

Abstract
The 5-HT3 receptor, a pentameric ligand-gated ion channel (pLGIC), is an important therapeutic target. During a recent fragment screen, 6-chloro-N-methyl-2-(4-methyl-1,4-diazepan-1-yl)quinazolin-4-amine (1) was identified as a 5-HT3R hit fragment. Here we describe the synthesis and structure–activity relationships (SAR) of a series of (iso)quinoline and quinazoline compounds that were synthesized and screened for 5-HT3R affinity using a [3H]granisetron displacement assay. These studies resulted in the discovery of several high affinity ligands of which compound 22 showed the highest affinity (pKi > 10) for the 5-HT3 receptor. The observed SAR is in agreement with established pharmacophore models for 5-HT3 ligands and is used for ligand–receptor binding mode prediction using homology modeling and in silico docking approaches.
1. Introduction
5-HT3 receptor antagonists like alosetron and granisetron are in clinical use to prevent emesis during chemotherapy-induced, radiotherapy-induced and postoperative nausea and vomiting and to alleviate the effects of irritable bowel syndrome (IBS).1,2 Recent studies have also indicated that the 5-HT3 receptor is involved in depression3 and may play a role in a range of other indications such as schizophrenia, anxiety, substance abuse and addiction, bulimia and pruritus. Moreover, the 5-HT3 receptor is thought to modulate analgesia, inflammation and cognitive processes.4 The 5-HT3 receptor belongs to the Cys-loop receptor family of ion channels,5 which include nicotinic acetylcholine (nACh), GABAA, and glycine receptors. All of these receptors consist of five subunits that surround a central ion conducting pore.6 At present, no high resolution structures are available for Cys-loop receptors, but the availability of crystal structures of the closely related acetylcholine binding protein (AChBP)7 has significantly improved our understanding of the extracellular domain. Together with the results from mutagenesis studies8−15 it is now acknowledged that the ligand binding site is situated in the extracellular domain at the interface of two subunits. The principal subunit contributes three loops (A–C), while the complementary side contributes three beta-sheets (“loops” D–F) from the adjacent subunit (Figure 1a). 5-HT3 receptors can be homomeric (all A subunits) or heteromeric (A + B to E), but the exact subunit composition/stoichiometry of the latter type is not yet clear.16,17 As early as 1989, a ligand-based pharmacophore for 5-HT3 receptor ligands was constructed.18 During the last two decades, this pharmacophore has been continuously refined.19−25 The consensus is that 5-HT3 receptor ligands share three pharmacophore features: an aromatic part (A), a basic moiety (B) and an intervening hydrogen bond acceptor (C) moiety (Figure 1b). Interestingly, Jørgensen and co-workers recently published 5-HT3A receptor ligands that lack a positively charged basic moiety, and it is suggested that these compounds exert their effects via an allosteric site of the receptor.26
Figure 1.

(a) Extracellular domains of two adjacent subunits of the 5-HT3 receptor. (b) Pharmacophore features for 5-HT3 receptor antagonists. A, B and C indicate respectively an aromatic part, a basic moiety and a hydrogen bond acceptor. (c) Structure of hit fragment VUF10434 (1).
Recently, we performed a cell-based fragment screen of 1010 fragments on the human 5-HT3A receptor resulting in a total of 70 hits.27 Within this set, the higher affinity ligands contained bicyclic heteroaromatic structures, that is, quinoxalines, quinazolines or quinolines. In the literature, fused six-membered aromatic rings have been described as 5-HT3 receptor ligands, such as quipazine, which contains a quinoline scaffold.28 Quipazine analogues have also been described as multicyclic aryl piperazine 5-HT3R ligands by Anzini and co-workers.29−32 In our screen, the quinazoline scaffold, VUF10434 (Figure 1c) was identified as having the highest affinity (18 nM) and a ligand efficiency (LE) of 0.5 (calculated as the ratio of Gibbs free energy in kcal to the amount of non-hydrogen atoms). Here, we examine the SAR of VUF10434 (1) by designing, synthesizing and testing a range of new derivatives and exploring their putative water-mediated binding modes in a 5-HT3A receptor binding site homology model.
2. Chemistry
Quinazolines and (iso)quinolines were either obtained from our in-house compound library or were synthesized as described below. 2,4-Dichloroquinazoline (2) was reacted with methylamine to yield the corresponding 4-substituted intermediate (5) which was directly used without further purification in the substitution reaction with N-methylpiperazine to form compound 21 (R2 = NMe, R3 = H) (Scheme 1). 2,4,6-Trichloroquinazoline (3) was reacted with tributyltin hydride and tetrakis(triphenylphosphine)palladium(0) to yield 2,6-dichloroquinazoline (4) which in turn was substituted with N-methylpiperazine under microwave irradiation to yield 11 (R2 = H, R3 = 6-Cl).
Scheme 1. Preparation of Quinazoline Compounds 6–22.

Reagents: (a) amine (R2H) EtOH or EtOAc, rt; (b) Bu3SnH, Pd(PPh3)4, toluene, 100 °C; (c) N-methylpiperazine, EtOAc, 140 °C; (d) (i) MeNH2, EtOAc, DiPEA, rt, (ii) amine (R1H), EtOAc, DiPEA, 120 °C; (e) (i) MeNH2, N-methylpyrrolidone, DiPEA, rt, (ii) amine (R1H), N-methylpyrrolidone, DiPEA, 150 °C.
Starting from 2-aminobenzoic acid (23), 2-thioxo-2,3-dihydroquinazolin-4(1H)-one (24) was synthesized using a procedure described in literature33 (Scheme 2). This intermediate is allowed to react with methyl iodide to create a more reactive intermediate (25) that is used in the subsequent substitution reaction with N-methylpiperazine which results in the formation compound 26 (Scheme 2).
Scheme 2. Preparation of 2-(4-Methylpiperazin-1-yl)quinazolin-4(3H)-one (26).

Reagents: (a) SOCl2, reflux; (b) NH4SCN, acetone, rt; (c) CH3I, aqueous NaOH, rt; (d) N-methylpiperazine, 160 °C.
Compounds 30 and 31 (Scheme 3) were synthesized by reacting commercially available 2,4-dibromoquinoline (27) with either ammoniumhydroxide or methylamine followed by coupling of N-methylpiperazine. This synthesis route yielded in the first step mixtures of both the 2- as well as the 4-substituted regio isomers (for 28 and 29). Since these two regioisomers were difficult to separate, it was decided to use them as regioisomeric mixtures in the substitution reaction with N-methylpiperazine. The obtained regioisomeric mixtures (for 30 and 31) could be separated by column chromatography. Both compound 30 and 31 were obtained as pure regioisomers whose identity was confirmed by 2D 1H NMR (NOESY, see Supporting Information, Figure S2).
Scheme 3. Preparation of Quinoline Compounds 30 and 31.

Reagents: (a) Aqueous NH4OH, 140 °C; (b) MeNH2, DiPEA, EtOAc, 100 °C; (c) N-methylpiperazine, THF or neat, 160 °C.
2-Methoxynaphtalene (32) was substituted with N-methylpiperazine using n-butyllithium as the base to create compound 33 in a good yield (Scheme 4).
Scheme 4. Preparation of 1-Methyl-4-(naphthalen-2-yl)piperazine (33).

Reagents: (a) N-Methylpiperazine, n-BuLi, 0 °C, then 2-methoxynaphthalene (32), THF, rt.
To investigate the role of the position of the heteroatom in the aromatic ring, 34 was reacted with deprotonated N-methylpiperazine in order to obtain 35 (Scheme 5).
Scheme 5. Preparation of 3-(4-Methylpiperazin-1-yl)quinoline (35).

Reagents: (a) NaNH2, t-BuOH, THF, 0 °C, N-methylpiperazine, 40 °C, then 3-bromoquinoline (34), 0 °C to rt.
For the synthesis of compounds 39 and 40 a similar approach was used as for the synthesis of compounds 30 and 31. First, the commercially available 1,3-dichloroisoquinoline (36) was reacted with either ammoniumhydroxide or methylamine to yield 37 or 38 respectively as the single 1-substituted regioisomers. Both intermediates were then used without purification in the reaction with N-methylpiperazine to yield 39 and 40 as the single regioisomers (Scheme 6).
Scheme 6. Preparation of isoquinoline compounds 39 and 40.

Reagents: (a) NH3 in MeOH, 120 °C; (b) MeNH2, DiPEA, EtOH, 100 °C; (c) N-methylpiperazine, 220 °C.
Compound 41 was reacted with a mixture of potassium nitrate and sulfuric acid that in situ yields the reactive nitric acid. This yielded a mixture of regioisomers with the 5- and 8-nitro-compounds (42, 43) as the main products. These regioisomers were separated by column chromatography and each individual regioisomers was subsequently reacted with N-methylpiperazine to yield nitro analogs 44 and 45. These intermediates in turn were reduced to the corresponding anilines (46, 47) with palladium on carbon and hydrogen gas (Scheme 7).
Scheme 7. Preparation of Quinolines 46 and 47.

