

Abstract
Chronically altered levels of network activity lead to changes in the morphology and functions of neurons. However, little is known of how changes in neuronal activity alter the intracellular signaling pathways mediating neuronal survival. Here, we use primary cultures of rat hippocampal neurons to show that elevated neuronal activity impairs phosphorylation of the serine/threonine kinase, Erk1/2, and the activation of signal transducer and activator of transcription 3 (STAT3) by phosphorylation of serine 727. Chronically stimulated neurons go through apoptosis when they fail to activate another serine/threonine kinase, Akt. Gain- and loss-of-function experiments show that STAT3 plays the key role directly downstream from Erk1/2 as the alternative survival pathway. Elevated neuronal activity resulted in increased expression of a tumor suppressor, p53, and its target gene, Bax. These changes are observed in Kv4.2 knock-out mouse hippocampal neurons, which are also sensitive to the blockade of TrkB signaling, confirming that the alteration occurs in vivo. Thus, this study provides new insight into a mechanism by which chronic elevation of activity may cause neurodegeneration.
Introduction
Neurons can detect global changes in input levels and can adjust their own levels of excitability to maintain an appropriate range of spiking activity (Davis, 2006; Turrigiano, 2007). Although the molecular details of synaptic scaling mechanisms that ensure this homeostatic plasticity have been studied extensively (Beattie et al., 2002; Shepherd et al., 2006; Cingolani et al., 2008; Seeburg et al., 2008; Cohen et al., 2011), little is known of the impact these mechanisms exert on the survival signaling of neurons.
Recently, we reported that neurons in newborn rodent hippocampus, while they are under selection through a process of developmental death, depend on neurotrophin for survival. Neurotrophins activate signaling pathways through the tropomyosin receptor kinase (Trk) (Zweifel et al., 2005). Interestingly, we found that the direct activation of these pathways by TrkB was not sufficient to promote survival of neurons. Rather, brain-derived neurotrophic factor (BDNF) recruits neurons into active networks, and this activity, together with integrin signals, mediates sustained Akt activation and promotes neuronal survival (Murase et al., 2011a). Whether or not this sustained Akt activation remains critical for the survival of mature neurons has not been tested.
Signal transducer and activator of transcription (STAT) proteins are transcription factors that respond to cytokines such as ILs (interleukins) (Levy and Darnell, 2002). STAT3-mediated cytokine signaling regulates gliogenesis as well as neurogenesis during brain development (Deverman and Patterson, 2009). Although neuroprotective roles of STAT3 have been reported in the context of injury (Schweizer et al., 2002; Dziennis et al., 2007; Jung et al., 2009), roles of STAT3 in the survival of neurons under healthy conditions have not been explored.
Here, we report that, after the vulnerable period, neurons lose their dependence on neurotrophins unless they are challenged by elevated activity. In this more mature state, elevation of network activity restores a dependence on neurotrophin-induced survival signals. Gain- and loss-of-function experiments identify STAT3 as a key mediator of the second survival pathway that mature neurons acquire. This study shows chronic elevation of activity may cause neurodegeneration through a mechanism similar to developmental death.
Materials and Methods
Reagents.
2-(4-Morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002), 2-(2-amino-3-methoxyphenyl)-4H-chromen-4-one (PD98059), rapamycin, and roscovitine were purchased from Calbiochem. Fluo-4 AM was purchased from Invitrogen. 4-Aminopyridine (4-AP), tetrodotoxin (TTX), picrotoxin (PTX), 4′,6-diamidino-2-phenylindole (DAPI), and K252a were purchased from Sigma-Aldrich. BDNF was purchased from R&D Systems.
Antibodies.
Antibodies were used at the following dilutions: polyclonal rabbit anti-phospho-Ser-727 STAT3 antibody, anti-phospho-Tyr-705 STAT3 antibody, anti-STAT3 antibody, anti-p53 antibody, and anti-Bax antibody (Santa Cruz Biotechnology), 1:500; anti-Erk1/2 antibody, anti-phospho-Erk1/2 antibody, anti-phospho-Ser-743 Akt antibody, cleaved caspase3 (c-cas3) antibody, and monoclonal mouse anti-STAT3 antibody (Cell Signaling Technology), 1:500; monoclonal mouse anti-cytochrome c antibody and NeuN antibody (BD Biosciences Pharmingen), 1:1000; and monoclonal mouse anti-β-actin antibody and anti-MAP2 antibody (Sigma-Aldrich), 1:10,000 and 1:1000, respectively. Polyclonal goat anti-TrkB antibody (R&D Systems) was used at various concentrations for function-blocking experiments.
Dissociated primary hippocampal culture.
Culture was prepared as described previously (Murase and McKay, 2006). Hippocampi from embryonic day 18 Sprague Dawley rat embryos of either sex were used for both astrocyte (plated at a density of 80,000 cells/ml) and neuron (density, 200,000 cells/ml) cultures. Astrocytes were cultured in Neurobasal (Invitrogen) with 5% FBS in 5% CO2 at 37°C for 14 d. Medium was changed completely twice weekly. Neurons were plated on confluent astrocyte beds and cultured in Neurobasal and B27 in 5% CO2 at 37°C. One-half of the medium was changed every 2 d. Experiments were performed using 14 d in vitro (DIV14) neurons.
Calcium imaging.
Cultures were incubated with 2 μm fluo-4 AM for 15 min. Images were obtained with a BX51W1 microscope (Olympus America) equipped with a 10× objective lens. The cultures were imaged in HEPES-buffered saline (110 mm NaCl, 5.4 mm KCl, 1.8 mm CaCl2, 0.8 mm MgCl2, 10 mm d-glucose, 10 mm HEPES-NaOH, pH 7.4, 290 mOsm) at 37°C using a CCD camera (QImaging) at 20 Hz for 40 s. Typically, 20 neurons in one experiment were used for analyses.
Immunoprecipitation.
Rat hippocampal culture was incubated with lysis buffer (150 mm NaCl, 1% NP-40, 50 μm Tris-HCl, pH 8.0) containing a protease inhibitor mixture (Roche) for 20 min on ice (60 μl per one 24-well culture dish). Lysate was centrifuged at 12,000 rpm for 5 min at 4°C. The supernatant was preabsorbed with 10% (v/v) of protein A-Sepharose beads (Pierce) for 1 h, and incubated with 3 μg/ml anti-STAT3 antibody for 2 h followed by incubation with 10% (v/v) of protein A-Sepharose beads for 1 h at 4°C. The beads were then rinsed three times with lysis buffer with protease inhibitor at 4°C before elution with an equal volume of 1× SDS loading buffer by boiling for 5 min.
Western blot.
Samples from dissociated culture were collected with 1× SDS loading buffer (60 μl per one 24-well culture dish). Hippocampi from wild-type C57BL/6 mice and Kv4.2−/− mice were homogenized in 300% (v/w) lysis buffer with protease inhibitor on ice. The homogenates were diluted with 2× SDS loading buffer. The samples were boiled for 5 min, and then applied to a 4–10% gradient SDS gel (Bio-Rad). The proteins were transferred to a nitrocellulose membrane. The membranes were blocked with 4% skim milk in PBS for 30 min. Incubation with antibodies was performed in the blocking solution. Membranes were washed with Tris-buffered saline with 0.05% Tween 20. The proteins were visualized with SuperSignal West Pico System (Pierce) and detected and analyzed with a BioChemi System (UVP BioImaging Systems). Mean ± SEM are plotted.
Immunocytochemistry.
