

Abstract
It is not known why people are more susceptible to bacterial infections such as non-Typhoid Salmonella (NTS) during and after a malaria infection but, in mice, malarial hemolysis impairs resistance to NTS by impairing the neutrophil oxidative burst. This acquired neutrophil dysfunction is a consequence of induction of the cytoprotective, heme degrading enzyme heme oxygenase-1 (HO-1) in neutrophil progenitors in bone marrow. In this study, we assessed whether neutrophil dysfunction occurs in humans with malaria and how this relates to hemolysis. We evaluated neutrophil function in 58 Gambian children with Plasmodium falciparum malaria (55 (95%) with uncomplicated disease), and examined associations with erythrocyte count, haptoglobin, hemopexin, plasma heme, expression of receptors for heme uptake, and HO-1 induction. Malaria caused the appearance of a dominant population of neutrophils with reduced oxidative burst activity, which gradually normalized over 8 weeks of follow-up. The degree of neutrophil impairment correlated significantly with markers of hemolysis and HO-1 induction. HO-1 expression was increased in blood during acute malaria, but at a cellular level HO-1 expression was modulated by changes in surface expression of the haptoglobin receptor (CD163). These findings demonstrate that neutrophil dysfunction occurs in P. falciparum malaria and support the relevance of the mechanistic studies in mice. Furthermore, they suggest the presence of a regulatory pathway to limit HO-1 induction by hemolysis in the context of infection, and indicate new targets for therapeutic intervention to abrogate the susceptibility to bacterial infection in the context of hemolysis in humans.
Introduction
Plasmodium falciparum malaria caused an estimated 655,000 deaths and 216 million cases globally in 2010 (1), but this almost certainly underestimates the indirect health burden (2) which includes increased susceptibility to Gram negative bacterial infections (3-4), particularly non-Typhoidal Salmonella (NTS) (3, 5-6). In areas with high malaria transmission, these indirect effects of malaria infection may explain more than half of the child mortality (2) and community acquired bacteremia (4). The incidence of NTS closely reflects that of malaria (4, 6-7) and there is compelling evidence that P. falciparum malaria increases susceptibility to NTS bacteremia in humans. In The Gambia, the incidence of NTS bacteremia has declined dramatically over the past thirty years, mirroring the decline in the incidence of malaria (7); this observation has since been confirmed in Kenya (4). In the pre-antibiotic era, malaria therapy for treatment of neurosyphilis was frequently complicated by NTS bacteremia even when NTS infection was otherwise very rare (8), and quinine alone often cured endemic malaria-NTS co-infection (9). NTS bacteremia incidence was found to be more closely related to malaria incidence than to stool carriage of NTS (6), and in Kenyan children sickle cell trait was found to have a protective effect against bacteremia, which was dependent on the protection it affords against malaria (4). Several studies have shown that susceptibility to NTS is greatest in the context of severe malarial anemia (5-6), while others have found that the greatest risk occurred in children with recent rather than current malaria infection (10-11).
Most mechanisms that have been proposed to account for the susceptibility to NTS that occurs in malaria involve monocyte and macrophage dysfunction. Malaria may impair monocyte and macrophage function through direct adhesion of infected RBCs (12), through the accumulation of hemozoin within these cells (13), or by impairment of systemic IL-12 production (14). However, other studies in mice have demonstrated that hemolysis - caused by malaria or in any other way - increases susceptibility to NTS and some other bacterial infections, whereas blood loss alone does not (15-17). We have recently shown in a mouse model of malarial anemia that resistance to S. typhimurium is impaired as a result of neutrophil dysfunction (rather than monocyte/macrophage dysfunction) caused by liberation of heme during hemolysis and by induction of the cytoprotective heme catabolising enzyme heme oxygenase-1 (HO-1) (18). In this model system, HO-1 induction in myeloid progenitor cells in the bone marrow leads to production of granulocytes with reduced oxidative burst activity, and their mobilization into the blood is enhanced by both hemolysis-derived heme and the response to bacterial co-infection. This results in the accumulation of functionally impaired granulocytes in the circulation which are able to phagocytose S. typhimurium but not able to kill the bacteria effectively, providing a new niche for bacterial replication. We found that normal resistance to S. typhimurium was restored by inhibition of heme oxygenase with the competitive inhibitor tin protoporphyrin, a drug that can be used to treat hyperbilirubinemia in newborns (19), suggesting that HO inhibitors might represent a novel therapeutic intervention to abrogate the susceptibility to NTS induced by malaria.
