

Abstract
OBJECTIVES:
Linaclotide is a minimally absorbed guanylate cyclase-C agonist. The objective of this trial was to determine the efficacy and safety of linaclotide in patients with irritable bowel syndrome with constipation (IBS-C).
METHODS:
This phase 3, double-blind, parallel-group, placebo-controlled trial randomized IBS-C patients to placebo or 290 μg oral linaclotide once daily in a 12-week treatment period, followed by a 4-week randomized withdrawal (RW) period. There were four primary end points, the Food and Drug Administration's (FDA's) primary end point for IBS-C (responder: improvement of ≥30% in average daily worst abdominal pain score and increase by ≥1 complete spontaneous bowel movement (CSBM) from baseline (same week) for at least 50% of weeks assessed) and three other primary end points, based on improvements in abdominal pain and CSBMs for 9/12 weeks. Adverse events (AEs) were monitored.
RESULTS:
The trial evaluated 800 patients (mean age=43.5 years, female=90.5%, white=76.9%). The FDA end point was met by 136/405 linaclotide-treated patients (33.6%), compared with 83/395 placebo-treated patients (21.0%) (P<0.0001) (number needed to treat: 8.0, 95% confidence interval: 5.4, 15.5). A greater percentage of linaclotide patients, compared with placebo patients, reported for at least 6/12 treatment period weeks, a reduction of ≥30% in abdominal pain (50.1 vs. 37.5%, P=0.0003) and an increase of ≥1 CSBM from baseline (48.6 vs. 29.6%, P<0.0001). A greater percentage of linaclotide patients vs. placebo patients were also responders for the other three primary end points (P<0.05). Significantly greater improvements were seen in linaclotide vs. placebo patients for all secondary end points (P<0.001). During the RW period, patients remaining on linaclotide showed sustained improvement; patients re-randomized from linaclotide to placebo showed return of symptoms, but without worsening of symptoms relative to baseline. Diarrhea, the most common AE, resulted in discontinuation of 5.7% of linaclotide and 0.3% of placebo patients.
CONCLUSIONS:
Linaclotide significantly improved abdominal pain and bowel symptoms associated with IBS-C for at least 12 weeks; there was no worsening of symptoms compared with baseline following cessation of linaclotide during the RW period.
INTRODUCTION
Irritable bowel syndrome (IBS) is a common gastrointestinal disorder characterized by frequent and intermittent episodes of abdominal pain and abdominal discomfort that are associated with altered bowel habits (1,2). The symptoms of IBS not only adversely affect a patient's health-related quality of life (3) but also place a significant financial burden on society due to reduced work productivity and increased use of healthcare-related resources (4,5). IBS with constipation (IBS-C) affects approximately one-third of IBS patients (3), occurs more commonly in women than men (6), and frequently includes additional symptoms, such as abdominal bloating, hard stools, straining, and sensation of incomplete evacuation (7,8).
Traditional therapies for IBS-C, generally directed towards the patient's predominant symptoms (9), are frequently associated with patient dissatisfaction (10). More recent therapies, including tegaserod and lubiprostone, have been shown to improve global symptoms of IBS-C (9). Tegaserod, a 5-HT4 partial agonist approved by the Food and Drug Administration (FDA) for the short-term treatment of women with IBS-C, was removed from the market in 2007 due to increased cardiovascular events in patients receiving the medication. Lubiprostone, a chloride channel activator that was approved by the FDA for the treatment of women with IBS-C in 2008, has shown efficacy using a global end point (global symptom relief) (11). Given the limited treatments currently available for patients with IBS-C, additional therapeutic options would be of value.
Linaclotide, a minimally absorbed 14-amino-acid peptide structurally related to the endogenous guanylin peptide family of hormones that regulate fluid and electrolyte homeostasis in the intestine, binds to and activates GCC (guanylate cyclase-C) on the luminal surface of the intestinal epithelium. Activation of GCC results in the generation of cyclic guanosine monophosphate (cGMP), which is increased in both the intracellular and extracellular compartments. The increase in cGMP within intestinal epithelial cells triggers a signal transduction cascade activating the cystic fibrosis transmembrane conductance regulator (12). This activation causes secretion of chloride and bicarbonate into the intestinal lumen; sodium ions and water follow, resulting in increased luminal fluid secretion and a reflex acceleration of intestinal transit. Extracellular cGMP, actively transported out of intestinal epithelial cells, is believed to reduce visceral hyperalgesia by modulating the activity of afferent pain fibers (13). In animal models, linaclotide treatment accelerated gastrointestinal transit and reduced visceral nociception (14); in human phase 2 clinical studies, it accelerated colonic transit (15) and improved abdominal pain and constipation associated with IBS-C (16). Likewise, in two large phase 3 trials in patients with chronic constipation, linaclotide significantly improved bowel and abdominal symptoms over 12 weeks (17).
The objective of this phase 3 clinical trial was to assess the efficacy and safety of linaclotide administered once daily as an oral capsule at a dose of 290 μg vs. placebo to patients with IBS-C. A 4-week randomized withdrawal (RW) period was included in this trial to assess the effect of discontinuing treatment with linaclotide.
METHODS
Trial design
This multicenter, randomized, double-blind, placebo-controlled, parallel-group trial was conducted at 118 outpatient clinical research centers (111 in the United States, 7 in Canada) from 14 July 2009 (first patient enrolled) to 12 July 2010 (last patient completed). The protocol and all trial procedures were approved by an Institutional Review Board, and the trial was designed, conducted, and reported in accordance with the principles of Good Clinical Practice guidelines. All patients gave written informed consent before their participation in the trial.
After a screening period of up to 21 days followed by a pretreatment baseline period of 14–21 days, eligible patients were randomly assigned with the use of an interactive voice-response system (IVRS) to receive once daily an oral capsule of either linaclotide 290 μg or placebo, in a 1:1 ratio. Patients who completed all 12 weeks of the double-blind treatment period were eligible to enter the double-blind 4-week RW period in which patients initially randomized to linaclotide were re-randomized (1:1) to linaclotide 290 μg or placebo, and patients previously randomized to placebo were assigned to receive linaclotide 290 μg once a day. Randomization assignments were generated in blocks of four and stratified according to trial center. All sponsor staff involved in the trial, trial center personnel, and patients were blinded to the allocation of trial treatment. Trial visits were conducted at screening, at the start of the pretreatment baseline period, at randomization (day 1), throughout the treatment period (weeks 2, 4, 8, and 12), and at the beginning and end of the RW period (weeks 13 and 16 (end of trial)). Patients made daily calls to the IVRS to report their symptoms throughout the trial.