Cultures were fixed with 4% paraformaldehyde, permeabilized in 0.5% Triton X-100, and blocked with PBS containing 5% NGS (Vector Laboratories). Primary and secondary antibodies were diluted with the blocking solution. Samples were incubated for 2 h with antibodies.
For the in vivo injection analyses, C57BL/6 and Kv4.2−/− mice were perfused with 4% PFA 2 d after the injection. Consecutive coronal slices of 50 μm thickness were made by a Leica VT100S vibrating microtome (Leica) and were immunostained with a neuronal marker, NeuN, and the apoptotic marker, c-cas3. Slices were compared with respect to distance from the injection site. Four consecutive slices per animal and three animals per condition were combined for the analyses. The analysis was done blind with respect to the content of the injections.
Cell quantification.
Neurons were visualized by immunostaining against neuron-specific microtubule-associated protein 2 (MAP2) (Izant and McIntosh, 1980). Fluorescent images were taken with a Zeiss confocal microscope (LSM-510) equipped with 10× lens or a 25× lens. Z-stacked images from eight sections (1 μm intervals) were used for the analyses. All experiments were repeated in at least three independent culture preparations. Image analyses were done using ImageJ. Images were taken from five fields; one from the center of the coverslip, and two vertically and two horizontally 400–3000 μm from the center. Because the densities of neurons were higher in the rim of coverslips than in other regions, we avoided sampling the edge of coverslips. Each coverslip was defined as an individual culture. Numbers represent mean ± SEM. All analyses were done blind.
Transfection.
Transfection was performed using Lipofectamine 2000 (Invitrogen). Cells were transfected with 1.6 μg/ml pEGFPC1 vector (Clontech), and/or 8 μg/ml activated Akt1/pUSE vector (Millipore), or 30 pm rat STAT3 siRNA (Santa Cruz) or wild-type and mutant STAT3 IRES EGFP/pMX plasmids (gift from Dr. Y. Gotoh, University of Tokyo, Tokyo, Japan) in OPTI-MEM (Invitrogen) for 30 min, and then the medium was replaced with Neurobasal medium. Transfection was performed 4 d before the experiments.
RT-PCR.
Hippocampi from C57BL/6 and Kv4.2−/− mice were homogenized in 300% (v/w) lysis buffer on ice. Rat hippocampal culture was incubated with lysis buffer with protease inhibitor mixture for 20 min on ice (60 μl per one 24-well culture dish). RNA was isolated from the homogenates using TriPure Isolation Reagent (Roche). RT-PCR was performed using SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen). Using 5 μg of total RNA, first-strand cDNA synthesis reaction by reverse transcriptase was done using oligo-dT12–18 as primers. PCR was performed using Taq polymerase (Roche). The sequences of the primers are the following: 5′-CCACACTTTCTACAATGAGC-3′ and 5′-CCGTCAGGATCTTCATGAGG-3′ for rat β-actin, 5′-CTACTAAGGTCGTGAGACGCTGCC-3′ and 5′-TCAGCATACAGGTTTCCTTCCACC-3′ for rat p53, 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′ for mouse GAPDH, and 5′-GATGGTGATGGCCTGGCTCC-3′ and 5′-GGTCGGCGGTTCATGCCCCC-3′ for mouse p53. Conditions for PCRs are as follows: 44 cycles of 95°C (15 s), 60°C (20 s), 72°C (15 s) for rat p53; and 35 cycles of 95°C (30 s), 62°C (30 s), 72°C (30 s) for rat β-actin, mouse p53, and GAPDH. The PCR products were separated in 2% agarose gel.
Chromatin immunoprecipitation.
Chromatin immunoprecipitation (ChIP) assays were performed as described by Ballas et al. (2001). C57BL/6 and Kv4.2−/− mice were perfused with 4% PFA. Hippocampi were homogenized with cell lysis buffer (CLB) (5 mm HEPES, pH 8, 85 mm KCl, and 0.5% Triton X-100) containing 1 mm phenylmethylsulfonyl fluoride (PMSF) using a glass tissue grinder on ice. The homogenate was centrifuged at 3000 rpm for 2 min at 4°C, and the pellet was resuspended in CLB with PMSF and centrifuged at 3000 rpm for 2 min at 4°C two times. The pellet was then resuspended in nuclear lysis buffer (NLB) (50 mm Tris-HCl, pH 8, 10 mm EDTA, 1% SDS) with 1 mm PMSF and was sonicated to yield 100–1000 bp DNA on ice, and was centrifuged at 12,000 rpm for 15 min at 4°C. The nuclear lysate was preabsorbed with recombinant protein G-agarose (rProtein G-agarose; Invitrogen) preincubated with 200 μg/ml yeast tRNA and 200 μg/ml salmon sperm (Invitrogen) for 1 h at 4°C. The chromatin suspension was diluted with ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 16.7 mm Tris-HCl, pH 8, 167 mm NaCl), and then immunoprecipitated with 5 μg/ml monoclonal mouse anti-STAT3 overnight at 4°C. The chromatin suspension was incubated with rProtein G-agarose pretreated with 3% BSA and yeast tRNA and salmon sperm for 4 h at 4°C. Agarose beads were washed with series of solutions as following at room temperature: ChIP dilution buffer, dialysis buffer (2 mm EDTA, 50 mm Tris-HCl, pH 8, 0.2% Sarkosyl), TSE-500 (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8, 500 mm NaCl), LiCl detergent (100 mm Tris, pH 8, 500 mm LiCl, 1% Triton X-100, 1% deoxycholic acid), and TE (10 mm Tris-HCl, pH 8, 1 mm EDTA). To change the solution, the beads were centrifuged at 3000 rpm for 1 min, and the supernatant was aspirated. The samples were eluted from the beads with 300 μl of elution buffer (50 mm NaHCO3, 1% SDS). Samples were incubated overnight at 65°C to reverse PFA cross-links following the addition of 20 μl of 5 m NaCl. DNA was then purified from the eluted samples using QIAGEN PCR purification kit (QIAGEN). PCR was performed to analyze the STAT3 binding site in the p53 promoter using the following DNA primers: 5′-GGGCCCGTFTTGGTTCATCC-3′ and 5′-CCGCGAGACTCCTGGCACAA-3′. Conditions for PCRs were as follows: 30 cycles of 94°C (30 s), 60°C (30 s), 72°C (1 min). The PCR products were separated in 1.5% agarose gel.
In vivo injection.
The C57BL/6 and Kv4.2KO mice (8–9 weeks of age) of either sex were anesthetized by ketamine/xylazine mixture (3.33 ml/kg) via intraperitoneal injection, before the surgery. The delivery of reagents to CA1 was done using the following stereotaxic coordinates from bregma: anteroposterior, 2.4 mm; mediolateral, ±2.28 mm; ventrodorsal, 1.6 mm. Reagents (0.5 μl) were delivered at a rate of 0.12 μl/min using a Hamilton needle and syringe attached with a microsyringe pump controller (World Precision Instruments). Anti-TrkB (1.0 mg/ml) and vehicle (PBS) alone were injected, one to each side of the brain. The incision was closed using a polyglycolic acid suture (CP Medical). Animals were allowed to recover at 37°C for 1–2 h.
Statistical analyses.
Statistical significance between two groups was determined with a two-tailed paired Student's t test. For multiple groups, statistical comparisons were made by ANOVA followed by individual group tests with the Bonferroni correction made for multiple comparisons.
