

Abstract
The peroxisome proliferator-activated receptor-gamma (PPARγ) is a member of the hormone-activated nuclear receptor superfamily. PPARγ can be activated by a diverse group of agents, such as endogenous polyunsaturated fatty acids, 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), and thiazolidinedione (TZD) drugs. PPARγ induces antiproliferative, antiangiogenic, and prodifferentiation pathways in several tissue types, thus making it a highly useful target for downregulation of carcinogenesis. These TZD-derived novel therapeutic agents, alone or in combination with other anticancer drugs, have translational relevance in fostering effective strategies for cancer treatment. TZDs have been proven for antitumor activity in a wide variety of experimental cancer models, both in vitro and in vivo, by affecting the cell cycle, inducing cell differentiation and apoptosis, as well as by inhibiting tumor angiogenesis. Angiogenesis inhibition mechanisms of TZDs include direct inhibition of endothelial cell proliferation and migration, as well as reduction in tumor cell vascular endothelial growth factor production. In prostate cancer, PPARγ ligands such as troglitazone and 15d-PGJ2 have also shown to inhibit tumor growth. This paper will focus on current discoveries in PPARγ activation, targeting prostate carcinogenesis as well as the role of PPARγ as a possible anticancer therapeutic option. Here, we review PPARγ as an antitumor agent and summarize the antineoplastic effects of PPARγ agonists in prostate cancer.
1. Introduction
Prostate cancer, an androgen-dependent disease, is one of the leading causes of cancers among men worldwide [1]. While the majority of men are diagnosed at early stages of the disease, a subset develop recurrent and eventually metastatic form of the disease [2]. Conventional chemopreventive measures such as surgical resection or radiotherapy are potentially curative for localized disease; however, it has shown to be of limited effectiveness [3]. On the other hand, advanced prostate cancer is associated with a poor prognosis. Tumor growth is originally androgen dependent. Androgens exert their effects through activation of the androgen receptor (AR), a member of the hormone nuclear receptor superfamily. In the mature prostatic gland, the AR regulates the expression of genes involved in cell division and proliferation of the epithelial cells [4]. For more than 60 years ago, androgen deprivation was established as a form of treatment for advanced incurable prostate cancer. Androgen ablation is a hormonal deprivation therapy where the circulating levels of androgen in the body are reduced [3]. The blocking of androgen stimulation often leads to either a partial or full remission; however, subsequent relapse often occurs and the disease reemerges within a few years in a poorly differentiated, androgen-independent form. Furthermore, androgen ablation therapy results in the development of more aggressive forms of prostate cancer which are independent of androgens for growth [3]. The response rates for the treatment are initially high (70–80%); however, almost all patients relapse and develop hormone-refractory prostate cancer (HRPC), resulting in increased morbidity and death [2]. Therefore, there is currently no effective treatment for such androgen-independent forms of prostate cancer. As a result, there is a great interest in identifying more effective treatment options for prostate cancer and alternative therapeutic strategies [3]. Recent studies have shown participation of the nuclear hormone receptor PPARγ in pathophysiology of prostate cancer and its potential in the development of improved anticancer strategies.
Peroxisome proliferator-activated receptor (PPAR) belongs to the nuclear hormone receptor superfamily of transcription factors that includes 48 human transcription factors whose activity is regulated by direct binding of steroid and thyroid hormones, vitamins, lipid metabolites, and xenobiotics [5, 6]. PPARs function as transactivation factors that heterodimerize with retinoid X receptors (RXRs) upon activation and bind to specific response elements (PPREs) in the target DNA of various target genes [7–10]. PPRE consists of direct repeat (DR) hexameric sequences (AGGTCA), separated by one or two nucleotides (DR-1 and DR-2 element) [11]. Distinct areas such as the DNA binding and the ligand-binding transactivation domains have been identified, and these domains influence the transduction of the PPAR-induced response.
PPARs have a subfamily of three different isoforms: PPARα, PPARβ/δ, and PPARγ. In particular, PPARγ plays an important role in the regulation of lipid homeostasis, adipogenesis, insulin resistance, and in the development of various organs. Apart from the established metabolic actions, PPARγ has also been shown to be overexpressed in several types of human cancers, including breast, colon, bladder, and prostate cancer. It was also suggested to induce apoptosis in several malignant cell lineages [12]. In addition, loss-of-function mutations of PPARγ have been found in some human colon and thyroid carcinomas [13]. In vitro and in vivo studies have demonstrated antiproliferative and proapoptotic actions of PPARγ agonists such as 15d-PGJ2 and thiazolidinediones (TZDs) thus suggesting that PPARγ could be a promising therapeutical target for the treatment of cancer [11, 14]. Binding of agonist ligands to PPARγ triggers a conformational change that attracts transcriptional coactivators, including members of the steroid receptor coactivator (SRC) family [5, 15]. Once activated, PPARγ heterodimerizes with retinoid X receptor and signal antiproliferative, antiangiogenic, and prodifferentiation pathways in several tissue types, thus making it a highly useful target for prevention and reduction of carcinogenesis (Figure 1). The synthetic PPARγ ligands, which have been used earlier for the treatment of insulin resistance in type II diabetes mellitus, have been shown to be potential candidates as drugs not only for prevention but also for treatment of prostate cancer [16].
Figure 1.
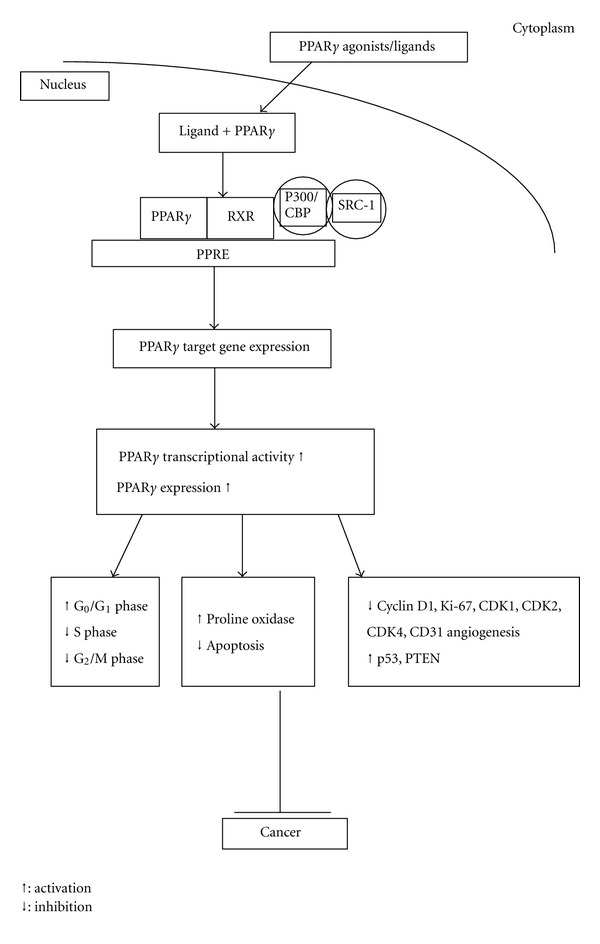
PPARγ activity. Upon ligand binding PPARγ, binds to RXR in the nucleus and associates with coactivators to bind PPRE located on target genes that control various activities.
The peroxisome proliferator-activated receptor-gamma has been the focus of intense research during the past decade because ligands for this receptor have emerged as potent insulin sensitizers used in the treatment of type II diabetes. Increased levels of circulating free fatty acids and lipid accumulation in nonadipose tissue have been implicated in the development of insulin resistance. This situation is improved by PPARγ ligands, which promote fatty acid storage in fat depots and regulate the expression of adipocyte-secreted hormones that impact on glucose homeostasis. Insulin resistance is a major defect in type II diabetes mellitus. Synthetic PPARγ agonists, the thiazolidinediones (TZDs), are used in type II diabetes therapy as insulin sensitizers [17] to overcome insulin resistance in target tissues. PPARγ acts as a transcriptional activator of many adipocyte-specific genes involved in lipid synthesis, handling and storage of lipids, growth regulation, insulin signaling, and adipokine production. Thus, PPARγ plays an important role as a major regulator in the differentiation of adipocytes. Thiazolidinediones are characterized by their ability to decrease insulin resistance by specifically targeting at PPARγ to mediate its activity and have been suggested to slow down the progression of insulin resistance [18].
Adipocyte differentiation is a highly regulated process taking place from birth throughout adult. Adipose tissue is composed of adipocytes, which store energy in the form of triglycerides and release it as free fatty acids [19]. Together with muscle, adipose tissue is the major regulator of energy balance of the body. Excessive accumulation of adipose tissue leads to obesity, whereas its absence is associated with lipodystrophic syndromes. PPARγ is highly expressed in the adipose tissue and is required for its development. During adipocyte differentiation, which ensues from PPARγ activation, expression of numerous genes specific for fatty acid metabolism is induced. In fact, functional PPREs have been identified in several genes implicated in adipocyte differentiation, with most of them being involved in lipid storage and control of metabolism. Cell proliferation and differentiation are considered to be mutually exclusive events. However, a close relationship has been established between both cell processes during the adipocyte differentiation program. One of the early events occurring during adipogenesis is the reentry into cell cycle of growth-arrested preadipocytes following hormonal induction. After several rounds of clonal expansion, cells arrest proliferation again and undergo terminal adipocyte differentiation. In the early stage of adipocyte differentiation, an increase in the E2F activity has been observed. E2Fs are transcription factors which regulate the expression of genes involved in DNA synthesis [20]. Consequently, expression of these genes, such as cyclin D1, c-Myc, or cyclin E, is increased in the early stages of adipogenesis [21].
It should be noted that neovascularization and adipogenesis are temporally and spatially coupled processes during prenatal life and they continue to reciprocally interact via paracrine signaling systems throughout adult life [22]. Moreover, Biscetti et al. showed that the activation of PPARα and PPARγ leads to endothelial tube formation in an endothelial/interstitial cell coculture assay. This effect was associated with increased production of the angiogenic cytokine vascular endothelial growth factor (VEGF). Neovascularization also occurred in vivo, when PPARα and PPARγ agonists were used in the murine corneal angiogenic model. It was concluded that PPARα- and PPARγ-induced angiogenesis is associated with local VEGF production. These findings demonstrate that PPARα and PPARγ activation stimulates neoangiogenesis through a VEGF-dependent mechanism. Neoangiogenesis is a crucial pathological event in type II diabetes. The ability of PPARα and PPARγ agonists to induce neoangiogenesis therefore has important implications for the clinical and therapeutic management of type II diabetes [23]. Thus, it is evident that PPARγ might contribute to protumorigenic process by inducing angiogenesis during the therapy for type II diabetes. However, in this paper we will focus on the anticancer role of PPARγ in aspect of the prevention of prostate cancer.