Trial patients
Female and male patients were eligible to participate if they were at least 18 years of age, and met modified Rome II criteria for IBS (18). In the 12 months before the screening visit, eligible patients were to have for at least 12 weeks, which need not be consecutive, abdominal pain, or abdominal discomfort that had ≥2 of these three features: (i) relieved with defecation, (ii) onset associated with a change in frequency of stool, and (iii) onset associated with a change in form (appearance) of stool, before starting chronic treatment with tegaserod or lubiprostone (if patients had taken these medications); and <3 spontaneous bowel movements (SBMs) per week (SBM=a bowel movement (BM) occurring in the absence of any laxative, suppository, or enema use during the preceding 24 h), and had at least one additional bowel symptom (straining, lumpy or hard stools, and sensation of incomplete evacuation during >25% of BMs), before starting chronic treatment with tegaserod, lubiprostone, polyethylene glycol 3350, or any laxative (if patients had taken these medications). In addition, patients had to report an average score ≥3.0 for daily abdominal pain at its worst (11-point NRS (numerical rating scale)) as well as an average of <3 complete SBMs (CSBMs) per week (CSBM=an SBM associated with a sense of complete evacuation, as reported by the patient) and ≤5 SBMs per week during the 14 days immediately before randomization (i.e., the baseline period).
Patients were excluded if they reported loose (mushy) or watery stools for >25% of their BMs during the 12 weeks before screening or, during the baseline period, a BSFS (Bristol Stool Form Scale) (19) score of 7 (watery, no solid pieces) for any SBM, or a BSFS score of 6 (fluffy pieces with ragged edges, a mushy stool) for >1 SBM. Other key exclusion criteria included history of cathartic colon, laxative or enema abuse, ischemic colitis, or pelvic floor dysfunction (unless successful treatment had been documented by a normal balloon expulsion test); bariatric surgery for treatment of obesity or surgery to remove a segment of the gastrointestinal tract at any time before the screening visit, surgery of the abdomen, pelvis, or retroperitoneal structures during the 6 months before the screening visit, appendectomy or cholecystectomy during the 60 days before the screening visit, or other major surgery during the 30 days before the screening visit; history of diverticulitis or any chronic condition that could be associated with abdominal pain or discomfort and could confound the assessments in the trial (e.g., inflammatory bowel disease, chronic pancreatitis, polycystic kidney disease, ovarian cysts, endometriosis, lactose intolerance); family history of a familial form of colorectal cancer. In general, patients were excluded if they were taking drugs that could cause constipation (e.g., narcotics); however, patients taking certain drugs for IBS that might be constipating (e.g., tricyclic antidepressants) were eligible provided that they were on a stable dose for at least 30 days before the screening visit and there was no plan to change the dose after the screening visit. Colonoscopy requirements were based on the American Gastroenterological Association guidelines (20). Women of childbearing potential were required to use contraceptives and have a negative serum pregnancy test. Patients were asked to refrain from making any major lifestyle changes (e.g., starting a new diet or changing their exercise pattern) during the trial.
Rescue medication (bisacodyl 5 mg tablet or 10 mg suppository) was allowed for severe constipation (i.e., 72 h after the patient's previous BM or when symptoms became intolerable). Use of rescue medication was not allowed on the day before, the day of, and the calendar day after the randomization visit. Patients on a stable, continuous regimen of fiber, bulk laxatives, stool softeners, or probiotics during the 30 days before the screening visit were allowed to continue, provided they maintained a stable dosage throughout the trial.
Efficacy assessments and end points
Daily reports by patients to IVRS included symptom ratings of worst abdominal pain, abdominal discomfort, abdominal cramping, abdominal fullness, and abdominal bloating (all abdominal symptoms were measured using an 11-point NRS), as well as the number of BMs and whether rescue medication was used. Each BM was assessed for sensation of complete bowel emptying (yes/no), stool consistency (7-point BSFS with 1=“separate hard lumps like nuts” to 7=“watery, no solid pieces”), and severity of straining (5-point ordinal scale). Weekly IVRS assessments included IBS severity and constipation severity (both using a 5-point ordinal scale), degree of IBS relief (7-point balanced scale), and adequate relief of IBS-C symptoms (yes/no). Assessment of satisfaction with the trial-medication's ability to relieve IBS symptoms (5-point ordinal scale) was captured at all study visits following randomization.
Primary end points
There were four prespecified primary end points in the trial, which were all responder end points. One of the four primary end points was based on the FDA recommendations for IBS-C trial design and end points in the recently finalized guidance for IBS clinical trials (May 2012) (21); a responder for this end point (to be referred to hereafter as “FDA end point”) was defined as a patient who met both of the following criteria in the same week for at least 6 of the 12 weeks of the treatment period: (i) an improvement of ≥30% from baseline in the average of the daily worst abdominal pain scores (to be referred to hereafter as “abdominal pain”) and (ii) an increase of ≥1 CSBM from baseline. This combined end point was added after the initiation of the trial, but before completion of enrollment and database lock, with a protocol amendment (no unblinding had occurred). The other three primary end points also required patients to meet weekly responder definitions, but for at least 9 of the 12 weeks of the treatment period. These weekly responder definitions were (i) an improvement of ≥30% in abdominal pain, (ii) ≥3 CSBMs and an increase of ≥1 CSBM from baseline, and (iii) a combined end point that defined a responder as a patient who met criteria for both i and ii in the same week.
Secondary end points
The secondary end points included 12-week change from baseline in abdominal pain, abdominal discomfort, abdominal bloating, stool frequency (CSBM and SBM weekly rates), stool consistency (BSFS), and severity of straining; secondary responder end points included abdominal pain and CSBM responders (using the individual components of the FDA end point). A number of other additional end points were also assessed, including 12-week change from baseline in abdominal fullness and abdominal cramping, IBS symptom severity, constipation severity, adequate relief of IBS-C symptoms, degree of relief of IBS symptoms, and treatment satisfaction.
Safety assessments
At each scheduled study visit, all patients were asked an open-ended question regarding adverse events (AEs). Patients reported AEs by recalling instances since the prior visit. The site investigator assessed all patient-reported AEs and judged each event for severity and relationship to the blinded trial medication. Other safety evaluations included physical examinations, electrocardiogram recordings, vital sign measurements, and standard clinical laboratory tests.