Results
Hippocampal neurons become BDNF dependent during elevated activity
As we reported previously, spontaneous network activity develops during the first 12 d in our primary cultures of hippocampal neurons (Murase et al., 2011a). After the conclusion of this period, on DIV14, we elevated the activity of the network by increasing the excitability of neurons by application of 4-AP, a drug that blocks the current mediated by A-type voltage-gated potassium channels (Zagotta et al., 1988). We monitored spontaneous network activity with the calcium indicator fluo-4 AM (Fig. 1A) and found treatment with 5 mm 4-AP increased the mean frequency of calcium responses (>100%; Fig. 1A).
Figure 1.
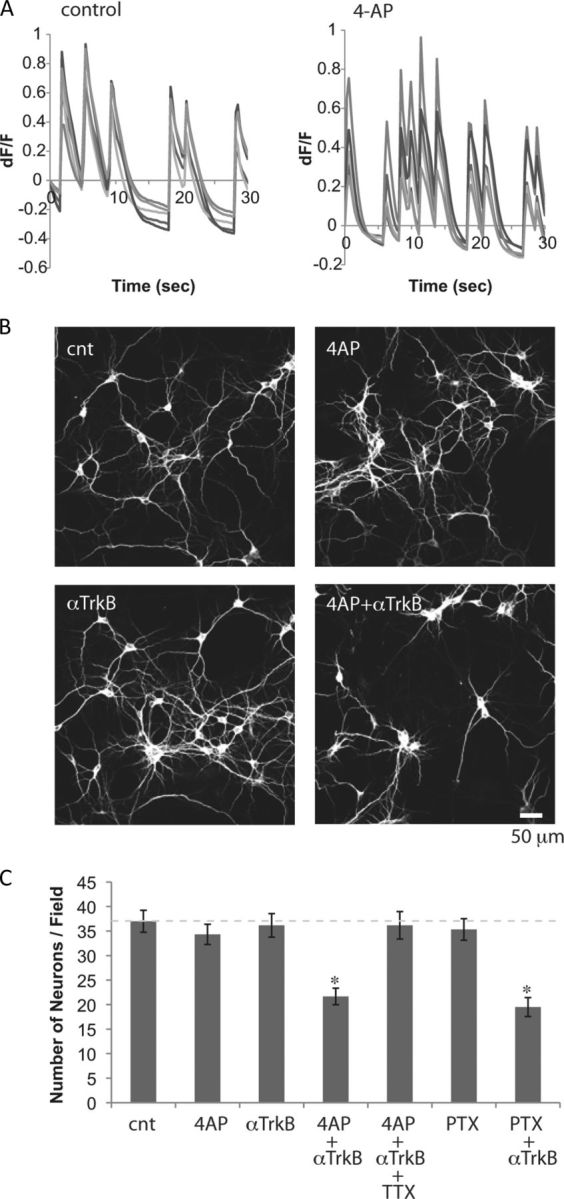
Neurons become TrkB dependent when network activity is elevated. A, Spontaneous activity of primary hippocampal neurons (DIV14), monitored by calcium responses using fluo-4 AM, were compared before and after application of 5 mm 4-AP. Ratio of change in fluorescence intensity (dF) to the intensity at t = 0 (F) is plotted. Responses of five simultaneously monitored neurons before and after application of 4-AP are shown as examples. B, Immunostaining for the neuronal marker, MAP2. Hippocampal neurons (DIV14) were exposed to 5 mm 4-AP and/or 5 μg/ml anti-TrkB antibody for 2 d. C, Numbers of neurons after the 2 d treatments. One micromolar TTX and 50 μm PTX were used (*p < 0.05, one-way ANOVA; n = 6). Error bars indicate SEM.
To test whether these neurons required neurotrophin for survival, we first showed that, in neurons (at DIV14), robust phosphorylation of Trk receptor followed exogenous application of BDNF. This effect was blocked by preincubation with a function-blocking anti-TrkB antibody (data not shown). Using this antibody, we found that blocking BDNF-TrkB signaling alone did not decrease the number of surviving neurons (Fig. 1B,C). This is strikingly different from the dramatic loss of immature neurons caused by this treatment during the developmental death period (Murase et al., 2011a). In mature neurons, elevation of network activity with 4-AP alone also did not affect the number of surviving neurons (Fig. 1B,C). However, elevating activity in the presence of the anti-TrkB antibody resulted in a significant decrease in neuron number (Fig. 1B,C). Blocking neuronal activity by applying the voltage-gated sodium channel blocker, TTX, attenuated 4-AP-induced TrkB dependency (Fig. 1C). Blocking phosphorylation of Trk receptors with 200 nm K252a, the specific inhibitor of Trk (Berg et al., 1992), also induced a loss of neurons but only when 4-AP was coapplied with the inhibitor (K252a alone, 100.5 ± 3.4% of control; K252a and 4-AP, 65.1 ± 7.2% of control; n = 6; p < 0.05). Elevating network activity by applying PTX, the GABAA receptor antagonist, also induced TrkB dependency (Fig. 1C). These results suggest that elevation of activity led to changes in intracellular signaling, causing the neurons to become, once again, dependent on BDNF-TrkB signaling.
Akt is necessary and sufficient for survival of chronically stimulated neurons
Activation of PI3K and recruitment of the serine-threonine kinase Akt to the plasma membrane initiates cell survival responses in many types of cells (Toker and Newton, 2000; Jacinto et al., 2006). Moreover, we recently reported that BDNF-triggered Akt activation is critical for the survival of immature neurons during the death period (Murase et al., 2011a). To determine whether Akt activation was sensitive to Trk activation, we exposed mature neurons to the function-blocking anti-TrkB antibody. The results show levels of phospho-Ser 473 Akt in neurons at DIV14 were severely reduced by the function-blocking anti-TrkB antibody (Fig. 2A), suggesting BDNF continues to play a major role in activating Akt signaling in these neurons. However, our new data suggest that the survival of these mature cells is not sensitive to neurotrophin-reduced Akt activation.
Figure 2.
