2, 0.05% NP-40, 10% glycerol) and 20 mM Tris-Cl (pH 7.5), followed by elution with 300 μg/ml FLAG peptide in TB (20 mM Tris [pH 7.9], 40 mM KCl, 20 μM ZnCl2, 5 mM DTT, 7 mM MgCl2, 0.75 μg/μl BSA). Semipurified ECs were then incubated with Anti-HA Affinity Matrix (Roche) for 1 hr at 4°C with shaking to isolate di-ECs, followed by repeated washes with EC-WB and 20 mM Tris-Cl (pH 7.5).
Ubiquitylation of Elongation Complexes
Unpurified ECs were incubated with yeast Uba1, Ubc5, and Rsp5 ± ubiquitin (with all lysine residues mutated) for 90 min at 30°C prior to transcription. Ubiquitylation efficiency was assayed by western blot with 4H8 Rpb1-CTD antibody.
Transcription Analysis
ECs in TB were incubated with 600 μM NTPs (either AUC or AUCG) for 5 min (or as specified). Transcription was stopped with STOP buffer (20 mM Tris [pH 7.9], 40 mM KCl, 20 μM ZnCl2, 5 mM DTT, 20 mM EDTA [pH 8.0], ± 1 mg/ml proteinase K) and, if proteinase K was added, incubated at 37°C for 30 min. Samples were either phenol-chloroform extracted and the RNA ethanol precipitated for analysis of transcripts by 8.3 M urea denaturing PAGE (6% polyacrylamide), or resuspended in loading buffer (3% glycerol, 2.5 mM 2-mercaptoethanol, 0.75 μg/μl BSA) for native agarose electrophoresis on a 0.7% agarose gel (with 5 mM 2-mercaptoethanol, 0.1 mM MgCl2, 10 μM ZnCl2).
Purification of RNA from the agarose gel was performed by staining in a 1:10,000 dilution of Sybr Gold, allowing visualization and excision of the band corresponding to the mono- or di-ECs, followed by maceration, phenol-chloroform extraction, butanol extraction, and ethanol precipitation (Saeki and Svejstrup, 2009). Transcripts were then analyzed by denaturing PAGE and exposure to phosphor imager screens or Kodak BioMax MR film.
Chromatin Immunoprecipitation
ChIP was performed with WT or elc1Δ strains containing GAL10-GAL7 or GAL10-GAL7ΔTATA. For the steady-state ChIP (Figure 4), cells were crosslinked directly for 20 min at room temperature with 1% formaldehyde. For the ChIP in Figure 5, the 0 min time point was taken after 2 hr in galactose and crosslinked (using conditions stated above). Glucose was added (2% final concentration) and time points taken every 2 min (up to 10 min) and crosslinked. Crosslinking was quenched with 200 mM glycine prior to resuspension and cell lysis in FA Lysis buffer (50 mM Tris [pH 7.5], 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-Deoxycholate, 1× protease inhibitors [Otero et al., 1999]). The chromatin was sonicated to a fragment length of 200–500 bp and then incubated with 2.2 μg of 4H8 (anti-CTD antibody) or 2.2 μg mouse IgG (where appropriate) for 2 hr prior to incubation with Protein G Agarose (Pierce) for 2 hr. Beads were washed three times for 3 min at room temperature in FA Lysis, once in FA500 (FA lysis with 500 mM NaCl), once in ChIP wash buffer (10 mM Tris [pH 8.0], 250 mM LiCl, 0.5% NP-40, 0.5% Na-Deoxycholate, 1 mM EDTA), and once in TES (10 mM Tris [pH 7.5], 1 mM EDTA, 100 mM NaCl). Finally, 100 μl ChIP elution buffer (50 mM Tris [pH 8.0], 10 mM EDTA, 1% SDS) was added and the samples were incubated at 65°C for 10 min. Thirty-five micrograms of RNase A (Sigma) was added to the eluate for 30 min at 37°C, followed by addition of 20 μg Proteinase K (Roche) for 2 hr at 42°C. DNA-protein crosslinks were reversed by incubating at 65°C for 6 hr, and the DNA was isolated using a PCR Purification Kit (QIAGEN).