Humans and mice with genetic deficiency of subunits of the phagocytic NADPH oxidase, a complex enzyme that catalyzes the generation of superoxide radicals in phagocytic cells, are known to be susceptible to NTS infection (20-21), and the importance of the neutrophil oxidative burst for killing of serum opsonized Salmonella by blood leukocytes from African children has been demonstrated in vitro (22). Impairment of the neutrophil oxidative burst in humans with malaria would thus be a compelling explanation for susceptibility to NTS bacteremia. In the current study we investigated whether the same mechanism may apply in humans, by examining neutrophil function in a cohort of children with predominantly uncomplicated malaria. Despite the fact that this population would not be considered at high risk of NTS co-infection, we found that malaria caused a marked abnormality of function in a large proportion of neutrophils, with impairment of oxidative burst capacity but not degranulation. The severity of the impairment of the neutrophil oxidative burst was strongly associated with hemolysis and prior induction of HO-1, but the duration of impairment was much longer than expected, lasting up to 8 weeks after infection.
Materials and Methods
Study subjects and procedures
The study and all procedures were approved by the Gambian Government / Medical Research Council Laboratories Joint Ethics Committee, and the London School of Hygiene and Tropical Medicine Ethics Committee. All human samples were collected with written informed consent from the participant or from the parent or legal guardian of child participants. Between September and December 2010, 58 Gambian children with P. falciparum malaria (defined by compatible clinical symptoms and >5000 asexual parasites/μL blood) were recruited within a longitudinal study investigating clinical, immunological and parasitological factors in mild and severe malaria, details of which have been published (23). Briefly, subjects were recruited, without selection for disease severity, from three peri-urban health centres: The Medical Research Council Gate Clinic, Brikama Health Centre and The Jammeh Foundation for Peace Hospital, Serekunda. Initial parasitemia (to determine eligibility for inclusion in the study) was estimated from Field’s stained thick blood films and subsequently accurately counted from 50 fields on Giemsa stained thin blood smears. All children underwent full clinical examination and were managed in accordance with Gambian government guidelines, treated with artemether-lumefantrine for three days. Severe malaria was defined using modified WHO criteria (24): severe anemia, defined as hemoglobin < 6g/dL; lactic acidosis defined as blood lactate > 7mmol/L; cerebral malaria defined as a Blantyre coma score ≤ 2 in the absence of hypoglycaemia, with the coma lasting at least for 2 hours; severe prostration (SP) defined as inability to sit unsupported (children > 6 months) or inability to suck (children ≤ 6 month).
Children suspected to have concomitant bacterial infections were excluded. For this study children underwent standardized assessment on the day of presentation (day 0) and days 7, 28 and 56, and blood was collected for: thick blood film, full blood count (EDTA), immunological assays (sodium heparin), and RNA (PaxGene tube) (days 0 and 28). On day 0 a thin blood film was prepared and sickle cell status, blood lactate, and glucose were determined. Blood samples were stored on ice, transported, to the laboratory within 2 hours of sample collection, and processed within 4 hours of collection. Full blood count was performed using a Medonic instrument (Clinical Diagnostics Solutions, Inc). Sickle cell status was determined by metabisulfite test and confirmed on cellulose acetate electrophoresis. The proportion of immature neutrophils, and hemozoin (malaria pigment) containing neutrophils and monocytes was determined from the day 0 Giemsa-stained thin blood film. Heparinised whole blood was used for assessment of neutrophil oxidative burst and degranulation (350μL), intracellular and cell surface flow cytometry (400μL), and for neutrophil isolation (1.25mL). On some occasions there was insufficient blood available to perform all assays. Single blood samples (handled as above) were obtained from 6 healthy Gambian children and 10 healthy Gambian adults, all without current or recent malaria, recruited from Brefet village where malaria transmission is now extremely low (25).
Laboratory reagents
All reagents were obtained from Sigma unless specified otherwise. GFP-expressing Salmonella enterica serovar Typhimurium pfpv 12023 (S. typhimurium) was a gift from Prof. David Holden (Imperial College London, UK), grown to late log-phase in Luria Bertani (LB) broth supplemented with ampicillin, and kept as frozen stock at −70°C in 10% glycerol.
Neutrophil oxidative burst and degranulation assays
The neutrophil oxidative burst was assessed in minimally manipulated whole blood using a modification of the assay described by Richardson et al. (26). Briefly, 50μL aliquots of blood were mixed with 50 μL of PMA (final concentration 1μM) or PBS (as control) and incubated for 15 min at 37°C in a water bath. Next 25 μL of PBS (unstained sample) or staining cocktail (Dihydrorhodamine 123, PECy7 anti-CD11b (eBioscience, ICRF44), and APC anti-CD15 (Miltenyi Biotec, VIMC6)(unstimulated and stimulated samples)) was added and incubated for 5 min at 37°C in the dark. Ammonium chloride RBC lysis buffer was added for 5 min at room temperature, shielded from light, before washing in PBS and resuspending cells in 1% paraformaldehyde in PBS. Samples were stored at 4°C protected from light, and analysed on the day of collection, using a 3 laser/9 channel CyAnTM ADP flowcytometer with Summit 4.3 software (Dako), after calibration of the FL-1 voltage with fluorescent beads (Spherotech). Data were analysed in FlowJo 7.6 (Tree Star, Inc, OR). The magnitude of the oxidative burst was quantified by the rhodamine median fluorescence intensity (MFI), and degranulation was quantified by the fold increase in surface CD11b MFI from the unstimulated to the stimulated sample. Neutrophils were divided into rhodamineHi and rhodamineLo populations at the mid-point of the nadir between peaks.