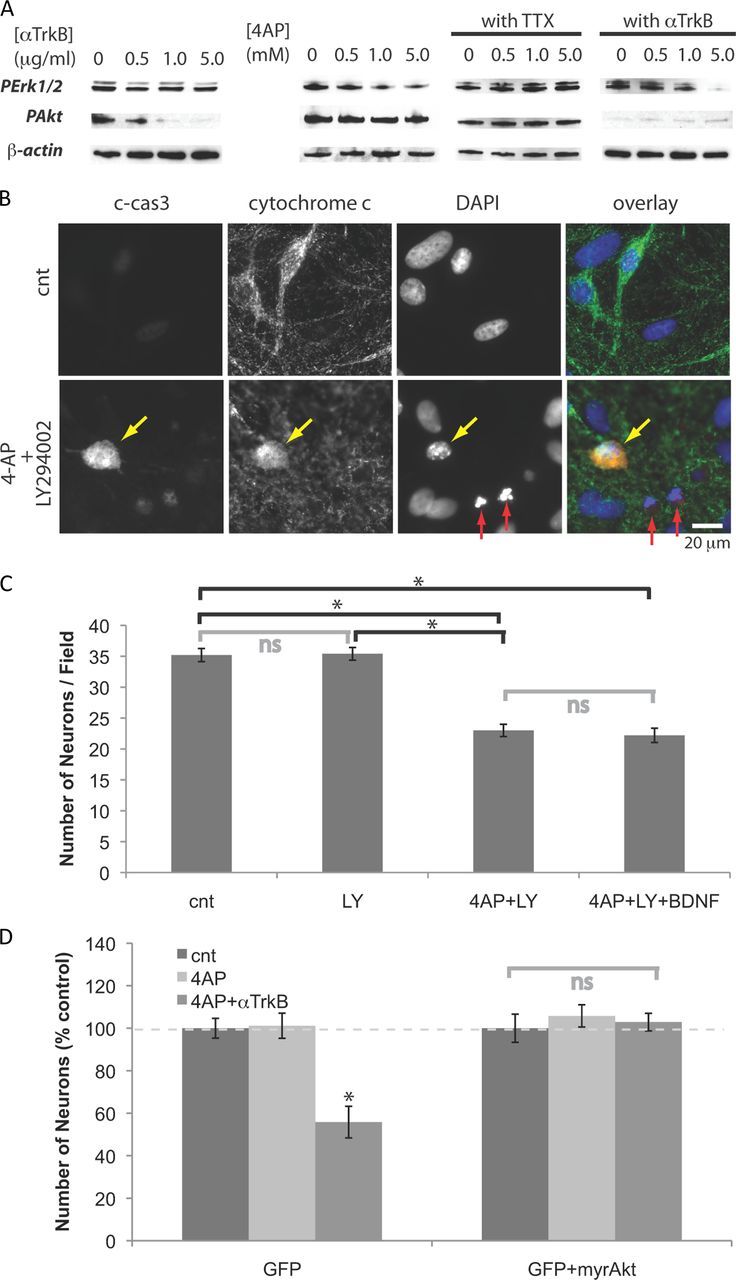
Neurons become dependent on Akt signaling when treated with 4-AP. A, Western blot analyses. Neurons were treated for 18 h with 5 mm 4-AP, and/or 5 μg/ml anti-TrkB antibody, or 1 mm TTX. B, Immunostaining for c-cas3 and cytochrome c, and DAPI staining. Neurons were treated for 2 d (from DIV12 to DIV14) with 5.0 mm 4-AP and/or 25 μm LY294002. The arrows indicate apoptotic neurons with (yellow) or without (red) activation of caspase 3. C, Numbers of neurons after 2 d treatments. Five millimolar 4-AP, 25 μm LY294002, 10 ng/ml BDNF were used (n = 4). *p < 0.05; ns, p > 0.05 (one-way ANOVA). D, Neurons (DIV9) were cotransfected with plasmids containing cDNAs of GFP and myristoylated Akt, and then incubated with 5 mm 4-AP (and 5 μg/ml anti-TrkB) for 2 d (from DIV12 to DIV14). Transfected neurons were detected by GFP fluorescence. Numbers of neurons remaining after 2 d treatments (n = 6). *p < 0.05. Error bars indicate SEM.
Other serine/threonine kinases, Erk1/2, are activated by many receptor tyrosine kinases, including TrkB, and are also often implicated in survival responses (Huang and Reichardt, 2003). Incubation with anti-TrkB antibody, however, did not affect levels of phospho-Erk1/2 without affecting Akt levels (Fig. 2A). Addition of TTX attenuated the effect of 4-AP, confirming that impairment of Erk1/2 signaling was due to the elevation of spiking activity (Fig. 2A). When neurons were treated with 4-AP and anti-TrkB antibody, both Akt and Erk1/2 signaling pathways were impaired (Fig. 2A).
To test whether blocking Akt signals can have the same effect as applying anti-TrkB antibody, we then added LY294002, the PI3K inhibitor. When treated with both 4-AP and LY294002, many neurons showed: (1) activation of caspase 3 (c-cas3), a protein that plays a central role in apoptosis (Fernandes-Alnemri et al., 1994); (2) a diffuse pattern of cytochrome c, a protein that initiates apoptosis (Liu et al., 1996); and (3) fragmented nuclei revealed by DAPI staining (Fig. 2B). These results suggest that, in overactivated neurons, Akt signals mediate survival. Consistent with these observations, treatment with 4-AP and LY294002 decreased the number of neurons, whereas LY294002 alone did not affect the number of neurons (Fig. 2C). Moreover, exogenous BDNF did not rescue these neurons from death caused by coapplication of 4-AP and LY294002 (Fig. 2C), suggesting Akt is critical for BDNF-mediated neuronal survival.
Phosphorylation triggers Akt to translocate to the plasma membrane, a step critical for its activation. Akt tagged with a myristoylation site is constitutively active (Kohn et al., 1996). When neurons were transfected with a control plasmid expressing only green fluorescent protein (GFP), numbers of transfected neurons decreased upon coapplication of 4-AP and anti-TrkB, whereas neurons cotransfected with a plasmid encoding myristoylated Akt were not affected by the treatment (Fig. 2D). Thus, constitutively active Akt is sufficient to block the induction of death caused by the 4-AP and anti-TrkB treatment. Together, these results suggest Erk1/2 signal is impaired during elevated neural activity, causing neurons to become dependent on Akt signals mediated mainly by TrkB activation.
STAT3 provides the alternative survival signal
When studying potential mediators of the Erk1/2 survival signal, we found that levels of phospho-Ser-727 STAT3 were severely impaired by treatment with 4-AP (Fig. 3A). This treatment caused no change in levels of phospho-Tyr-705 STAT3 (data not shown). Blocking network activity by applying TTX completely abolished this effect of 4-AP (Fig. 3A). Inhibiting Erk1/2 with PD98059 effectively suppressed the phosphorylation of Ser-727 STAT3 and occluded the effect of 4-AP (Fig. 3A). Neither the treatments with anti-TrkB nor LY294002 affected the phosphorylation (Fig. 3A). Inhibition of mTOR by rapamycin (Brown et al., 1994), cyclin-dependent kinase 5 (Cdk5) by roscovitine (Meijer et al., 1997), however, resulted in a minor reduction of phosphorylation (Fig. 3B). Next, we analyzed the interaction between Erk1/2 and STAT3 with an immuno-coprecipitation experiment. When the lysate from hippocampal culture was precipitated with anti-STAT3, we observed it coprecipitated Erk1/2, suggesting that there is an interaction between Erk1/2 and STAT3 (Fig. 3C). These results suggest that STAT3 may be phosphorylated at Ser-727 by Erk1/2.
Figure 3.

Phosphorylation of STAT3 is impaired following treatment with 4-AP. A, Western blot analyses of PSer-727 STAT3 and total STAT3. Neurons were treated for 18 h with the indicated concentrations of 4-AP and 1 μm TTX, 50 μm PD98059, or the indicated concentrations of anti-TrkB antibody and LY294002. B, Effect of inhibition of various serine/threonine kinases on PSer-727 STAT3. Neurons were incubated for 2 h with 20 ng/ml rapamycin, 50 μm roscovitine, or 50 μm PD98059. C, Immunoprecipitation of STAT3. Shown are Western blot analyses of STAT3 and Erk1/2.
To directly test whether phosphorylation of STAT3 plays a role in the survival of neurons, we overexpressed the phosphomimetic Ser727Glu STAT3 (SE-STAT3). Neurons expressing only GFP declined significantly in number after treatment with 4-AP in the presence of the anti-TrkB antibody or LY294002, but the survival of neurons expressing Ser727Glu STAT3 was not affected by these treatments (Fig. 4A,B). These results suggest that STAT3 signaling mediates neuronal survival and the role of TrkB in neuronal survival can only be assessed when the Erk-STAT3 pathway is inhibited.
Figure 4.