Pharmacokinetic assessments
During the treatment period, a subset of patients had blood samples taken at the randomization and week 4 visits to determine if linaclotide or its active metabolite, MM 419447, could be detected at quantifiable levels in the plasma.
Statistical methods and data analysis
The overall family-wise type I error rate for testing the primary and secondary efficacy end points was controlled at the 0.05 significance level using a five-step serial gate-keeping, multiple-comparison procedure. Based on this multiple-comparison procedure and the results of a previous phase 2b study (16), a sample size of 400 patients per treatment arm was selected to provide >85% overall power to simultaneously detect a difference between the placebo and linaclotide groups for the primary end points.
Responder end points were analyzed using a Cochran–Mantel–Haenszel (CMH) test controlling for geographic region. Continuous change-from-baseline end points were analyzed using an ANCOVA (analysis of covariance) model with fixed-effect terms for treatment group and geographic region and the corresponding baseline value as a covariate. Least-squares means (i.e., means adjusted for the other effects) from the ANCOVA model based on patients' overall average scores (except for SBMs and CSBMs, for which the overall weekly rates were calculated) are presented. Geographic region was used as a factor in the analyses rather than individual trial centers due to the potential for very small numbers of patients at some trial centers.
If a patient dropped out of the trial or otherwise did not report efficacy data for a particular treatment-period week (patients were required to complete at least four IVRS calls during a treatment week), the patient was not considered a responder for that week. An observed-cases approach to missing data was applied to the change-from-baseline secondary end points, such that if a patient dropped out of the trial or otherwise did not report data, the average of the non-missing data over the 12 weeks of the treatment period was the patient's value. Patients were assumed to have not had BMs nor taken rescue medication if the corresponding daily question was not answered. For the analysis of adequate relief, degree of relief of IBS symptoms, and treatment satisfaction, a last observation carried forward method was used. All P values were based on two-sided tests.
All randomized patients who took at least one dose of trial medication were included in safety analyses (safety population). Efficacy analyses were based on the ITT (intent-to-treat) population, which included all patients in the safety population who had at least one post-randomization entry of the primary efficacy assessment (i.e., IVRS assessment of abdominal pain or CSBMs).
RESULTS
Patient disposition, demographics, and baseline characteristics
Of the 2,424 patients who were screened for participation in this trial, 803 (33%) were randomized to treatment (Figure 1). Two patients were randomized at more than one trial center but only data from the trial center in which they were first randomized were included in statistical analyses. Of the 802 patients who received double-blind trial medication (safety population), 800 patients had at least one post-randomization entry of the primary efficacy assessment (ITT population). The demographics of the ITT population are shown in Table 1. Following completion of the treatment period, a total of 647 (81%) ITT patients entered the RW period of the trial, of which 645 received at least one dose of trial medication and were included in the RW population. Mean compliance with the trial-medication dosing (assessed by counting pills returned at trial visits) up to trial discontinuation/completion of the 12-week treatment period was 95 and 94% for the placebo and linaclotide groups, respectively. Compliance with the daily IVRS call-in (patients who completed ≥80% of scheduled calls) during the treatment period was 73 and 71% for placebo- and linaclotide-treated patients, respectively. During the pretreatment baseline period, 88% of patients experienced abdominal pain every day and 76% of patients had no CSBMs.
Figure 1.

Patient flow through the study.
Table 1. Summary of patient demographic and baseline characteristics (ITT population).
| Placebo, N=395 | Linaclotide 290 μg, N=405 | |
|---|---|---|
| Demographic data | ||
| Age (years), mean (range) | 43.7 (18–84) | 43.3 (19–81) |
| ≥65 years, n (%) | 26 (6.6) | 19 (4.7) |
| Sex, n (%) | ||
| Female | 357 (90.4) | 367 (90.6) |
| Male | 38 (9.6) | 38 (9.4) |
| Race, n (%) | ||
| White | 301 (76.2) | 314 (77.5) |
| Black | 75 (19.0) | 78 (19.3) |
| Other | 19 (4.8) | 13 (3.2) |
| BMI, mean (s.d.) | 27.6 (6.2) | 28.3 (6.4) |
| Abdominal symptoms, mean (s.d.) | ||
| Abdominal paina | 5.6 (1.7) | 5.7 (1.7) |
| Abdominal discomforta | 6.0 (1.7) | 6.2 (1.6) |
| Abdominal bloatinga | 6.5 (1.9) | 6.7 (1.8) |
| Abdominal fullnessa | 6.5 (1.8) | 6.8 (1.7) |
| Abdominal crampinga | 5.4 (1.9) | 5.4 (1.9) |
| Bowel symptoms, mean (s.d.) | ||
| CSBMs/week | 0.2 (0.5) | 0.2 (0.5) |
| SBMs/week | 1.9 (1.4) | 1.9 (1.4) |
| Stool consistencyb | 2.4 (1.0) | 2.3 (1.0) |
| Strainingc | 3.4 (0.8) | 3.6 (0.8) |
| Constipation severityd | 3.7 (0.6) | 3.8 (0.6) |
| IBS severityd | 3.7 (0.6) | 3.7 (0.6) |
BMI, body mass index; CSBM, complete SBM; IBS, irritable bowel syndrome; ITT, intent-to-treat; SBM, spontaneous bowel movement.
Assessed using an 11-point Numerical Rating Scale: 0=none; 10=very severe.
Assessed using the BSFS: 1=separate hard lumps, like nuts (hard to pass); 2=sausage-shaped, but lumpy; 3=like a sausage but with cracks on its surface; 4=like a sausage or snake, smooth and soft; 5=soft blobs with clear cut edges (passed easily); 6=fluffy pieces with ragged edges, a mushy stool; 7=watery, no solid pieces (entirely liquid).
Assessed using a 5-point ordinal scale: 1=not at all; 2=a little bit; 3=a moderate amount; 4=a great deal; 5=an extreme amount.
Assessed using a 5-point ordinal scale: 1=none; 2=mild; 3=moderate; 4=severe; 5=very severe.
All demographic characteristics were similar between treatment groups. For baseline clinical characteristics, significant differences were observed for abdominal fullness (P=0.011), stool consistency (P=0.046), and straining (P=0.020).
Efficacy results
For all primary and secondary efficacy end points, the linaclotide 290-μg group demonstrated statistically significant improvement compared with the placebo group, controlling for multiplicity.