Quantitative PCR was performed using 18 μl reaction buffer (0.2 μM forward primer, 0.2 μM reverse primer, 1× iQ Custom SYBR Green SuperMix [BioRad]) and 2 μl DNA. Primers targeted to the distal cassette of the GAL10-GAL7 construct were used (forward 5′GAGGGGATATGGAAAGGGAA3′; reverse 5′CCGGTGATTTCTTGTCTGCT3′) as well as control primers directed to the telomeres of chromosome 6 (forward 5′TAACAAGCGGCTGGACTACTTT3′; reverse 5′GATAACTCTGAACTGTGCATCC3′) and primers targeted to endogenous GAL1 (forward 5′ACGAGTCTCAAGCTTCTTGC3′; reverse 5′TATAGACAGCTGCCCAATGC3′).
The steady-state ChIP (Figure 4) values were divided by the input, telomere, and IgG control values and normalized to the signal for GAL10-GAL7ΔTATA (= 1). Values obtained for the ChIP in Figure 5 were divided by the input and telomere signal and normalized to the 0 min time point (= 100%). Columns on the graphs represent the mean value and bars show standard error, calculated from three biological replicates.
ChIP-Seq
WT and elc1Δ cells were grown in YPD to mid-log phase and crosslinked as indicated earlier; however, cells were resuspended in FA500 before lysis. Sonicated chromatin was incubated for 2 hr with 2.2 μg 4H8 or mouse IgG prior to incubation with Protein G Agarose (Pierce) for 1 hr. Beads were washed three times for 3 min with FA500, twice with FA lysis, twice with ChIP wash buffer, and twice with TES. Elution, RNase treatment, and reverse crosslinking were performed as described for standard ChIP. DNA was purified by two rounds of phenol-chloroform extraction followed by ethanol precipitation before being subjected to standard library preparation techniques (Illumina) and Advanced Sequencing on an Illumina GAIIx DNA sequencer.
RNA-Seq
WT and elc1Δ cells were grown in YPD to mid-log phase prior to total RNA extraction (QIAGEN RNeasy Kit). RNA was subjected to standard library preparation techniques (Illumina), including Ribo-Zero hybrid-selection (Epicenter Biotechnologies), and Advanced Sequencing on an Illumina GAIIx sequencer.
Bioinformatic Analysis
Short read sequences from ChIP-Seq and RNA-Seq were aligned to the S288c reference genome (Version 20110326 downloaded from the Saccharomyces Genome Database [SGD] [Cherry et al., 1998]) using the Novoalign (http://www.novocraft.com) software. Reads at each position along the genome were extracted. In ChIP-Seq data analysis, ChIP signals (Log2 values) were divided by the relative IgG control for each genomic position excluding the ones with < 5 reads coverage. Quantile normalization between samples was then performed. Normalized ChIP-Seq values for the regions flanking the middle positions of intergenic regions (± 500 bp) between convergent or divergent gene pairs were extracted, and the median values of the signal in each position in these gene pairs were calculated and plotted.
RNA-Seq coverage at each position along the genome was divided by the total number of reads mapped in each sample. Regions flanking the end positions of every gene (± 500 bp) were taken, and normalized RNA-Seq signals were extracted for every gene. The mean values of RNA-Seq signals at each position in all genes were calculated and plotted.
Acknowledgments
This work was supported by grants from Cancer Research UK and European Research council (ERC) (to J.Q.S.), the National Institutes of Health (to L.M.S.), and by a Boehringer Ingelheim Fonds Fellowship (to D.J.H.). We thank Nick Proudfoot (Oxford University) for the kind gift of plasmids and Michael Ranes for assistance and useful discussions. We also thank the Fermentation Service, the Advanced Sequencing Facility, and the Bioinformatics and Biostatistics Service at Cancer Research UK London Research Institute for help and advice. Members of the Svejstrup laboratory are thanked for comments on the manuscript.
