

Abstract
BACKGROUND
The upcoming reauthorization of the Prescription Drug User Fee Act focuses on improving the review process for new drug applications at the Food and Drug Administration (FDA).
METHODS
Using publicly available information from the FDA, the European Medicines Agency (EMA), and Health Canada, we compared the time for completion of the first review and the total review time for all applications involving novel therapeutic agents approved by the three regulatory agencies from 2001 through 2010 and determined the geographic area in which each novel therapeutic agent was first approved for use.
RESULTS
There were 510 applications for novel therapeutic agents approved from 2001 through 2010 — 225 by the FDA, 186 by the EMA, and 99 by Health Canada; among the applications, there were 289 unique agents. The median length of time for completion of the first review was 303 days (interquartile range, 185 to 372) for applications approved by the FDA, 366 days (interquartile range, 310 to 445) for those approved by the EMA, and 352 days (interquartile range, 255 to 420) for those approved by Health Canada (P<0.001 for the comparison across the three agencies). The median total review time was also shorter at the FDA than at the EMA or Health Canada (P = 0.002). Among the 289 unique novel therapeutic agents, 190 were approved in both the United States and Europe (either by the EMA or through the mutual recognition process), of which 121 (63.7%) were first approved in the United States; similarly, 154 were approved in both the United States and Canada, of which 132 (85.7%) were first approved in the United States.
CONCLUSIONS
For novel therapeutic agents approved between 2001 and 2010, the FDA reviewed applications involving novel therapeutics more quickly, on average, than did the EMA or Health Canada, and the vast majority of these new therapeutic agents were first approved for use in the United States. (Funded by the Pew Charitable Trusts.)
The Prescription Drug User Fee Act (PDUFA) of 1992 was enacted to augment the resources of the Food and Drug Administration (FDA) that are devoted to reviewing applications for drugs for humans and to ensuring drug efficacy and safety.1 In exchange for meeting clear performance standards, the FDA is authorized by PDUFA to collect user fees for each new application to support the infrastructure needed for review, such as the hiring of additional staff. Although PDUFA has contributed to a substantial reduction in the review times for drug applications over the past two decades,2,3 meeting these performance standards has also been associated with higher rates of drug withdrawals and blackbox warnings.3-5
PDUFA is subject to reauthorization every 5 years, and previous renewals have identified specific areas of emphasis. For example, the 2007 PDUFA IV renewal focused on drug safety and postmarketing surveillance. In anticipation of the 2012 PDUFA V renewal, the FDA has worked with industry to develop a framework of future performance goals, including a program to “improve the efficiency and effectiveness” of the review process for new-drug applications involving new molecular entities or novel biologic drugs,6,7 hereafter referred to as “novel therapeutics.” By enhancing transparency and communication between applicants and the FDA, this program is designed to increase the number of novel therapeutics approved after a single regulatory review cycle, reflecting a response to criticism that the FDA has focused on safety at the expense of timely reviews.8,9
Novel therapeutics are central to any debate about PDUFA reauthorization, since these therapies most likely represent advances in care, although there is variation in the degree of innovation among novel therapeutics, ranging from first-in-class therapeutics that address unmet public health needs to “me too” drugs. However, little objective information is currently available about the length of time that the FDA takes for the review of applications for novel therapeutics, particularly as compared with the length of time for the review of applications by regulatory agencies serving countries with populations similar to the population of the United States. Previous studies have examined the review process for applications submitted during earlier PDUFA periods but have not assessed the agency’s performance under PDUFA IV to inform the PDUFA V reauthorization process.10-12 More recently, a study that focused only on novel cancer drugs approved between 2003 and 2010 showed that the drug-approval process was faster at the FDA than at the European Medicines Agency (EMA), the primary regulator serving the European Union.13 Another relevant issue is the geographic area in which new medications are “first to market” (i.e., first approved for use). We compared review times for novel therapeutics among the FDA, the EMA, and Health Canada, the regulator serving Canada, during the most recent PDUFA periods. We also evaluated review times across PDUFA periods to explore the potential effect of PDUFA IV on review times at the FDA.