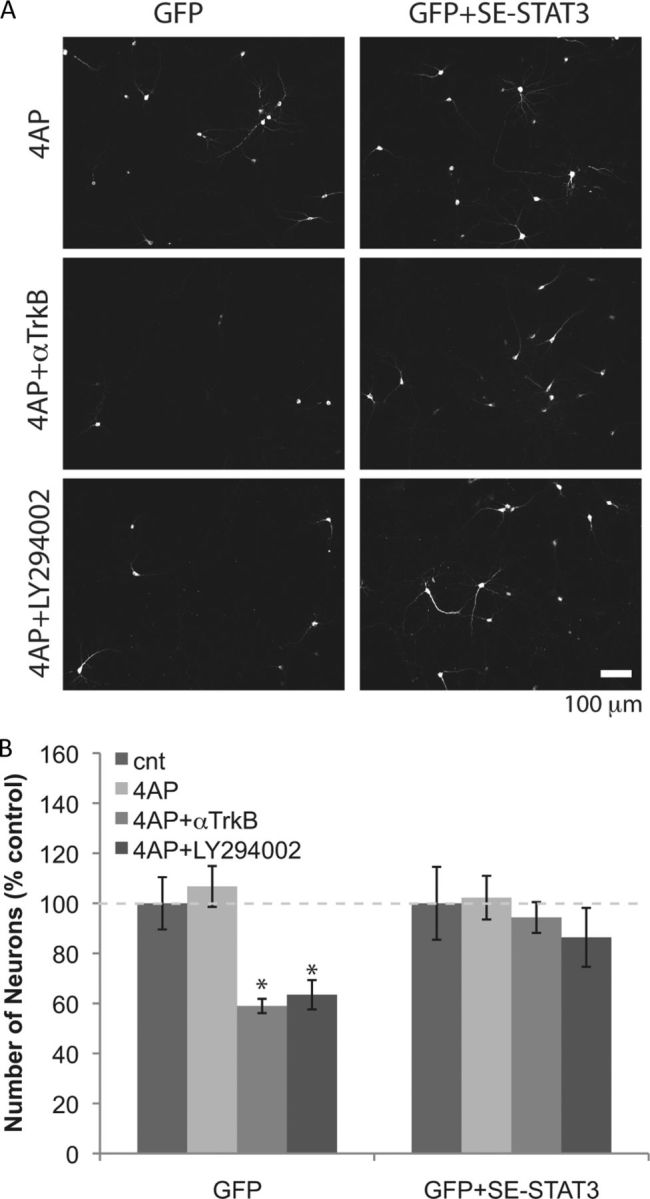
Ser727Glu STAT3 prevents neurons from becoming TrkB and Akt dependent during elevated activity. A, Neurons (DIV9) were cotransfected with plasmids containing cDNAs of GFP and Ser727Glu STAT3, and then incubated with 5 mm 4-AP (and 5 μg/ml anti-TrkB, or 25 μm LY294002) for 2 d (from DIV12 to DIV14). Transfected neurons were detected by GFP fluorescence. B, Numbers of neurons remaining after 2 d treatments. *p < 0.05, n = 8 (one-way ANOVA). Error bars indicate SEM.
Consistent with a central role for STAT3 in regulating the alternate survival pathway, neurons expressing Ser727Ala STAT3, which cannot be phosphorylated, were vulnerable to anti-TrkB or LY294002 treatment, which does not change activity of Erk phosphorylation (Fig. 5). Neurons expressing GFP only or coexpressing wild-type STAT3 were not affected by these treatments (Fig. 5). These results show that serine phosphorylation of STAT3 is necessary and sufficient for the survival of mature hippocampal neurons.
Figure 5.

Ser727Ala STAT3 induces TrkB and Akt dependency in neurons. Neurons (DIV9) were cotransfected with plasmids containing cDNAs for GFP, wild-type STAT3, or Ser727Ala STAT3, and then incubated with 5 μg/ml anti-TrkB, or 25 μm LY294002 for 2 d (from DIV12 to DIV14). Numbers of neurons in control were taken as 100% (n = 10). *p < 0.05, one-way ANOVA. Error bars indicate SEM.
Finally, to confirm that loss of STAT3 signaling is responsible for TrkB and Akt dependency, we examined the effect of knocking down endogenous STAT3 expression. We cotransfected neurons after the death period (DIV9) with STAT3 siRNA and GFP vector. Application of STAT3 siRNA significantly downregulated STAT3 levels and upregulated a tumor suppressor, p53, and Bax, a proapoptotic target of p53 (Deckwerth et al., 1996) (Fig. 6A). Numbers of siRNA-containing neurons dramatically decreased upon treatment with anti-TrkB or LY294002 (Fig. 6A,B). Application of BDNF did not rescue the effect of LY294002 on STAT3 knockdown neurons, indicating BDNF provides its survival effect mainly through Akt. These results are consistent with our observation that endogenous STAT3 is required for resistance against neurotrophin deprivation.
Figure 6.
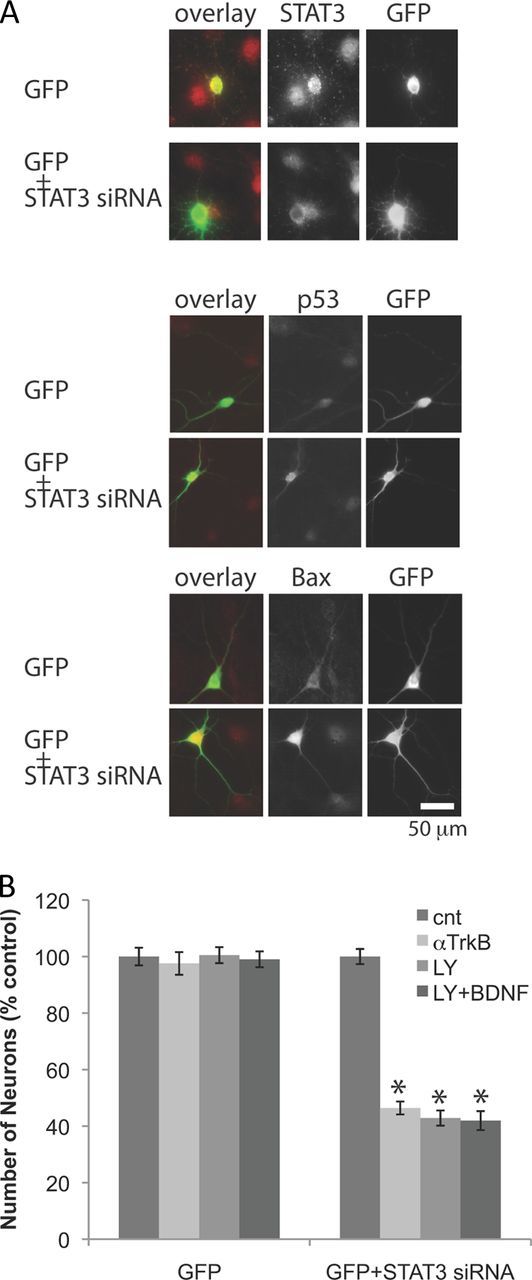
STAT3 blockade by siRNA induces TrkB and Akt dependency in neurons. A, Immunostaining of GFP, STAT3, p53, and Bax. Neurons (DIV9) were cotransfected with a plasmid containing cDNA of GFP and STAT3 siRNA. B, Number of neurons after incubation with 5 μg/ml anti-TrkB, or 25 μm LY294002 (and 10 ng/ml BDNF) for 2 d (DIV12 to DIV14). Numbers of neurons in control were taken as 100% (n = 5). *p < 0.05. Error bars indicate SEM.