Published online: October 4, 2012
Footnotes
Supplemental Information includes four figures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2012.08.027.
Accession Numbers
Raw sequencing data are available for download from NCBI Gene Expression Omnibus under accession number GSE38384.
Supplemental Information
References
- Berretta J., Morillon A. Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO Rep. 2009;10:973–982. doi: 10.1038/embor.2009.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P. RNA dust: where are the genes? DNA Res. 2010;17:51–59. doi: 10.1093/dnares/dsq006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Sun M., Kent W.J., Huang X., Xie H., Wang W., Zhou G., Shi R.Z., Rowley J.D. Over 20% of human transcripts might form sense-antisense pairs. Nucleic Acids Res. 2004;32:4812–4820. doi: 10.1093/nar/gkh818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry J.M., Adler C., Ball C., Chervitz S.A., Dwight S.S., Hester E.T., Jia Y., Juvik G., Roe T., Schroeder M. SGD: Saccharomyces Genome Database. Nucleic Acids Res. 1998;26:73–79. doi: 10.1093/nar/26.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer P., Bushnell D.A., Kornberg R.D. Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science. 2001;292:1863–1876. doi: 10.1126/science.1059493. [DOI] [PubMed] [Google Scholar]
- García-Rubio M.L., Aguilera A. Topological constraints impair RNA polymerase II transcription and causes instability of plasmid-borne convergent genes. Nucleic Acids Res. 2012;40:1050–1064. doi: 10.1093/nar/gkr840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnatt A.L., Cramer P., Fu J., Bushnell D.A., Kornberg R.D. Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 A resolution. Science. 2001;292:1876–1882. doi: 10.1126/science.1059495. [DOI] [PubMed] [Google Scholar]
- Hanawalt P.C., Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- Harreman M., Taschner M., Sigurdsson S., Anindya R., Reid J., Somesh B., Kong S.E., Banks C.A., Conaway R.C., Conaway J.W., Svejstrup J.Q. Distinct ubiquitin ligases act sequentially for RNA polymerase II polyubiquitylation. Proc. Natl. Acad. Sci. USA. 2009;106:20705–20710. doi: 10.1073/pnas.0907052106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquier A. The complex eukaryotic transcriptome: unexpected pervasive transcription and novel small RNAs. Nat. Rev. Genet. 2009;10:833–844. doi: 10.1038/nrg2683. [DOI] [PubMed] [Google Scholar]
- Kettenberger H., Armache K.J., Cramer P. Complete RNA polymerase II elongation complex structure and its interactions with NTP and TFIIS. Mol. Cell. 2004;16:955–965. doi: 10.1016/j.molcel.2004.11.040. [DOI] [PubMed] [Google Scholar]
- Kireeva M.L., Komissarova N., Waugh D.S., Kashlev M. The 8-nucleotide-long RNA:DNA hybrid is a primary stability determinant of the RNA polymerase II elongation complex. J. Biol. Chem. 2000;275:6530–6536. doi: 10.1074/jbc.275.9.6530. [DOI] [PubMed] [Google Scholar]
- Kornberg R.D. The molecular basis of eukaryotic transcription. Proc. Natl. Acad. Sci. USA. 2007;104:12955–12961. doi: 10.1073/pnas.0704138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue N.F., Flanagan P.M., Sugimoto K., Kornberg R.D. Initiation by yeast RNA polymerase II at the adenoviral major late promoter in vitro. Science. 1989;246:661–664. doi: 10.1126/science.2510298. [DOI] [PubMed] [Google Scholar]
- Ma N., McAllister W.T. In a head-on collision, two RNA polymerases approaching one another on the same DNA may pass by one another. J. Mol. Biol. 2009;391:808–812. doi: 10.1016/j.jmb.2009.06.060. [DOI] [PubMed] [Google Scholar]