METHODS
DATA SOURCES
We obtained information about regulatory decisions made by the FDA, the EMA, and Health Canada from publicly accessible databases that catalogue approved drugs and regulatory actions. All the authors assume full responsibility for the accuracy and completeness of the data. For the FDA, documents available through Drugs@FDA provide information on regulatory actions (e.g., approvals of new therapeutic agents, new indications for current therapeutic agents, and drug labeling changes), including the dates for each of these actions, for all prescription therapeutic agents currently approved in the United States; regulatory actions involving unapproved drugs are not included.14 The EMA maintains European public assessment reports, which summarize scientific reviews and list notable regulatory events for all prescription drugs currently approved by the EMA, as well as for some rejected and withdrawn drugs.15 The EMA represents the major, but not the only, pathway to drug approval in Europe; in certain circumstances, drug makers have the option of submitting their applications to national regulators.16 The Drug Product Database of Health Canada lists basic information about all prescription, nonprescription, and veterinary drugs approved by Health Canada17; the Summary Basis of Decision documents contain information similar to that in the EMA European public assessment reports for all novel pharmaceutical agents approved since 2005.18 The databases of all three regulatory agencies were downloaded between July 6 and July 15, 2011.
STUDY SAMPLE
We identified a sample of applications involving novel therapeutics that received regulatory approval from each agency between January 1, 2001, and December 31, 2010 (Fig. 1). We excluded applications involving reformulations of drugs, combination therapies, and nontherapeutic agents, such as diagnostic contrast agents, and removed duplicate records; we also excluded applications in which key regulatory dates were unavailable (Table S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org).
Figure 1. Identification of the Sample of Novel Therapeutic Applications for the Current Analysis.
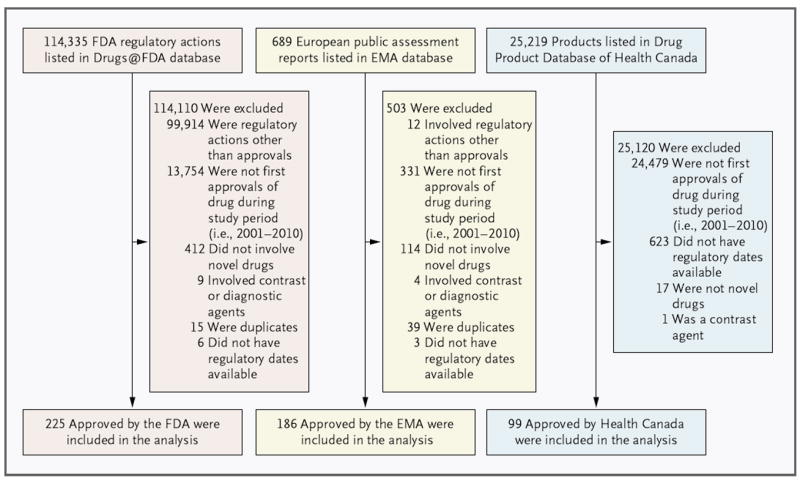
The analysis includes applications approved by the Food and Drug Administration (FDA), the European Medicines Agency (EMA), and Health Canada from 2001 through 2010. Health Canada has posted Summary Basis of Decision documents, which list the dates needed to calculate review time, only for therapeutics approved since 2005. As a result, 623 applications were excluded from the analysis because regulatory dates were not available from Summary Basis of Decision documents.
REGULATORY REVIEW TIME
The first two authors manually abstracted key regulatory dates using the documents described above14,15,18 and determined the number of regulatory review cycles required to approve each application. A new cycle was defined as any company-initiated regulatory review, including the initial submission, resubmission of an unapproved or withdrawn application, and appeal of a regulatory decision. For 5% of the applications, we validated the information by means of repeat abstraction; agreement rates were very high, with only one instance of disagreement.
For all applications, the first-review time was defined as the number of days between submission of the application and completion of the first review (the date on which the regulatory agency notified the applicant of the decision). For the EMA, the completion date for the first review was the date the agency issued an initial recommendation. We did not use the date of the final authorization by the European Commission to market the drug, because this is an administrative action that is taken without regulatory review. For Health Canada, the completion date for the first review was the date the agency issued a first decision, provided within Summary Basis of Decision documents, which marks the end of the regulatory review process and publicly announces the decision of the agency. We did not use the date recorded in the Drug Product Database, which reflects the date on which the drug was launched to market.
Since first-review time does not always capture the full length of the regulatory review in the case of applications requiring multiple review cycles, we also calculated “total review time” — the combined length of all the regulatory review cycles required for approval. In the case of a drug approved after one review cycle, this variable equals the first-review time; in other cases, it is the sum of the time required for each review cycle.