Flow cytometry for cell surface receptors and intracellular HO-1 expression
Whole blood was subjected to ammonium chloride RBC lysis and, after washing, cell pellets were resuspended in surface marker antibody cocktail (FITC anti-CD91 (AbD Serotec, A2Mr α-2), PE anti-CD16b (BD Pharmingen, CLB-gran11.5), PERCP anti-CD14 (BD Pharmingen, MφP9), APC anti-CD163 (R&D Systems, 215927)) or a similar cocktail instead containing the corresponding manufacturer-matched isotype-control antibodies for CD91 (Mouse IgG1) and CD163 (Mouse IgG1, 11711). Cells were permeabilized with CytoFix/CytoPerm (BD) before intracellular staining with polyclonal anti-HO-1 (Assay Designs, SPA-895) or an equivalent concentration of polyclonal control rabbit serum (Covance), followed by PE-Cy7 conjugated secondary antibody (F(ab’)2 anti-rabbit IgG, Santa Cruz Biotechnology). The expression of HO-1, CD163 and CD91 were quantified as the ratio of MFI to the respective isotype control antibody for the same sample.
Neutrophil isolation and Salmonella phagocytosis and killing assays
CD15+ cells were isolated from whole blood, after red blood cell lysis and labelling with APC anti-CD15, using anti-CD15 magnetic beads and MS columns (all from Miltenyi Biotec) according to the manufacturer’s instructions. CD15+ cells were resuspended in RPMI + 2mM L-Glutamine at a concentration of 107/ml. GFP-expressing S. typhimurium (concentration confirmed by serial dilution) were opsonised in 10% pooled healthy Gambian adult serum (derived from 10 donors, as has been described by others (22)) for 20 min in the dark at room temperature. Neutrophils and S. typhimurium were mixed continuously at a ratio of 50:1 at 60 rpm at 37°C. Bacterial counts were assessed at time 0 and 120 min by 10-fold dilutions of aliquots of the neutrophil-S. typhimurium suspension in 1% Triton, and plating onto LB agar, with colony forming units counted 16-18 h later. Bacterial killing was quantified as the percentage reduction in bacterial count between time 0 and 120 min. Phagocytosis was assessed after 15 min of incubation, by removing the neutrophil-S. typhimurium suspension directly into PBS 4% paraformaldehyde and analysing by flow cytometry. To control for autofluorescence and surface binding of bacteria without phagocytosis, control samples were prepared in an identical manner except that neutrophils and S. typhimurium were both fixed with 4% formaldehyde before mixing together. The proportion of cells phagocytosing bacteria was determined by subtraction of the proportion of GFP+ cells in the fixed-control samples from that in the respective unfixed sample.
ELISAs
Plasma levels of P. falciparum histidine rich protein-2 (PfHRP-2) (Cellabs), hemopexin, haptoglobin (both Genway), C-Reactive protein (CRP, R&D systems) and HO-1 (Enzo Life Sciences) were measured by ELISA. All ELISA assays were performed according to the manufacturer’s instructions and samples for each assay were performed in a single batch.
Heme Assay
Total plasma heme (that is plasma hemoglobin plus free- and protein bound-heme) was measured using a colorimetric heme assay kit (QuantiChrom heme, BioAssay Systems).
Quantitative RT-PCR
Total RNA was extracted from PAX tubes using PAXgene blood RNA kits (Qiagen) according to the manufacturer’s instructions, and converted into cDNA using a reverse transcription reagent kit (Invitrogen). HMOX1 (141250) gene expression was determined by qRT-PCR on a DNA Engine Opticon® (MJ Research) using a TaqMan® Probe kit with primers (all Metabion) as described by Hirai et al. (27). 18S rRNA was used as an endogenous reference gene since its expression has been shown to be stable in acute and convalescent samples from malaria cases regardless of disease severity (23), and was amplified with a commercial kit (rRNA primers and VIC labeled probe, Applied Biosystems). Data were analysed using Opticon Monitor 3TM analysis software (BioRad). HMOX1 expression was quantified as the ratio of the transcript number of the HMOX1 to 18S rRNA.
Estimation of total parasite biomass
Total parasite biomass was calculated from plasma PfHRP-2 concentration using the method of Dondorp et al. (28). This assumes that PfHRP-2 concentration is an integral of all PfHRP-2 released in preceding rounds of schizogeny (when infected erythrocytes rupture to release merozoites), and is therefore a reliable indicator of cumulative hemolysis since the start of the infection (28). We modified the calculation to account for the relatively higher blood volume at lower body weight in small children (29). To account for variation in size of children, parasite biomass was expressed as parasites per kg body weight.