PPARγ activation has been implicated to inhibit the proliferation of malignant cells from different lineages such as liposarcoma [24], breast adenocarcinoma [12], prostate carcinoma [25], colorectal carcinoma [26], non-small-cell lung carcinoma [27], pancreatic carcinoma [28], bladder cancer cells [29], and gastric carcinoma cells [30]. Several clinical trials have been initiated that incorporate TZDs for prevention of head and neck cancer or lung cancer. One phase II trial studying the effectiveness of pioglitazone in preventing head and neck cancer in individuals with oral leukoplakia showed that 71% of individuals treated with pioglitazone had complete or partial response, 10% had stable disease, and 19% had progressive disease. Similarly, another clinical trial evaluating the chemopreventive effect of pioglitazone in subjects at risk for lung cancer is currently recruiting participants [31]. Although PPARγ activators have been proven to contribute to anticancer actions during many in vitro studies, their advancement into human cancer clinical trials has met with limited success. We will provide an overview of the current findings on PPARγ activation and the targeting in prostate carcinogenesis prevention with the respect of applying PPARγ activators as cancer chemoprevention strategies [32]. There is a demand for safe agents that target high-risk conditions such as preexisting intraepithelial neoplasia, a high-risk prostate cancer precursor. PPARγ-targeted strategies may help to fulfill this demand. As a consequence, PPARγ may be considered an important molecular target for anticancer drug development.
2. PPARγ Ligands (Synthetic and Natural)
There are a variety of endogenous ligands for the PPARγ such as long-chain polyunsaturated fatty acids, arachidonic acid metabolites derived from the cyclooxygenase and lipoxygenase pathways, and fatty acid derived components of oxidized low density lipoproteins (OxLDL) (e.g., 9-hydroxyoctadecadienoic acid and 13-hydroxyoctadecadienoic acid) [33, 34]. The antidiabetic thiazolidinedione (TZD) class of drugs including troglitazone (TGZ), rosiglitazone (BRL49653), pioglitazone, and ciglitazone are synthetic ligands of PPARγ. In addition, nonthiazolidinedione derivatives, such as 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO), CDDO-imidazolide (CDDO-Im), GW-7845, JTT-501, KRP-297, L-764406, MCC-555, GW-0072, and GW-0207 are also synthetic ligands of PPARγ [33]. Most TZDs are selective for PPARγ over the PPARα and PPARβ/δ subtypes, but there are some exceptions. Thiazolidinedione KRP-297 is a potent PPARγ agonist and a weak PPARα 127 agonist [35]. The PPARγ LBD consists of 13 α-helices and a 4-strand β-sheet and provides a large binding pocket (1300 Å) that allows access to a number of structurally diverse ligands [36]. The processes of PPARγ activation include interaction with a heat-shock protein as well as cellular signaling that alters the phosphorylation status of PPARγ and the interaction of ligands of both pharmacological and physiological origin. The activation by ligands is the dominant active pathway. After activation, PPARγ forms heterodimer with the RXR and then binds to specific recognition sites, the peroxisome proliferator response elements (PPREs) in the target gene, and regulates transcription of specific genes [34]. Other synthetic compounds that function as ligands include nonsteroidal anti-inflammatory drugs (NSAIDs), such as indomethacin, ibuprofen, flufenamic acid, and fenoprofen. Moreover, there are naturally occurring molecules that function as the endogenous ligands for PPARγ. Among these, the cyclopentenone prostaglandin 15d-PGJ2 was found to be the most potent [37]. The natural PPARγ ligand, 15d-PGJ2, activates PPARγ at micromolar concentrations in humans in vivo [38]. It has been shown that 15d-PGJ2 upregulates the expression, transcriptional activity, and DNA-binding activity of PPARγ.
In addition to synthetic ligands, some natural potent PPARγ agonists from several medicinal plants such as saurufuran A from Saururus chinensis (Saururaceae), flavonoids such as chrysin and kaempferol, and phenolic compounds from Glycyrrhiza uralensis (Fabaceae) have also been found [39, 40]. Studies have correlated the consumption of soy-rich diets with a decreased risk of developing hormone-dependent cancers, including prostate cancer [41, 42]. Genistein is a prostate cancer preventive phytochemical found at high levels in soybean and soy foods. To better understand the molecular mechanisms underlying the beneficial effects of genistein on prostate cancer prevention, a DNA microarray approach to examine the effects of genistein at concentrations in the physiologic range on global gene expression patterns in androgen-responsive cancer cells was used. Microarray analyses were performed on androgen-responsive LNCaP human prostate cancer cells exposed to 0, 1, 5, or 25 μM genistein. A concentration-dependent modulation of multiple cellular pathways that are important in prostate carcinogenesis was revealed. The androgen-receptor- (AR-) mediated pathways, in particular, appeared to be modulated by genistein at the lowest concentrations. Furthermore, Lee et al. conducted in silico screening for human peroxisome proliferator-activated receptor-gamma (hPPARγ) by performing an automated docking study with 450 flavonoids. 3,6-dihydroxyflavone increased the binding between PPARγ and steroid receptor coactivator-1 (SRC-1) by approximately 5-fold. The 6-hydroxy group of the A-ring of 3,6-dihydroxyflavone participated in hydrogen-bonding interactions with the side chain of Tyr327, His449, and Tyr473. The B-ring formed a hydrophobic interaction with Leu330, Leu333, Val339, Ile341, and Met364. Therefore, 3,6-dihydroxyflavone can be regarded as a potent agonist of hPPAR with cytotoxic effects on human prostate cancer cells [43].
Currently, because of the antitumor and antiangiogenic properties as well as the low toxicity profile, the TZD members have been tested in clinical trials for treatment of human cancers expressing high levels of PPARγ [44]. Evidence indicates that troglitazone and ciglitazone block BH3 domain-mediated interactions between the antiapoptotic Bcl-2 (B-cell leukemia/lymphoma 2) members. Moreover, these TZDs facilitate the degradation of cyclin D1 and caspase-8-related FADD-like IL-l-converting enzyme (FLICE) inhibitory protein through proteasome-mediated proteolysis and downregulate the gene expression of prostate-specific antigen gene expression by inhibiting androgen activation of the androgen response elements in the promoter region [45]. Moreover, TZDs have been proposed in differentiation-mediated therapy of various human carcinomas associated with high levels of PPARγ. Although PPARγ is involved in differentiation and cellular metabolic changes after ligand binding, PPARγ-independent effects have also been described for PPARγ agonists. It has been shown that the naturally occurring PPARγ ligand 15d-PGJ2 inhibits the secretion of tumor necrosis factor-α (TNFα) and interleukin 6 in macrophages stimulated by bacterial lipopolysaccharide and directly blocks activity of the IκB kinase complex in a PPARγ-independent way through inhibition of IκB kinases. Furthermore, it has also been demonstrated that PPARγ ligand troglitazone inhibits cholesterol biosynthesis independently of PPARγ [46]. TZDs have been reported to exert receptor-dependent as well as receptor-independent effects [47]. TZDs can exert receptor-independent effects on several criteria, including that (1) concentrations needed to observe TZD actions were much greater than reported EC50 values; (2) the rank order of efficacy for TZDs (troglitazone > pioglitazone > rosiglitazone) to elicit a response was inverse to their known binding affinities (measured EC50 values) for PPARγ; (3) effects occurred in the absence of PPARγ expression or of PPAR DNA binding elements (PPRE) in gene promoters. Examples of the receptor-independent effects reported include suppression of inflammatory gene expression, modification of energy and fuel metabolism, suppression of cell proliferation and induction of cytotoxicity, and perturbation of mitochondrial function. Receptor-independent effects of TZDs were also reported in human leukemia (HL) cell lines. In HL-60 cells [48], troglitazone induced cell arrest and subsequent cell death and was associated with downregulation of c-myc, c-myb, and cyclin D2 expression. Since these genes lack a PPRE in their promoter regions, the effect could not directly be PPARγ mediated. Similar findings in a human basophilic leukemia cell line [49] suggested that troglitazone suppressed cell growth independently of PPARγ via a decrease in cyclin E levels and hyperphosphorylation of retinoblastoma tumor suppressor gene product. Moreover, receptor-knockout studies indicate direct evidence that the cytotoxicity induced by TZDs is independent of PPARγ. In PPARγ (−/−) and PPARγ (+/+), mouse embryonic stem cells inhibition of tumor growth by two TZDs, troglitazone and ciglitazone, was independent of PPARγ activity [50]. These compounds blocked the G1/S transition by inhibiting translation initiation as a consequence of partial depletion of intracellular calcium stores and resulting activation of PKR, a kinase that phosphorylates the alpha subunit of eukaryotic initiation factor 2, thus rendering it inactive [51].
Xin et al. first showed that TZDs were potent inhibitors of angiogenesis in vitro [52]. The findings indicated that PPARγ is expressed in endothelial cells and its ligands can inhibit endothelial cell proliferation induced by growth factors or cause their apoptosis in vitro [52–54]. Furthermore, TZDs inhibit vascular endothelial growth factor (VEGF) and leptin-induced endothelial cell migration [55]. However, clinical studies suggest that TZDs are largely ineffective as monotherapeutic agents in treating prostate carcinoma [56]. Because cancer cells modify several transduction pathways to achieve continuous progression and survival [57], application of multiple drug-strategies appears important to achieve effective treatment. Such a strategy grants synergistic antiproliferative effects and/or permit the use of lower drug doses that might otherwise be less effective when used as monotherapy [58].
In addition, activation of PPARγ by pioglitazone caused arrest of exponential growth of fibroblasts and SV40 large T-antigen transformed adipocytes [59]. Examination of the properties of PPARγ in adipocytes suggests that it may be possible to selectively modulate PPARγ activity in an analogous fashion [5]. For example, PPARγ is required for the expression of the adipocyte-specific fatty-acid binding protein aP2 [60]. In mature adipocytes, even in the absence of a pharmaceutical ligand, PPARγ binds to the aP2 promoter along with coactivator proteins [61].