FIRST TO MARKET
To determine the geographic area in which each novel therapeutic was first approved for use, we compared the approval dates across the three agency jurisdictions (i.e., the United States, Europe, and Canada) for therapeutics approved by multiple regulatory agencies. For this determination, we examined all regulatory decisions, including those that were made outside our study period (i.e., before 2001), since some therapeutics approved by one agency may have been approved by another before 2001. The dates of approval by the FDA and the EMA were obtained from Drugs@FDA and EMA European public assessment reports, respectively. For therapeutics approved by Health Canada before 2005, only the launch date listed in the Drug Product Database was publicly available. We imputed approval dates by subtracting the median difference between the launch date and the approval date among therapeutics approved by Health Canada after 2005 (for which both launch and approval dates were available) from the launch date. In addition, because the EMA is not the only pathway to drug approval in Europe, we searched the Heads of Medicines Agencies Mutual Recognition Product Index to determine whether drugs approved by the FDA or Health Canada, but not the EMA, had been approved in Europe through the alternative mutual recognition process.19
OTHER CHARACTERISTICS OF APPLICATIONS
We categorized applications according to several characteristics. First, we determined the PDUFA period during which each application was submitted. Applications submitted before October 1, 2002, were classified in the PDUFA I–II period, applications submitted between October 1, 2002, and September 30, 2007, were classified in the PDUFA III period, and applications submitted after September 30, 2007, were classified in the PDUFA IV period. Using information from the databases of the regulatory agencies, we categorized applications according to orphan or nonorphan status of the drug (in the case of FDA and EMA), priority or standard review (in the case of the FDA and Health Canada), and designation of the drug as a small-molecule or biologic agent. Since Health Canada does not classify small-molecule or biologic agents, we used FDA and EMA classifications when possible and manually classified all others. We also categorized applications according to the classification of the drug in one of seven therapeutic classes, on the basis of the Anatomical Therapeutic Chemical codes of the World Health Organization (WHO), which are provided for EMA and Health Canada approvals. When possible, we used these classifications to assign codes to therapeutics reviewed by the FDA. Otherwise, we determined Anatomical Therapeutic Chemical codes by accessing information on the WHO website or by manual review.20
STATISTICAL ANALYSIS
Using descriptive statistics, we characterized the applications submitted to each agency according to key characteristics. Next, we calculated median first-review and total review times for all three agencies and compared them using Kruskal–Wallis tests. We then used Wilcoxon tests to conduct pairwise comparisons of review times between the FDA and the EMA, between the FDA and Health Canada, and between the EMA and Health Canada. These analyses were then repeated with stratification according to characteristics of the applications (PDUFA period and priority vs. standard review) and of the therapeutics (small-molecule vs. biologic agent, therapeutic class, and orphan vs. nonorphan status). We subsequently repeated these analyses among subsamples of novel therapeutics approved by either two of the three regulatory agencies or all three agencies. In addition, we used Kruskal–Wallis tests to compare first-review times and total review times within each agency across PDUFA periods. Finally, we characterized the geographic jurisdiction in which the therapeutics were first approved. All analyses were performed with the use of JMP 7.0.1 software (SAS Institute), and all statistical tests were two-tailed. A type I error rate of 0.02 was used for Wilcoxon tests to account for multiple pairwise comparisons.
RESULTS
We examined the regulatory review time for 510 applications for novel therapeutic agents that were approved by one or more of the three agencies: 225 that were approved by the FDA, 186 by the EMA, and 99 by Health Canada (Table S2 in the Supplementary Appendix); among these applications, there were 289 unique agents, 72 of which were approved by all three regulatory agencies (Fig. S1 in the Supplementary Appendix). There were no significant differences among the regulatory agencies with respect to type of drug reviewed, the therapeutic class of the drug reviewed, the use of priority review. However, the EMA approved a larger proportion of orphan-designated therapeutics than did the FDA (28.0% vs. 16.9%, P = 0.007) (Table 1).
Table 1.