For the individual components of the FDA end point, a significantly greater percentage of linaclotide-treated patients, compared with placebo-treated patients, reported a reduction of ≥30% in abdominal pain for at least 6 out of the 12 weeks of the treatment period (50.1 vs. 37.5%, P=0.0003 (Figure 2)) or an increase of ≥1 CSBM from baseline for at least 6 out of the 12 weeks of the treatment period (48.6 vs. 29.6%, P<0.0001 (Figure 2)). A total of 136 of 405 patients (33.6%) receiving linaclotide compared with 83 of 395 patients (21.0%) receiving placebo (odds ratio: 1.9, 95% confidence interval: 1.4, 2.7; P<0.0001) met the FDA end point ( Table 2; Figure 2). A significantly greater percentage of linaclotide-treated patients than placebo-treated patients also met the responder requirements for the other three primary end points, which required improvement in abdominal pain (i.e., a reduction of ≥30% in abdominal pain), CSBM rate (i.e., ≥3 CSBMs and an increase of ≥1 CSBM), or both for at least 9 of the 12 weeks of the treatment period ( Table 2). The NNT (number needed to treat) for the primary end points ranged from 7.6 to 14.3.
Figure 2.

FDA end point and components. FDA end point: ≥30% abdominal pain reduction and increase ≥1 CSBM from baseline in the same week for ≥6/12 weeks. ****P value <0.0001, ***<0.001 for linaclotide vs. placebo (Cochran–Mantel–Haenszel (CMH) test). P values met the criterion for statistical significance based on the multiple-comparison procedure. CSBM, complete spontaneous bowel movement; FDA, Food and Drug Administration; Lin, linaclotide; NNT, number needed to treat.
Table 2. Primary efficacy parameter results (ITT population).
|
Primary efficacy parameters |
Placebo responder (N=395), n (%) |
Linaclotide responder (N=405), n (%) |
Difference |
Odds ratio (95% CI) |
P valuea |
NNT (95% CI) |
| FDA end point (each week, ≥30% decrease in worst abdominal pain+an increase ≥1 CSBM from baseline for at least 6/12 weeks) | 83 (21.0) | 136 (33.6) | 12.6 | 1.9 (1.4, 2.7) | <0.0001 | 8.0 (5.4, 15.5) |
| ≥30% Decrease in worst abdominal pain (each week, ≥30% decrease in abdominal pain from baseline for at least 9/12 weeks) | 107 (27.1) | 139 (34.3) | 7.2 | 1.4 (1.0, 1.9) | 0.0262 | 13.8 (7.4, 116.1) |
| ≥3 CSBMs and an increase of ≥1 CSBM (each week, ≥3 CSBM+an increase ≥1 CSBM from baseline for at least 9/12 weeks) | 25 (6.3) | 79 (19.5) | 13.2 | 3.7 (2.3, 5.9) | <0.0001 | 7.6 (5.6, 11.6) |
| Combined responder (each week ≥30% decrease in worst abdominal pain+≥3 CSBM+an increase ≥1 CSBM from baseline for at least 9/12 weeks) | 20 (5.1) | 49 (12.1) | 7.0 | 2.6 (1.5, 4.5) | 0.0004 | 14.2 (9.2, 31.3) |
CI, confidence interval; CSBM, complete spontaneous bowel movement; FDA, Food and Drug Administration; ITT, intent-to-treat; NNT, number needed to treat.
P values were based on a comparison of linaclotide vs. the placebo group using the Cochran–Mantel–Haenszel test.
Linaclotide-treated patients also experienced statistically significantly greater improvements compared with placebo-treated patients for the secondary and additional end points ( Table 3). During the first week of treatment and for each subsequent week of treatment, linaclotide-treated patients reported greater improvements in worst abdominal pain and CSBM frequency compared with placebo-treated patients (P<0.001; Figure 3). At week 12, the mean decrease from baseline in worst abdominal pain was 2.4% for linaclotide vs. 1.5% for placebo (P<0.0001), and the mean increase from baseline in the weekly CSBM rate was 2.4 and 0.9 for linaclotide and placebo, respectively (P<0.0001). At the end of the Treatment Period (week 12), 52% of linaclotide-treated patients were either “very satisfied” or “quite satisfied” with treatment compared with 23% of placebo-treated patients (P<0.0001).
Table 3. Other efficacy parameter results (ITT population).
| Placebo, N=395 | Linaclotide 290 μg, N=405 | Difference | P value | NNT (95% CI) | |
|---|---|---|---|---|---|
| Worst abdominal pain | |||||
| Mean (11-point NRS scale) | 4.4 | 3.7 | |||
| aChange from baseline, meanb,c | −1.1 | −1.9 | −0.7 | <0.0001 | |
| a% of patients with ≥30% decrease in worst abdominal pain for at least 6/12 weeksd | 37.5 | 50.1 | 12.7 | 0.0003 | 7.9 (5.1, 17.1) |
| Abdominal discomfort | |||||
| Mean (11-point NRS scale) | 4.7 | 4.1 | |||
| aChange from baseline, meanb,c | −1.2 | −2.0 | −0.7 | <0.0001 | |
| % of patients with ≥30% decrease in abdominal discomfort for at least 6/12 weeksd | 37.0 | 48.1 | 11.2 | 0.0013 | 8.9 (5.6, 22.8) |
| Abdominal bloating | |||||
| Mean (11-point NRS scale) | 5.3 | 4.6 | |||
| aChange from baseline, meanb,c | −1.1 | −1.9 | −0.8 | <0.0001 | |