It has been reported that binding to helix 12 (H12) of the ligand-binding domain of PPARγ is required for full agonist activity [62]. Previously, the degree of stabilization of the activation function 2 (AF-2) surfaces was thought to correlate with the degree of agonism and transactivation. However, it was observed that the structurally similar PPARγ TZD full agonists rosiglitazone (Avandia) and pioglitazone (Actos) had different clinical adverse events. This indicates that subtle changes in ligand receptor interaction can lead to different pharmacological responses to these agents. As a result, the emphasis has shifted to the development of “selective PPARγ modulators” or SPPARMs. SPPARMs are PPARγ modulators that exhibit potent insulin sensitization activity but are antiadipogenic in animal models of type II diabetes [63–65]. Partial agonists displayed reduced transcriptional activity in reporter assays, and, in animal models of type II diabetes, they demonstrated the SPPARM phenotype. Selective recruitment of transcriptional coactivators has been implicated in partial agonist and SPPARM phenotype. The binding of agonist to the receptor's LBD induces structural changes that facilitate dissociation of repressor molecules (e.g., NCoR and SMRT) and association of activator proteins known to be coactivators to the receptor [66, 67]. These transcriptional coactivators bind to the receptor complex, modify local chromatin structure, and recruit the transcription machinery to target gene promoters [68]. Partial agonists have been shown to have decreased recruitment of CBP (CREB-binding protein) and SRC1 (steroid receptor coactivator-1) coactivators but retain association with PGC1α (PPARgamma coactivator-1alpha) [69]. The structure of a partial-agonist-bound PPARγ showed no direct interactions between ligand and H12 [70], supporting the idea that this structural feature is key to maximal transactivation potency of PPARγ. These studies implicated that the degree of H12 stabilization is proportional to the degree of agonism and transcriptional output for full agonists [62].
In prostate cancer, some investigators reported that Telmisartan, an angiotensin II receptor, induced early apoptosis and DNA fragmentation with treatment of 100 μM Telmisartan. These findings indicate that Telmisartan, which is also a selective PPARγ modulator, may mediate potent antiproliferative effects against prostate cancer cells through PPARγ pathway. Thus, Telmisartan could be a potent target for prevention and treatment in prostate cancer [71], while the available data has clearly suggested that PPARγ ligands exhibit potent antiproliferative actions on a wide variety of neoplastic cells. Thus, partial agonists can selectively modulate PPARγ activity by creating interfaces that affect coactivator binding qualitatively as well as quantitatively. In addition, as the corepressors and coactivators interface with overlapping surfaces on the receptor [72], partial agonism may also result from ligand inducing an intermediate conformation that is recognized by both classes of cofactors. Moreover, partial agonists may have full transrepression activity [73], thereby providing a distinct mechanism of selective modulation of PPARγ activity.
Furthermore, MCC-555, a unique partial agonist of PPARγ as an antidiabetic drug, inhibited the growth of prostate cancer cells both in vitro and in vivo [74–76]. Several previous reports indicate that, for some types of the PPARγ ligands, their anticancer effects might be independent of PPARγ activation. For example, 15d-PGJ2 exerts inhibitory effect against cancer cell proliferation both with and without subjecting to PPARγ activation. In the PPARγ-indpendent mechanism, repression activity of 15d-PGJ2 on NFκB-related gene expression was reported, and the action was through covalent modification of critical cysteine residues in IκB kinase and thus preventing the nuclear translocation of NFκB [77]. In addition, some studies revealed that PPARγ classical agonists TZDs (e.g., troglitazone) exhibit anticancer effects via a PPARγ-independent pathway and some non-PPARγ targets such as extracellular signal-regulated kinases, c-Jun N-terminal protein kinase, p38, and Bcl-2 members have been implicated [45, 78].
In vitro studies using various solid and hematological tumor cell lines showed that RWJ-241947 had antiproliferative activity against prostate cancer cells, with the strongest effect against the androgen-independent PC-3 prostate cancer cells. RWJ-241947 belongs to the TZD family, and it is established as an antidiabetic drug in animal models of type II diabetes. Like other TZDs, RWJ-241947 binds to PPARγ and exerts transcriptional activities. However, its binding affinity for PPARγ is less than 10% of that of rosiglitazone. Its transcriptional properties are unique because it can function as a full or partial agonist or antagonist, depending on the cell type or DNA-binding site. RWJ-241947 increased expression of cyclin-dependent kinase inhibitor p21WAF1, deceased cyclin E, and induced apoptosis in PC-3 cells. On the other hand, RWJ-241947 increased E-cadherin and lowered protein expression of prostate-specific antigen without downregulating the androgen receptor in androgen-dependent LNCaP prostate cancer cells. Reporter gene assays showed that this PPARγ ligand inhibited androgen activation of the androgen receptor response elements of the prostate-specific antigen gene. In vivo treatment of male beige/nude/X-linked immunodeficient (BNX) mice with RWJ-241947 profoundly suppressed growth of PC-3 prostate cancer xenografts with prominent apoptosis, as well as fibrosis, including inflammatory and giant cell reaction in the remaining tumor tissue [76].
3. Effect of PPARγ Ligands on the Proliferation of Prostate Cancer Cells
Inhibition of cell proliferation can occur through regulations in cell cycle and/or apoptosis [3]. c-Myc, a protooncogene product, plays an important role in cell cycle progression, and apoptosis. The c-Myc protein is a basic/helix-loop-helix/leucine zipper transcription factor. It dimerizes with Max and binds specific E-box sequences within DNA and regulates gene transcription. Furthermore, c-Myc regulates expressions of several gene products that are involved in cell proliferation and apoptosis. Expressions of the cyclin dependent kinase (CDK) inhibitors p21 and p27 were downregulated by c-Myc [79, 80]. CDK inhibitors block progression of the cell cycle by inactivating the formation of cyclin/CDK complexes, which are crucial for phosphorylation of retinoblastoma protein when complexed with E2F. Two proteins which promote cell proliferation, cyclin-dependent kinase 4 (CDK4), and the phosphatase CDC25A are also positively regulated by c-Myc [80, 81]. In addition, c-Myc also regulates expressions of proteins that control apoptosis. Expression of the proapoptotic proteins BAD in the rat frontal cortex and BAX in glioblastomas has been shown to be regulated by c-Myc [82]. Akinyeke and Stewart showed that troglitazone not only suppresses prostate cancer cell growth but also decreases c-Myc protein expression [3]. These results suggest that inhibition of c-Myc expression through activation of PPARγ promotes prostate cancer cells to restore characteristics of normal prostate cells phenotype.
PPARγ agonists upregulate CDK inhibitors therefore inducing arrest of the cell cycle. Arrest in G1 phase through PPARγ activation has been described in different tumor cell lines [7]. Alterations in p21 and cyclin D1 expression can reduce the phosphorylation of retinoblastoma (Rb) protein, resulting in G1 cell cycle arrest. Troglitazone (TGZ) showed a dose- and time-dependent inhibition of the PC-3 cells as examined by MTT assays. In addition, TGZ produced a dose-dependent cell cycle arrest in G0/G1 of PC-3 cells lines by increasing the distribution of PC-3 cells into the G0/G1 and sub-G1 phase [83]. These discoveries demonstrated that the PPARγ-induced growth inhibition was linked to the G1 phase cell cycle arrest through the upexpression of the cyclin-dependent kinase inhibitors p21 and p27 and/or repression of cyclin D1 expression.
Furthermore, to investigate the role of PPARγ in human prostate cancer cells, several ligands of different potency and selectivity were applied to the cell line DU145 and the cell viabilities were assessed after continuous treatment with the drugs. Troglitazone caused a suppression of the growth of these cells, in comparison with vehicle-treated cells. The decrease occurred in a time- and dose-dependent manner, with a 50% inhibition detected at 7 days, at 10 μM concentration [84]. In addition, Yoshimura et al. examined the effects of PPARγ ligands on cell proliferation in prostatic carcinoma (PC) cell lines and investigated the inhibitory effect of troglitazone and 15d-PGJ2 on PC-derived cell lines using MTT assay and Hoechst staining. These PPAR-γ ligands induced the reduction of cell viability with the half-maximal concentration of growth inhibition of PC cell lines. Furthermore, counting cells at days 1, 2, and 3 clearly showed marked inhibitory effects of PPARγ ligands on cell proliferation [85].
Campbell et al. examined the positive association between total fat intake and increased risk for prostate cancer in a case-control study with 175 cases and 233 controls (odds ratio = 1.8, 95% CI = 0.9–3.4) [86, 87]. This study investigated the likelihood that γ-tocopherol (GT) could induce growth arrest in PC-3 prostate cancer cells through the regulation of fatty acid metabolism. The findings indicated that GT treatment resulted in upregulation of the PPARγ protein expression at concentrations as low as 5 μM and continued to affect the expression of the PPARγ protein at 40 μM. Growth arrest (40%) and upregulations in PPARγ mRNA and protein expressions were achieved with exposure to GT within 6 h [88]. In addition, expressions of proteins downstream of the PPARγ pathway were also examined. Cyclin D1, cyclin D3, Bcl-2, and NFκB proteins were found to be downregulated following GT treatment. These data demonstrated that the growth arrest mediated by GT follows a PPARγ-dependent mechanism [89].
Additionally, GSK-3β expression and NFκB activity have important roles in prostate cancer development [90]. To investigate the mechanisms of the PPARγ agonist-induced prostate cancer cell growth inhibition, the authors examined the effect of troglitazone on the expression of PPARγ and GSK-3β, activity of NFκB, as well as on the prostate cancer cell growth. Troglitazone induced the expression of PPARγ in the nucleus of PC-3 cells, but not in LNCaP cells. Troglitazone (0–16 μM) inhibited cancer cell growth in both cells accompanied by the induction of cell cycle arrest in G0/G1 phase and an increased apoptotic cell death in concentration-dependent manner. Troglitazone inhibited the constitutive expression of GSK-3β and activation of NFκB. Cotreatment of troglitazone with a GSK-3β inhibitor (AR-a014418) or GSK-3β siRNA significantly augmented the inhibitory effect of troglitazone on the NFκB activity and on prostate cancer cell growth inhibition and apoptotic cell death. These results suggest that PPARγ agonist, troglitazone, inhibits prostate cancer cell growth through inactivation of NFκB via suppression of GSK-3β expression.