Characteristics of Novel Therapeutic Applications for Drugs Approved by the Food and Drug Administration (FDA), the European Medicines Agency (EMA), and Health Canada from 2001 through 2010.*
| Variable | FDA (N = 225) |
EMA (N = 186) |
Health Canada (N = 99) |
|---|---|---|---|
| no. of applications (%) | |||
| PDUFA period† | |||
| I–II | 76 (33.8) | 46 (24.7) | 6 (6.1) |
| III | 104 (46.2) | 97 (52.2) | 60 (60.6) |
| IV | 45 (20.0) | 43 (23.1) | 33 (33.3) |
| Type of drug | |||
| Small molecule | 189 (84.0) | 142 (76.3) | 81 (81.8) |
| Biologic | 36 (16.0) | 44 (23.7) | 18 (18.2) |
| Therapeutic class | |||
| Cardiovascular, diabetes, or endocrine | 29 (12.9) | 27 (14.5) | 13 (13.1) |
| Hematologic, oncologic, or immune-modulating | 59 (26.2) | 68 (36.6) | 34 (34.3) |
| Musculoskeletal or pain-reducing | 13 (5.8) | 7 (3.8) | 4 (4.0) |
| Psychiatric or central nervous system | 24 (10.7) | 14 (7.5) | 10 (10.1) |
| Respiratory or gastrointestinal | 25 (11.1) | 22 (11.8) | 9 (9.1) |
| Antiinfective | 31 (13.8) | 24 (12.9) | 14 (14.1) |
| Other | 44 (19.6) | 24 (12.9) | 15 (15.2) |
| Orphan status†‡ | |||
| Orphan designation | 38 (16.9) | 52 (28.0) | — |
| Nonorphan designation | 187 (83.1) | 134 (72.0) | — |
| Review status§ | |||
| Priority | 79 (35.1) | — | 31 (31.3) |
| Standard | 146 (64.9) | — | 68 (68.7) |
| Review cycles† | |||
| 1 | 139 (61.8) | 179 (96.2) | 68 (68.7) |
| 2 | 82 (36.4) | 5 (2.7) | 24 (24.2) |
| ≥3 | 4 (1.8) | 2 (1.1) | 7 (7.1) |
Information on application approvals by Health Canada is available only for 2005 and later. Applications submitted before October 1, 2002, were classified in the Prescription Drug User Fee Act (PDUFA) I–II period; applications submitted between October 1, 2002, and September 30, 2007, were classified in the PDUFA III period; and applications submitted after September 30, 2007, were classified in the PDUFA IV period.
P≤0.05 for the comparison across the three agencies.
Health Canada does not designate orphan versus nonorphan status.
Status with respect to priority or standard review was not analyzed for EMA because the EMA Accelerated Assessment procedure, which is similar to priority review at the FDA, was not established until 2007.
REVIEW CYCLES
The majority of the applications for novel therapeutics approved by each regulatory agency, including virtually all those approved by the EMA, involved a single review cycle. A total of 139 (61.8%) of the novel therapeutics approved by the FDA were approved after one cycle, and 86 (38.2%) required two or more cycles. The corresponding values for the EMA were 179 (96.2%) and 7 (3.8%), and those for Health Canada were 68 (68.7%) and 31 (31.3%) (P<0.001 for the comparison across the three agencies).
FIRST-REVIEW TIME
The median length of time for the first review differed among the three agencies, with reviews completed 50 to 60 days more quickly by the FDA than by the EMA or Health Canada. The median length of time for a first review by the FDA was 303 days (interquartile range, 185 to 372), as compared with 366 days (interquartile range, 310 to 445) for the EMA and 352 days (interquartile range, 255 to 420) for Health Canada (P<0.001 for the comparison across the three agencies). After stratification according to key characteristics, including the PDUFA period during which the application was submitted, the drug type, the therapeutic class (except anti-infective agents), priority or standard review, and orphan or nonorphan status, the median length of time for the first review by the FDA was shorter than the median times for the first review by the EMA and by Health Canada, although not all the comparisons were significant (Fig. 2, and Table S3 in the Supplementary Appendix).
Figure 2. Median First-Review Times and Total Review Times for Applications for Novel Therapeutics Approved by the FDA, the EMA, and Health Canada.