Reagents: (a) H2SO4, KNO3, 0 °C to rt; (b) N-methylpiperazine, 140 °C; (c) H2, Pd/C, MeOH, rt.
3. Biochemical Evaluation and SAR Studies
Target compounds were evaluated using competition with the 5-HT3-specific radioligand [3H]granisetron34 and most compounds displayed high affinity (expressed here as pKi) for the 5-HT3A receptor (Tables 1–5). With the quinazoline scaffold of compound 1 as a starting point, the role of the basic moiety as a key feature in the 5-HT3 pharmacophore first was explored. A series of N-methylhomopiperazine replacements were investigated (Table 1) by introducing several other cyclic amines. The N-methylpiperazine analogue 6 shows a 16-fold increase in affinity. All other compounds, including rigid tertiary amine 7, rigid secondary amines (8, 9) and flexible tertiary amine 10, show reduced affinity for the 5-HT3A receptor. Thus, for this series of compounds, the N-methyl piperazine moiety at the R1 position results in the highest affinity.
Table 1. 5-HT3A Receptor Binding Affinities (pKi) of Compounds 1, 6–10.
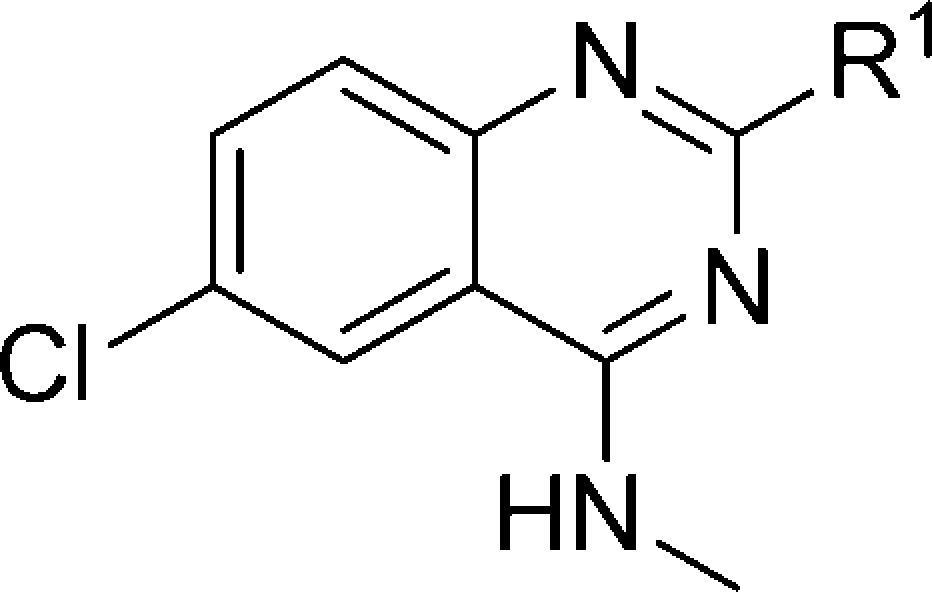

Determined by radioligand competition using [3H]granisetron.35
Table 5. 5-HT3A Receptor Binding Affinities (pKi) of Different (Methyl)aniline Compounds.

Determined by radioligand competition using [3H]granisetron.
Next, keeping the N-methylpiperazine moiety at the R1-position, we explored the SAR associated with the R2 position (Table 2). Removal of the substituent in the R2 position (i.e., R2 = H), leading to compound 11, results in a ∼10-fold reduction in affinity. The same reduction in affinity is observed for the removal or addition of a methyl group on the aniline nitrogen atom (compounds 12, 13). Interesting is the ∼500-fold lower affinity of compound 48 (Table 2). This compound can adopt distinct tautomeric states (Figure 2) and consequently nitrogen atom N3 of compound 48b is no longer capable of interacting as a HBA.
Table 2. 5-HT3A Receptor Binding Affinities (pKi) of Compounds with Different Substituents at R2.
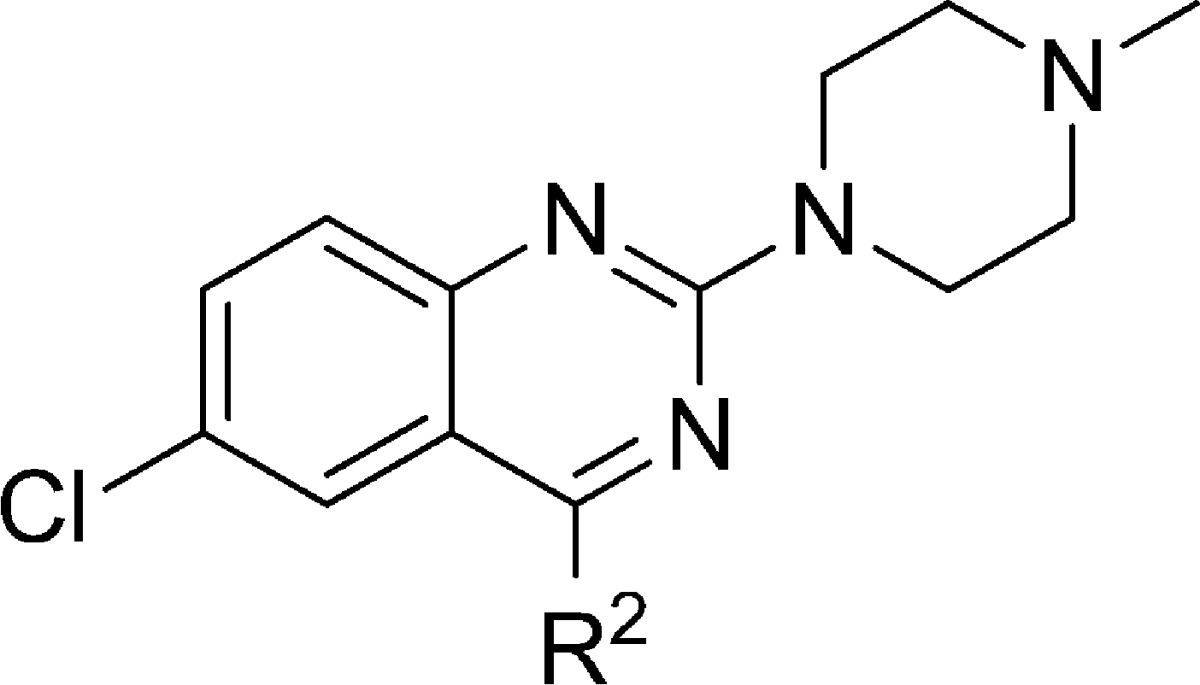

Determined by radioligand competition using [3H]granisetron
Figure 2.

Tautomers of compound 48.
Analogs that contain more bulky groups at position R2, including saturated ring systems (15) and aromatic moieties (17), all show lower affinities than the lead compound (Table 2). Introduction of a large polar group results in high affinity 5-HT3 compounds 18 and 20. The SAR at both the R1 and R2 position follow similar trends, as observed for the histamine H4R,35−37 and agrees with our earlier reports that fragment library screening indicates a remarkable degree of binding site similarities of the 5-HT3AR and H4R proteins.38
In Table 3, the effect of the 6-chloro atom on 5-HT3 receptor affinity is shown. Replacement of the 6-chloro atom by a hydrogen atom results in a ∼5-fold reduction in affinity (compare 6 and 21 respectively) when R2 = NMe. Replacement of R3 = 6-Cl by R3 = H results in analogs that have a higher affinity for the 5-HT3A receptor when R2 = H, OH or NH2 (the affinity increases ∼5, ∼10, and ∼100 fold, respectively). Interestingly, for the H4R the latter modification leads to a reduction in affinity of more than 10-fold, marking a clear difference in SAR for 5-HT3 and H4R receptors.35 Ultimately, the SAR for these receptors is different, as can be deduced by comparing the different affinities of compounds 6, 8–10, 12–20, 22 and 48–5235−37 which have been synthesized, evaluated for H4R affinity and published by our group earlier. Importantly, compound 22(35) is now identified as a 5-HT3 ligand with subnanomolar affinity and ∼40 000 fold selectivity for the 5-HT3A receptor over the H4R.
Table 3. Effect of R3 = Cl or R3 = H on the 5-HT3AR Affinity.
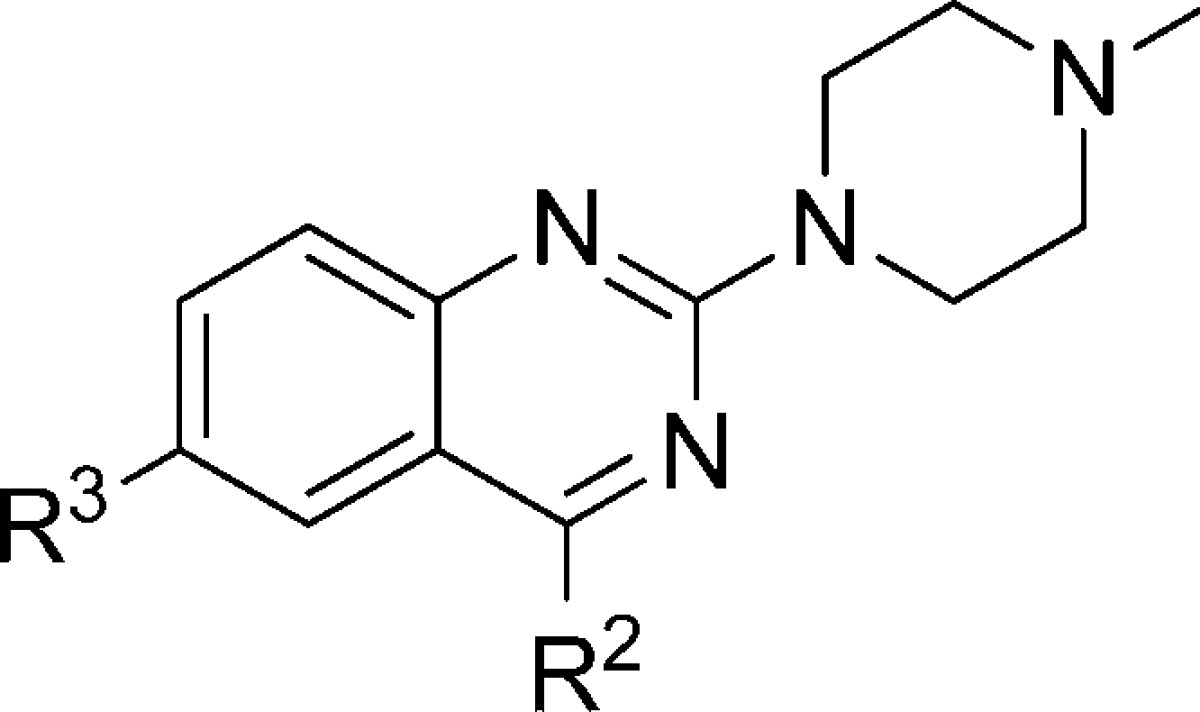
| # | R2 | R3 | pKia |
|---|---|---|---|
| 11 | H | Cl | 8.13 ± 0.27 |
| 49 | H | H | 8.87 ± 0.10 |
| 48 | OH | Cl | 6.24 ± 0.24 |
| 26 | OH | H | 7.33 ± 0.12 |
| 12 | NH2 | Cl | 7.92 ± 0.09 |
| 22 | NH2 | H | 10.29 ± 0.15 |
| 6 | NMe | Cl | 8.95 ± 0.05 |
| 21 | NMe | H | 8.53 ± 0.05 |
Determined by radioligand competition using [3H]granisetron.
The role of the H-bond acceptor moiety was explored using the ligands in Table 4. As already illustrated by the pharmacophore model in Figure 1B, a distance of ∼5 Å between the basic moiety and the H-bond acceptor is essential. This is in line with our present findings, as the distance between these pharmacophore features in the minimal global energy conformation of our new compounds is calculated to be ∼5 Å. The highest affinity compounds are those where the heteroaromatic nitrogen atom is positioned next to the N-methylpiperazine group. These are compounds 49 (N1 = 4.96 Å, N3 = 4.97 Å), 51 (4.71 Å) and 52 (4.75 Å), with the highest affinity compound (51) having a pKi > 9. The affinity of compound 50 is 3–10 fold lower than compounds 49, 51 and 52; here the distance of N1 to the basic nitrogen atom is 4.79 Å while the distance for N4 is 6.42 Å. The distance between the basic nitrogen atom and the H-bond acceptor in compound 35 is 6.53 Å, and affinity drops >250 fold when compared to (iso)quinolines 51 and 52. The importance of this pharmacophore feature is supported by the fact that compound 33, which has no H-bond acceptor in the ring, shows similar affinity as compound 35.
Table 4. 5-HT3A Receptor Binding Affinities (pKi) of Compounds 33, 35 and 49–52.