4. Effect of PPAR-γ Ligands on the Apoptosis of Prostate Cancer Cells
It has been shown that PPARγ agonists induce apoptosis. In glioma cells, Zander and coworkers [91] described an upregulation of the proapoptotic proteins BAX and BAD and a functional role of BAX upregulation for the induction of apoptotic cell death. Upregulated expressions of BAD and BAX cause apoptosis by the release of cytochrome C and subsequent activation of several effector caspases [92]. In line with this, PPARγ activation leads to increased caspase 3 activity [7]. Several studies have shown that PPARγ activation leads to inhibition of growth of prostate-cancer cell lines, which is accompanied by morphological changes such as prominent enlarged cytoplasmic vacuoles [7]. TZDs were suggested to exhibit anti-tumor apoptotic effects in human prostate carcinoma (PC) cell lines. Likewise, pharmacological inhibitors of fatty acid synthase (FASN), a metabolic enzyme highly expressed in PC, induce apoptosis in prostate and other cancer cells. A positive correlation between PPARγ and FASN protein in PC cell lines was established, and the synergism between TZDs and FASN blockers in PC cell viability reduction and apoptosis induction was demonstrated. It was concluded that combined treatment of TZDs and FASN has enhanced anti-tumor properties in both androgen-dependent LNCaP and androgen-independent PC-3 and DU-145 cells, when compared with single drug exposure [56].
It has also been shown that the tumor suppressor PTEN (phosphatase and tensin homolog at chromosome ten) possesses PPRE on its promoter and is a PPARγ-targeted gene [93, 94]. PTEN encodes for a phosphatase that dephosphorylates and inactivates kinase PI3K. PI3K through activation of AKT kinase (also known as protein kinase B) inhibits apoptosis. Moreover PPARγ antisense oligonucleotide treatment resulted in significant decrease in caspase 9 activity, further demonstrating the proapoptotic action of PPARγ through a PTEN/caspase mechanism.
Exposure to the troglitazone has been shown to induce apoptosis in LNCaP, C4-2, and PC-3 prostate cancer cells [3, 95]. The ability of TZDs to promote apoptosis and cell cycle arrest appears to be associated with alterations in protein expressions and activities of antiapoptotic genes. In PC-3 and C4-2 cells, ciglitazone, rosiglitazone, and pioglitazone increased the level of the cyclin-dependent kinase inhibitor p21 [96]. TZDs treatment also stimulated proteasomal degradation of cyclin D1 and β-catenin within human prostate cancer cells [96]. In addition, studies have shown decreased phosphorylation and subsequent inactivation of retinoblastoma protein (Rb) in PC-3 cells exposed to ciglitazone. Data from Shiau et al. indicated that troglitazone induces apoptosis in PC-3 cells by reducing the activity of the anti-apoptotic proteins Bcl-2 and Bcl-xL [95].
5. Effect of PPARγ Ligands on the Invasion/Metastasis of Prostate Cancer Cells
Metastasis in cancer involves the process of spreading cancerous cells from the original organ or part to another nonadjacent organ or part. PPARγ ligands have been shown to affect endothelial cell proliferation as well as migration and hence regulate angiogenesis [97]. Annicotte et al. have shown that PPARγ activity was repressed by histone deacetylases (HDACs) and enhanced in the presence of HDAC inhibitors. E-cadherin is one of the major factors that inhibit metastasis and invasion of prostate cancer cells through maintenance of the adherens junctions important for epithelial cell-cell adhesion and inhibition of epithelial-to-mesenchymal transition required for cancer progression. Downregulation of E-cadherin expression contributes to oncogenesis [28], and it has been observed to occur in 50% of prostate cancers. Their observations demonstrated that a combination treatment using HDAC inhibitors and PPARγ agonists inhibits invasion of prostate cancer cells in vivo, through upregulation of E-cadherin expression [98].
Deacetylation of histones has been correlated with a transcriptionally silent state of chromatin. Inhibition of HDAC activity by natural or synthetic compounds results in the reversion of the phenotype of tumoral cells into normal cells or apoptosis in cancer cells [99]. Numerous studies demonstrated that HDAC3 when complexed with PPARγ in the promoters of PPARγ-targeted genes results in gene repression. In PC3 cell line, mRNA and protein expression of the cell cycle inhibitors p19, p21, and p27 were increased in response to pioglitazone, valproic acid, and to a higher extent in the combination treatment. Moreover, cyclin D1 mRNA and protein levels were decreased upon treatment with pioglitazone alone or in combination with valproic acid.
Moreover, HDAC inhibitors, such as valproic acid or sodium butyrate (NaBu), had a synergistic effect with thiazolidinediones in the activation of PPARγ-targeted genes [100]. This suggests that a cotreatment of HDAC inhibitors and PPARγ agonists potentiates the effects in the arrest of proliferation, increases apoptosis, and decreases the invasion potential of prostate cancer cells.
6. Effect of PPARγ Ligands on the Angiogenesis of Prostate Cancer Cells
Angiogenesis is a physiological process that involves the growth of new blood vessels from preexisting capillaries. Dysregulated angiogenesis can cause many abnormal disorders such as cancer, obesity, arthritis, and blindness. Angiogenesis is regulated by numerous angiogenic factors and mediators. As a major mediator of angiogenesis, vascular endothelial growth factor (VEGF) induces angiogenesis in ischemic or inflamed tissues, wound healing, rheumatoid arthritis, or diabetic retinopathy as well as during carcinogenesis.
High concentrations of PPARγ are found in tumor endothelium and in healthy skin endothelial cells, and PPARγ activation can induce PPARγ expression in tumor endothelial cells [101]. Earlier study has shown that activation of PPARγ by TZDs inhibits angiogenesis and neovascularization both in vitro and in vivo and blocks the release of VEGF from smooth muscle cells. Consistent with these data, it has been observed that leptin-induced migration of endothelial cells which is essential for generation of new vessels is inhibited by PPARγ agonists [7]. Interleukin-8 (IL8/CXCL8) is a key effector in prostate cancer progression and contributes to the resistance to standard chemotherapeutic drugs. IL8 belongs to the ELR + CXC chemokine subfamily. It stimulates angiogenesis and has been described as a potent attractant for granulocytic immune cells [2, 102–104]. Like other chemokines that recognize and bind G-protein-coupled receptors, IL8 acts through two receptors, CXC receptor 1 and 2 (CXCR1-2). IL8 has been shown to be involved in prostate cancer progression. Normal prostate epithelial cells and tissues produce low amounts of IL8, whereas prostate cancer cells from primary and metastatic tumors have higher levels of IL8 productions [105–109]. This effect is caused by progressive increases in activation of NFκB transcription factor [110] and correlated to an elevated adherence of the prostate tumor cells to the endothelium [111]. At the cell level, IL8 promotes the transition of prostate cancer to the hormone-refractory prostate cancer (HRPC) state via induction of androgen receptor expression and activation. It stimulates proliferation, invasion, and chemotaxis of HRPC cells through CXCR2 [112]. IL8 has also been involved in PC3 cell tumorigenicity [107], implying that this factor may represent a new molecular target for prostate cancer treatment.
Also hypoxia-induced angiogenesis can be targeted by PPARγ ligands in cancer therapy, even if the precise mechanisms still remain unclear and require further investigation. As angiogenesis is a crucial aspect for tumor development and metastasis, modulation of angiogenesis by PPARγ ligands would contribute significant clinical benefits in future prostate cancer therapy [97].
It is well known that angiogenesis plays an important role in the pathophysiology of ischemic and neoplastic disorders, especially cancer. 15d-PGJ2 is involved in regulation of angiogenic mediators including vascular endothelial growth factor and hence participates in the blood vessel formation by means of angiogenesis. 15d-PGJ2 inhibits angiogenesis via suppression of proinflammatory enzymes and cytokines, while it also stimulates angiogenesis via induction of heme oxygenase-1, endothelial nitric-oxide synthase, and hypoxia-inducible factor-1α [33].
7. Chemopreventive Effects of PPARγ Ligands in Prostate Cancer Mice Models
Considerable interest has been focusing on TZDs as potential chemopreventive agents in oncology, encouraging observations on the potential anticancer effect of these drugs in several in vitro experimental models. Interesting results from animal models studies and in pilot clinical trials have been obtained.
Kubota et al. examined that in vivo treatment of PC-3 tumors grown in male BNX triple immunodeficient mice with oral troglitazone (500 mg/kg/day) produced significant inhibition of tumor growth (P = 0.01). However, the only objective side effect of troglitazone in mice was the elevation of serum transaminases. Short-term culture of four surgically obtained human prostate cancer tumors with troglitazone (10 microM, 4 days) produced marked and selective necrosis of the cancer cells (about 60%), but not the adjacent normal prostate cells. Taken together, these results suggest that troglitazone may be a useful therapeutic agent for the treatment of prostate cancer, especially in the setting of low disease burden [25].
Moreover, troglitazone exhibited a powerful antiproliferative effect on aggressive, androgen-independent PC-3 prostate cancer cells, both in vitro and in vivo using a murine model. Forty male BNX nu/nu nude mice at 8 weeks of age were used for the experiment. PC-3 cells in 0.1 mL of Matrigel were injected subcutaneously into bilateral sides of each mouse, forming two tumors per mouse. The histological analysis of tumors treated with troglitazone revealed cytological changes of apoptosis including nuclear and cytoplasmic shrinkage and formation of nuclear fragments and apoptotic bodies. In order to determine whether the dramatic effects on PC-3 by troglitazone were mediated through activation of PPARγ, the author also evaluated other PPARγ ligands such as BRL49653, 15dPGJ2, ciglitizone, and indomethacin on prostate cancer cell lines. BRL49653 was more potent than troglitazone in growth inhibition. In addition, 15dPGJ2 had a similar potency as troglitazone, and the other two ligands were slightly less potent than troglitazone when treated against PC-3 cells [25]. The TZDs rosiglitazone and troglitazone also reduced growth of LNCaP and PC-3 tumors in nude mouse xenograft models [25, 96, 101].
PPARγ has been proven to enhance p53 expression, by binding to the NFκB-responsive element, located in the promoter region of p53 [17, 113]. A recent work by Yu et al. implicated an inhibitory role of PPARγ in hepatocarcinogenesis [114, 115]. In this case, an animal model was used to genetically ablate PPARγ expression on one allele (PPARγ +/−), which decreased PPARγ expression, but was not lethal during embryogenesis as observed in total PPARγ knockout (PPARγ −/−) mice [35, 36]. Using a diethylnitrosamine- (DEN-) induced hepatocarcinoma cell (HCC) model, the authors showed that activation of PPARγ by rosiglitazone blocked tumor development in PPARγ wild-type (PPARγ +/+) littermates, whereas it did not alter tumor formation in PPARγ +/− mice. To elucidate the underlying mechanism, the authors transduced the human hepatoma cell line Hep3B with a PPARγ-expressing adenovirus. In these transduced cells, PPARγ overexpression induced a G2/M arrest and apoptosis, mediated by extrinsic (Fas and TNFα) and intrinsic (caspases 3, 7, 9, and PARP) pathways. Both cell cycle arrest and cell death were enhanced in response to rosiglitazone-mediated PPARγ activation [116–118].