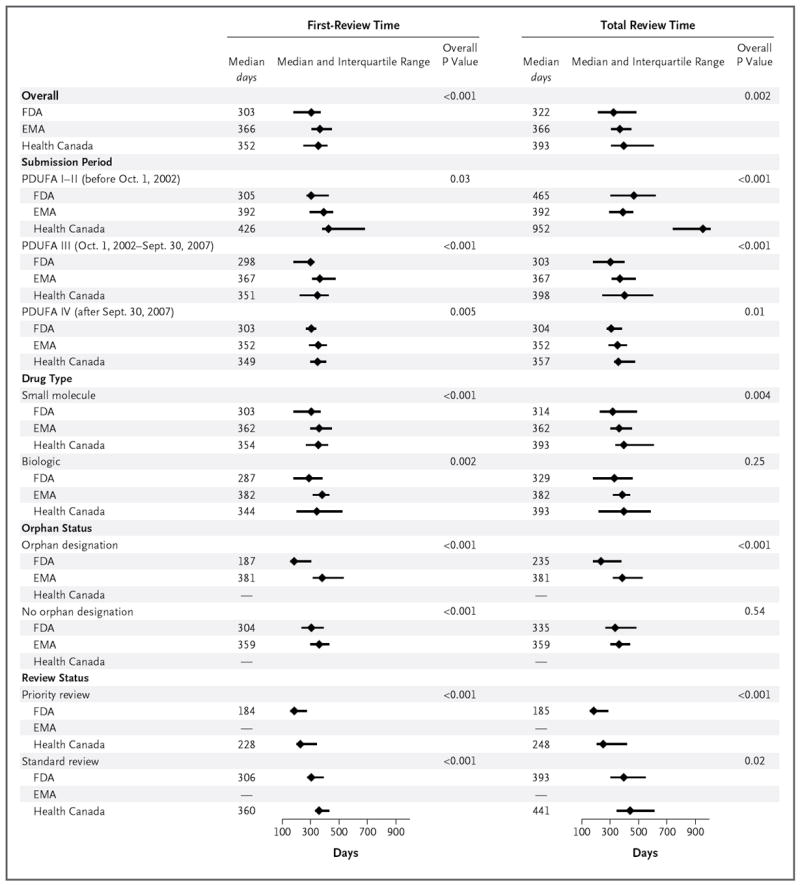
Information on application approvals by Health Canada is available only from 2005 on. Status with respect to priority or standard review was not analyzed for the EMA because the EMA Accelerated Assessment procedure, which is similar to priority review at the FDA, was not established until 2007. The overall P value is for the comparison across the three agencies. P<0.02 for all of the following comparisons: overall first-review time between the FDA and the EMA and between the FDA and Health Canada and overall total review time between the FDA and Health Canada; total review time during PDUFA I–II between the FDA and Health Canada and between the EMA and Health Canada; first-review and total review times during PDUFA III between the FDA and the EMA and between the FDA and Health Canada; first-review time during PDUFA IV between the FDA and the EMA and between the FDA and Health Canada and total review time during PDUFA IV between the FDA and Health Canada; and first-review time for small-molecule drugs between the FDA and the EMA and between the FDA and Health Canada, total review time for small-molecule drugs between the FDA and Health Canada, and first-review time for biologic agents between the FDA and the EMA.
In addition, in the case of the FDA and the EMA, the median length of time for the first review was significantly shorter for applications approved after one review cycle than for applications requiring multiple review cycles: 278 days (interquartile range, 183 to 307) as compared with 320 days (interquartile range, 301 to 456) for applications approved by the FDA (P<0.001) and 359 days (interquartile range, 304 to 435) as compared with 503 days (interquartile range, 478 to 566) for applications approved by the EMA (P = 0.002). However, there was no corresponding significant difference with respect to applications approved by Health Canada: 351 days (interquartile range, 243 to 424) for applications approved after one review cycle as compared with 356 days (interquartile range, 297 to 420) for applications requiring multiple review cycles (P = 0.82).
Among 72 therapeutics approved by all three regulatory agencies, the median length of time for the first review was approximately 100 days shorter at the FDA than at the other two agencies: 254 days (interquartile range, 182 to 307) at the FDA, as compared with 356 days (interquartile range, 302 to 410) at the EMA and 346 days (interquartile range, 228 to 424) at Health Canada (P<0.001). This finding was consistent among subsamples of therapeutics approved by two of three regulatory agencies (Table S4 in the Supplementary Appendix). Finally, the median length of time for the first review by the FDA was shortest for applications submitted during the PDUFA III period (P = 0.004), but there was no significant difference in the length of time for the first review by the EMA or by Health Canada according to the PDUFA period in which the application was submitted (P = 0.11 for both comparisons).
TOTAL REVIEW TIME
Similar to our results for first-review time, the median total review time differed among the three agencies, with the FDA reviews completed 44 to 71 days more quickly than those completed by the other two agencies (P = 0.002). The median total review time at the FDA was 322 days (interquartile range, 218 to 484), as compared with 366 days (interquartile range, 310 to 447) at the EMA and 393 days (interquartile range, 310 to 603) at Health Canada. Again, the shorter total review time at the FDA as compared with the EMA and Health Canada was consistent even after stratification according to most characteristics (Fig. 2, and Table S3 in the Supplementary Appendix).