- Mason P.B., Struhl K. Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol. Cell. 2005;17:831–840. doi: 10.1016/j.molcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- Mourier T., Willerslev E. Does selection against transcriptional interference shape retroelement-free regions in mammalian genomes? PLoS ONE. 2008;3:e3760. doi: 10.1371/journal.pone.0003760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero G., Fellows J., Li Y., de Bizemont T., Dirac A.M.G., Gustafsson C.M., Erdjument-Bromage H., Tempst P., Svejstrup J.Q. Elongator, a multisubunit component of a novel RNA polymerase II holoenzyme for transcriptional elongation. Mol. Cell. 1999;3:109–118. doi: 10.1016/s1097-2765(00)80179-3. [DOI] [PubMed] [Google Scholar]
- Prescott E.M., Proudfoot N.J. Transcriptional collision between convergent genes in budding yeast. Proc. Natl. Acad. Sci. USA. 2002;99:8796–8801. doi: 10.1073/pnas.132270899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribar B., Prakash L., Prakash S. Requirement of ELC1 for RNA polymerase II polyubiquitylation and degradation in response to DNA damage in Saccharomyces cerevisiae. Mol. Cell. Biol. 2006;26:3999–4005. doi: 10.1128/MCB.00293-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeki H., Svejstrup J.Q. Stability, flexibility, and dynamic interactions of colliding RNA polymerase II elongation complexes. Mol. Cell. 2009;35:191–205. doi: 10.1016/j.molcel.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson S., Dirac-Svejstrup A.B., Svejstrup J.Q. Evidence that transcript cleavage is essential for RNA polymerase II transcription and cell viability. Mol. Cell. 2010;38:202–210. doi: 10.1016/j.molcel.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somesh B.P., Reid J., Liu W.F., Søgaard T.M., Erdjument-Bromage H., Tempst P., Svejstrup J.Q. Multiple mechanisms confining RNA polymerase II ubiquitylation to polymerases undergoing transcriptional arrest. Cell. 2005;121:913–923. doi: 10.1016/j.cell.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Svejstrup J.Q. Mechanisms of transcription-coupled DNA repair. Nat. Rev. Mol. Cell Biol. 2002;3:21–29. doi: 10.1038/nrm703. [DOI] [PubMed] [Google Scholar]
- Svejstrup J.Q. Contending with transcriptional arrest during RNAPII transcript elongation. Trends Biochem. Sci. 2007;32:165–171. doi: 10.1016/j.tibs.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Thomas B.J., Rothstein R. Elevated recombination rates in transcriptionally active DNA. Cell. 1989;56:619–630. doi: 10.1016/0092-8674(89)90584-9. [DOI] [PubMed] [Google Scholar]
- Vallon-Christersson J., Staaf J., Kvist A., Medstrand P., Borg A., Rovira C. Non-coding antisense transcription detected by conventional and single-stranded cDNA microarray. BMC Genomics. 2007;8:295. doi: 10.1186/1471-2164-8-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Bushnell D.A., Westover K.D., Kaplan C.D., Kornberg R.D. Structural basis of transcription: role of the trigger loop in substrate specificity and catalysis. Cell. 2006;127:941–954. doi: 10.1016/j.cell.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woudstra E.C., Gilbert C., Fellows J., Jansen L., Brouwer J., Erdjument-Bromage H., Tempst P., Svejstrup J.Q. A Rad26-Def1 complex coordinates repair and RNA pol II proteolysis in response to DNA damage. Nature. 2002;415:929–933. doi: 10.1038/415929a. [DOI] [PubMed] [Google Scholar]
- Xu Z., Wei W., Gagneur J., Perocchi F., Clauder-Münster S., Camblong J., Guffanti E., Stutz F., Huber W., Steinmetz L.M. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009;457:1033–1037. doi: 10.1038/nature07728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Wei W., Gagneur J., Clauder-Münster S., Smolik M., Huber W., Steinmetz L.M. Antisense expression increases gene expression variability and locus interdependency. Mol. Syst. Biol. 2011;7:468. doi: 10.1038/msb.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P., Ma D., Xu M. Nested genes in the human genome. Genomics. 2005;86:414–422. doi: 10.1016/j.ygeno.2005.06.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.