| % of patients with ≥30% decrease in abdominal bloating for at least 6/12 weeksd | 29.9 | 43.5 | 13.6 | <0.0001 | 7.4 (5.0, 14.3) |
| Abdominal fullness | |||||
| Mean (11-point NRS scale) | 5.3 | 4.6 | |||
| Change from baseline, meanb,c | −1.1 | −2.0 | −0.9 | <0.0001 | |
| % of patients with ≥30% decrease in abdominal fullness for at least 6/12 weeksd | 32.9 | 44.0 | 11.0 | 0.0012 | 9.1 (5.6, 23.0) |
| Abdominal cramping | |||||
| Mean (11-point NRS scale) | 4.1 | 3.5 | |||
| Change from baseline, meanb,c | −1.1 | −1.7 | −0.6 | <0.0001 | |
| % of patients with ≥30% decrease in abdominal cramping for at least 6/12 weeksd | 39.5 | 49.9 | 10.4 | 0.0029 | 9.6 (5.8, 28.3) |
| CSBMs | |||||
| Mean CSBMs/week | 1.0 | 2.6 | |||
| aChange from baseline, meanb,c | 0.7 | 2.3 | 1.6 | <0.0001 | |
| CSBM ≤24 h first dose (%)c | 13.2 | 32.3 | 19.2 | <0.0001 | 5.2 (4.0, 7.4) |
| a% of patients w/ CSBM rate increase ≥1 per week for at least 6/12 weeksd | 29.6 | 48.6 | 19.0 | <0.0001 | 5.3 (3.9, 8.1) |
| SBMs | |||||
| Mean SBMs/week | 3.2 | 6.0 | |||
| aChange from baselineb,c | 1.1 | 3.9 | 2.8 | <0.0001 | |
| SBM ≤24 h after first dose (%)c | 43.8 | 67.4 | 23.6 | <0.0001 | 4.2 (3.3, 5.9) |
| % of patients w/ SBM rate increase ≥2 per week from baseline for at least 6/12 weeksd | 29.4 | 57.5 | 28.2 | <0.0001 | 3.6 (2.9, 4.6) |
| Stool consistency | |||||
| Mean BSFS score (1–7) | 3.1 | 4.5 | |||
| aChange from baseline, meanb,c | 0.7 | 2.1 | 1.4 | <0.0001 | |
| Mean weekly % of SBMs without hard or lumpy stools (BSFS ≥3) | 60.7 | 79.4 | 18.7 | <0.0001 | |
| Straining | |||||
| Mean straining score (1–5) | 2.8 | 2.2 | |||
| aChange from baseline, meanb,c | −0.7 | −1.3 | −0.7 | <0.0001 | |
| Mean weekly % of SBMs without significant straining (i.e., score ≤3) | 71.7 | 85.3 | 13.6 | <0.0001 | |
| Constipation severity | |||||
| Mean constipation severity score (1–5) | 3.1 | 2.6 | |||
| Change from baseline, meanb,c | −0.6 | −1.2 | −0.6 | <0.0001 | |
| % of patients with decrease of ≥1 for at least 6/12 weeksd | 42.5 | 59.5 | 17.0 | <0.0001 | 5.9 (4.2, 9.9) |
| IBS severity | |||||
| Mean IBS severity score (1–5) | 3.1 | 2.7 | |||
| Change from baseline, meanb,c | −0.5 | −1.0 | −0.5 | <0.0001 | |
| % of patients with decrease of ≥1 for at least 6/12 weeksd | 37.5 | 56.3 | 18.8 | <0.0001 | 5.3 (3.9, 8.3) |
| Adequate relief | |||||
| % of patients reporting adequate relief of IBS symptoms for at least 75% of the weeks (i.e., 9/12 weeks)d | 21.3 | 36.8 | 15.5 | <0.0001 | 6.4 (4.6, 10.7) |
| % of patients reporting adequate relief of IBS symptoms for at least 50% of the weeks (i.e., 6/12 weeks)d | 34.2 | 48.9 | 14.7 | <0.0001 | 6.8 (4.7, 12.6) |
| Degree of reliefe | |||||
| % of patients reporting “Somewhat Relieved,” “Considerably Relieved,” or “Completely Relieved” for 100% of the weekly scores or “Considerably Relieved” or “Completely Relieved” for at least 50% of the weekly scoresd | 24.3 | 41.2 | 16.9 | <0.0001 | 5.9 (4.3, 9.5) |
BSFS, Bristol Stool Forms Scale; CI, confidence interval; CSBM, complete SBM; IBS, irritable bowel syndrome; ITT, intent-to-treat; NNT, number needed to treat; NRS, numerical rating scale; SBM, spontaneous bowel movement.
Secondary end point.
Changes from baseline are the least-squares means from the analysis of covariance (ANCOVA) model.
P values were based on a comparison of linaclotide vs. the placebo group using the ANCOVA model.
P values were based on a comparison of linaclotide vs. the placebo group using the Cochran–Mantel–Haenszel test.
Degree of Relief scale: 1=completely relieved; 2=considerably relieved; 3=somewhat relieved; 4=unchanged; 5=somewhat worse; 6=considerably worse; 7=as bad as I can imagine.
Figure 3.
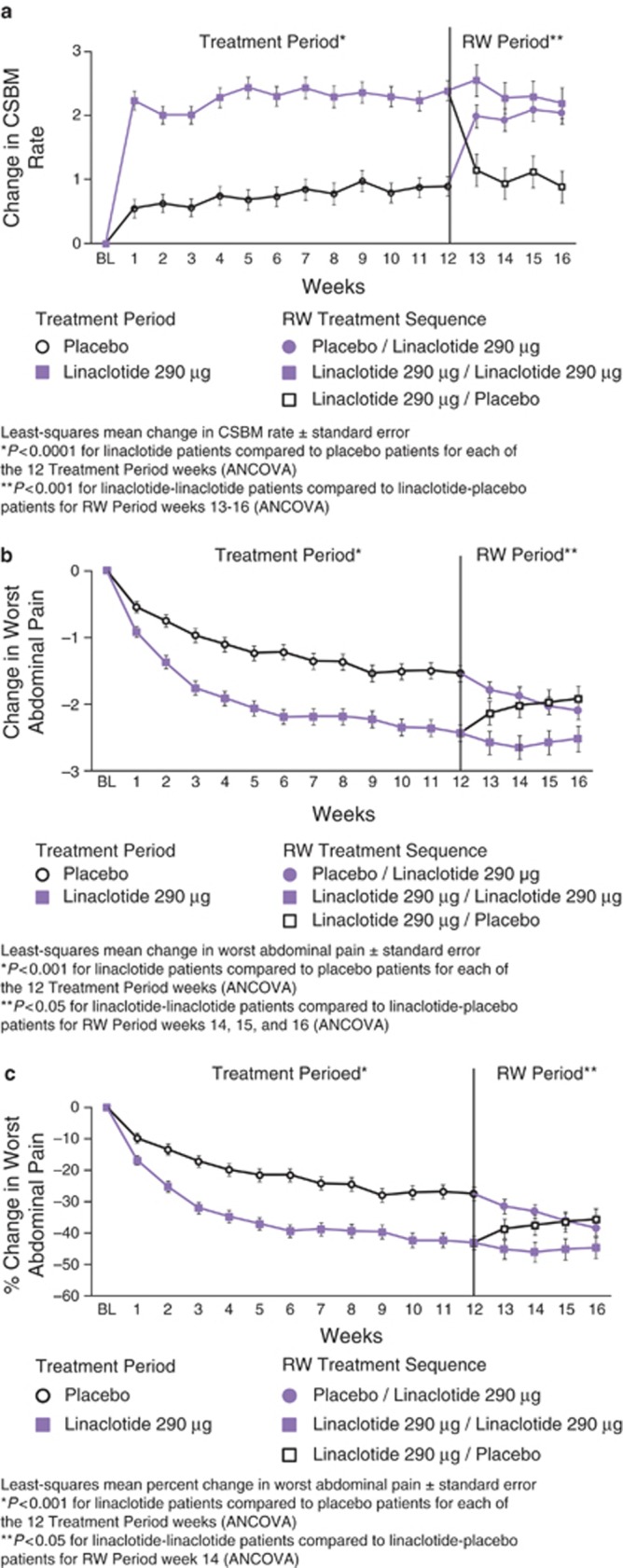
Weekly results for complete spontaneous bowel movement (CSBM) frequency (a, *P<0.0001 for linaclotide patients compared with placebo patients for each of the 12 Treatment-Period weeks, **P<0.001 for linaclotide–linaclotide patients compared with linaclotide–placebo patients for RW Period weeks 13–16); reduction in worst abdominal pain (b, *P<0.001 for linaclotide patients compared with placebo patients for each of the 12 Treatment Period weeks, **P<0.05 for linaclotide–linaclotide patients compared with linaclotide–placebo patients for RW Period weeks 14–16); and percent reduction in worst abdominal pain (c,*P<0.001 for linaclotide patients compared with placebo patients for each of the 12 Treatment Period weeks, **P<0.05 for linaclotide–linaclotide patients compared with linaclotide–placebo patients for RW Period week 14). All P values were derived from an analysis of covariance model.