We reported previously that young neurons go through a well characterized period of developmental death, and during this period, neurons are dependent upon Akt signaling for their survival (Murase et al., 2011a). Our new findings reported here prompted us to investigate levels of STAT3 during these distinct phases of neuronal vulnerability. We found that the total level of STAT3 protein was much lower in young neurons (DIV7) than in mature neurons (DIV14). The levels of PSer STAT3 followed the same proportions (Fig. 7A). We also observed slightly lower levels of pErk1/2. In the neonatal hippocampus, p53 is a key regulator for the survival of neonatal neurons (Murase et al., 2011b). We found levels of p53 and Bax declined significantly after the developmental death period (Fig. 7A). Neurons cotransfected with GFP and wild-type STAT3-expressing plasmids showed lower levels of p53 and Bax than those transfected with GFP plasmid only (Fig. 7B). Furthermore, cotransfection with GFP and wild-type STAT3-expressing plasmids rescued the young neurons from death induced by anti-TrkB or LY294002, whereas S727A STAT3 coexpression failed to rescue the neurons (Fig. 7C). These results suggest immature neurons are vulnerable because they contain low levels of STAT3. Together, our results show levels of STAT3 signaling are regulated developmentally, effectively controlling the vulnerability of neurons.
Figure 7.

STAT3 is responsible for the change in vulnerability during development. A, Western blot analyses of pErk1/2, STAT3, p53, and Bax from hippocampal cultures (DIV7 and DIV14). B, Immunostaining of p53 and Bax in young neuron (DIV7) expressing GFP (and wild-type STAT3). C, Young neurons (DIV7) expressing GFP (and wild-type STAT3 or Ser727Ala STAT3) were incubated in 5 μg/ml anti-TrkB antibody or 25 μm LY294002 for 2 d. *p < 0.05; n = 10. Error bars indicate SEM.
Kv4.2 knock-out mouse hippocampus shows impairment of Erk1/2-STAT3 signaling
The finding that levels of p53 declined significantly after the developmental death period prompted us to check whether p53 levels increased in neurons challenged by elevated activity. Western blot analyses showed that the treatment of hippocampal neurons (DIV14) with 4-AP resulted in the upregulation of p53 (Fig. 8A). Bax also increased in the treated neurons (Fig. 8A). RT-PCR showed the amount of p53 mRNA had increased (Fig. 8A), suggesting that the transcription of p53 is stimulated by the treatment. These results suggest the increase in a proapoptotic protein, Bax, contributes to induction of vulnerability.
Figure 8.
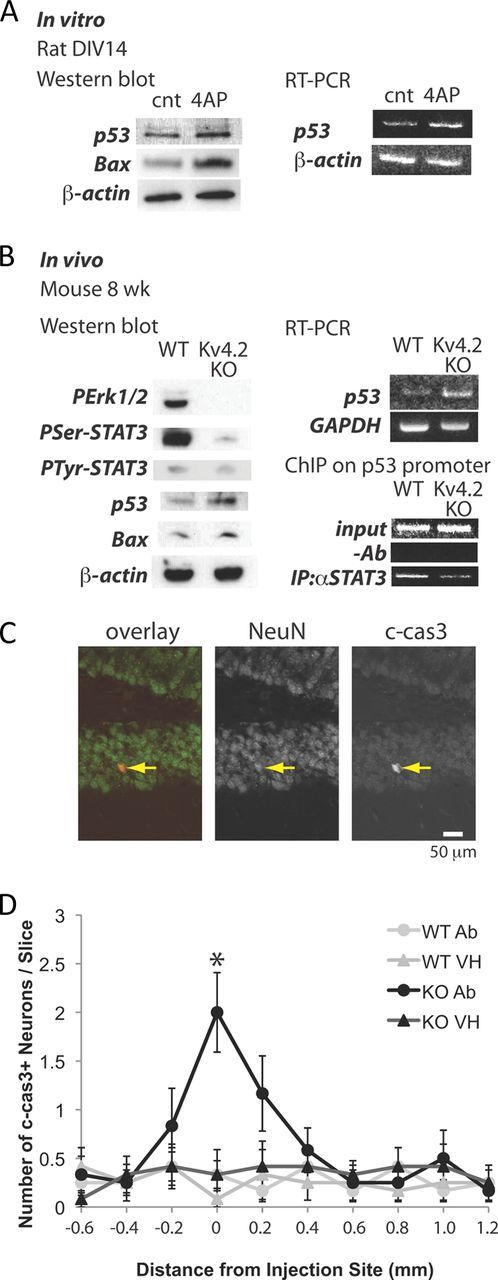
Elevated excitability increases levels of p53 and Bax in vitro and in vivo. A, Left panel, Western blot analyses of p53 and Bax from rat hippocampal culture (DIV14) treated with 5 mm 4-AP overnight. Right panel, RT-PCR analysis of p53. B, Left panel, Western blot analyses of PErk1/2, PSer-727 STAT3, p53, and Bax from wild-type and Kv4.2KO mouse hippocampus (8 weeks). Right top panel, RT-PCR analysis of p53. Right bottom panel, ChIP assay of STAT3 binding to the p53 promoter. C, Immunostaining of NeuN and c-cas3 in Kv4.2KO mouse hippocampus injected with anti-TrkB antibody. The arrow indicates an apoptotic neuron. D, Number of c-cas3+ neurons in wild-type and Kv4.2KO hippocampal slices injected with vehicle and anti-TrkB antibody. *p < 0.001; n = 12. Error bars indicate SEM.
We then checked whether these alterations observed in vitro also occur in vivo by comparing the hippocampi of Kv4.2 knock-out mice with those of wild-type mice. Kv4.2 is an A-type voltage-gated potassium channel subunit expressed in hippocampal neurons (Sheng et al., 1992), and the loss of Kv4.2 function results in dendritic excitability (Kim et al., 2007). Western blot analyses showed that genetic deletion of Kv4.2 resulted in loss of Erk1/2-STAT3 signaling, and upregulation of p53 and Bax (Fig. 8B). RT-PCR showed that the knock-out hippocampus contained higher levels of p53 mRNA than the wild type. To check whether loss of STAT3 signaling directly affects p53 expression, we performed a ChIP assay on wild-type and Kv4.2KO hippocampal neurons. Immunoprecipitation of STAT3 showed the binding of STAT3 to p53 promoter, which was impaired in Kv4.2KO neurons (Fig. 8B), suggesting that the change in STAT3 signaling directly affects p53 expression. To confirm that elevated excitability causes vulnerability in vivo, we then made bilateral injections to the wild type and Kv4.2KO CA1 in adult mice; the function-blocking anti-TrkB antibody was delivered to one side of the brain, and vehicle control was delivered to the other side. Neither the antibody nor vehicle injections caused notable neuronal death in the wild-type hippocampi (Fig. 8D). However, we observed an area of significantly increased numbers of apoptotic neurons centered around the antibody-injected region of Kv4.2KO hippocampi, but no increase in neuronal death on the contralateral side where vehicle alone had been injected (Fig. 8C,D), suggesting that Kv4.2KO hippocampal neurons were indeed sensitive to the blockade of TrkB signaling. Together, these results suggest that elevated excitability in vitro and in vivo inhibits the Erk1/2-STAT3 survival pathway that controls the survival of neurons through transcription of p53 (Fig. 9).
Figure 9.

Elevation of activity impairs Erk1/2-STAT3 signaling to control p53 expression. In mature neurons, Erk1/2 activates STAT3, and STAT3 binds and regulates p53 transcription, leading to resistance to the loss of neurotrophin-Akt survival signaling. This alternative survival pathway is impaired when neurons are challenged by elevated activity.