It has been reported that a large proportion of human prostate tumors (40%) carry hemizygous deletions of the PPARγ gene. These findings suggest a functional role of PPARγ as a tumor suppressor gene. Saez et al. concluded that neither hemizygous deletion of PPARγ nor complete ablation of PPARα influenced the development of prostate cancer. In order to elucidate the mechanism of PPARγ signaling in tumor development, strains of mice with defined loss-of-function mutations in the PPARγ gene were generated. Mice devoid of PPARγ gene died in utero whereas heterozygotes were viable [119].
8. Ongoing Clinical Trials with PPARγ Agonists
CS-7017, a Daiichi Sankyo compound, is an oral PPARγ agonist currently undergoing a single arm phase I evaluation [32, 120] for advanced metastatic cancer. A single-arm combination therapy phase I/II clinical trial employing a taxane and CS-7017, for anaplastic thyroid cancer, is also in progress (NCT00603941). There are two recently completed single-arm phase IIa leukoplakia reversal clinical trials, one employing rosiglitazone and the other employing pioglitazone [121]. The pioglitazone clinical trial showed leukoplakia reversal in most patients, and a randomized phase II clinical trial with pioglitazone in leukoplakia patients has been planned [122]. These ongoing or recently completed studies show considerable interest in the clinical study with PPARγ agonists as therapeutic agents.
Thymoquinone, an active ingredient isolated from Nigella sativa, was found to induce the activity of PPARγ and PPAR-β/δ in MCF-7 breast cancer cells [123, 124]. PPARγ has been reported by numerous studies to play a significant role in anticancer mechanisms. By using molecular docking analysis, thymoquinone was shown to interact with seven polar residues and six nonpolar residues in the PPARγ receptor [124]. Thymoquinone-induced apoptosis and decreased surviving levels in MCF-7 cells could be reversed by incubation with GW9662, an irreversible PPARγ inhibitor, suggesting the involvement of PPARγ activity in the anticancer activity of thymoquinone [124].
Recently, a clinical assessment of PPARγ agonists in patients with prostate carcinoma was conducted. In a phase II clinical study, patients with histologically confirmed advanced prostate cancer who had no symptoms of metastatic disease were treated with troglitazone (800 mg per day orally) and showed extended stabilization of prostate-specific antigen (PSA) concentrations, indicating disease stabilization. One patient showed a striking decrease in PSA concentration to almost undetectable amounts [84]. In an index case, a 75-year-old patient with occult recurrent prostate cancer showed a decrease in PSA after oral treatment with troglitazone (600–800 mg per day for 1.5 years) [125]. Segawa and coworkers [126] analyzed prostate tissue from 203 patients and found that PPARγ immunoreactivity was significantly higher in patients with prostate cancer and prostatic intraepithelial neoplasia than in those with benign prostate hyperplasia and in men with healthy prostates. In summary, PPARγ expression is upregulated in prostate cancer and the induction of PPARγ activity provides an additional therapeutic option for treatment of prostate cancer in the near future.
In conclusion, many earlier studies have shown that TZDs inhibit growth of human prostate cancer cells both in vitro and in vivo [38]. Two clinical trials treatment with the TZD troglitazone slowed the progression of prostate cancer within patients [84, 125] suggesting that TZDs may serve as effective therapeutic agents for prostate cancer.
9. Conclusion and Future Perspectives
Nearly 10 years have passed since the first PPAR subtype was identified. Since then, intense research has led to the development of clinical approaches and synthetic ligands of this nuclear hormone receptor, while some of which are now undergoing clinical trials. Several antineoplastic effects such as induction of apoptosis and differentiation have been described as a result of ligand activation of PPARγ both in vitro and in vivo.
The mechanisms by which the PPARγ agonists promote apoptosis in cancer cells remain to be fully elucidated. The first few clinical trials to make use of the anti-neoplastic effects mediated by PPARγ have shown conflicting results. On one hand, some studies indicated beneficial effects with PPARγ ligand treatment [42, 43, 80]; however, some other studies could not detect significant anti-neoplastic effects of PPARγ agonists, and the repression effects on tumor growth remain limited [127–129]. Another criticism is that drug concentrations in studies on humans have not been identified. In addition, treatment with a PPARγ agonist might be effective in the prevention of tumor development or might be successful in the treatment of massive tumor growth, if combined with known chemotherapeutics and radiation [7].
Clearly, risk stratification and the targeting of these agents to specific intraepithelial neoplastic conditions will be important in the future testing of these promising chemoprevention drugs. The antiproliferative, prodifferentiation effects of PPARγ activators (TZDs) suggest that these compounds might be useful in slowing the proliferation of undifferentiated tumor cells. Prostate cancers express abundant and higher constitutive levels of PPARγ than do normal prostate cells and are inhibited by ligand activation of PPARγ [130]. These findings have critical implications for the application of PPARγ agonists as potential therapeutic or preventive agents that will spare normal tissue while acting on malignant or premalignant tissue. It is anticipated that PPARγ ligands will provide not only useful mechanistic pathway information but also open a new era of therapeutic options for sufferers of prostate cancer.
Acknowledgments
This work was supported by grants from the National Medical Research Council of Singapore (Grant R-713-000-124-213), National Kidney Foundation (Grant R-713-000-138-592) and Cancer Science Institute of Singapore, Experimental Therapeutics I Program (Grant R-713-001-011-271) to A. P. Kumar. A. P. Kumar is also supported by the John Nott Cancer Fellowship 2012 from Cancer Council Western Australia. G. Sethi was supported by grants from the NUS Academic Research Fund (R-184-000-207-112) and National Medical Research Council of Singapore (R-184-000-211-213).
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer Journal for Clinicians. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Hirsch J, Johnson CL, Nelius T, Kennedy R, Riese WD, Filleur S. PEDF inhibits IL8 production in prostate cancer cells through PEDF receptor/phospholipase A2 and regulation of NFκB and PPARγ . Cytokine. 2011;55(2):202–210. doi: 10.1016/j.cyto.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 3.Akinyeke TO, Stewart LV. Troglitazone suppresses c-Myc levels in human prostate cancer cells via a PPARγ-independent mechanism. Cancer Biology and Therapy. 2011;11(12):1046–1058. doi: 10.4161/cbt.11.12.15709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson PS, Clegg N, Arnold H, et al. The program of androgen-responsive genes in neoplastic prostate epithelium. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(18):11890–11895. doi: 10.1073/pnas.182376299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lehrke M, Lazar MA. The many faces of PPARγ . Cell. 2005;123(6):993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 6.Huang THW, Razmovski-Naumovski V, Kota BP, Lin DSH, Roufogalis BD. The pathophysiological function of peroxisome proliferator-activated receptor-γ in lung-related diseases. Respiratory Research. 2005;6(1, article 102) doi: 10.1186/1465-9921-6-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grommes C, Landreth GE, Heneka MT. Antineoplastic effects of peroxisome proliferator-activated receptor γ agonists. The Lancet Oncology. 2004;5(7):419–429. doi: 10.1016/S1470-2045(04)01509-8. [DOI] [PubMed] [Google Scholar]
- 8.Celi FS, Shuldiner AR. The role of peroxisome proliferator-activated receptor gamma in diabetes and obesity. Current Diabetes Reports. 2002;2(2):179–185. doi: 10.1007/s11892-002-0078-2. [DOI] [PubMed] [Google Scholar]
- 9.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83(6):841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 10.Koeffler HP. Peroxisome proliferator-activated receptor γ and cancers. Clinical Cancer Research. 2003;9(1):1–9. [PubMed] [Google Scholar]
- 11.Venkatachalam G, Kumar AP, Sakharkar KR, Thangavel S, Clement MV, Sakharkar MK. PPARγ disease gene network and identification of therapeutic targets for prostate cancer. Journal of Drug Targeting. 2011;19(9):781–796. doi: 10.3109/1061186X.2011.568062. [DOI] [PubMed] [Google Scholar]
- 12.Elstner E, Müller C, Koshizuka K, et al. Ligands for peroxisome proliferator-activated receptory and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(15):8806–8811. doi: 10.1073/pnas.95.15.8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarraf P, Mueller E, Smith WM, et al. Loss-of-function mutations in PPARγ associated with human colon cancer. Molecular Cell. 1999;3(6):799–804. doi: 10.1016/s1097-2765(01)80012-5. [DOI] [PubMed] [Google Scholar]
- 14.Pignatelli M, Sánchez-Rodríguez J, Santos A, Perez-Castillo A. 15-Deoxy-Δ12,14-prostaglandin J2 induces programmed cell death of breast cancer cells by a pleiotropic mechanism. Carcinogenesis. 2005;26(1):81–92. doi: 10.1093/carcin/bgh308. [DOI] [PubMed] [Google Scholar]
- 15.McKenna NJ, O’Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108(4):465–474. doi: 10.1016/s0092-8674(02)00641-4. [DOI] [PubMed] [Google Scholar]