Statistics
The study was designed to detect a 30% difference in neutrophil oxidative burst activity between samples at day 0 and day 28 with 80% power at the 0.05 significance level, allowing for 15% loss to follow up. Statistical analysis was performed using PASW Statistics 18 (SPSS Inc). Variables were examined for normality of distribution, and most were found to be non-normal. Two-tailed non-parametric tests for paired (Wilcoxon matched pairs test) or related repeated measures (Friedman’s two way ANOVA) were used to compare longitudinal data at different time points, and correlation was tested with Spearman’s rho correlation. In order to normalize distribution for general linear model analysis, some variables were Log10 transformed or converted to binary variables, as described in the text. Haptoglobin concentrations showed a bimodal distribution, and were thus converted to a binary variable (<0.349 mg/mL, the lowest value observed in healthy control samples, or ≥0.349 mg/mL). Sample volumes did not allow for some assays to be performed at time points after day 0, in which case values from six healthy control children were presented for comparison, but not for formal statistical analysis.
Results
Subjects
58 children with P. falciparum malaria were recruited to the study, 55 (94.8%) of whom had uncomplicated malaria (Table I). Four children had recurrent episodes of parasitemia during the course of follow-up and were excluded from longitudinal analyses; another 13 children were lost to follow-up or withdrew consent. Thus, at days 7, 28, and 56 the number (%) of children in follow-up were 52 (89.6%), 46 (79.3%), and 41 (70.1%) respectively.
Table I.
Demographic, clinical and laboratory characteristics at recruitment.
| Variable | Category | n*(%) | Median (IQR) |
Normal Range |
|---|---|---|---|---|
| Sex | Male | 36 (62) | ||
| Female | 22 (38) | |||
| Ethnicity | Mandingo | 25 (43) | ||
| Fula | 10 (17) | |||
| Wolof | 7 (12) | |||
| Manjago | 5 (9) | |||
| Jola | 5 (9) | |||
| Serere | 4 (7) | |||
| Aku | 1 (2) | |||
| Fanti | 1 (2) | |||
| Age (years) | 58 | 8 (4-12) | ||
| Severity | Uncomplicated | 55 (95) | ||
| Severe | 3 (5) | |||
| prostration | 2 | |||
| lactic acidosis | 1 | |||
| Plasmodium species | P. falciparum | 55 (95) | ||
| P. falciparum and | 3 (5) | |||
| P. malariae | ||||
| Sickle cell screen | Negative | 53 (91) | ||
| AS | 1 (2) | |||
| Not done | 4 (7) | |||
| Hemoglobin, g/dl | 58 | 11.5 | 9.5-14.4† | |
| (9.98-12.5) | ||||
| Erythrocyte count,×1012 /L | 58 | 4.26 (3.83-4.66) |
||
| Mean corpuscular volume, fL | 56 | 76.9 (73.5-80.7) |
67.8-90.0† | |
| Leukocyte count, x109/L | 57 | 8.50 (6.74-10.3) |
4.1-11.1† | |
| Percent neutrophils | 58 | 74.8 (66.3-83.0) |
35-75‡ | |
| Percent bands§ | 58 | 1.39 (0.57-1.39) |
0-10‡ | |
| Percent immature§¶ | 58 | 5.32 (2.71-10.8) |
0‡ | |
| Percent pigmented§ | 58 | 1.22 (0.42-4.08) |
||
| Percent of monocytes with pigment |
58 | 20 (0-50) |
||
| Parasite density, parasites/μL | 57 | 92800 (28200-219000) |
||
| Parasite biomass, parasites/kg | 55 | 1.23×1010 (5.21×109-2.14×1010) |
||
| C-reactive protein, mg/L | 46 | 106.4 (64.5-234.3) |
||
| Lactate, mmol/L | 42 | 2.0 (1.6-2.45) |
Data were not available for every variable for every subject.
90% reference interval for Gambian children aged 6-12 years (30), or
general pediatric reference interval (31), where available, for laboratory parameters.
Percentage of all neutrophils.
Neutrophil precursors less mature than band forms. IQR, Interquartile range.
Prolonged impairment of the neutrophil oxidative burst
We assessed the PMA-stimulated oxidative burst of neutrophils using a whole blood flow cytometric assay based on the oxidation of dihydrorhodamine 123 to its fluorescent derivative rhodamine, where the magnitude of the oxidative burst is quantified by the rhodamine fluorescence intensity (26). The assay was modified to allow simultaneous assessment of degranulation based on upregulation of CD11b (32), and surface staining of CD15 to identity neutrophils. We found that neutrophils (Fig. 1A) from subjects with acute malaria (day 0) showed an abnormal, bimodal, distribution of oxidative burst activity (Fig. 1B), with distinct populations of rhodamineHi and rhodamineLo cells, whereas CD11b expression showed a unimodal distribution (Fig. 1C). Overall, neutrophil rhodamine median fluorescence intensity (MFI) increased significantly over time (Fig. 1D), but in view of the bimodal distribution of neutrophil rhodamine fluorescence, we also compared the proportion of cells that were rhodamineLo and the rhodamine MFI of the rhodamineLo cells over time. The proportion of rhodamineLo cells decreased significantly during the convalescent period (Fig. 1E) but remained above that of healthy controls for at least 56 days; the rhodamine MFI of the rhodamineLo cells also significantly increased over time (Fig. 1F). In contrast, there was no evidence of abnormalities in neutrophil degranulation as assessed by CD11b expression (Fig. 1G). Of interest, the rhodamine MFI of the rhodamineHi cells was higher on days 0 and 7 after presentation (P=0.04, and P<0.001 respectively, Wilcoxon matched pairs test), than on day 56 (Fig. 1H), suggesting that the oxidative burst is primed in these neutrophils (33). These findings are consistent with our observations in mice that hemolysis and infection can prime the oxidative burst of mature, circulating neutrophils whilst simultaneously mobilizing immature neutrophils with impaired oxidative burst activity from the bone marrow (18). However the duration of these neutrophil abnormalities following P. falciparum infection was longer than we expected. We assessed whether a similar abnormality was present in monocytes in peripheral blood (Supplementary Fig. 1A), but found that the oxidative burst appeared to be slightly enhanced on day 0 compared with other time points (Supplementary Fig. 1B,C).