Discussion
It remains unclear how neurons integrate signals from different extracellular cues to initiate events that support survival. Previously, we reported that, during the period of developmental neuronal death, neurons require neurotrophin-induced Akt signaling for their survival (Murase et al., 2011a). Neurons that survive this vulnerable period, the subjects of the current study, no longer depend on Akt signaling for continued viability. Here, we show that elevation of network activity induces neurotrophin dependency in neurons by inhibiting a survival-signaling pathway that inhibits activation of STAT3. This inhibition is caused by the impairment of Erk1/2 and, as a consequence, these neurons become dependent on Trk-mediated Akt activation. Erk1/2 and Akt can be simultaneously activated by many tyrosine receptor kinases including Trk (Huang and Reichardt, 2003), and these kinases share common substrates; one is cAMP response element-binding protein (Tan et al., 1996; Du and Montminy, 1998), which plays an important role in the survival of neurons (Finkbeiner et al., 1997). The response to elevated activity, a severe reduction in levels of PErk1/2 with little change in the levels of PAkt, suggests that Erk1/2 and Akt are activated independently.
A variety of serine/threonine kinases has been reported to phosphorylate STAT3 at Ser-727 (Chung et al., 1997; Oh et al., 1998; Jain et al., 1999; Lim and Cao, 2001; Fu et al., 2004). However, as we show here, in hippocampal neurons, Ser phosphorylation of STAT3 largely depends on Erk1/2. Overexpression of the phosphomimetic mutant Ser727Glu STAT3 completely attenuated neurotrophin dependency in chronically activated neurons, suggesting STAT3 is the key molecule downstream from the Erk1/2 survival signal. Ser727Ala STAT3, however, acts as a dominant-negative species. Studies have shown different effects of Ser phosphorylation on STAT3 activity (Decker and Kovarik, 2000): Originally, the dominant role was assigned to PTyr-705, and PSer-727 was reported to be required for maximum activity (Wen et al., 1995; Decker and Kovarik, 2000). Others observed transcriptional activity in the absence of detectable PTyr-705 (Ceresa and Pessin, 1996; Ng et al., 2006). In some cases, PSer-727 inhibited PTyr-705 and STAT3 activity (Chung et al., 1997; Jain et al., 1998; Lim and Cao, 1999). Only low levels of PTyr-705 were detected in our study, and the elevation of network activity in vivo and in vitro did not affect the levels of Tyr phosphorylation. These contradictory findings may reflect differences of cell type and cellular conditions.
Previously, we reported that young neurons, during the developmental death period, require neurotrophin and Akt signaling for their survival (Murase et al., 2011a). Notably, neurons that survive this period of vulnerability have outgrown their dependency on Akt signaling for continued viability. Similar changes in vulnerability to death have been observed in developing sympathetic neurons (Easton et al., 1997). Several lines of evidence suggest STAT3 signaling controls the vulnerability of neurons: (1) during development, STAT3 levels increase dramatically as neurons become resistant to neurotrophin deprivation; (2) overexpression of STAT3 in immature neurons attenuates death caused by the blockade of Trk-Akt signaling; (3) expression of dominant-negative (Ser727Ala) STAT3 as well as knockdown of endogenous STAT3 in older neurons (DIV14) induces Trk-Akt dependency. Following chronic elevation of network activity, these mature neurons once again became dependent on Trk-Akt signaling caused by the loss of STAT3 signaling. Thus, these mature neurons revert to features characteristic of immature neurons.
Our new results indicate that chronically activated neurons in vitro and in vivo express higher levels of p53 and its proapoptotic target, Bax. We found that this change in p53 expression occurs at its transcription level. These results are consistent with a previous study that demonstrated p53 transcription is negatively regulated by STAT3 (Niu et al., 2005). During the developmental death period, p53 serves as a key regulator to control survival of hippocampal neurons (Murase et al., 2011b). The increased p53 expression in chronically activated neurons reflects this feature of immature neurons.
The identification of STAT3 phosphoserine modification as a critical link between neuronal activity and survival raises many further questions. Among the most interesting is how STAT3 activation is quantitatively linked to the appropriate level of activity. This mechanism is of potential clinical interest as an altered balance of excitation/inhibition (E/I) is thought to contribute to many diseases (Heinemann, 2004; Belforte et al., 2010; Rubenstein, 2010; Yizhar et al., 2011). In these pathological conditions, neurons are subject to chronically elevated activity. Although neurons can use compensatory synaptic scaling mechanisms to adjust their net input (Davis, 2006; Turrigiano, 2007), our results suggest neurons can undergo dramatic changes in vulnerability that may contribute to the degeneration of dendritic morphology and neuronal death observed in neurological diseases characterized by altered E/I balance (Paul et al., 1981; Garey et al., 1998; Boda et al., 2010). As STAT3 has been reported to modulate the expression of synaptic proteins (Lund et al., 2008), it would be interesting to determine whether STAT3 also plays a role in homeostatic plasticity.
Footnotes
This work was supported by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke–National Institutes of Health. We thank Dr. Yukiko Gotoh for wild-type and mutant STAT3 IRES EGFP/pMX plasmids.
The authors declare no competing financial interests.
References
- Ballas N, Battaglioli E, Atouf F, Andres ME, Chenoweth J, Anderson ME, Burger C, Moniwa M, Davie JR, Bowers WJ, Federoff HJ, Rose DW, Rosenfeld MG, Brehm P, Mandel G. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31:353–365. doi: 10.1016/s0896-6273(01)00371-3. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg MM, Sternberg DW, Parada LF, Chao MV. K-252a inhibits nerve growth factor-induced trk proto-oncogene tyrosine phosphorylation and kinase activity. J Biol Chem. 1992;267:13–16. [PubMed] [Google Scholar]
- Boda B, Dubos A, Muller D. Signaling mechanisms regulating synapse formation and function in mental retardation. Curr Opin Neurobiol. 2010;20:519–527. doi: 10.1016/j.conb.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- Ceresa BP, Pessin JE. Insulin stimulates the serine phosphorylation of the signal transducer and activator of transcription (STAT3) isoform. J Biol Chem. 1996;271:12121–12124. doi: 10.1074/jbc.271.21.12121. [DOI] [PubMed] [Google Scholar]
- Chung J, Uchida E, Grammer TC, Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol. 1997;17:6508–6516. doi: 10.1128/mcb.17.11.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA, Thalhammer A, Yu LM, Catalano M, Ramos T, Colicos MA, Goda Y. Activity-dependent regulation of synaptic AMPA receptor composition and abundance by beta3 integrins. Neuron. 2008;58:749–762. doi: 10.1016/j.neuron.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JE, Lee PR, Chen S, Li W, Fields RD. MicroRNA regulation of homeostatic synaptic plasticity. Proc Natl Acad Sci U S A. 2011;108:11650–11655. doi: 10.1073/pnas.1017576108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628–2637. doi: 10.1038/sj.onc.1203481. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. 2009;64:61–78. doi: 10.1016/j.neuron.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998;273:32377–32379. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]