- 16.Ito K, Utsunomiya H, Yaegashi N, Sasano H. Biological roles of estrogen and progesterone in human endometrial carcinoma—new developments in potential endocrine therapy for endometrial cancer. Endocrine Journal. 2007;54(5):667–679. doi: 10.1507/endocrj.kr-114. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt MV, Brüne B, Von Knethen A. The nuclear hormone receptor PPARγ as a therapeutic target in major diseases. The Scientific World Journal. 2010;10:2181–2197. doi: 10.1100/tsw.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larsen TM, Toubro S, Astrup A. PPARgamma agonists in the treatment of type II diabetes: is increased fatness commensurate with long-term efficacy? International Journal of Obesity. 2003;27(2):147–161. doi: 10.1038/sj.ijo.802223. [DOI] [PubMed] [Google Scholar]
- 19.Fajas L, Fruchart JC, Auwerx J. Transcriptional control of adipogenesis. Current Opinion in Cell Biology. 1998;10(2):165–173. doi: 10.1016/s0955-0674(98)80138-5. [DOI] [PubMed] [Google Scholar]
- 20.Nevins JR. E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science. 1992;258(5081):424–429. doi: 10.1126/science.1411535. [DOI] [PubMed] [Google Scholar]
- 21.Reichert M, Eick D. Analysis of cell cycle arrest in adipocyte differentiation. Oncogene. 1999;18(2):459–466. doi: 10.1038/sj.onc.1202308. [DOI] [PubMed] [Google Scholar]
- 22.Cao Y. Angiogenesis modulates adipogenesis and obesity. Journal of Clinical Investigation. 2007;117(9):2362–2368. doi: 10.1172/JCI32239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biscetti F, Gaetani E, Flex A, et al. Selective activation of peroxisome proliferator-activated receptor (PPAR)α and PPARγ induces neoangiogenesis through a vascular endothelial growth factor-dependent mechanism. Diabetes. 2008;57(5):1394–1404. doi: 10.2337/db07-0765. [DOI] [PubMed] [Google Scholar]
- 24.Tontonoz P, Singer S, Forman BM, et al. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor γ and the retinoid X receptor. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(1):237–241. doi: 10.1073/pnas.94.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kubota T, Koshizuka K, Williamson EA, et al. Ligand for peroxisome proliferator-activated receptor γ (Troglitazone) has potent antitumor effect against human prostate cancer both in vitro and in vivo . Cancer Research. 1998;58(15):3344–3352. [PubMed] [Google Scholar]
- 26.Brockman JA, Gupta RA, Dubois RN. Activation of PPARγ leads to inhibition of anchorage-independent growth of human colorectal cancer cells. Gastroenterology. 1998;115(5):1049–1055. doi: 10.1016/s0016-5085(98)70072-1. [DOI] [PubMed] [Google Scholar]
- 27.Chang TH, Szabo E. Induction of differentiation and apoptosis by ligands of peroxisome proliferator-activated receptor γ in non-small cell lung cancer. Cancer Research. 2000;60(4):1129–1138. [PubMed] [Google Scholar]
- 28.Cano A, Pérez-Moreno MA, Rodrigo I, et al. The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nature Cell Biology. 2000;2(2):76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 29.Guan YF, Zhang YH, Breyer RM, Davis L, Breyer MD. Expression of peroxisome proliferator-activated receptor γ (PPARγ) in human transitional bladder cancer and its role in inducing cell death. Neoplasia. 1999;1(4):330–339. doi: 10.1038/sj.neo.7900050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato H, Ishihara S, Kawashima K, et al. Expression of peroxisome proliferator-activated receptor (PPAR)γ in gastric cancer and inhibitory effects of PPARγ agonists. British Journal of Cancer. 2000;83(10):1394–1400. doi: 10.1054/bjoc.2000.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Weiser-Evans MC, Nemenoff R. Anti- and protumorigenic effects of PPARgamma in lung cancer progression: a double-edged sword. PPAR Research. 2012;2012:12 pages. doi: 10.1155/2012/362085.362085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ondrey F. Peroxisome proliferator-activated receptor γ pathway targeting in carcinogenesis: implications for chemoprevention. Clinical Cancer Research. 2009;15(1):2–8. doi: 10.1158/1078-0432.CCR-08-0326. [DOI] [PubMed] [Google Scholar]
- 33.Kim EH, Surh YJ. The role of 15-deoxy-Δ12,14-prostaglandin J2, an endogenous ligand of peroxisome proliferator-activated receptor γ, in tumor angiogenesis. Biochemical Pharmacology. 2008;76(11):1544–1553. doi: 10.1016/j.bcp.2008.07.043. [DOI] [PubMed] [Google Scholar]
- 34.Wang T, Xu J, Yu X, Yang R, Han ZC. Peroxisome proliferator-activated receptor γ in malignant diseases. Critical Reviews in Oncology/Hematology. 2006;58(1):1–14. doi: 10.1016/j.critrevonc.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 35.Houseknecht KL, Cole BM, Steele PJ. Peroxisome proliferator-activated receptor gamma (PPARγ) and its ligands: a review. Domestic Animal Endocrinology. 2002;22(1):1–23. doi: 10.1016/s0739-7240(01)00117-5. [DOI] [PubMed] [Google Scholar]
- 36.Murakami K, Tobe K, Ide T, et al. A novel insulin sensitizer acts as a coligand for peroxisome proliferator-activated receptor-α (PPAR-α) and PPAR-γ. Effect of PPAR-α activation on abnormal lipid metabolism in liver of Zucker fatty rats. Diabetes. 1998;47(12):1841–1847. doi: 10.2337/diabetes.47.12.1841. [DOI] [PubMed] [Google Scholar]
- 37.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-deoxy-Δ12,14-prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ . Cell. 1995;83(5):803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 38.Bolden A, Bernard L, Jones D, Akinyeke T, Stewart LV. The PPAR gamma agonist troglitazone regulates Erk 1/2 phosphorylation via a PPAR?-Independent, MEK-dependent pathway in human prostate cancer cells. PPAR Research. 2012;2012:9 pages. doi: 10.1155/2012/929052.929052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li MY, Lee TW, Yim APC, Chen GG. Function of PPARγ and its ligands in lung cancer. Critical Reviews in Clinical Laboratory Sciences. 2006;43(2):183–202. doi: 10.1080/10408360600552587. [DOI] [PubMed] [Google Scholar]
- 40.Kuroda M, Mimaki Y, Sashida Y, et al. Phenolics with PPAR-γ ligand-Binding activity obtained from licorice (Glycyrrhiza uralensis Roots) and ameliorative effects of glycyrin on genetically diabetic KK-Ay mice. Bioorganic and Medicinal Chemistry Letters. 2003;13(24):4267–4272. doi: 10.1016/j.bmcl.2003.09.052. [DOI] [PubMed] [Google Scholar]
- 41.Steiner C, Arnould S, Scalbert A, Manach C. Isoflavones and the prevention of breast and prostate cancer: new perspectives opened by nutrigenomics. British Journal of Nutrition. 2008;99(supplement 1):ES78–ES108. doi: 10.1017/S0007114508965788. [DOI] [PubMed] [Google Scholar]
- 42.Takahashi Y, Lavigne JA, Hursting SD, et al. Using DNA microarray analysis to elucidate the effects of genistein in androgen-responsive prostate cancer cells: identification of novel targets. Molecular Carcinogenesis. 2004;41(2):108–119. doi: 10.1002/mc.20045. [DOI] [PubMed] [Google Scholar]
- 43.Lee JY, Kim JK, Cho MC, et al. Cytotoxic flavonoids as agonists of peroxisome proliferator-activated receptor γ on human cervical and prostate cancer cells. Journal of Natural Products. 2010;73(7):1261–1265. doi: 10.1021/np100148m. [DOI] [PubMed] [Google Scholar]
- 44.Palakurthi SS, Aktas H, Grubissich LM, Mortensen RM, Halperin JA. Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor γ and mediated by inhibition of translation initiation. Cancer Research. 2001;61(16):6213–6218. [PubMed] [Google Scholar]
- 45.Weng JR, Chen CY, Pinzone JJ, Ringel MD, Chen CS. Beyond peroxisome proliferator-activated receptor γ signaling: the multi-facets of the antitumor effect of thiazolidinediones. Endocrine-Related Cancer. 2006;13(2):401–413. doi: 10.1677/erc.1.01182. [DOI] [PubMed] [Google Scholar]
- 46.Wang M, Wise SC, Leff T, Su TZ. Troglitazone, an antidiabetic agent, inhibits cholesterol biosynthesis through a mechanism independent of peroxisome proliferator-activated receptor-γ . Diabetes. 1999;48(2):254–260. doi: 10.2337/diabetes.48.2.254. [DOI] [PubMed] [Google Scholar]
- 47.Lea MA, Sura M, Desbordes C. Inhibition of cell proliferation by potential peroxisome proliferator-activated receptor (PPAR) gamma agonists and antagonists. Anticancer Research. 2004;24(5):2765–2771. [PubMed] [Google Scholar]
- 48.Laurora S, Pizzimenti S, Briatore F, et al. Peroxisome proliferator-activated receptor ligands affect growth-related gene expression in human leukemic cells. Journal of Pharmacology and Experimental Therapeutics. 2003;305(3):932–942. doi: 10.1124/jpet.103.049098. [DOI] [PubMed] [Google Scholar]
- 49.Abe A, Kiriyama Y, Hirano M, et al. Troglitazone suppresses cell growth of KU812 cells independently of PPARγ . European Journal of Pharmacology. 2002;436(1-2):7–13. doi: 10.1016/s0014-2999(01)01577-1. [DOI] [PubMed] [Google Scholar]
- 50.Palakurthi SS, Aktas H, Grubissich LM, Mortensen RM, Halperin JA. Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor γ and mediated by inhibition of translation initiation. Cancer Research. 2001;61(16):6213–6218. [PubMed] [Google Scholar]
- 51.Feinstein DL, Spagnolo A, Akar C, et al. Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochemical Pharmacology. 2005;70(2):177–188. doi: 10.1016/j.bcp.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 52.Xin X, Yang S, Kowalski J, Gerritsen ME. Peroxisome proliferator-activated receptor γ ligands are potent inhibitors of angiogenesis in vitro and in vivo . Journal of Biological Chemistry. 1999;274(13):9116–9121. doi: 10.1074/jbc.274.13.9116. [DOI] [PubMed] [Google Scholar]
- 53.Marx N, Bourcier T, Sukhova GK, Libby P, Plutzky J. PPARγ activation in human endothelial cells increases plasminogen activator inhibitor type-1 expression: PPARγ as a potential mediator in vascular disease. Arteriosclerosis, Thrombosis, and Vascular Biology. 1999;19(3):546–551. doi: 10.1161/01.atv.19.3.546. [DOI] [PubMed] [Google Scholar]