During the 4-week RW Period, patients who were re-randomized from linaclotide to placebo showed an increase in worst abdominal pain and a decrease in CSBMs to levels similar to those observed in the placebo group during the Treatment Period. The patients who continued to take linaclotide showed sustained improvement in worst abdominal pain and CSBMs similar to that previously observed during the Treatment Period. These improvements were statistically significant compared to patients re-randomized to placebo for weeks 13–16 for CSBMs (P<0.001) and weeks 14–16 for worst abdominal pain (P<0.05). Patients who switched from placebo to linaclotide showed levels of improvement similar to those experienced by linaclotide-treated patients during the Treatment Period (Figure 3).
Safety
A total of 228 of 406 linaclotide-treated patients (56.2%) reported at least one treatment-emergent AE (TEAE) compared with 210 of 396 placebo-treated patients (53.0%) in the 12-week treatment period ( Table 4). Most TEAEs were mild or moderate in severity (93.8%, linaclotide; 98.1%, placebo). The incidences of diarrhea (P<0.0001), flatulence (P=0.0084), and abdominal pain (P=0.0462) TEAEs were significantly greater in the linaclotide-treated patients compared with placebo-treated patients. The most common TEAE in the 12-week treatment period was diarrhea, experienced by 19.5% of linaclotide-treated patients compared with 3.5% of placebo-treated patients. The occurrences of diarrhea were reported to be mild or moderate in 71 of 79 linaclotide-treated patients (89.9%) and 13 of 14 placebo-treated patients (92.9%) who experienced diarrhea. There were no SAEs of diarrhea reported during the trial. None of the patients who reported diarrhea experienced clinically significant sequelae (e.g., orthostatic hypotension or dehydration). More than half of linaclotide-treated patients who experienced diarrhea had onset within the first 2 weeks of treatment. Diarrhea was the most common AE resulting in treatment discontinuation in linaclotide-treated patients (5.7 vs. 0.3% in placebo-treated patients); overall, AEs resulted in the premature discontinuation of 32 patients (7.9%) and 11 patients (2.8%) taking linaclotide and placebo, respectively, in the treatment period.
Table 4. Treatment-emergent adverse events (safety population).
| Adverse event (preferred term) | Placebo (N=396), n (%) | Linaclotide 290 μg (N=406), n (%) | P value |
|---|---|---|---|
| Patients with at least 1 TEAE | 210 (53.0) | 228 (56.2) | 0.3949 |
| Diarrhea | 14 (3.5) | 79 (19.5) | <0.0001 |
| Abdominal pain | 10 (2.5) | 22 (5.4) | 0.0462 |
| Flatulence | 6 (1.5) | 20 (4.9) | 0.0084 |
| Headache | 14 (3.5) | 20 (4.9) | 0.3825 |
| Abdominal distension | 3 (0.8) | 9 (2.2) | 0.1434 |
Treatment-emergent adverse events (TEAEs) reported in ≥2% of linaclotide-treated patients and at an incidence greater than reported in placebo-treated patients during the treatment period.
P value was based on a Fisher's exact test comparing linaclotide and placebo.
Rates of serious AEs (SAEs) did not differ between linaclotide and placebo groups (two patients in each group (0.5%)). In the linaclotide group, the SAEs consisted of one patient who experienced asthma and a second patient who experienced pericardial effusion and pericarditis leading to withdrawal from the trial. In the placebo group, the SAEs consisted of one patient who experienced chronic cholecystitis and a second patient who experienced duodenitis, gastroenteritis, hiatal hernia, esophagitis, renal cyst, and urinary tract infection. There were no deaths during the treatment period; one screened patient died as a result of cardiorespiratory arrest and ventricular fibrillation due to a possible drug overdose, but this patient died before randomization and did not receive trial medication.
There were no clinically significant differences between the linaclotide and placebo groups in the incidence of abnormal laboratory parameters, vital signs, or electrocardiogram parameters. Serum bicarbonate levels were below the lower limit of normal at the end of treatment in seven patients receiving linaclotide compared with one patient receiving placebo. None of these patients reported diarrhea as an AE or other AEs that were considered to be related to low bicarbonate levels.
In the subset of patients who were assessed for linaclotide exposure, no quantifiable plasma levels of linaclotide were detected following trial-medication dosing at the randomization and week 4 trial visits. All patients tested (72 placebo and 64 linaclotide) had levels lower than the limit of quantification for linaclotide (<0.2 ng/ml) and its primary metabolite, MM-419447 (<2.0 ng/ml).