Determined by radioligand competition using [3H]granisetron.
For quinazolines (Table 3) R2 = NH2 in combination with R3 = H results in a significant increase in affinity (22). Therefore, the same derivatization strategy was applied to the corresponding (iso)quinolines (Table 5). Here, 4-NMe substitution of the quinoline scaffold (31) gives a ∼30-fold drop in affinity. In addition, the 4-NH2 analogue (30) also shows a loss of affinity although to a lesser extent. In order to explore the effect of the aniline moiety when positioned on the second aromatic ring of the quinoline moiety, two additional compounds were synthesized. The compound that has the aniline functionality at the 5-position of the quinoline scaffold resulted in compound 46. The 5-HT3AR affinity dropped ∼1000-fold and ∼100-fold when compared to compounds 51 and 30, respectively. The 8-aniline (47) derivative shows a comparable affinity to compound 46. Finally, a similar approach for the isoquinoline scaffold results in compounds 40 and 39. Here, addition of a 2-NMe moiety (compound 40) results in a ∼5-fold lower affinity. The 2-NH2 analogue (compound 39), however, shows a 140-fold decrease in affinity when compared to the parent isoquinoline compound 52.
To reassure that the most active compound (22) has no cross-target affinities, this compound was subjected to a broader pharmacological screening panel at a concentration of 1000 times its Ki for 5-HT3AR. Compound 22 shows no affinity for other closely related receptors, except for nACh (α7) and 5-HT2B receptors for which 31% and 43% inhibition is observed respectively (Table 6 and Supporting Information, Tables S1–S3).
Table 6. Cross-target Pharmacology of Compound 22 at a Concentration of 0.1 μM.
| target | radioligand | % inhibition of control specific binding ± s.e.m.a |
|---|---|---|
| GABAA1 (α1,β2,γ2)b | [3H]muscimol | <15 |
| Glycinec | [3H]strychnine | <15 |
| nACh (α4β2)b | [3H]cytisine | <15 |
| nACh (α7)b | [3H]epibatidine | 31 ± 1 |
| 5-HT1Ab | [3H]8-OH-DPAT | <15 |
| 5-HT1Bc | [125I]CYP (+30 μM isoproterenol) | <15 |
| 5-HT1Dc | [3H]serotonin | <15 |
| 5-HT2Ab | [125I](±)DOI | <15 |
| 5-HT2Bb | [125I](±)DOI | 43 ± 0 |
| 5-HT2Cb | [125I](±)DOI | <15 |
| 5-HT4Eb | [3H]GR113808 | <15 |
| 5-HT6b | [3H]LSD | <15 |
| 5-HT7b | [3H]LSD | <15 |
Results are expressed as percent inhibition of control specific binding obtained in the presence of compound 22. Results showing an inhibition <15% are considered non binding and are displayed as <15 (see Supporting Information for more details).
Human receptor.
Rat receptor.
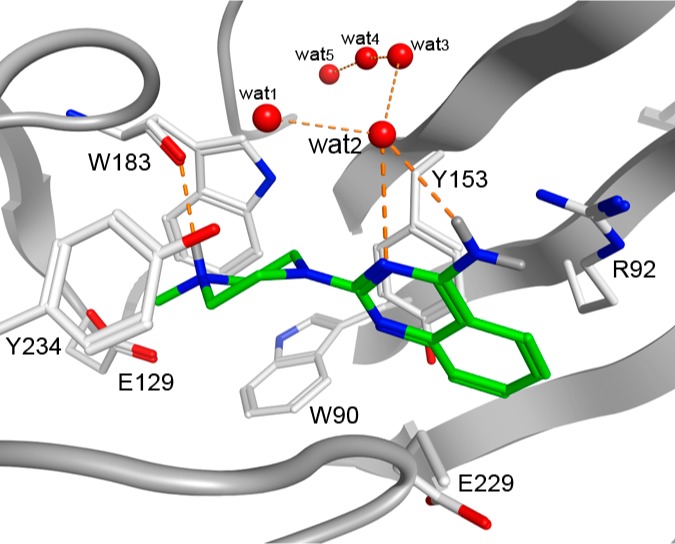
Although protein structural information of cys-loop receptors is very limited, the emerging crystallographic data on AChBP structures, in combination with 5-HT3R site directed mutagenesis studies allow some preliminary considerations with respect to protein–ligand interactions. A homology model of the 5-HT3A receptor binding site was constructed using the tropisetron bound AChBP crystal structure (PDB ID: 2WNC) as a template.39 The derived binding orientation of tropisetron is in agreement with published site-directed mutagenesis studies. The basic tropane moiety of tropisetron interacts with the carbonyl backbone of W183 and is positioned in an aromatic cavity consisting of W18311,40,41 and Y2348,42 from the principle subunit, and W9013,43 of the complementary subunit. The basic nitrogen atom of tropisetron is positioned at 4.5 Å from E129,9 a residue that is critical for both serotonin and granisetron binding. The indole ring of tropisetron is in close proximity to R92,13,43 which can interact with this moiety through a cation-π interaction, and has been previously identified to interact with the indazole ring of granisetron.15,43 Finally, five water molecules from the template cocrystal structure (wat1–wat5) were included; these form a structural water network that interacts with both the carbonyl moiety of tropisetron and the receptor.39 We found that these water molecules have a high level of conservation across the AChBP structures that are cocrystallized with small antagonists (Figure 3a).44 Compounds were docked into the homology model with GOLD45 using standard settings and the resulting poses were scored with GOLDscore.46
Figure 3.