- 54.Gralinski MR, Rowse PE, Breider MA. Effects of troglitazone and pioglitazone on cytokine-mediated endothelial cell proliferation in vitro . Journal of Cardiovascular Pharmacology. 1998;31(6):909–913. doi: 10.1097/00005344-199806000-00015. [DOI] [PubMed] [Google Scholar]
- 55.Goetze S, Eilers F, Bungenstock A, et al. PPAR activators inhibit endothelial cell migration by targeting Akt. Biochemical and Biophysical Research Communications. 2002;293(5):1431–1437. doi: 10.1016/S0006-291X(02)00385-6. [DOI] [PubMed] [Google Scholar]
- 56.Mansour M, Schwartz D, Judd R, et al. Thiazolidinediones/PPARγ agonists and fatty acid synthase inhibitors as an experimental combination therapy for prostate cancer. International Journal of Oncology. 2011;38(2):537–546. doi: 10.3892/ijo.2010.877. [DOI] [PubMed] [Google Scholar]
- 57.Karayi MK, Markham AF. Molecular biology of prostate cancer. Prostate Cancer and Prostatic Diseases. 2004;7(1):6–20. doi: 10.1038/sj.pcan.4500697. [DOI] [PubMed] [Google Scholar]
- 58.Hatton JL, Yee LD. Clinical use of PPARγ ligands in cancer. PPAR Research. 2008;2008:13 pages. doi: 10.1155/2008/159415.159415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Panigrahy D, Shen LQ, Kieran MW, Kaipainen A. Therapeutic potential of thiazolidinediones as anticancer agents. Expert Opinion on Investigational Drugs. 2003;12(12):1925–1937. doi: 10.1517/13543784.12.12.1925. [DOI] [PubMed] [Google Scholar]
- 60.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell. 1994;79(7):1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 61.Chui PC, Guan HP, Lehrke M, Lazar MA. PPARγ regulates adipocyte cholesterol metabolism via oxidized LDL receptor 1. Journal of Clinical Investigation. 2005;115(8):2244–2256. doi: 10.1172/JCI24130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bruning JB, Chalmers MJ, Prasad S, et al. Partial agonists activate PPARγ using a helix 12 independent mechanism. Structure. 2007;15(10):1258–1271. doi: 10.1016/j.str.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 63.Berger JP, Petro AE, Macnaul KL, et al. Distinct properties and advantages of a novel peroxisome proliferator-activated protein [gamma] selective modulator. Molecular Endocrinology. 2003;17(4):662–676. doi: 10.1210/me.2002-0217. [DOI] [PubMed] [Google Scholar]
- 64.Rangwala SM, Lazar MA. The dawn of the SPPARMs? Science"s STKE. 2002;2002(121):p. PE9. doi: 10.1126/stke.2002.121.pe9. [DOI] [PubMed] [Google Scholar]
- 65.Rocchi S, Picard F, Vamecq J, et al. A unique PPARγ ligand with potent insulin-sensitizing yet weak adipogenic activity. Molecular Cell. 2001;8(4):737–747. doi: 10.1016/s1097-2765(01)00353-7. [DOI] [PubMed] [Google Scholar]
- 66.Shiau AK, Barstad D, Loria PM, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95(7):927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 67.Nagy L, Kao HY, Love JD, et al. Mechanism of corepressor binding and release from nuclear hormone receptors. Genes and Development. 1999;13(24):3209–3216. doi: 10.1101/gad.13.24.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes and Development. 2006;20(11):1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 69.Fujimura T, Sakuma H, Konishi S, et al. FK614, a novel peroxisome proliferator-activated receptor γ modulator, induces differential transactivation through a unique ligand-specific interaction with transcriptional coactivators. Journal of Pharmacological Sciences. 2005;99(4):342–352. doi: 10.1254/jphs.fp0050578. [DOI] [PubMed] [Google Scholar]
- 70.Oberfield JL, Collins JL, Holmes CP, et al. A peroxisome proliferator-activated receptor gamma ligand inhibits adipocyte differentiation. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(11):6102–6106. doi: 10.1073/pnas.96.11.6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Funao K, Matsuyama M, Kawahito Y, et al. Telmisartan is a potent target for prevention and treatment in human prostate cancer. Oncology Reports. 2008;20(2):295–300. [PubMed] [Google Scholar]
- 72.Hu X, Lazar MA. The CoRNR motif controls the recruitment of compressors by nuclear hormone receptors. Nature. 1999;402(6757):93–96. doi: 10.1038/47069. [DOI] [PubMed] [Google Scholar]
- 73.Pascual G, Fong AL, Ogawa S, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ . Nature. 2005;437(7059):759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zou G, Gao Z, Wang J, et al. Deoxyelephantopin inhibits cancer cell proliferation and functions as a selective partial agonist against PPARγ . Biochemical Pharmacology. 2008;75(6):1381–1392. doi: 10.1016/j.bcp.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 75.Reginato MJ, Bailey ST, Krakow SL, et al. A potent antidiabetic thiazolidinedione with unique peroxisome proliferator-activated receptor γ-activating properties. Journal of Biological Chemistry. 1998;273(49):32679–32684. doi: 10.1074/jbc.273.49.32679. [DOI] [PubMed] [Google Scholar]
- 76.Kumagai T, Ikezoe T, Gui D, et al. RWJ-241947 (MCC-555), a unique peroxisome proliferator-activated receptor-γ ligand with antitumor activity against human prostate cancer in vitro and in Beige/Nude/X-Linked immunodeficient mice and enhancement of apoptosis in myeloma cells induced by arsenic trioxide. Clinical Cancer Research. 2004;10(4):1508–1520. doi: 10.1158/1078-0432.ccr-0476-03. [DOI] [PubMed] [Google Scholar]
- 77.Straus DS, Pascual G, Li M, et al. 15-deoxy-Δ12,14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(9):4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gouni-Berthold I, Berthold HK, Weber AA, et al. Troglitazone and rosiglitazone induce apoptosis of vascular smooth muscle cells through an extracellular signal-regulated kinase-independent pathway. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2001;363(2):215–221. doi: 10.1007/s002100000352. [DOI] [PubMed] [Google Scholar]
- 79.Keller H, Givel F, Perroud M, Wahli W. Signaling cross-talk between peroxisome proliferator-activated receptor/retinoid X receptor and estrogen receptor through estrogen response elements. Molecular Endocrinology. 1995;9(7):794–804. doi: 10.1210/mend.9.7.7476963. [DOI] [PubMed] [Google Scholar]
- 80.Gelman L, Fruchart JC, Auwerx J. An update on the mechanisms of action of the peroxisome proliferator-activated receptors (PPARs) and their roles in inflammation and cancer. Cellular and Molecular Life Sciences. 1999;55(6-7):932–943. doi: 10.1007/s000180050345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhu Y, Qi C, Calandra C, Rao MS, Reddy JK. Cloning and identification of mouse steroid receptor coactivator-1 (mSRC-1), as a coactivator of peroxisome proliferator-activated receptor gamma. Gene Expression. 1996;6(3):185–195. [PMC free article] [PubMed] [Google Scholar]
- 82.Jeon WJ, Kim SH, Seo MS, et al. Repeated electroconvulsive seizure induces c-Myc down-regulation and Bad inactivation in the rat frontal cortex. Experimental and Molecular Medicine. 2008;40(4):435–444. doi: 10.3858/emm.2008.40.4.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhu H, Pan X, Qi H, et al. Troglitazone attenuates epidermal growth factor receptor signaling independently of peroxisome proliferatoractivated receptor in PC-3 cells. Oncology Reports. 2011;25(1):81–90. [PubMed] [Google Scholar]
- 84.Mueller E, Smith M, Sarraf P, et al. Effects of ligand activation of peroxisome proliferator-activated receptor γ in human prostate cancer. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(20):10990–10995. doi: 10.1073/pnas.180329197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yoshimura R, Yoshimura R, Matsuyama M, Hase T, et al. The effect of peroxisome proliferator-activated receptor-gamma ligand on urological cancer cells. International Journal of Molecular Medicine. 2003;12(6):861–865. [PubMed] [Google Scholar]
- 86.Campbell SE, Musich PR, Whaley SG, et al. Gamma tocopherol upregulates the expression of 15-S-HETE and induces growth arrest through a PPAR gamma-dependent mechanism in PC-3 human prostate cancer cells. Nutrition and Cancer. 2009;61(5):649–662. doi: 10.1080/01635580902825654. [DOI] [PubMed] [Google Scholar]
- 87.Deneo-Pellegrini H, De Stefani E, Ronco A, Mendilaharsu M. Foods, nutrients and prostate cancer: a case-control study in Uruguay. British Journal of Cancer. 1999;80(3-4):591–597. doi: 10.1038/sj.bjc.6690396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.De Marzo AM, Marchi VL, Epstein JI, Nelson WG. Proliferative inflammatory atrophy of the prostate: implications for prostatic carcinogenesis. The American Journal of Pathology. 1999;155(6):1985–1992. doi: 10.1016/S0002-9440(10)65517-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cooney RV, Franke AA, Harwood PJ, Hatch-Pigott V, Custer LJ, Mordan LJ. γ-Tocopherol detoxification of nitrogen dioxide: superiority to α- tocopherol. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(5):1771–1775. doi: 10.1073/pnas.90.5.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ban JO, Kwak DH, Oh JH, et al. Suppression of NF-κB and GSK-3β is involved in colon cancer cell growth inhibition by the PPAR agonist troglitazone. Chemico-Biological Interactions. 2010;188(1):75–85. doi: 10.1016/j.cbi.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 91.Zander T, Kraus JA, Grommes C, et al. Induction of apoptosis in human and rat glioma by agonists of the nuclear receptor PPARγ . Journal of Neurochemistry. 2002;81(5):1052–1060. doi: 10.1046/j.1471-4159.2002.00899.x. [DOI] [PubMed] [Google Scholar]
- 92.Cho WH, Choi CH, Park JY, Kang SK, Kim YK. 15-Deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) induces cell death through caspase-independent mechanism in A172 human glioma cells. Neurochemical Research. 2006;31(10):1247–1254. doi: 10.1007/s11064-006-9157-0. [DOI] [PubMed] [Google Scholar]