During the RW period, TEAEs occurred in 22.2% of linaclotide–linaclotide patients, 22.1% of linaclotide–placebo patients, and 30.6% of placebo–linaclotide patients. With the exceptions of diarrhea and abdominal pain, the incidence of TEAEs was similar across the three treatment sequences. The incidence of diarrhea was 1.9, 0.6, and 11.7%, in linaclotide–linaclotide, linaclotide–placebo, and placebo–linaclotide patients, respectively. The incidence of abdominal pain was 1.3% in the linaclotide–linaclotide patients and 2.4% in the placebo–linaclotide patients; there were no TEAEs of abdominal pain in the linaclotide–placebo patients. There was no evidence of “rebound” (i.e., worsening in IBS-C symptoms compared with the baseline period in the linaclotide–placebo patients). No SAEs were reported during the RW period.
DISCUSSION
In this large phase 3 clinical trial, a greater percentage of IBS-C patients who were treated with linaclotide achieved statistically significant improvement in the key symptoms of IBS-C, including abdominal pain and constipation, compared with placebo. Four primary outcomes measures were assessed, including the FDA end point. This end point required that patients experience a benefit of at least 30% when compared with baseline in abdominal pain and an increase of ≥1 CSBM from baseline in the same week for at least 6 out of the 12 weeks of the treatment period. In spite of the rigor of this end point, 33.6% of linaclotide-treated patients were responders compared with 21.0% of placebo-treated patients (P<0.0001). Furthermore, statistically significant differences in responder rates were also demonstrated for the three other primary end points, which required (i) a decrease in abdominal pain of ≥30%, (ii) both an absolute value of ≥3 CSBMs and an increase of ≥1 CSBM from baseline, and (iii) both abdominal pain and CSBM criteria for at least 9 of the 12 weeks of the treatment period.
Although IBS is a disorder with multiple symptoms, abdominal pain is one of the cardinal manifestations and strongly correlates with IBS severity (22) and utilization of healthcare resources (23). Also, an improvement in abdominal pain of ≥30% has been shown to be clinically important in IBS patients (23), and in patients reporting pain relief in general (24). In this trial, more than half of linaclotide-treated patients reported an improvement in abdominal pain of ≥30% for at least 6 out of 12 weeks compared with 37.5% of placebo-treated patients, for an NNT of 7.9. Improvement in abdominal pain began within the first week of therapy, and once reaching maximum at 6–8 weeks, was sustained throughout the remainder of the treatment period. By the last week of the treatment period (week 12), linaclotide-treated patients reported a mean improvement of 43.2% in abdominal pain compared with 27.5% for placebo-treated patients. During the 4-week RW period, patients re-randomized to remain on linaclotide had continued relief of abdominal pain, showing durability of response, while those re-randomized from linaclotide to placebo showed a gradual worsening of abdominal pain symptoms to the level experienced by patients receiving placebo during the treatment period, but without signs of a “rebound” or worsening of symptoms relative to baseline.
In addition to abdominal pain, linaclotide improved several other important abdominal symptoms that are frequently reported by IBS-C patients, including abdominal bloating and abdominal discomfort, beginning during the first week of treatment and continuing throughout the 12-week treatment period. Linaclotide also improved bowel function, including SBM and CSBM frequency, straining, stool consistency, and constipation severity. However, in contrast to the gradual improvement in abdominal symptoms, improvement in bowel function occurred more rapidly. Most linaclotide-treated patients experienced an SBM within 24 h of the first dose of linaclotide (67.4 vs. 43.8% for placebo, P<0.0001); maximal improvement in bowel function usually occurred within the first week. Thus, improvement with linaclotide in abdominal (sensory) symptoms such as abdominal pain may be attributable to more than improvement in bowel function alone. Preclinical data suggest that cGMP, which is released intra- and extracellularly following GCC activation by linaclotide, can reduce the firing of pain-sensing visceral afferent fibers (13). Further studies are under way that may provide a better understanding of the mechanisms by which linaclotide exerts its beneficial effects directly on abdominal sensory symptoms.
Diarrhea was the most common TEAE in linaclotide-treated patients and appears to be an extension of linaclotide's pharmacological effects. Although diarrhea was reported in 19.5% of linaclotide-treated patients, only 2% reported that they had severe diarrhea and only 5.7% discontinued the drug due to diarrhea. The incidence of SAEs was similar between linaclotide- and placebo-treated patients (n=2 patients in each group); diarrhea was not reported as an SAE.
In conclusion, linaclotide significantly improved abdominal and bowel symptoms in this phase 3 trial (12-week treatment period+4-week RW period).
STUDY HIGHLIGHTS

Acknowledgments
Annie Neild, PhD, of Ironwood Pharmaceuticals, provided editorial assistance.
Guarantor of the article: Jeffrey M. Johnston, MD.
Specific author contributions: Wrote the initial draft of the manuscript, assisted in the interpretation of data, and provided critical revision: Satish Rao and Anthony J. Lembo; designed the trial: Jeffrey M. Johnston, Bernard J. Lavins, Harvey A. Schneier, and Steven J. Shiff; assisted in the interpretation of data and critical revision of the manuscript for important intellectual content: Jeffrey M. Johnston, Bernard J. Lavins, Caroline B. Kurtz, Mark G. Currie, Harvey A. Schneier, and Steven J. Shiff; provided statistical design, analyses, and interpretation: James E. MacDougall, Xinwei D. Jia, Kelvin Shi, and James Z. Shao; coordinated acquisition of data and trial supervision: Paul Eng and Susan M. Fox.
Financial support: This trial was funded by Forest Research Institute and Ironwood Pharmaceuticals, Inc.
Potential competing interests: Jeffrey M. Johnston, Caroline B. Kurtz, James E. MacDougall, James Z. Shao, Bernard J. Lavins, and Mark G. Currie are employees of Ironwood Pharmaceuticals and own stock/stock options in Ironwood Pharmaceuticals. Harvey A. Schneier, Steven J. Shiff, Paul Eng, Susan M. Fox, Xinwei D. Jia, and Kelvin Shi are employees of Forest Laboratories and own stock/stock options in Forest Laboratories. Anthony J. Lembo and Satish Rao are paid consultants to Ironwood Pharmaceuticals and Forest Research Institute.
Footnotes
Portions of this manuscript were presented as an oral presentation at Digestive Disease Week, May 2011, Chicago, IL.