- Dziennis S, Jia T, Rønnekleiv OK, Hurn PD, Alkayed NJ. Role of signal transducer and activator of transcription-3 in estradiol-mediated neuroprotection. J Neurosci. 2007;27:7268–7274. doi: 10.1523/JNEUROSCI.1558-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton RM, Deckwerth TL, Parsadanian AS, Johnson EM., Jr Analysis of the mechanism of loss of trophic factor dependence associated with neuronal maturation: a phenotype indistinguishable from Bax deletion. J Neurosci. 1997;17:9656–9666. doi: 10.1523/JNEUROSCI.17-24-09656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Litwack G, Alnemri ES. CPP32, a novel human apoptotic protein with homology to Caenorhabditis elegans cell death protein Ced-3 and mammalian interleukin-1 beta-converting enzyme. J Biol Chem. 1994;269:30761–30764. [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME. CREB: a major mediator of neuronal neurotrophin responses. Neuron. 1997;19:1031–1047. doi: 10.1016/s0896-6273(00)80395-5. [DOI] [PubMed] [Google Scholar]
- Fu AK, Fu WY, Ng AK, Chien WW, Ng YP, Wang JH, Ip NY. Cyclin-dependent kinase 5 phosphorylates signal transducer and activator of transcription 3 and regulates its transcriptional activity. Proc Natl Acad Sci U S A. 2004;101:6728–6733. doi: 10.1073/pnas.0307606100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM, Barnes TR, Hirsch SR. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry. 1998;65:446–453. doi: 10.1136/jnnp.65.4.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann U. Basic mechanisms of partial epilepsies. Curr Opin Neurol. 2004;17:155–159. doi: 10.1097/00019052-200404000-00012. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Izant JG, McIntosh JR. Microtubule-associated proteins: a monoclonal antibody to MAP2 binds to differentiated neurons. Proc Natl Acad Sci U S A. 1980;77:4741–4745. doi: 10.1073/pnas.77.8.4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Jain N, Zhang T, Fong SL, Lim CP, Cao X. Repression of Stat3 activity by activation of mitogen-activated protein kinase (MAPK) Oncogene. 1998;17:3157–3167. doi: 10.1038/sj.onc.1202238. [DOI] [PubMed] [Google Scholar]
- Jain N, Zhang T, Kee WH, Li W, Cao X. Protein kinase C delta associates with and phosphorylates Stat3 in an interleukin-6-dependent manner. J Biol Chem. 1999;274:24392–24400. doi: 10.1074/jbc.274.34.24392. [DOI] [PubMed] [Google Scholar]
- Jung JE, Kim GS, Narasimhan P, Song YS, Chan PH. Regulation of Mn-superoxide dismutase activity and neuroprotection by STAT3 in mice after cerebral ischemia. J Neurosci. 2009;29:7003–7014. doi: 10.1523/JNEUROSCI.1110-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jung SC, Clemens AM, Petralia RS, Hoffman DA. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron. 2007;54:933–947. doi: 10.1016/j.neuron.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn AD, Takeuchi F, Roth RA. Akt, a pleckstrin homology domain containing kinase, is activated primarily by phosphorylation. J Biol Chem. 1996;271:21920–21926. doi: 10.1074/jbc.271.36.21920. [DOI] [PubMed] [Google Scholar]
- Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Lim CP, Cao X. Serine phosphorylation and negative regulation of Stat3 by JNK. J Biol Chem. 1999;274:31055–31061. doi: 10.1074/jbc.274.43.31055. [DOI] [PubMed] [Google Scholar]
- Lim CP, Cao X. Regulation of Stat3 activation by MEK kinase 1. J Biol Chem. 2001;276:21004–21011. doi: 10.1074/jbc.M007592200. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Lund IV, Hu Y, Raol YH, Benham RS, Faris R, Russek SJ, Brooks-Kayal AR. BDNF selectively regulates GABAA receptor transcription by activation of the JAK/STAT pathway. Sci Signal. 2008;1:ra9. doi: 10.1126/scisignal.1162396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, Inagaki M, Delcros JG, Moulinoux JP. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem. 1997;243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- Murase S, McKay RD. A specific survival response in dopamine neurons at most risk in Parkinson's disease. J Neurosci. 2006;26:9750–9760. doi: 10.1523/JNEUROSCI.2745-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murase S, Owens DF, McKay RD. In the newborn hippocampus, neurotrophin-dependent survival requires spontaneous activity and integrin signaling. J Neurosci. 2011a;31:7791–7800. doi: 10.1523/JNEUROSCI.0202-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murase S, Poser SW, Joseph J, McKay RD. p53 controls neuronal death in the CA3 region of the newborn mouse hippocampus. Eur J Neurosci. 2011b;34:374–381. doi: 10.1111/j.1460-9568.2011.07758.x. [DOI] [PubMed] [Google Scholar]
- Ng YP, Cheung ZH, Ip NY. STAT3 as a downstream mediator of Trk signaling and functions. J Biol Chem. 2006;281:15636–15644. doi: 10.1074/jbc.M601863200. [DOI] [PubMed] [Google Scholar]
- Niu G, Wright KL, Ma Y, Wright GM, Huang M, Irby R, Briggs J, Karras J, Cress WD, Pardoll D, Jove R, Chen J, Yu H. Role of Stat3 in regulating p53 expression and function. Mol Cell Biol. 2005;25:7432–7440. doi: 10.1128/MCB.25.17.7432-7440.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H, Fujio Y, Kunisada K, Hirota H, Matsui H, Kishimoto T, Yamauchi-Takihara K. Activation of phosphatidylinositol 3-kinase through glycoprotein 130 induces protein kinase B and p70 S6 kinase phosphorylation in cardiac myocytes. J Biol Chem. 1998;273:9703–9710. doi: 10.1074/jbc.273.16.9703. [DOI] [PubMed] [Google Scholar]
- Paul LA, Fried I, Watanabe K, Forsythe AB, Scheibel AB. Structural correlates of seizure behavior in the mongolian gerbil. Science. 1981;213:924–926. doi: 10.1126/science.7256289. [DOI] [PubMed] [Google Scholar]
- Rubenstein JL. Three hypotheses for developmental defects that may underlie some forms of autism spectrum disorder. Curr Opin Neurol. 2010;23:118–123. doi: 10.1097/WCO.0b013e328336eb13. [DOI] [PubMed] [Google Scholar]
- Schweizer U, Gunnersen J, Karch C, Wiese S, Holtmann B, Takeda K, Akira S, Sendtner M. Conditional gene ablation of Stat3 reveals differential signaling requirements for survival of motoneurons during development and after nerve injury in the adult. J Cell Biol. 2002;156:287–297. doi: 10.1083/jcb.200107009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeburg DP, Feliu-Mojer M, Gaiottino J, Pak DT, Sheng M. Critical role of CDK5 and Polo-like kinase 2 in homeostatic synaptic plasticity during elevated activity. Neuron. 2008;58:571–583. doi: 10.1016/j.neuron.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Tsaur ML, Jan YN, Jan LY. Subcellular segregation of two A-type K+ channel proteins in rat central neurons. Neuron. 1992;9:271–284. doi: 10.1016/0896-6273(92)90166-b. [DOI] [PubMed] [Google Scholar]
- Shepherd JD, Rumbaugh G, Wu J, Chowdhury S, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron. 2006;52:475–484. doi: 10.1016/j.neuron.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Rouse J, Zhang A, Cariati S, Cohen P, Comb MJ. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J. 1996;15:4629–4642. [PMC free article] [PubMed] [Google Scholar]
- Toker A, Newton AC. Cellular signaling: pivoting around PDK-1. Cell. 2000;103:185–188. doi: 10.1016/s0092-8674(00)00110-0. [DOI] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic signaling: the positive side of negative feedback. Curr Opin Neurobiol. 2007;17:318–324. doi: 10.1016/j.conb.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Wen Z, Zhong Z, Darnell JE., Jr Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P, Deisseroth K. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Brainard MS, Aldrich RW. Single-channel analysis of four distinct classes of potassium channels in Drosophila muscle. J Neurosci. 1988;8:4765–4779. doi: 10.1523/JNEUROSCI.08-12-04765.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel LS, Kuruvilla R, Ginty DD. Functions and mechanisms of retrograde neurotrophin signalling. Nat Rev Neurosci. 2005;6:615–625. doi: 10.1038/nrn1727. [DOI] [PubMed] [Google Scholar]