- 93.Voutsadakis IA. Peroxisome proliferator-activated receptor γ (PPARγ) and colorectal carcinogenesis. Journal of Cancer Research and Clinical Oncology. 2007;133(12):917–928. doi: 10.1007/s00432-007-0277-y. [DOI] [PubMed] [Google Scholar]
- 94.Patel L, Pass I, Coxon P, Downes CP, Smith SA, Macphee CH. Tumor suppressor and anti-inflammatory actions of PPARγ agonists are mediated via upregulation of PTEN. Current Biology. 2001;11(10):764–768. doi: 10.1016/s0960-9822(01)00225-1. [DOI] [PubMed] [Google Scholar]
- 95.Shiau CW, Yang CC, Kulp SK, et al. Thiazolidenediones mediate apoptosis in prostate cancer cells in part through inhibition of Bcl-xL/Bcl-2 functions independently of PPARγ . Cancer Research. 2005;65(4):1561–1569. doi: 10.1158/0008-5472.CAN-04-1677. [DOI] [PubMed] [Google Scholar]
- 96.Lyles BE, Akinyeke TO, Moss PE, Stewart LV. Thiazolidinediones regulate expression of cell cycle proteins in human prostate cancer cells via PPARγ-dependent and PPARγ-independent pathways. Cell Cycle. 2009;8(2):268–277. doi: 10.4161/cc.8.2.7584. [DOI] [PubMed] [Google Scholar]
- 97.Schweitzer A, Knauer SK, Stauber RH. Nuclear receptors in head and neck cancer: current knowledge and perspectives. International Journal of Cancer. 2010;126(4):801–809. doi: 10.1002/ijc.24968. [DOI] [PubMed] [Google Scholar]
- 98.Annicotte JS, Culine S, Fajas L. Role of PPARγ in the control of prostate cancer growth: a new approach for therapy. Bulletin du Cancer. 2007;94(2):135–137. [PubMed] [Google Scholar]
- 99.Annicotte JS, Iankova I, Miard S, et al. Peroxisome proliferator-activated receptor γ regulates E-cadherin expression and inhibits growth and invasion of prostate cancer. Molecular and Cellular Biology. 2006;26(20):7561–7574. doi: 10.1128/MCB.00605-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fajas L, Egler V, Reiter R, et al. The retinoblastoma-histone deacetylase 3 complex inhibits PPARγ and adipocyte differentiation. Developmental Cell. 2002;3(6):903–910. doi: 10.1016/s1534-5807(02)00360-x. [DOI] [PubMed] [Google Scholar]
- 101.Panigrahy D, Singer S, Shen LQ, et al. PPARγ ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. Journal of Clinical Investigation. 2002;110(7):923–932. doi: 10.1172/JCI15634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vandercappellen J, Van Damme J, Struyf S. The role of CXC chemokines and their receptors in cancer. Cancer Letters. 2008;267(2):226–244. doi: 10.1016/j.canlet.2008.04.050. [DOI] [PubMed] [Google Scholar]
- 103.Kobayashi Y. Neutrophil infiltration and chemokines. Critical Reviews in Immunology. 2006;26(4):307–315. doi: 10.1615/critrevimmunol.v26.i4.20. [DOI] [PubMed] [Google Scholar]
- 104.Bizzarri C, Beccari AR, Bertini R, Cavicchia MR, Giorgini S, Allegretti M. ELR+ CXC chemokines and their receptors (CXC chemokine receptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacology and Therapeutics. 2006;112(1):139–149. doi: 10.1016/j.pharmthera.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 105.Lu Y, Cai Z, Xiao G, et al. Monocyte chemotactic protein-1 mediates prostate cancer-induced bone resorption. Cancer Research. 2007;67(8):3646–3653. doi: 10.1158/0008-5472.CAN-06-1210. [DOI] [PubMed] [Google Scholar]
- 106.Begley LA, Kasina S, Mehra R, et al. CXCL5 promotes prostate cancer progression. Neoplasia. 2008;10(3):244–254. doi: 10.1593/neo.07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Moore BB, Arenberg DA, Stoy K, et al. Distinct CXC chemokines mediate tumorigenicity of prostate cancer cells. The American Journal of Pathology. 1999;154(5):1503–1512. doi: 10.1016/S0002-9440(10)65404-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Araki S, Omori Y, Lyn D, et al. Interleukin-8 is a molecular determinant of androgen independence and progression in prostate cancer. Cancer Research. 2007;67(14):6854–6862. doi: 10.1158/0008-5472.CAN-07-1162. [DOI] [PubMed] [Google Scholar]
- 109.Seaton A, Scullin P, Maxwell PJ, et al. Interleukin-8 signaling promotes androgen-independent proliferation of prostate cancer cells via induction of androgen receptor expression and activation. Carcinogenesis. 2008;29(6):1148–1156. doi: 10.1093/carcin/bgn109. [DOI] [PubMed] [Google Scholar]
- 110.Shen H, Lentsch AB. Progressive dysregulation of transcription factors NF-κB and STAT1 in prostate cancer cells causes proangiogenic production of CXC chemokines. American Journal of Physiology. 2004;286(4):C840–C847. doi: 10.1152/ajpcell.00335.2003. [DOI] [PubMed] [Google Scholar]
- 111.Engl T, Relja B, Blumenberg C, et al. Prostate tumor CXC-chemokine profile correlates with cell adhesion to endothelium and extracellular matrix. Life Sciences. 2006;78(16):1784–1793. doi: 10.1016/j.lfs.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 112.Reiland J, Furcht LT, McCarthy JB. CXC-chemokines stimulate invasion and chemotaxis in prostate carcinoma cells through the CXCR2 receptor. The Prostate. 1999;41(2):78–88. doi: 10.1002/(sici)1097-0045(19991001)41:2<78::aid-pros2>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 113.Bonofiglio D, Gabriele S, Aquila S, et al. Peroxisome proliferator-activated receptor gamma activates fas ligand gene promoter inducing apoptosis in human breast cancer cells. Breast Cancer Research and Treatment. 2009;113(3):423–434. doi: 10.1007/s10549-008-9944-1. [DOI] [PubMed] [Google Scholar]
- 114.Yu J, Shen B, Chu ESH, et al. Inhibitory role of peroxisome proliferator-activated receptor gamma in hepatocarcinogenesis in mice and in vitro . Hepatology. 2010;51(6):2008–2019. doi: 10.1002/hep.23550. [DOI] [PubMed] [Google Scholar]
- 115.Yu J, Qiao L, Zimmermann L, et al. Troglitazone inhibits tumor growth in hepatocellular carcinoma in vitro and in vivo . Hepatology. 2006;43(1):134–143. doi: 10.1002/hep.20994. [DOI] [PubMed] [Google Scholar]
- 116.Baek SJ, Kim JS, Jackson FR, Eling TE, McEntee MF, Lee SH. Epicatechin gallate-induced expression of NAG-1 is associated with growth inhibition and apoptosis in colon cancer cells. Carcinogenesis. 2004;25(12):2425–2432. doi: 10.1093/carcin/bgh255. [DOI] [PubMed] [Google Scholar]
- 117.Chen YL, Lin PC, Chen SP, et al. Activation of nonsteroidal anti-inflammatory drug-activated gene-1 via extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase revealed a isochaihulactone-triggered apoptotic pathway in human lung cancer A549 cells. Journal of Pharmacology and Experimental Therapeutics. 2007;323(2):746–756. doi: 10.1124/jpet.107.126193. [DOI] [PubMed] [Google Scholar]
- 118.Liu T, Bauskin AR, Zaunders J, et al. Macrophage inhibitory cytokine 1 reduces cell adhesion and induces apoptosis in prostate cancer cells. Cancer Research. 2003;63(16):5034–5040. [PubMed] [Google Scholar]
- 119.Saez E, Olson P, Evans RM. Genetic deficiency in Pparg does not alter development of experimental prostate cancer. Nature Medicine. 2003;9(10):1265–1266. doi: 10.1038/nm928. [DOI] [PubMed] [Google Scholar]
- 120.Khuri FR, Lee JJ, Lippman SM, et al. Randomized phase III trial of low-dose isotretinoin for prevention of second primary tumors in stage I and II head and neck cancer patients. Journal of the National Cancer Institute. 2006;98(7):441–450. doi: 10.1093/jnci/djj091. [DOI] [PubMed] [Google Scholar]
- 121.'Shaughnessy JA O, Kelloff GJ, Gordon GB, et al. Treatment and prevention of intraepithelial neoplasia: an important target for accelerated new agent development. Clinical Cancer Research. 2002;8(2):314–346. [PubMed] [Google Scholar]
- 122.Evans RM. The nuclear receptor superfamily: a Rosetta stone for physiology. Molecular Endocrinology. 2005;19(6):1429–1438. doi: 10.1210/me.2005-0046. [DOI] [PubMed] [Google Scholar]
- 123.Woo CC, Kumar AP, Sethi G, Tan KH. Thymoquinone: potential cure for inflammatory disorders and cancer. Biochemical Pharmacology. 2012;1583(4):443–451. doi: 10.1016/j.bcp.2011.09.029. [DOI] [PubMed] [Google Scholar]
- 124.Woo CC, Loo SY, Gee V, et al. Anticancer activity of thymoquinone in breast cancer cells: possible involvement of PPAR-γ pathway. Biochemical Pharmacology. 2011;82(5):464–475. doi: 10.1016/j.bcp.2011.05.030. [DOI] [PubMed] [Google Scholar]
- 125.Hisatake JI, Ikezoe T, Carey M, Holden S, Tomoyasu S, Koeffler HP. Down-regulation of prostate-specific antigen expression by ligands for peroxisome proliferator-activated receptor γ in human prostate cancer. Cancer Research. 2000;60(19):5494–5498. [PubMed] [Google Scholar]
- 126.Segawa Y, Yoshimura R, Hase T, et al. Expression of peroxisome proliferator-activated receptor (PPAR) in human prostate cancer. The Prostate. 2002;51(2):108–116. doi: 10.1002/pros.10058. [DOI] [PubMed] [Google Scholar]
- 127.Saez E, Tontonoz P, Nelson MC, et al. Activators of the nuclear receptor PPARγ enhance colon polyp formation. Nature Medicine. 1998;4(9):1058–1061. doi: 10.1038/2042. [DOI] [PubMed] [Google Scholar]
- 128.Lefebvre AM, Chen I, Desreumaux P, et al. Activation of the peroxisome proliferator-activated receptor γ promotes the development of colon tumors in C57BL/6J-APC(Min)/+ mice. Nature Medicine. 1998;4(9):1053–1057. doi: 10.1038/2036. [DOI] [PubMed] [Google Scholar]
- 129.Kulke MH, Demetri GD, Sharpless NE, et al. A phase II study of troglitazone, an activator of the PPARγ receptor, in patients with chemotherapy-resistant metastatic colorectal cancer. The Cancer Journal. 2002;8(5):395–399. doi: 10.1097/00130404-200209000-00010. [DOI] [PubMed] [Google Scholar]
- 130.Subbarayan V, Sabichi AL, Kim J, et al. Differential peroxisome proliferator-activated receptor-γ isoform expression and agonist effects in normal and malignant prostate cells. Cancer Epidemiology Biomarkers and Prevention. 2004;13(11):1710–1716. [PubMed] [Google Scholar]