FIGURE 1.

P. falciparum malaria causes prolonged impairment of the neutrophil oxidative burst. (A) Representative FACS plots showing the gating of the neutrophil population based on forward scatter and side scatter characteristics followed by selection of single cells based on pulse width and forward scatter, and selection of the CD15+ population. (B,C) Rhodamine (B) and CD11b (C) fluorescence of unstimulated (filled histogram) and PMA-stimulated neutrophils (unfilled histogram) on days 0, 7, 28 and 56 after presentation with P. falciparum malaria. Representative plots from a healthy control child are also shown for comparison. RhodamineHi and rhodamineLo populations of neutrophils were defined for each sample by partition at the nadir of the bimodal distribution, and percentages of the total cells in each population are shown (B). (D-H) Longitudinal analyses of neutrophil function, compared using Friedman’s two way ANOVA for all subjects with valid data at all time points. Healthy controls are also shown for comparison, but not included in the statistical analysis (D,E,G). Horizontal lines represent medians. n stated for valid data at every time point. (D) Rhodamine MFI for all neutrophils (rhodamineHi and rhodamineLo considered together as a single population); n=32. (E) Proportion of neutrophils that are rhodamineLo; n=28. (F) Rhodamine MFI of rhodamineLo neutrophils; n=28. (G) Degranulation of all neutrophils, assessed by fold change in surface CD11b (PMA stimulated CD11b MFI: unstimulated CD11b MFI); n=29. (H) Rhodamine MFI of rhodamineHi neutrophils; n=32.
Hemolysis and neutrophil dysfunction
We have previously shown that hemolysis-derived heme impairs neutrophil function during malaria infection in mice through two related mechanisms: mobilization of functionally immature neutrophils from bone marrow, and impairment of the oxidative burst capacity of developing neutrophils due to HO-1 induction in bone marrow progenitors (18). RBC destruction in malaria is multifactorial, but the severity of intravascular hemolysis can be inferred from levels of the plasma proteins haptoglobin and hemopexin (34), which provide sequential lines of defense against heme-mediated toxicity by binding cell-free hemoglobin and cell-free heme respectively (35). Only once haptoglobin is depleted do levels of hemopexin begin to fall, indicating that heme is being released from cell-free hemoglobin (36).
To assess the extent of hemolysis in study participants we measured erythrocyte count and total parasite biomass (Table I), total plasma heme (Fig. 2A), haptoglobin (Fig. 2B) and hemopexin (Fig. 2C), and examined their correlation with the proportion and function of rhodamineLo neutrophils (Table 2). Although only 13 children (22.4%) were actually anemic by Gambian reference standards (Hb<9.5 g/dL) (30), total plasma heme levels were significantly greater on day 0 than on day 28 (Fig. 2A). Levels of haptoglobin showed a bimodal distribution (Fig. 2B), consistent with the expected depletion of haptoglobin by hemolysis in some subjects (36), but also increased production of haptoglobin as part of the acute phase response in other subjects (37-38). Hemopexin levels, however, were relatively normally distributed and very similar to healthy controls (Fig. 2C), suggesting that in these subjects with predominantly uncomplicated malaria, hemolysis does not liberate sufficient cell-free heme to deplete plasma hemopexin (35-36).
FIGURE 2.