Among 72 products approved by all three regulatory agencies, the median total review time was approximately 90 to 100 days shorter at the FDA than at the other two agencies: 268 days (interquartile range, 182 to 384) at the FDA, as compared with 356 days (interquartile range, 302 to 419) at the EMA and 366 days (interquartile range, 255 to 588) at Health Canada (P<0.001). This finding was also consistent among subsamples of therapeutics approved by two of three regulatory agencies (Table S4 in the Supplementary Appendix). Finally, at both the FDA and Health Canada, the median total review time was shortest for applications submitted during the PDUFA III and PDUFA IV periods (P<0.001 for the comparison among PDUFA periods for each agency), but there was no significant difference in total review times at the EMA according to the PDUFA period in which the application was submitted (P = 0.10).
FIRST TO MARKET
Among 289 unique novel therapeutic agents, 190 were approved in both the United States and Europe (either by the EMA or through the mutual recognition process), 154 in both the United States and Canada, and 137 in both Europe and Canada (Fig. 3). Among those approved in both the United States and Europe, 121 (63.7%) were first approved in the United States, with the drugs available a median of 96 days (interquartile range, −205 to 410) earlier in the United States. Similarly, among those approved in the United States and Canada, 132 (85.7%) were first approved in the United States, with the drugs available a median of 355 days (interquartile range, 125 to 786) earlier in the United States.
Figure 3. Geographic Areas in Which Novel Therapeutics Approved in Multiple Markets Were First Approved for Use.
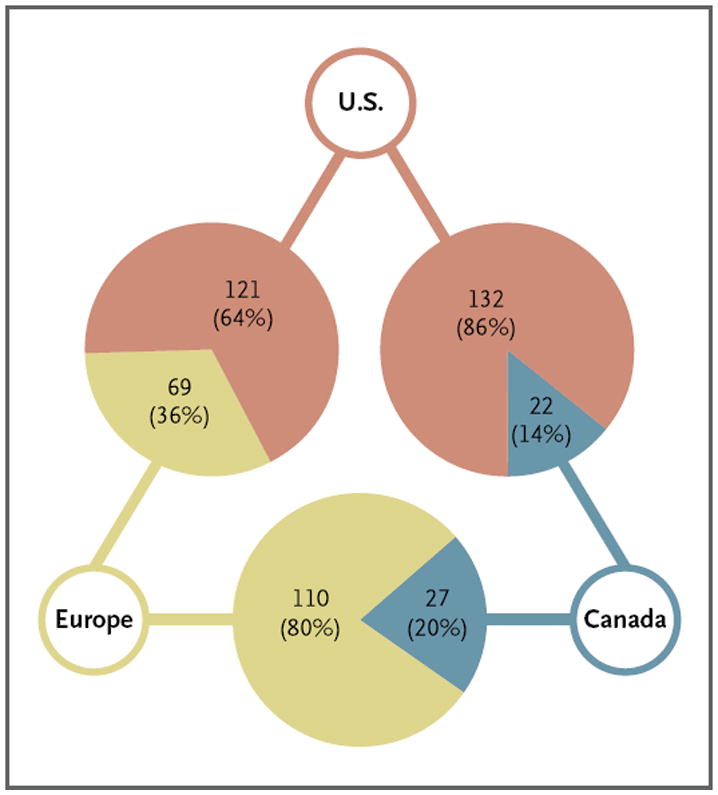
The data for Europe include both drugs approved by the EMA and those approved through the mutual recognition process.
DISCUSSION
Our analysis of novel therapeutics approved between 2001 and 2010 shows that the FDA has provided more rapid reviews of applications involving novel therapeutics than have the EMA and Health Canada and that the vast majority of the novel therapeutics first received approval for use in the United States. Furthermore, both firstreview and total review times at the FDA were essentially the same for applications submitted during the PDUFA III and PDUFA IV periods. These findings contradict recent criticisms of the speed of review by the FDA and question whether review speed is justified as an emphasis for PDUFA V, particularly since the FDA continues to outpace its European and Canadian peers.