AChBP-based homology model of the human 5-HT3A receptor binding site (protein carbon atoms and cartoon representation in white). (a) Overlay of the waters participating in the structural water network in several different AChBP crystal structures (red: 2WNC; blue: 2BYR; cyan: 2PGZ; green: 2BYS; yellow: 2XYT). (b–d) Binding poses for the 5-HT3 receptor antagonists. (b) Tropisetron (gold sticks). (c) Compound 22 (green sticks). (d) Compound 35 (green sticks).
From the observed docking poses of compounds 22 and 35 (Figure 3c–d) and other novel ligands (data not shown) we speculate that the basic moiety of these ligands interacts with the same residues as observed for the basic moiety of the reference ligand tropisetron (Figure 3b). In the binding model, heteroaromatic HBA (N3) of the quinazoline ring of compound 22 can interact with a structural water molecule (wat2) in the binding site (Figure 3c) similar to the carbonyl oxygen atom of tropisetron. This interaction is also possible for other ligands where the distance between the basic nitrogen atom and the HBA is ∼5 Å. Therefore, this protein–ligand model is able to accommodate the classic ligand-based 5-HT3R pharmacophore (Figure 1b) by suggesting that the hydrogen bond acceptor feature (C) of the ligands binds to the protein via conserved water molecules, thereby explaining the subtle differences in SAR with respect to this pharmacophore feature. Future crystallization studies of 5-HT3R ligands with AChBP might lead to additional insights with respect to binding mode, although the ultimate proof via crystallization studies of the actual 5-HT3R remains a scientific challenge. Nevertheless, careful SAR studies and ligand-based design approaches in combination the use of AChBP derived structural information and site-directed mutagenesis might lead to insights that enable structure-based drug design approaches that have proven to be efficient for water-soluble drug targets.
Conclusion
In summary, optimization of fragment hit (1) has led to the identification of several novel 5-HT3 receptor ligands with (sub)nanomolar affinities that are comparable to some of the most potent 5-HT3 ligands described to date. These ligands match the known pharmacophore descriptors for 5-HT3 ligands. We found that the N-methylpiperazine group at the R1 position is favorable, and that a hydrogen bond acceptor at ∼5 Å from the basic nitrogen is essential for high affinity binding. Compound 22 was identified as the ligand with the highest 5-HT3 receptor affinity in this study (pKi = 10.29) and showed at least a-1000 fold selectivity over related targets. The high affinity and previously reported receptor specificity of closely related compounds47 suggests that some of these compounds will make ideal candidates for future in vivo studies.
Experimental Section
Chemistry
Chemicals and solvents were purchased from Aldrich and used as received. Unless indicated otherwise, all reactions were carried out under an inert atmosphere of dry N2. TLC analyses were performed with Merck F254 alumina silica plates using UV visualization or staining. Column purifications were carried out automatically using the Biotage equipment. All HRMS spectra were recorded on Bruker microTOF mass spectrometer using ESI in positive ion mode. 1H NMR spectra were recorded on a Bruker 250 (250 MHz) or a Bruker 500 (500 MHz) spectrometer. Data are reported as follows: chemical shift, integration, multiplicity (s = singlet, d = doublet, t = triplet, br = broad, m = multiplet), and coupling constants (Hz). Chemical shifts are reported in ppm with the natural abundance of deuterium in the solvent as the internal reference (CHCl3 in CDCl3: δ 7.26 and CH3OH in CH3OD: δ 3.31, (CH3)2SO in (CD3)2SO: δ 2.50). 13C NMR spectra were recorded on a Bruker 500 (126 MHz) spectrometer with complete proton decoupling. Chemical shifts are reported in ppm with the solvent resonance resulting from incomplete deuteration as the internal reference (CDCl3: δ 77.16, CH3OD: δ 49.00, (CD3)2SO: δ 39.52). Systematic names for molecules according to IUPAC rules were generated using the Chemdraw AutoNom program. Purity was determined using a Shimadzu HPLC/MS workstation with a LC-20AD pump system, SPD-M20A diode array detection, and a LCMS-2010 EV liquid chromatography mass spectrometer. The column used is an Xbridge C18 5 μm column (100 mm ×4.6 mm). Compound purities were calculated as the percentage peak area of the analyzed compound by UV detection at 230 nm. Solvents used were the following: solvent B = MeCN 0.1% Formic Acid; solvent A = water 0.1%. The analysis was conducted using a flow rate of 1.0 mL/min, start 5% B, linear gradient to 90% B in 4.5 min, then 1.5 min at 90% B, linear gradient to 5% B in 0.5 min and then 1.5 min at 5% B, total run time of 8 min. Compounds 6, 8–10, 12–20, 22 and 48–52 were synthesized by our group as described by Smits et al.35−37
2-Chloro-N-methylquinazolin-4-amine (5)
Methanamine in ethanol (40%, w/v, 0.24 mL, 2.76 mmol) and DiPEA (357 mg, 2.76 mmol) were added to a suspension of 2,4-dichloroquinazoline (2) (500 mg, 2.51 mmol) in EtOH (20 mL) and stirred at rt. After 4.5 h the reaction mixture was concentrated under reduced pressure to a volume of 1 mL. The resulting mixture was diluted with water (25 mL) and extracted with EtOAc. The combined organic extracts were dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to yield 444 mg of 5 (2.29 mmol, 91%) as a white solid. 1H NMR (250 MHz, CDCl3) δ ppm 7.86–7.61 (m, 3H), 7.44 (m, 1H), 6.11 (br s, 1H), 3.22 (d, J = 4.9, 3H).
6-Chloro-2-(3-(dimethylamino)azetidin-1-yl)-N-methylquinazolin-4-amine (7)
Methanamine in ethanol (33%, w/v, 0.11 mL, 0.86 mmol) and DiPEA (0.61 mL, 3.5 mmol) were added to a suspension of 2,4,6-trichloroquinazoline (3) (200 mg, 0.86 mmol) in EtOAc (2 mL) and stirred at rt until TLC indicated complete conversion. Then N,N-Dimethyl-3-azetidinamine dihydrochloride (150 mg, 0.87 mmol) was added and the resulting suspension was heated by microwave irradiation at 120 °C for 30 min. The resulting mixture was diluted with water and extracted with EtOAc. The combined organic extracts were washed with brine and dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude oil was purified over SiO2 (EtOAc/MeOH/Et3N = 90/5/5, v/v/v) to yield 98 mg of 7 (2.29 mmol, 91%) as a beige solid. 1H NMR (500 MHz, CDCl3) δ ppm 7.47–7.38 (m, 3H), 5.54 (br s, 1H), 4.23–4.16 (m, 2H), 4.06–3.99 (m, 2H), 3.24–3.14 (m, 1H), 3.11 (d, J = 4.8 Hz, 3H), 2.22 (s, 6H); 13C NMR (126 MHz, CDCl3) δ ppm 160.56, 159.70, 150.40, 132.90, 127.22, 125.65, 120.25, 111.31, 55.99, 54.31, 41.91, 28.01; LCMS: ret. time 2.06 min, purity >99%, [M + H]+ 292.05; HRMS m/z: [M + H]+ calcd for C14H19ClN5: 292.1323, found: 292.1312.
6-Chloro-2-(4-methylpiperazin-1-yl)quinazoline (11)
Tributyltin hydride (312 mg, 1.07 mmol) was added dropwise to a round-bottom flask containing 2,4,6-trichloroquinazoline (3) (250 mg, 1.07 mmol) in dry toluene (5 mL). Then tetrakis (triphenylphosphine)palladium(0) (60 mg, 0.05 mmol) was added and the reaction mixture was stirred at 100 °C for 1 h. Next, toluene was removed under reduced pressure, the residue was dissolved in DCM (5 mL) and hydrolyzed with a saturated solution of potassium fluoride. The mixture was stirred vigorously for 30 min, filtered over a pad of Celite and washed with DCM. The aqueous phase was extracted with DCM and the combined organic extracts were dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure. The residue was purified over SiO2 (Hept/DCM = 50/50 to 0/100, v/v) yielding 30 mg of 4. This crude intermediate was then added to a microwave tube containing N-methylpiperazine (1 mL, 9.0 mmol) and EtOAc (3 mL). The resulting mixture was heated at 140 °C for 30 min under microwave irradiation. The solvent and excess of N-methylpiperazine were removed under reduced pressure and the residue was purified over SiO2 (KPNH) (DCM/EtOAc = 90/10 to 60/40, v/v) yielding 20 mg of 11 (0.08 mmol, 7% over two steps) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ ppm 8.92 (s, 1H), 7.62 (d, J = 2.2 Hz, 1H), 7.59–7.56 (m, 1H), 7.50 (d, J = 9.0 Hz, 1H), 4.02–3.93 (m, 4H), 2.56–2.47 (m, 4H), 2.36 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 160.47, 159.23, 150.89, 134.80, 127.40, 127.37, 126.01, 119.87, 55.01, 46.22, 43.92; LCMS: ret. time 2.71 min, purity 97%, [M + H]+ 263.05, HRMS m/z: [M + H]+ calcd for C13H16ClN4: 263.1058, found: 263.1048.
N-Methyl-2-(4-methylpiperazin-1-yl)quinazolin-4-amine (21)