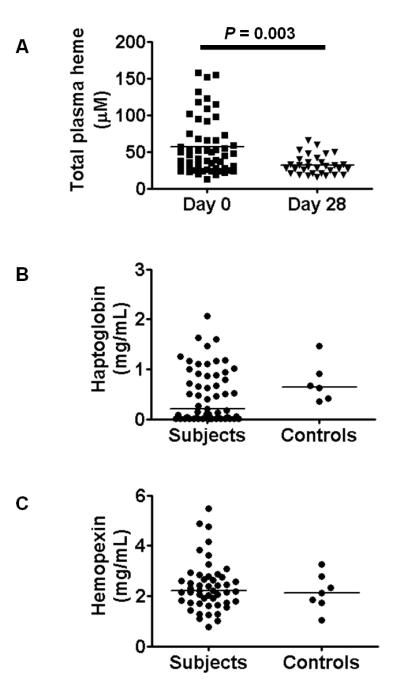
Indicators of hemolysis in subjects with P. falciparum malaria. (A) Total plasma heme on day 0 and day 28, compared using Wilcoxon matched pairs test for those with data at both time points (n=32). (B) Distribution of plasma haptogobin levels at day 0, n=57. For comparison levels in six healthy control children are also shown. (C) Distribution of plasma hemopexin levels at day 0, n=49. For comparison levels in six healthy control children are also shown.
Table II.
Association of neutrophil dysfunction with hemolysis on day 0.
| % RhodamineLo Neutrophils |
Rhodamine MFI of RhodamineLo Neutrophils |
|||||
|---|---|---|---|---|---|---|
| Variable | n* | Correlation coefficient |
P | n* | Correlation coefficient |
P |
| Parasite biomass/kg | 53 | 0.208 | 0.134 | 53 | −0.539 | <0.001 |
| Erythrocyte count | 56 | −0.001 | 0.992 | 56 | 0.280 | 0.037 |
| Haptoglobin | 55 | 0.004 | 0.976 | 55 | 0.292 | 0.031 |
| Total plasma heme | 54 | 0.222 | 0.106 | 54 | −0.268 | 0.050 |
| C-reactive protein | 44 | 0.350 | 0.020 | 44 | −0.452 | 0.002 |
| Immature neutrophil % | 56 | 0.232 | 0.085 | 56 | 0.031 | 0.822 |
| Pigmented neutrophil % | 56 | −0.017 | 0.898 | 56 | 0.180 | 0.185 |
Indicators of hemolysis, inflammation and neutrophil characteristics were assessed for correlation with the proportion of rhodamineLo neutrophils and with the rhodamine MFI of rhodamineLo neutrophils on day 0 using Spearman’s correlation.
Data were not available for every variable for every subject.
Since rhodamineLo cells may be similar to the functionally immature granulocytes released into the circulation during malaria and NTS infection in mice (18), we assessed whether their frequency was associated with markers of inflammation, hemolysis and neutrophil characteristics (Table II). The proportion of rhodamineLo cells was significantly correlated with C-reactive protein (CRP) concentration, which would be consistent with their mobilization as part of an inflammatory response (39), but did not correlate directly with measures of hemolysis or hemozoin uptake. There was a non-significant positive correlation between the proportion of immature neutrophils and the proportion of rhodamineLo cells (Table II), suggesting that the rhoadmineLo population is not only due to morphologically immature neutrophils, but must also include morphologically mature, but functionally abnormal neutrophils. By contrast, the magnitude of the oxidative burst among rhodamineLo neutrophils (rhodamineLo MFI) showed significant univariate correlations with erythrocyte count and haptoglobin concentration, and significant negative correlations with total parasite biomass, CRP, and total plasma heme. Since these variables are likely to be highly correlated with each other, we analysed their effects on rhodamineLo neutrophil MFI using a general linear model. After stepwise elimination of the least significant variables in the model, only parasite biomass remained significantly associated with rhodamineLo neutrophil MFI (F=16.036, P < 0.001). Overall these results are consistent with the acute phase inflammatory response being the primary determinant of release of rhodamineLo neutrophils into the circulation in children with uncomplicated malaria, but parasite burden and consequent hemolysis being the major determinant of the impairment of the oxidative burst in rhodamineLo neutrophils. Neither variable was associated with the proportion of pigment containing neutrophils.
Factors associated with HO-1 induction
In malaria-infected mice we found that heme-mediated HO-1 induction in neutrophil progenitors in bone marrow was necessary to impair the oxidative burst of developing neutrophils (18). Since there were no clinical indications for bone marrow aspiration in any of the study subjects, our analysis of the HO-1 pathway was restricted to parameters measurable in peripheral blood, namely plasma HO-1, whole blood HMOX1 gene expression and HO-1 protein expression in peripheral blood cells. Also, since the induction of cellular HO-1 by haptoglobin-hemoglobin or heme-hemopexin complexes depends on the presence of surface receptors for their uptake (CD163 and CD91 respectively) (37, 40-41), we examined CD163 and CD91 expression on monocytes and neutrophils.
As previously reported in mice (18) and humans (42), HO-1 expression (assessed by fluorescence intensity) in circulating neutrophils was not increased in acute malaria infection compared with convalescence (data not shown). In contrast, monocyte HO-1 expression was higher on day 0 than on days 7 or 28 (Fig. 3A). Plasma HO-1 was higher in subjects on day 0 than in healthy control children (Fig. 3B), and as we have previously reported (42), whole blood HMOX1 expression was significantly higher on day 0 than following recovery on day 28 (Fig. 3C).
FIGURE 3.