References
- Longstreth GF, Thompson WG, Chey WD, et al. Functional bowel disorders. Gastroenterology. 2006;130:1480–1491. doi: 10.1053/j.gastro.2005.11.061. [DOI] [PubMed] [Google Scholar]
- Hellstrom PM, Saito YA, Bytzer P, et al. Characteristics of acute pain attacks in patients with irritable bowel syndrome meeting Rome III criteria. Am J Gastroenterol. 2011;106:1299–1307. doi: 10.1038/ajg.2011.78. [DOI] [PubMed] [Google Scholar]
- Drossman DA, Camilleri M, Mayer EA, et al. AGA technical review on irritable bowel syndrome. Gastroenterology. 2002;123:2108–2131. doi: 10.1053/gast.2002.37095. [DOI] [PubMed] [Google Scholar]
- Paré P. Health-related quality of life, work productivity, and health care resource utilization of subjects with irritable bowel syndrome: baseline results from LOGIC (Longitudinal Outcomes Study of Gastrointestinal Symptoms in Canada), a naturalistic study. Clin Ther. 2006;28:1726–1735. doi: 10.1016/j.clinthera.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Cash B, Sullivan S, Barghout V. Total costs of IBS: employer and managed care perspective. Am J Manag Care. 2005;11:S7–S16. [PubMed] [Google Scholar]
- Mayer EA. Clinical practice: irritable bowel syndrome. N Engl J Med. 2008;358:1692–1699. doi: 10.1056/NEJMcp0801447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hungin AP, Chang L, Locke GR, et al. Irritable bowel syndrome in the United States: prevalence, symptom patterns and impact. Aliment Pharmacol Ther. 2005;21:1365–1375. doi: 10.1111/j.1365-2036.2005.02463.x. [DOI] [PubMed] [Google Scholar]
- Hungin AP, Whorwell PJ, Tack J, et al. The prevalence, patterns and impact of irritable bowel syndrome: an international survey of 40,000 subjects. Aliment Pharmacol Ther. 2003;17:643–650. doi: 10.1046/j.1365-2036.2003.01456.x. [DOI] [PubMed] [Google Scholar]
- Brandt LJ, Chey WD, Foxx-Orenstein AE, et al. An evidence-based position statement on the management of irritable bowel syndrome. Am J Gastroenterol. 2009;104 (Suppl 1:S1–S35. doi: 10.1038/ajg.2008.122. [DOI] [PubMed] [Google Scholar]
- Hulisz D. The burden of illness of irritable bowel syndrome: current challenges and hope for the future. J Manag Care Pharm. 2004;10:299–309. doi: 10.18553/jmcp.2004.10.4.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drossman DA, Chey WD, Johanson JF, et al. Clinical trial: lubiprostone in patients with constipation-associated irritable bowel syndrome—results of two randomized, placebo-controlled studies. Aliment Pharmacol Ther. 2009;29:329–341. doi: 10.1111/j.1365-2036.2008.03881.x. [DOI] [PubMed] [Google Scholar]
- Bryant AP, Busby RW, Bartolini WP, et al. Linaclotide is a potent and selective guanylate cyclase C agonist that elicits pharmacological effects locally in the gastrointestinal tract. Life Sci. 2010;8:19–20. doi: 10.1016/j.lfs.2010.03.015. [DOI] [PubMed] [Google Scholar]
- Castro J, Martin C, Hughes PA, et al. A novel role of cyclic GMP in colonic sensory neurotransmission in healthy and TNBS-treated mice. Gastroenterology. 2011;140:S–538. [Google Scholar]
- Eutamene H, Bradesi S, Larauche M, et al. Guanylate cyclase C-mediated antinociceptive effects of linaclotide in rodent models of visceral pain. Neurogastroenterol Motil. 2010;22:312–322. doi: 10.1111/j.1365-2982.2009.01385.x. [DOI] [PubMed] [Google Scholar]
- Andresen V, Camilleri M, Busciglio IA, et al. Effect of 5 days linaclotide on transit and bowel function in females with constipation-predominant irritable bowel syndrome. Gastroenterology. 2007;133:761–768. doi: 10.1053/j.gastro.2007.06.067. [DOI] [PubMed] [Google Scholar]
- Johnston JM, Kurtz CB, Macdougall JE, et al. Linaclotide improves abdominal pain and bowel habits in a phase IIb study of patients with irritable bowel syndrome with constipation. Gastroenterology. 2010;139:1877–1886. doi: 10.1053/j.gastro.2010.08.041. [DOI] [PubMed] [Google Scholar]
- Lembo AJ, Schneier HA, Shiff SJ, et al. Two randomized trials of linaclotide for chronic constipation. N Engl J Med. 2011;365:527–536. doi: 10.1056/NEJMoa1010863. [DOI] [PubMed] [Google Scholar]
- Thompson WG, Longstreth GF, Drossman DA, et al. Functional bowel disorders and functional abdominal pain. Gut. 1999;45 (Suppl 2:II43–II47. doi: 10.1136/gut.45.2008.ii43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SJ, Heaton KW. Stool form scale as a useful guide to intestinal transit time. Scand J Gastroenterol. 1997;32:920–924. doi: 10.3109/00365529709011203. [DOI] [PubMed] [Google Scholar]
- Winawer S, Fletcher R, Rex D, et al. Colorectal cancer screening and surveillance: clinical guidelines and rationale-update based on new evidence. Gastroenterology. 2003;124:544–560. doi: 10.1053/gast.2003.50044. [DOI] [PubMed] [Google Scholar]
- Guidance for industry: irritable bowel syndrome – clinical evaluation of drugs for treatment Food and Drug Administration 2012. Available at: URL: : http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/ Guidances/UCM205269.pdf Accessed 4 June 2012.
- Lembo A, Ameen VZ, Drossman DA. Irritable bowel syndrome: toward an understanding of severity. Clin Gastroenterol Hepatol. 2005;3:717–725. doi: 10.1016/s1542-3565(05)00157-6. [DOI] [PubMed] [Google Scholar]
- Spiegel B, Bolus R, Harris LA, et al. Measuring irritable bowel syndrome patient-reported outcomes with an abdominal pain numeric rating scale. Aliment Pharmacol Ther. 2009;30:1159–1170. doi: 10.1111/j.1365-2036.2009.04144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrar JT, Young JP, LaMoreaux L, et al. Clinical importance of changes in chronic pain intensity measured on an 11-point numerical pain rating scale. Pain. 2001;94:149–158. doi: 10.1016/S0304-3959(01)00349-9. [DOI] [PubMed] [Google Scholar]