5 (400 mg, 2.07 mmol) was added to a microwave tube containing N-methylpiperazine (1.6 mL, 14.5 mmol) and EtOAc (5 mL). The resulting mixture was heated at 140 °C for 120 min under microwave irradiation. The reaction mixture was diluted with H2O (10 mL) and extracted with EtOAc. The combined organic extracts were dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure and purified over SiO2 (EtOAc/Hept/Et3N = 80/15/5, v/v) yielding 330 mg of compound 21 (1.28 mmol, 62%) as a white solid. 1H NMR (250 MHz, CDCl3) δ ppm 7.60–7.33 (m, 3H), 7.03–6.96 (m, 1H), 5.74 (br s, 1H), 4.09–3.80 (m, 4H), 3.07 (d, J = 4.8 Hz, 3H), 2.60–2.36 (m, 4H), 2.31 (s, 3H); 13C NMR (126 MHz, CDCl3) δ ppm 160.46, 159.09, 151.99, 132.38, 125.69, 120.85, 120.78, 110.66, 55.23, 46.29, 43.84, 27.97; LCMS: ret. time 1.49 min, purity >99%, [M + H]+ 258.05; HRMS m/z: [M + H]+ calcd for C14H20N5: 258.1713, found: 258.1681.
2-Thioxo-2,3-dihydroquinazolin-4(1H)-one (24)
A suspension of anthranilic acid (23) (10.0 g, 73.0 mmol) in thionyl chloride (40.0 mL) was heated at reflux for 2 h. The thionyl cloride was removed under reduced pressure, DCM (20 mL) was added to the residue and was also removed under reduced pressure (repeated three times). Next, the crude acid chloride was dissolved in acetone (20 mL) and added dropwise to a suspension of NH4SCN (5.75 g, 75.5 mmol) in acetone (10 mL). The reaction mixture was stirred at rt for 45 min and the solid was collected by filtration over a Büchner filter. This solid was then suspended in an aqueous solution of NaOH (10% w/w) (70 mL) and filtered over a Büchner filter. Water (70 mL) was added to the residue and the resulting mixture was acidified to pH 2 with aqueous 2N HCl, the resulting solid was collected via filtration over a Büchner funnel. After washing with a H2O/MeO (50:50) solution (100 mL) and subsequent drying under vacuum at 35 °C. The product, 10.0 g of 24, was obtained as a beige solid and used in the next step without further purification.
2-(Methylthio)quinazolin-4(3H)-one (25)
A solution of methyliodide (17.9 g, 126 mmol) in MeOH (80 mL) was added to a suspension of 24 (10.0 g, 56.2 mmol) in an aqueous solution of NaOH (1% w/w) (80 mL). After 1 h at rt the pH was adjusted to pH 7 with an aqueous solution of HCl (1 M) and the solvents were removed under reduced pressure. A total of 7.0 g 25 was obtained as an off white solid and used in the synthesis of 26 without further purification.
2-(4-Methylpiperazin-1-yl)quinazolin-4(3H)-one (26)
Compound 25 (2.00 g, 10.4 mmol) was added to a microwave tube containing N-methylpiperazine (10.0 mL, 111 mmol). The resulting mixture was heated at 160 °C under microwave irradiation for 30 min. The excess of N-methylpiperazine was removed under reduced pressure and the residue was suspended in H2O (30 mL). The insoluble material was dissolved by addition of an aqueous NaOH solution (10% w/w, 10 mL). After filtration over a Büchner filter to remove insolubles, the filtrate was concentrated under reduced pressure and purified over SiO2 (KPNH) (EtOAc) yielding 1.08 g of 26 (4.43 mmol, 43%) as an off white solid. 1H NMR (250 MHz, MeOD) δ ppm 8.04 (dd, J = 8.0, 1.2 Hz, 1H), 7.70–7.60 (m, 1H), 7.46–7.39 (m, 1H), 7.33–7.22 (m, 1H), 3.85–3.73 (m, 4H), 2.65–2.55 (m, 4H), 2.39 (s, 3H). 13C NMR (63 MHz, CDCl3) δ ppm 165.68, 151.35, 150.44, 134.97, 126.25, 125.35, 122.68, 116.85, 54.79, 46.20, 45.14; LCMS: ret. time 2.04 min, purity >99%, [M + H]+ 244.95; HRMS m/z: [M + H]+ calcd for C13H17N4O: 245.1397, found: 245.1401.
2-Bromoquinolin-4-amine and 4-bromoquinolin-2-amine (28)
(mixture of both regio isomers). 2,4-Dibromoquinoline (27) (860 mg, 3.00 mmol) was added to a microwave tube containing an aqueous solution of ammonium hydroxide (28–30%, w/v, 10 mL). The resulting mixture was heated at 140 °C under microwave irradiation for 120 min. The reaction mixture was diluted with water (15 mL) and subsequently extracted with DCM (3 × 15 mL). The combined organic extracts were dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure. The resulting crude product was purified over SiO2 (Hept/EtOAc = 50/50, v/v) yielding 378 mg (1.70 mmol, 57%) of a regioisomeric mixture containing 2-(4-methylpiperazin-1-yl)quinolin-4-amine and 4-(4-methylpiperazin-1-yl)quinolin-2-amine.
2-(4-Methylpiperazin-1-yl)quinolin-4-amine (30)
The regioisomeric mixture (28) (378 mg, 1.70 mmol) was added to a microwave tube containing THF (4 mL) and N-methylpiperazine (1.20 g, 12.0 mmol). The resulting mixture was heated at 160 °C for 90 min under microwave irradiation. The reaction mixture was quenched with a saturated aqueous solution of NaHCO3 (4 mL) and extracted with EtOAC (100 mL). The combined organic extracts were then washed with H2O. The combined organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified over SiO2 (KPNH) (DCM/MeOH = 95/5, v/v) yielding 100 mg (0.41 mmol) of 30 as an off white solid. 1H NMR (250 MHz, CDCl3) δ ppm 7.66 (d, J = 7.8 Hz, 1H), 7.55 (d, J = 8.2 Hz, 1H), 7.52–7.45 (m, 1H), 7.19–7.13 (m, 1H), 6.16 (s, 1H), 4.51 (br s, 2H), 3.72–3.63 (m, 4H), 2.57–2.45 (m, 4H), 2.34 (s, 3H); 13C NMR (63 MHz, CDCl3) δ ppm 158.65, 150.48, 148.63, 129.63, 127.23, 121.30, 119.91, 115.41, 90.34, 55.13, 46.24, 45.14; LCMS: ret. time 5.24 min, purity >99%, [M + H]+ 243.10; HRMS m/z: [M + H]+ calcd for C14H19N4: 243.1604, found: 243.1596.
N-Methyl-2-(4-methylpiperazin-1-yl)quinolin-4-amine (31)
Methanamine in MeOH (40%, w/v, 0.6 mL, 7.00 mmol) was added to a microwave tube containing 1,4-dibromoquinoline (27) (100 mg, 0.35 mmol), DiPEA (50 mg, 0.38 mmol) and EtOAc (2 mL). The resulting mixture was heated at 100 °C under microwave irradiation for 6 h. The reaction mixture was concentrated under reduced pressure, dissolved in N-methylpiperazine (5 mL) and heated at 160 °C under microwave irradiation for 1 h. Then the mixture was diluted with H2O (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organic extracts were washed with water, dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure and purified over SiO2 (EtOAc/MeOH/Et3N = 96/2/2, v/v/v) yielding 46 mg of 31 (0.18 mmol, 51%) as a white solid. 1H NMR (500 MHz, CDCl3) δ ppm 7.65 (d, J = 8.4 Hz, 1H), 7.52–7.43 (m, 2H), 7.20–6.93 (m, 1H), 5.92 (s, 1H), 4.89 (s, 1H), 3.79–3.69 (m, 4H), 3.00 (d, J = 5.0 Hz, 3H), 2.58–2.51 (m, 4H), 2.35 (s, 3H); 13C NMR (126 MHz, CDCl3) δ ppm 158.99, 151.51, 148.11, 129.21, 127.30, 121.08, 118.92, 115.39, 85.14, 55.21, 46.27, 45.25, 30.06. LCMS ret. time 1.83 min, purity 97%, [M + H]+ 257.05; HRMS m/z: [M + H]+ calcd for C15H21N4: 257.1761, found: 257.1767.
1-Methyl-4-(naphthalen-2-yl)piperazine (33)
A solution of n-butyllithium in hexanes (2.5 N, 8.8 mL, 22.0 mmol) was added dropwise to a stirred solution of N-methylpiperazine (2.0 mL, 18.1 mmol) in THF while maintaining a temperature around 0 °C. The reaction mixture was kept at 0 °C for another 30 min and subsequently stirred at rt for 1 h. Then 2-methoxynaphtalene (32) (3.16 g, 20.0 mmol) was added and the reaction mixture was stirred at rt for another 16 h. The reaction mixture was poured over an aqueous HCl solution (10%, 100 mL), basidified with an aqueous solution of NaOH (2.5 M) and extracted with DCM. The combined organic extracts were dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure and purified over SiO2 (EtOAc/Et3N = 95/5, v/v) yielding 3.56 g (15.7 mmol, 79%) of 33 as a white solid. 1H NMR (250 MHz, CDCl3) δ ppm 7.79–7.59 (m, 3H), 7.43–7.31 (m, 1H), 7.30–7.18 (m, 2H), 7.10 (d, J = 2.4 Hz, 1H), 3.36–3.21 (m, 4H), 2.69–2.49 (m, 4H), 2.36 (s, 3H); 13C NMR (63 MHz, CDCl3) δ ppm 149.16, 134.66, 128.71, 128.55, 127.45, 126.77, 126.28, 123.38, 119.38, 110.27, 55.19, 49.51, 46.20. LCMS: ret. time 3.01 min, purity >99%, [M + H]+ 226.95; HRMS m/z: [M + H]+ calcd for C15H19N2: 227.1543, found: 227.1536.
3-(4-Methylpiperazin-1-yl)quinoline (35)
t-BuOH (1.48 g, 20.0 mmol) in THF (6 mL) was slowly added to a stirred suspension of NaNH2 (1.56 g, 40.0 mmol) in THF (6 mL) at 0 °C. The resulting suspension was stirred at 40 °C for 2 h. N-methylpiperazine (2.0 g, 20.0 mmol) was added and the resulting suspension was stirred at 40 °C for 2 h. Then, the reaction mixture was cooled to 0 °C and a solution of 3-bromoquinoline (34) (2.08 g, 10.0 mmol) in THF (5 mL) was added. The resulting mixture was stirred at rt for 1 h. Next, the reaction mixture was cooled to 0 °C and hydrolyzed with H2O (20 mL). The resulting mixture was extracted with DCM, the combined organic extracts were dried over Na2SO4, filtered, concentrated under reduced pressure and purified over SiO2 (EtOAc/MeOH = 99/1 to 90/10, v/v) yielding 890 mg (3.91 mmol, 19%) of 35 as an off white solid. 1H NMR (250 MHz, CDCl3) δ ppm 8.85 (d, J = 2.8, 1H), 8.08–7.97 (m, 1H), 7.72 (m, 1H), 7.60–7.45 (m, 2H), 7.40 (d, J = 2.8, 1H), 3.48–3.32 (m, 4H), 2.79 – 2.65 (m, 4H), 2.46 (s, 3H); 13C NMR (63 MHz, CDCl3) δ ppm 144.96, 144.79, 142.99, 128.96, 128.84, 126.92, 126.59, 126.39, 116.73, 54.87, 49.14, 46.13. LCMS: ret. time 2.18 min, purity 99%, [M + H]+ 227.95; HRMS m/z: [M + H]+ calcd for C14H18N3: 228.1495, found: 228.1483.