Factors associated with heme oxygenase-1 expression in P. falciparum malaria. (A-C): heme oxygenase-1 induction. (A) Representative FACS analysis of HO-1 induction in moncoytes showing exclusion of red blood cells, gating on single cells, and subsequent definition of monocytes as CD14+. Histograms show fluorescence of monocytes stained intracellularly with control antibody (filled) or anti-HO-1 antibody (unfilled) followed by a secondary detection antibody. Quantitative data for HO-1 expression in monocytes (ratio of anti-HO-1 to control antibody fluorescence) for all subjects on days 0, 7 and 28 are shown in the right hand panel, compared using Friedman’s two way ANOVA (n=20). (B) The distribution of Plasma HO-1 on day 0, n=57. For comparison levels in six healthy control children are also shown. (C) Whole blood HMOX1 RNA expression, measured by qRT-PCR, compared between samples on day 0 and day 28 using Wilcoxon matched pairs test for those with data at both time points (n=42). (D-H) CD163 and CD91 expression on monocytes and neutrophils. (D) Representative flow cytometry analysis of healthy control subject neutrophils (CD14−CD16b+) and monocytes (CD14+CD16b−), showing staining with respective control (filled histogram) and anti-CD91 or anti-CD163 antibodies (unfilled histograms). Quantitative data for expression (ratio to control antibody fluorescence) of CD163 in monocytes (E) and neutrophils (F), and CD91 in monocytes (G) and neutrophils (H), compared using Friedman’s two way ANOVA for all subjects with valid data at all time points (CD163, n=19; CD91 n=20). Horizontal bars represent medians. n stated for those with valid data at all time points.
In control subjects, CD163 and CD91 were expressed on the surface of monocytes but were not detectable on neutrophils (Fig. 3D). In children with malaria, monocyte CD163 expression was significantly lower at day 0 than on days 7 and 28 (Fig. 3E), whereas CD163 remained undetectable on neutrophils from most subjects (Fig. 3F). CD91 expression did not change significantly over time on monocytes (Fig. 3G) or neutrophils (Fig. 3H), although when all subjects were considered together, there did appear to be very low level CD91 expression on neutrophils at all time points.
To further explore the likely pathways of HO-1 induction during malaria infection, we constructed a simple conceptual model (Fig. 4), beginning with the malaria parasite as the cause of hemolysis, inflammation, and tissue hypoxia/ischemia (43) - all of which may induce HO-1 expression (44) - and analysed univariate correlations between the various measures of HO-1 induction. As expected, parasite biomass was strongly correlated with total plasma heme and CRP. Plasma HO-1 correlated much more strongly with CRP and lactate than with plasma heme, supporting the idea that it may be released in response to harmful and inflammatory stimuli. Surprisingly, however, neither whole blood HMOX1 expression nor monocyte specific HO-1 correlated significantly with total plasma heme, CRP, or lactate. As the haptoglobin receptor CD163 has been reported to be down regulated during inflammation (45), and to be shed from the cell surface during acute, uncomplicated malaria infections (46), we explored whether monocyte specific HO-1 expression might be confounded by changes in expression of CD163, using a general linear model controlling for the effect of CD163 expression. This revealed a significant interaction between total plasma heme and CD163 expression, but strong independent associations between monocyte HO-1 and total plasma heme (F=15.1, P < 0.001) and monocyte HO-1 and CD163 expression (F=9.378, P = 0.003). In other words, monocyte HO-1 induction would closely correlate with total plasma heme, but down regulation of surface CD163 prevents the uptake of hemoglobin-haptoglobin complexes and hence limits HO-1 induction. This explains in part the discordance between different measures of the HO-1 induction pathway in blood. In summary this analysis demonstrates that HO-1 protein expression in myeloid cells can be increased by hemoglobin and heme liberated during malarial hemolysis, although this effect is limited by reductions in surface CD163 expression.
FIGURE 4.

A conceptual model of the pathways leading to HO-1 induction in acute P. falciparum malaria. The biomass of P. falciparum parasites within the subject was considered to be the quantifiable cause of hemolysis, inflammation and tissue hypoxia / ischemia, all of which are stimuli for induction of HO-1. The variable measured to quantify each step in the pathway is indicated in italics. Associations between variables were tested using Spearman’s correlation as indicated by lines with arrows showing the hypothesized relationship from cause to effect. Significant correlations are denoted by solid lines, and line thickness indicates the significance of the correlation (thin line 0.01 ≤ P <0.05, medium thickness line 0.001 ≤ P <0.01, heavy line P <0.001), whereas non-significant correlations are denoted by dashed lines. The strength of correlation is indicated by Spearman’s rho adjacent to significant correlation lines.