Our findings were consistent across analyses of both first-review and total review times. The FDA completed its first review 49 to 63 days, or approximately 15%, faster than did the EMA and Health Canada and completed its total review 44 to 71 days faster. The FDA total review time was faster despite the fact that the EMA nearly always approved applications after one review cycle. Most important, among a subsample of therapeutics approved by all three regulatory agencies, the total length of time for FDA review, including the total time for applications involving multiple review cycles, was approximately 90 to 100 days shorter than the total review time at the EMA and Health Canada. Furthermore, our estimates of EMA review time are conservative, since we used the date the initial recommendation was issued, rather than the date that marketing of the drug was authorized by the European Commission, for the date of completion of the review.
Regulatory agencies have dual, and sometimes opposing, responsibilities to ensure patient safety while providing timely access to promising new medicines.21,22 Although some patient advocacy groups and some pharmaceutical and biotechnology companies consider timely access to new medications to be the priority,8,9 it is difficult to identify the “right” speed with which to approve novel therapeutic applications. Contrasting the review time at the FDA against the review time at peer regulatory agencies serving similar populations suggests that the FDA reviews applications for novel therapeutics more quickly, but that does not imply that further improvements are not attainable. Our study reflects only the speed of the regulatory review process and does not attempt to compare the quality of regulatory decisions among the agencies with respect to safety or effectiveness. Future analyses should compare markers of regulatory safety, such as label changes, drug withdrawals, or black-box warnings, among the agencies.
There are other limitations of this study. First, we examined only applications that were approved, because information about unapproved applications is not publicly available. Because more than 80% of applications submitted to the FDA are eventually approved,23 the effect of unapproved applications on review time is likely to be minimal. Second, we focused on novel therapeutics — namely, new molecular entities and original biologic agents — which represent a fraction of the medical products that are reviewed by regulatory agencies. Future research should examine the other review responsibilities of the FDA, such as the review of applications for reformulated drugs and combination therapies, generic drugs, and medical devices, since serious concerns have been raised, in particular, about the approval processes for generic drugs and medical devices.24-26 Nevertheless, although we were unable to assess the novelty of the approved therapeutics, it is unlikely that differences in therapeutic novelty across the three agencies accounted for our findings. Analyses were consistent among subsamples of applications approved by either two of three or all three regulatory agencies; within these subsamples, the degree of innovation of the therapeutics was consistent among the agencies. Finally, we examined only applications approved between 2001 and 2010. Estimates of the speed of the review process may be biased by the inclusion of applications approved early in our sample period that had been under review for a lengthy period of time, as well as by applications approved late in our sample period that were under review for only a brief period of time.
In conclusion, we found that among novel therapeutics approved between 2001 and 2010, the FDA reviewed applications more quickly, on average, than did the EMA and Health Canada, and the vast majority of these novel therapeutics were first approved for use in the United States. Our findings contradict recent criticisms of the speed of review by the FDA and lead to questions about whether the speed of the review process is justified as an emphasis for PDUFA V, particularly since the FDA continues to outpace its European and Canadian peers.
Supplementary Material
Acknowledgments
Supported by the Pew Charitable Trusts. Dr. Krumholz is supported by a Cardiovascular Outcomes Center Award (1U01HL105270-02) from the National Heart, Lung, and Blood Institute. Dr. Ross is supported by a grant (K08 AG032886) from the National Institute on Aging and by the American Federation for Aging Research through the Paul B. Beeson Career Development Award Program.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Food and Drug Administration. Prescription Drug User Fee Act (PDUFA) http://www.fda.gov/ForIndustry/UserFees/PrescriptionDrugUserFee/default.htm.
- 2.Philipson T, Berndt E, Gottschalk A, Strobeck M. Assessing the safety and efficacy of the FDA: the case of the Prescription Drug User Fee Act. Cambridge, MA: National Bureau of Economic Research; 2005. [Google Scholar]
- 3.Effect of user fees on drug approval times, withdrawals, and other agency activities (GAO-02-958) Washington, DC: General Accounting Office; Sep, 2002. www.gao.gov/new.items/d02958.pdf. [Google Scholar]
- 4.Carpenter D, Zucker EJ, Avorn J. Drug-review deadlines and safety problems. N Engl J Med. 2008;358:1354–61. doi: 10.1056/NEJMsa0706341. [DOI] [PubMed] [Google Scholar]
- 5.Carpenter D, Chattopadhyay J, Moffitt S, Nall C. The complications of controlling agency time discretion: FDA review deadlines and postmarket drug safety. Am J Pol Sci. 2012;56:98–114. doi: 10.1111/j.1540-5907.2011.00544.x. [DOI] [PubMed] [Google Scholar]
- 6.Food and Drug Administration. Prescription Drug User Fee Act (PDUFA) reauthorization. http://www.accessdata.fda.gov/FDATrack/track-proj?program=cder&id=CDER-OPI-PDUFA-Reauthorization.