3-(4-Methylpiperazin-1-yl)isoquinolin-1-amine (39)
A solution of ammonia in methanol (7N, 10 mL, 70.0 mmol) was added to a microwave tube containing 1,3-dichloroisoquinoline (36) (400 mg, 2.02 mmol). The resulting mixture was heated at 120 °C for 6 h under microwave irradiation. The reaction mixture was concentrated under reduced pressure and the crude product (37) (180 mg) was added to a microwave tube containing N-methylpiperazine (5.0 mL, 45.0 mmol). The resulting mixture was heated at 220 °C for 2 h under microwave irradiation. Then the excess N-methylpiperazine was removed under reduced pressure and the crude product was purified over SiO2 (EtOAc/Et3N = 98/2, v/v) to yield 102 mg of 39 (0.42 mmol, 21% over 2 steps) as a dark green oil. 1H NMR (500 MHz, CDCl3) δ ppm 7.60 (d, J = 8.2 Hz, 1H), 7.48 (d, J = 8.2 Hz, 1H), 7.44–7.40 (m, 1H), 7.17–7.12 (m, 1H), 6.22 (s, 1H), 4.97 (s, 2H), 3.69–3.32 (m, 4H), 2.65–2.46 (m, 4H), 2.36 (s, 3H); 13C NMR (126 MHz, CDCl3) δ ppm 155.50, 155.21, 140.46, 130.05, 125.86, 122.63, 122.10, 112.78, 91.00, 55.02, 46.25, 46.19. LCMS: ret. time 1.86 min, purity >99%, [M + H]+ 243.00; HRMS m/z: [M + H]+ calcd for C14H19N4: 243.1604, found: 243.1604.
N-Methyl-3-(4-methylpiperazin-1-yl)isoquinolin-1-amine (40)
Methanamine in ethanol (33%, w/v, 1.3 mL, 15.0 mmol) was added to a microwave tube containing 1,3-dichloroisoquinoline (36) (500 mg, 2.52 mmol), DiPEA (522 mg, 4.04 mmol) and EtOH (5 mL). The resulting mixture was heated at 100 °C under microwave irradiation for 6 h and at rt for 16 h. The reaction mixture was concentrated under reduced pressure, diluted with H2O (10 mL) and extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with water and brine, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude product (38) (450 mg) was added to a microwave tube containing N-methylpiperazine (3.5 g, 35.0 mmol). The resulting mixture was heated at 220 °C for 20 min under microwave irradiation. Then the mixture was diluted with H2O (10 mL) and extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with water, dried over anhydrous sodium sulfate, filtered, concentrated under reduced pressure and purified over SiO2 (DCM/EtOAc/Et3N = 50/49/1, v/v/v) yielding 228 mg of 40 (0.89 mmol, 35% over 2 steps) as a light brown solid. 1H NMR (500 MHz, CDCl3) δ ppm 7.54 (d, J = 8.2 Hz, 1H), 7.46 (d, J = 8.2 Hz, 1H), 7.42 – 7.35 (m, 1H), 7.15 – 7.06 (m, 1H), 6.10 (s, 1H), 5.17 (s, 1H), 3.63 – 3.54 (m, 4H), 3.13 (d, J = 4.8 Hz, 3H), 2.63 – 2.53 (m, 4H), 2.37 (s, 3H); 13C NMR (126 MHz, CDCl3) δ ppm 155.53, 154.95, 140.16, 129.53, 125.88, 121.71, 121.34, 113.35, 88.60, 55.09, 46.33, 46.10, 28.68; LCMS: ret. time 1.94 min, purity 95%, [M+H]+ 257.10; HRMS m/z: [M+H]+ calcd for C15H21N4: 257.1761, found: 257.1757.
2-Chloro-5-nitroquinoline (42) and 2-Chloro-8-nitroquinoline (43)
2-chloroquinoline (41) (9.47 g, 57.9 mmol) was dissolved in sulfuric acid (62 mL, 1.16 mol) while stirring at 0 °C. After 15 min, potassium nitrate (6.23 g, 61.6 mmol) dissolved in sulfuric acid (20 mL) was slowly added to the solution. The mixture was stirred for 2 h while maintaining the temperature below 10 °C. Subsequently, the mixture was stirred overnight at rt and poured over ice (200 g). The resulting suspension was filtered over a Büchner funnel and washed twice with ice water (200 mL) and ethanol (40 mL). The residual solid was dissolved in dichloromethane (50 mL) and dried with anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The obtained 10.4 g crude product was purified by column chromatography over SiO2 (EtOAc/Hept = 5/95, v/v) to obtain 42 (2.70 g, 13 mmol, 22%) as a white solid and 43 (4.06 g, 19.5 mmol, 34%) as an off-white solid. 2-Chloro-5-nitroquinoline (42):1H NMR (250 MHz, CDCl3) δ ppm 8.99 (d, J = 9.2 Hz, 1H), 8.43–8.31 (m, 2H), 7.84 (t, J = 8.1 Hz, 1H), 7.64 (d, J = 9.2 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ ppm 152.47, 148.02, 145.43, 135.54, 134.95, 128.82, 125.45, 124.89, 119.91; LCMS: ret. time 4.56 min, purity >99%, [M + H]+ 220.00. 2-Chloro-8-nitroquinoline (43):1H NMR (250 MHz, CDCl3) δ ppm 8.20 (d, J = 8.7 Hz, 1H), 8.12–8.01 (m, 2H), 7.64 (t, J = 7.9 Hz, 1H), 7.54 (d, J = 8.7 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ ppm 153.56, 147.15, 138.98, 138.62, 131.75, 127.61, 125.78, 124.93, 124.55; LCMS: ret. time 4.35 min, purity >99%, [M + H]+ 208.90.
2-(4-Methylpiperazin-1-yl)-5-nitroquinoline (44)
2-chloro-5-nitroquinoline (42) (800 mg, 3.84 mmol) was dissolved in N-methylpiperazine (10 mL). The solution was stirred at 140 °C overnight. The reaction mixture was quenched with water and extracted with ethyl acetate. The red organic layer was collected, dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain a brown oil which was purified by column chromatography over SiO2 (EtOAc/Et3N = 96/4, v/v) to obtain 44 (892 mg, 3.28 mmol, 85%) as a light brown solid. 1H NMR (250 MHz, CDCl3) δ ppm 8.68 (d, J = 9.7 Hz, 1H), 8.00–7.90 (m, 2H), 7.55 (dd, J = 8.4, 7.8 Hz, 1H), 7.16 (d, J = 9.7 Hz, 1H), 3.86–3.78 (m, 4H), 2.59–2.50 (m, 4H), 2.37 (s, 3H); 13C NMR (126 MHz, CDCl3) δ ppm 157.07, 148.77, 145.73, 133.34, 133.02, 127.56, 119.82, 115.52, 112.26, 54.91, 46.21, 44.66; LCMS: ret. time 3.62 min, purity >99%, [M + H]+ 273.10; HRMS m/z: [M + H]+ calcd for C14H17N4O2: 273.1333, found: 273.1337.
2-(4-Methylpiperazin-1-yl)-8-nitroquinoline (45)
2-chloro-8-nitroquinoline (43) (800 mg, 3.84 mmol) was dissolved in N-methylpiperazine (10 mL). Potassium carbonate (530 mg, 3.84 mmol) was added. The solution was stirred overnight at 140 °C. The reaction mixture turned brown/dark red. The reaction mixture was quenched with water and extracted with ethyl acetate. The red organic layer was collected, dried over anhydrous sodium sulfate and concentrated under vacuum to obtain a brown oil which was purified by column chromatography (EtOAc/Et3N = 96/4, v/v) to obtain 45 (989 mg, 3.63 mmol, 95%) as a light brown solid. 1H NMR (250 MHz, CDCl3) δ ppm 7.93–7.87 (m, 2H), 7.74 (dd, J = 8.0, 1.3 Hz, 1H), 7.23–7.15 (m, 1H), 7.04 (d, J = 9.3 Hz, 1H), 3.85–3.78 (m, 4H), 2.56–2.48 (m, 4H), 2.35 (s, 3H); 13C NMR (126 MHz, CDCl3) δ ppm 157.56, 145.42, 139.94, 137.29, 131.48, 124.53, 124.17, 120.03, 110.60, 54.85, 46.16, 44.57; LCMS: ret. time 2.70 min, purity >99%, [M + H]+ 273.05; HRMS m/z: [M + H]+ calcd for C14H17N4O2: 273.1333, found: 273.1341.
2-(4-Methylpiperazin-1-yl)quinolin-5-amine (46)
2-(4-methylpiperazin-1-yl)-5-nitroquinoline (44) (206 mg, 0.76 mmol) was dissolved in methanol (50 mL). Pd/C 5 wt % (30 mg) was added to the solution and the resulting suspension was stirred overnight at rt under a hydrogen gas atmosphere. Hereafter, the mixture was filtered over Celite and concentrated under reduced pressure to obtain 46 (143 mg, 0.59 mmol, 81%) as a brown solid. 1H NMR (500 MHz, CDCl3) δ ppm 7.93 (d, J = 9.3 Hz, 1H), 7.35–7.30 (m, 1H), 7.18 (d, J = 8.4 Hz, 1H), 6.92 (d, J = 9.3 Hz, 1H), 6.53 (d, J = 7.4 Hz, 1H), 4.01 (s, 2H), 3.81–3.70 (m, 4H), 2.59–2.50 (m, 4H), 2.36 (s, 3H); 13C NMR (126 MHz, CDCl3) δ ppm 157.30, 148.82, 142.21, 130.97, 130.11, 117.84, 113.00, 107.81, 107.14, 55.05, 46.26, 45.02; LCMS: ret. time 0.96 min, purity 99%, [M + H]+ 243.10; HRMS m/z: [M + H]+ calcd for C14H19N4: 243.1604, found: 243.1599.
2-(4-Methylpiperazin-1-yl)quinolin-8-amine (47)
2-(4-methylpiperazin-1-yl)-8-nitroquinoline (43) (203 mg, 0.75 mmol) was dissolved in methanol (50 mL). Pd/C 5 wt % (30 mg) was added to the solution and the resulting suspension was stirred overnight at rt under a hydrogen gas atmosphere. Hereafter, the mixture was filtered over Celite and was concentrated over vacuum to obtain 47 (168 mg, 0.69 mmol, 93%) as a brown solid. 1H NMR (250 MHz, MeOD) δ ppm 7.93 (d, J = 9.1 Hz, 1H), 7.14 (d, J = 9.1 Hz, 1H), 7.07–7.00 (m, 2H), 6.94–6.89 (m, 1H), 3.87–3.76 (m, 4H), 2.88–2.77 (m, 4H), 2.54 (s, 3H); 13C NMR (126 MHz, CH3OH+D2O) δ ppm 156.95, 143.24, 139.14, 138.53, 124.87, 124.47, 117.51, 112.90, 111.07, 55.36, 45.36, 45.35; LCMS: ret. time 2.07 min, purity 98%, [M + H]+ 243.10; HRMS m/z: [M + H]+ calcd for C14H19N4: 243.1604, found: 243.1599.
Radioligand Binding
5-HT3AR
HEK293 cells stably expressing 5-HT3AR were scraped into 1 mL of ice-cold HEPES buffer (10 mM, pH 7.4) and frozen. After thawing, they were washed with HEPES buffer and homogenized using a fine-bore syringe. Fifty microliters of cell membranes were incubated in 0.5 mL HEPES buffer containing 0.7 nM [3H]granisetron (∼Kd) and differing concentrations of the test compound. Competition binding (8 point) was performed on at least three separate plates of transfected cells. Nonspecific binding was determined using 1 mM quipazine. Reactions were incubated for at least 24 h at 4 °C, to allow compounds with slow kinetics to equilibrate. Incubations were terminated by vacuum filtration using a Brandel cell harvester (Alpha Biotech Ltd., London, UK) onto GF/B filters presoaked in 0.3% polyethyleneimine. Radioactivity was determined by scintillation counting. Data were fit according to the equation:
where L is the concentration of ligand present; BL is the binding in the presence of ligand concentration L; Bmin is the binding when L = 0; Bmax is the binding when L = ∞, L50 is the concentration of L which gives a binding equal to (Bmax + Bmin)/2; and nH is the Hill coefficient. Ki values were estimated from IC50 values using the Cheng-Prusoff equation48Ki = IC50/(1+[L]/Kd) where Ki is the equilibrium dissociation constant for binding of the unlabeled antagonist, IC50 is the concentration of antagonist that blocks half the specific binding, [L] is the free concentration of radioligand and Kd is the equilibrium dissociation constant of the radioligand.
Non 5-HT3AR
Off-target binding of compound 22 was assessed at 13 additional targets (Table 6). With the exception of nACh(α7), all single point radioligand competition binding was performed by Cerep.49 The results are expressed as a percent inhibition of control specific binding (100-(measured specific binding/control specific binding)*100) obtained in the presence of compound 22 (see Supporting Information, Tables S1–S3 for more details).
Homology Modeling
A model of the 5-HT3A receptor binding site was constructed by homology modeling using MOE (version 2010.10, Chemical Computing Group Montreal),50 based on the tropisetron bound AChBP X-ray crystal structure determined at 2.20 Å resolution (PDB code: 2WNC).39 The sequence of the human 5-HT3A gene (Q7KZM7) was aligned with the Aplysia californica gene (Q8WSF8) using the “Protein Align” option in MOE (standard settings) and adjusted manually. The final sequence alignment is given in Supporting Information, Figure S2. Chains A and E of the original PDB structure were selected to serve as the template. Structural waters located in this binding pocket of the 2WNC crystal structure form a conserved protein–ligand H-bond interaction network in several other AChBP crystals containing small competitive antagonists (e.g., 2BYR, 2PGZ, 2BYS, 2XYT(44)), and were included in the 5-HT3A receptor model. The template backbone, the ligand and the water molecules were fixed and 10 preliminary receptor models were constructed based on the template backbone. During this construction, the ligand and waters were considered as an additional restraint using the “Environment” option within MOE. The structural quality of the models was checked using the evaluation modules in MOE. During this evaluation the focus was on the binding site region of the model. The protein geometry of receptor atoms was evaluated for their bond lengths, bond angles, atom clashes and contact energies. Ramachandran plots were used to check the Phi and Psi angles of all residues. Model 1 was selected for further refinement, hydrogen atoms were added, partial atomic charges were calculated and the protein was minimized around the fixed ligand and static water molecules using the Amber99 force field in MOE.
Molecular Docking
Ligands were protonated according to physiological pH using the MMFF94 force field and MOE software. Relevant tautomers of the ligands were created and subsequently the three-dimensional structures were energy minimized and converted into mol2 files using Molecular Network’s Corina.51 Docking studies were performed with the docking program GOLD (Version 5.0, CCDC, Cambridge, UK)45 using default settings. The protein binding site was defined by a radius of 12 Å around W183 (atom NE1) of the principle subunit. A total of 30 dockings were set up for each ligand run with a root-mean-square deviation (rmsd) tolerance of 1.5 Å for early termination. Docking scores were calculated with the GoldScore scoring function.
Acknowledgments
M.V. and I.d.E. would like to acknowledge EEC grant (Neurocypres) for financial support. This work was furthermore supported by COST Action BM0806COST. S.C.R.L. and A.J.T. are Wellcome Trust funded (081925). There are no competing interests.
Glossary
Abbreviations Used
- AChBP
acetylcholine binding protein
- pLGIC
pentameric ligand-gated ion channel
- IBS
irritable bowel syndrome
- 5-HT
5-hydroxy tryptamine
- nACh
nicotinic acetylcholine
- GABA
γ-aminobutyric acid
- (±) DOI
(±) 2,5-dimethoxy-4-iodoamphetamine
- 8-OH-DPAT
8-hydroxy-2-(di-N-propylamino)tetralin
- MeOH
methanol
- EtOH
ethanol
- EtOAc
ethyl acetate
- DiPEA
diisopropyl ethyl amine
- t-BuOH
tert-butoxide
- n-BuLi
n-butyllithium
- DCM
dichloromethane
- Hept
heptanes
- MeNH2
methylamine
- HBA
hydrogen bond acceptor
- ESI
electron-spray ionization
- UV
ultraviolet
- LCMS
liquid chromatography mass spectrometry
Supporting Information Available
Overview of the 2D NOESY 1H NMR interactions found for compounds 30 and 31. The sequence alignment between the human 5-HT3A gene (Q7KZM7) and the Aplysia californica gene (Q8WSF8). Radioligand binding studies for the receptors shown in Table 6. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
M.H.V. and A.J.T. contributed equally to this work.
The authors declare no competing financial interest.
Author Contributions
‡ These authors contributed equally to this work.
Supplementary Material
References
- Hsu E. S. A review of granisetron, 5-hydroxytryptamine3 receptor antagonists, and other antiemetics. Am. J. Therap. 2010, 17(5), 476–486. [DOI] [PubMed] [Google Scholar]
- Spiller R. C. Targeting the 5-HT3 receptor in the treatment of irritable bowel syndrome. Curr. Opin. Pharmacol. 2011, 11(1), 68–74. [DOI] [PubMed] [Google Scholar]
- Rajkumar R.; Mahesh R. The auspicious role of the 5-HT3 receptor in depression: a probable neuronal target?. J. Psychopharmacol. 2010, 24(4), 455–469. [DOI] [PubMed] [Google Scholar]
- Thompson A. J.; Lummis S. C. The 5-HT3 receptor as a therapeutic target. Expert Opin. Ther. Targets 2007, 11(4), 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maricq A. V.; Peterson A. S.; Brake A. J.; Myers R. M.; Julius D. Primary structure and functional expression of the 5HT3 receptor, a serotonin-gated ion channel. Science (New York, N.Y 1991, 254(5030), 432–437. [DOI] [PubMed] [Google Scholar]
- Unwin N. Acetylcholine receptor channel imaged in the open state. Nature 1995, 373(6509), 37–43. [DOI] [PubMed] [Google Scholar]
- Brejc K.; van Dijk W. J.; Klaassen R. V.; Schuurmans M.; van Der Oost J.; Smit A. B.; Sixma T. K. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 2001, 411(6835), 269–276. [DOI] [PubMed] [Google Scholar]
- Beene D. L.; Price K. L.; Lester H. A.; Dougherty D. A.; Lummis S. C. Tyrosine residues that control binding and gating in the 5-hydroxytryptamine3 receptor revealed by unnatural amino acid mutagenesis. J. Neurosci. 2004, 24(41), 9097–9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price K. L.; Bower K. S.; Thompson A. J.; Lester H. A.; Dougherty D. A.; Lummis S. C. R. A hydrogen bond in loop a is critical for the binding and function of the 5-HT3 receptor. Biochemistry 2008, 47(24), 6370–6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryanarayanan A.; Joshi P. R.; Bikadi Z.; Mani M.; Kulkarni T. R.; Gaines C.; Schulte M. K. The loop C region of the murine 5-HT3A receptor contributes to the differential actions of 5-hydroxytryptamine and m-chlorophenylbiguanide. Biochemistry 2005, 44(25), 9140–9149. [DOI] [PubMed] [Google Scholar]
- Thompson A. J.; Lochner M.; Lummis S. C. Loop B is a major structural component of the 5-HT3 receptor. Biophys. J. 2008, 5728–5736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. J.; Padgett C. L.; Lummis S. C. Mutagenesis and molecular modeling reveal the importance of the 5-HT3 receptor F-loop. J. Biol. Chem. 2006, 281(24), 16576–16582. [DOI] [PubMed] [Google Scholar]
- Thompson A. J.; Price K. L.; Reeves D. C.; Chan S. L.; Chau P. L.; Lummis S. C. Locating an antagonist in the 5-HT3 receptor binding site using modeling and radioligand binding. J. Biol. Chem. 2005, 280(21), 20476–20482. [DOI] [PubMed] [Google Scholar]
- Venkataraman P.; Venkatachalan S. P.; Joshi P. R.; Muthalagi M.; Schulte M. K. Identification of critical residues in loop E in the 5-HT3ASR binding site. BMC Biochem. 2002, 3, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D.; White M. M. Spatial orientation of the antagonist granisetron in the ligand-binding site of the 5-HT3 receptor. Mol. Pharmacol. 2005, 68(2), 365–371. [DOI] [PubMed] [Google Scholar]