- 7.Idem. PDUFA reauthorization performance goals and procedures, fiscal years 2013 through 2017. http://www.fda.gov/downloads/forindustry/userfees/prescriptiondruguserfee/ucm270412.pdf.
- 8.Fox JL. Interest groups jostle to influence PDUFA V. Nat Biotechnol. 2011;29:1062. doi: 10.1038/nbt1211-1062. [DOI] [PubMed] [Google Scholar]
- 9.Kronquist AR. The Prescription Drug User Fee Act: history and reauthorization issues for 2012. Washington, DC: The Heritage Foundation; Dec 21, 2011. http://www.heritage.org/research/reports/2011/12/the-prescription-drug-user-fee-act-history-and-reauthorization-issues-for-2012. [Google Scholar]
- 10.Rawson NSB. Time required for approval of new drugs in Canada, Australia, Sweden, the United Kingdom and the United States in 1996-1998. CMAJ. 2000;162:501–4. [PMC free article] [PubMed] [Google Scholar]
- 11.Hirako M, McAuslane N, Salek S, Anderson C, Walker S. A comparison of the drug review process at five international regulatory agencies. Drug Inf J. 2007;41:291–308. [Google Scholar]
- 12.Faden LB, Kaitin KI. Assessing the performance of the EMEA’s centralized procedure: a comparative analysis with the US FDA. Drug Inf J. 2007;42:45–56. [Google Scholar]
- 13.Roberts SA, Allen JD, Sigal EV. Despite criticism of the FDA review process, new cancer drugs reach patients sooner in the United States than in Europe. Health Aff (Millwood) 2011;30:1375–81. doi: 10.1377/hlthaff.2011.0231. [DOI] [PubMed] [Google Scholar]
- 14.Food and Drug Administration. Drugs@FDA: FDA approved drug products. http://www.accessdata.fda.gov/scripts/cder/drugsatfda.
- 15.European Medicines Agency. European public assessment reports. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/epar_search.jsp.
- 16.Idem. Applying for EU marketing authorisation: for medicinal products for human use. http://www.ema.europa.eu/docs/en_GB/document_library/Brochure/2011/03/WC500104233.pdf.
- 17.Health Canada. Drug Product Database (DPD) http://www.hc-sc.gc.ca/dhp-mps/prodpharma/databasdon/index-eng.php.
- 18.Idem. Summary Basis of Decision (SBD) documents: drugs. http://www.hc-sc.gc.ca/dhp-mps/prodpharma/sbd-smd/drug-med/index-eng.php.
- 19.Heads of Medicines Agencies. Human mutual recognition product index. http://www.hma.eu/mri.html.
- 20.WHO Collaborating Center for Drug Statistics Methodology. ATC/DDD index 2012. http://www.whocc.no/atc_ddd_index.
- 21.Eichler H-G, Pignatti F, Leufkens H, Breckenridge A. Balancing early market access to new drugs with the need for benefit/risk data: a mounting dilemma. Nat Rev Drug Discov. 2008;7:818–26. doi: 10.1038/nrd2664. [DOI] [PubMed] [Google Scholar]
- 22.Institute of Medicine. The future of drug safety: promoting and protecting the health of the public. Washington, DC: National Academy Press; 2006. [Google Scholar]
- 23.Food and Drug Administration. FY 2010 PDUFA performance report. http://www.fda.gov/AboutFDA/ReportsManualsForms/Reports/UserFeeReports/PerformanceReports/PDUFA/ucm243357.htm.
- 24.Generic Pharmaceutical Association. GPhA says cuts to FDA generic drug program would have ‘devastating impact’ on consumers and government. 2011 Mar 21; http://www.gphaonline.org/media/press-releases/2011/gpha-says-cuts-fda-generic-drug-program-would-have-%E2%80%98devastating-impact%E2%80%99-co.
- 25.Suter LG, Paltiel AD, Rome BN, et al. Medical device innovation — is “better” good enough? N Engl J Med. 2011;365:1464–6. doi: 10.1056/NEJMp1109285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Challoner DR, Vodra WW. Medical devices and health — creating a new regulatory framework for moderate-risk devices. N Engl J Med. 2011;365:977–9. doi: 10.1056/NEJMp1109150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.