- Barrera N. P.; Herbert P.; Henderson R. M.; Martin I. L.; Edwardson J. M. Atomic force microscopy reveals the stoichiometry and subunit arrangement of 5-HT3 receptors. Proc. Natl. Acad. Sci. U.S.A. 2005, 102(35), 12595–12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. J.; Price K. L.; Lummis S. C. R. Cysteine modification reveals which subunits form the ligand binding site in human heteromeric 5-HT3AB receptors. J. Physiol. 2011, 589(17), 4243–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A. W.; Peroutka S. J. Three-dimensional steric molecular modeling of the 5-hydroxytryptamine3 receptor pharmacophore. Mol. Pharmacol. 1989, 36(4), 505–511. [PubMed] [Google Scholar]
- Asagarasu A.; Matsui T.; Hayashi H.; Tamaoki S.; Yamauchi Y.; Sato M. Design and Synthesis of Piperazinylpyridine Derivatives as Novel 5-HT(1A) Agonists/5-HT(3) Antagonists for the Treatment of Irritable Bowel Syndrome (IBS). Chem. Pharm. Bull. (Tokyo) 2009, 57(1), 34–42. [DOI] [PubMed] [Google Scholar]
- Cappelli A.; Anzini M.; Vomero S.; Mennuni L.; Makovec F.; Doucet E.; Hamon M.; Menziani M. C.; De Benedetti P. G.; Giorgi G.; Ghelardini C.; Collina S. Novel potent 5-HT(3) receptor ligands based on the pyrrolidone structure: synthesis, biological evaluation, and computational rationalization of the ligand-receptor interaction modalities. Bioorg. Med. Chem. 2002, 10(3), 779–801. [DOI] [PubMed] [Google Scholar]
- Clark R. D.; Miller A. B.; Berger J.; Repke D. B.; Weinhardt K. K.; Kowalczyk B. A.; Eglen R. M.; Bonhaus D. W.; Lee C. H. 2-(Quinuclidin-3-yl)pyrido[4,3-b]indol-1-ones and isoquinolin-1-ones. Potent conformationally restricted 5-HT3 receptor antagonists. J. Med. Chem. 1993, 36(18), 2645–2657. [DOI] [PubMed] [Google Scholar]
- Evans S.; Galdes A.; Gall M. Molecular modeling of 5-HT3 receptor ligands. Pharmacol., Biochem. Behav. 1991, 40(4), 1033–1040. [DOI] [PubMed] [Google Scholar]
- Hibert M.; Hoffmann R.; Miller R.; Carr A. Conformation-activity relationship study of 5-HT3 receptor antagonists and a definition of a model for this receptor site. J. Med. Chem. 1990, 33(6), 1594–1600. [DOI] [PubMed] [Google Scholar]
- Mahesh R.; Perumal R. V.; Pandi P. V. Pharmacophore based synthesis of 3-chloroquinoxaline-2-carboxamides as serotonin3 (5-HT3) receptor antagonist. Biol. Pharm. Bull. 2004, 27(9), 1403–1405. [DOI] [PubMed] [Google Scholar]
- Thompson A. J.; Lummis S. C. 5-HT3 receptors. Curr. Pharm. Des 2006, 12(28), 3615–3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen C. G.; Frølund B.; Kehler J.; Jensen A. A. Discovery of benzamide analogues as a novel class of 5-HT3 receptor agonists. ChemMedChem 2012, 6(4), 725–736. [DOI] [PubMed] [Google Scholar]
- Thompson A. J.; Verheij M. H.; Leurs R.; De Esch I. J.; Lummis S. C. An efficient and information-rich biochemical method design for fragment library screening on ion channels. Biotechniques 2010, 49(5), 822–829. [DOI] [PubMed] [Google Scholar]
- Hong E.; Sancilio L. F.; Vargas R.; Pardo E. G. Similarities between the pharmacological actions of quipazine and serotonin. Eur. J. Pharmacol. 1969, 6(3), 274–280. [DOI] [PubMed] [Google Scholar]
- Anzini M.; Cappelli A.; Vomero S.; Giorgi G.; Langer T.; Hamon M.; Merahi N.; Emerit B. M.; Cagnotto A.; Skorupska M.; et al. Novel, potent, and selective 5-HT3 receptor antagonists based on the arylpiperazine skeleton: synthesis, structure, biological activity, and comparative molecular field analysis studies. J. Med. Chem. 1995, 38(14), 2692–2704. [DOI] [PubMed] [Google Scholar]
- Cappelli A.; Anzini M.; Vomero S.; Canullo L.; Mennuni L.; Makovec F.; Doucet E.; Hamon M.; Menziani M. C.; De Benedetti P. G.; Bruni G.; Romeo M. R.; Giorgi G.; Donati A. Novel potent and selective central 5-HT3 receptor ligands provided with different intrinsic efficacy. 2. Molecular basis of the intrinsic efficacy of arylpiperazine derivatives at the central 5-HT3 receptors. J. Med. Chem. 1999, 42(9), 1556–1575. [DOI] [PubMed] [Google Scholar]
- Cappelli A.; Anzini M.; Vomero S.; Mennuni L.; Makovec F.; Doucet E.; Hamon M.; Bruni G.; Romeo M. R.; Menziani M. C.; De Benedetti P. G.; Langer T. Novel potent and selective central 5-HT3 receptor ligands provided with different intrinsic efficacy. 1. Mapping the central 5-HT3 receptor binding site by arylpiperazine derivatives. J. Med. Chem. 1998, 41(5), 728–741. [DOI] [PubMed] [Google Scholar]
- Cappelli A.; Gallelli A.; Manini M.; Anzini M.; Mennuni L.; Makovec F.; Menziani M. C.; Alcaro S.; Ortuso F.; Vomero S. Further studies on the interaction of the 5-hydroxytryptamine3 (5-HT3) receptor with arylpiperazine ligands. development of a new 5-HT3 receptor ligand showing potent acetylcholinesterase inhibitory properties. J. Med. Chem. 2005, 48(10), 3564–3575. [DOI] [PubMed] [Google Scholar]
- Somers F.; Ouedraogo R.; Antoine M.-H.; de Tullio P.; Becker B.; Fontaine J.; Damas J.; Dupont L.; Rigo B.; Delarge J.; Lebrun P.; Pirotte B. Original 2-Alkylamino-6-halogenoquinazolin-4(3H)-ones and KATP Channel Activity. J. Med. Chem. 2001, 44(16), 2575–2585. [DOI] [PubMed] [Google Scholar]
- Nelson D. R.; Thomas D. R. [3H]-BRL 43694 (granisetron), a specific ligand for 5-HT3 binding sites in rat brain cortical membranes. Biochem. Pharmacol. 1989, 38(10), 1693–1695. [DOI] [PubMed] [Google Scholar]
- Smits R. A.; de Esch I. J.; Zuiderveld O. P.; Broeker J.; Sansuk K.; Guaita E.; Coruzzi G.; Adami M.; Haaksma E.; Leurs R. Discovery of quinazolines as histamine H4 receptor inverse agonists using a scaffold hopping approach. Journal of medicinal chemistry 2008, 51(24), 7855–7865. [DOI] [PubMed] [Google Scholar]
- Smits R. A.; Lim H. D.; Hanzer A.; Zuiderveld O. P.; Guaita E.; Adami M.; Coruzzi G.; Leurs R.; de Esch I. J. Fragment based design of new H4 receptor-ligands with anti-inflammatory properties in vivo. J. Med. Chem. 2008, 51(8), 2457–2467. [DOI] [PubMed] [Google Scholar]
- Smits R. A.; Lim H. D.; Stegink B.; Bakker R. A.; de Esch I. J.; Leurs R. Characterization of the histamine H4 receptor binding site. Part 1. Synthesis and pharmacological evaluation of dibenzodiazepine derivatives. J. Med. Chem. 2006, 49(15), 4512–4516. [DOI] [PubMed] [Google Scholar]
- Verheij M. H. P.; de Graaf C.; de Kloe G. E.; Nijmeijer S.; Vischer H. F.; Smits R. A.; Zuiderveld O. P.; Hulscher S.; Silvestri L.; Thompson A. J.; van Muijlwijk-Koezen J. E.; Lummis S. C. R.; Leurs R.; de Esch I. J. P. Fragment library screening reveals remarkable similarities between the G protein-coupled receptor histamine H4 and the ion channel serotonin 5-HT3A. Bioorg. Med. Chem. Lett. 2011, 21(18), 5460–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs R. E.; Sulzenbacher G.; Shi J.; Talley T. T.; Conrod S.; Kem W. R.; Taylor P.; Marchot P.; Bourne Y. Structural determinants for interaction of partial agonists with acetylcholine binding protein and neuronal alpha7 nicotinic acetylcholine receptor. EMBO J. 2009, 28, 3040–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beene D. L.; Brandt G. S.; Zhong W.; Zacharias N. M.; Lester H. A.; Dougherty D. A. Cation-pi interactions in ligand recognition by serotonergic (5-HT3A) and nicotinic acetylcholine receptors: the anomalous binding properties of nicotine. Biochemistry 2002, 41(32), 10262–10269. [DOI] [PubMed] [Google Scholar]
- Duffy N. H.; Lester H. A.; Dougherty D. A. Ondansetron and Granisetron binding orientation in the 5-HT(3) receptor determined by unnatural amino acid mutagenesis. ACS Chem. Biol. 2012, 10.1021/cb300246j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price K. L.; Lummis S. C. The role of tyrosine residues in the extracellular domain of the 5-hydroxytryptamine3 receptor. J. Biol. Chem. 2004, 279(22), 23294–23301. [DOI] [PubMed] [Google Scholar]
- Yan D.; Schulte M. K.; Bloom K. E.; White M. M. Structural features of the ligand-binding domain of the serotonin 5HT3 receptor. J. Biol. Chem. 1999, 274(9), 5537–5541. [DOI] [PubMed] [Google Scholar]
- PDB: 2BYR, 2PGZ, 2BYS, 2XYT.
- Verdonk M. L.; Chessari G.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Nissink J. W. M.; Taylor R. D.; Taylor R. Modeling water molecules in protein–ligand docking using GOLD. J. Med. Chem. 2005, 48(20), 6504–6515. [DOI] [PubMed] [Google Scholar]
- Verdonk M. L.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Taylor R. D. Improved protein-ligand docking using GOLD. Proteins 2003, 52(4), 609–623. [DOI] [PubMed] [Google Scholar]
- Thompson A. J.; Verheij M.; De Esch I.; Lummis S. C. R., VUF10166, a novel compound with differing activities at 5-HT3A and 5-HT3AB receptors. J. Pharmacol. Exp. Ther. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y.; Prusoff W. H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22(23), 3099–108. [DOI] [PubMed] [Google Scholar]
- Cerep, Le bois l’Evêque 86600 Celle l’Evescault France. www.cerep.fr.
- MOE, Version 2010.10; Chemical Computing Group, Inc.: Montreal, Canada, 2010.
- Corina, version 3.46; Molecular Networks GmbH: Erlangen, Germany, 2006.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.