Neutrophil oxidative burst and prior HO-1 induction
In mice we had observed that suppression of the oxidative burst of circulating neutrophils by hemolysis required the release of immature neutrophils from bone marrow, requiring either a lag time, or an additional stimulus (such as NTS infection), to cause these cells to rapidly enter the circulation (18). Having observed that an abnormal population of neutrophils was present for a prolonged period of time following P. falciparum, we looked for evidence of an association between HO-1 induction on day 0, and neutrophil oxidative burst on day 7. We used the ratio of day 0 to day 28 whole blood HMOX1(HMOX1day0/day28) expression as an indicator of induction in acute malaria. We found a significant negative correlation between HMOX1day0/day28 and the rhodamine MFI of rhodamineLo neutrophils on day 7 (Spearman’s correlation coefficient = −0.352, P = 0.028, n=39). There was no significant correlation with the proportion of rhodamineLo cells on day 7 (Spearman’s correlation coefficient = −0.101, P = 0.542, n=39). Although the kinetics of the process of HO-1 induction, suppression of oxidative burst capacity in developing neutrophils, and subsequent release of the functionally immature neutrophils into the circulation are unknown, the observed association between HO-1 induction during acute disease and neutrophil dysfunction during early convalescence is consistent with this sequence of events.
Salmonella phagocytosis and killing
Since we have observed that neutrophil killing (but not phagocytosis) of S. typhimurium is defective in malaria-infected mice (18), we assessed the ex-vivo killing and phagocytosis of serum-opsonised S. typhimurium by neutrophils isolated from whole blood of subjects and controls from whom sufficient blood remained after the preceding assays (Fig. 5A–C). Bacterial killing, calculated as the reduction in the viable bacterial count 2 hr after co-culture, was slightly higher on day 0 than at subsequent time points (Fig. 5A). There was no significant correlation between bacterial killing on day 0 and either the proportion of rhodamineLo neutrophils, the rhodamine MFI of the rhodamineLo neutrophils, total plasma heme or parasite biomass (data not shown). Bacterial phagocytosis, determined by flow cytometric analysis of the proportion of GFP+ neutrophils after 15 min co-culture (Fig. 5B), did not vary significantly over time following infection (Fig. 5C). However phagocytosis at day 0 was inversely correlated with parasite biomass (n=26, Spearman’s correlation coefficient = −0.512, P =0.008), and with total plasma heme (n=28, Spearman’s correlation coefficient = −0.441, P= 0.019) (Fig. 5D). Taken together with the neutrophil oxidative burst assay data (Fig. 1 F,H), these data indicate that although there may be some degree of (heme-mediated) priming of neutrophil function in children with acute malaria (day 0) which enhances bacterial killing during acute illness (47), phagocytosis of S. typhimurium by circulating neutrophils becomes increasingly impaired with increasing parasite burden and increasingly severe hemolysis, and that the ability of neutrophils to kill S. typhimurium once they are phagocytosed might also become impaired.
FIGURE 5.

Ex-vivo killing and phagocytosis of S. typhimurium by neutrophils from children with P. falciparum malaria. (A) Killing of S. typhimurium. Neutrophils isolated on days 0, 7, 28 and 56 after presentation with P. falciparum malaria were mixed with S. typhimurium at ratio of 50:1 and killing expressed as the percentage reduction in bacterial numbers after 2 h incubation. Statistical comparison using Friedman’s two way ANOVA for all subjects with valid data at all time points, n=18. Data from control subjects shown for comparison. (B) Representative flow cytometry plots showing phagocytosis of GFP+ S. typhimurium by neutrophils isolated on day 0. RBCs and debris were excluded based on forward scatter and side scatter characteristics, then single cells were selected based on pulse width and forward scatter characteristics (upper row). The proportion of GFP+CD15+ cells was determined in samples where both neutrophils and S. typhimurium were fixed in 4% formaldehyde prior to incubation (to control for surface binding without phagocytosis), and in unfixed samples (lower row). (C) Phagocytosis of S. typhimurium. Neutrophils isolated on days 0, 7, 28 and 56 after presentation with P. falciparum malaria were mixed with S. typhimurium at a ratio of 50:1 and phagocytosis expressed as the percentage GFP+ neutrophils after 15 min incubation. The percentage GFP+ cells was calculated by subtracting the proportion of GFP+ cells in formaldehyde fixed samples from that in unfixed samples. Statistical comparison using Friedman’s two way ANOVA for all subjects with valid data at all time points, n=7. Data from control subjects shown for comparison. (D) Correlation of phagocytosis (on day 0) with parasite biomass on day 0 (left hand panel, n=26) and total plasma heme on day 0 (right hand panel, n=28).
Discussion
Although the association between malaria infection and susceptibility to NTS bacteremia has been recognized for almost a century (9), the mechanism has been elusive. The strongest association is with severe malarial anemia (5-6) and at least two other conditions associated with hemolytic anemia - sickle cell disease (48) and acute bartonellosis (49) - also predispose to NTS bacteremia. This is likely a result of both the nature of the defect in host-defense induced by malaria, and also the prevailing epidemiology of invasive bacterial infection: NTS is one of the most common causes of bacteremia in Sub-Saharan Africa (50-51). In a mouse model we recently showed that hemolysis due to malaria or phenylhydrazine treatment impaired resistance to S. typhimurium, which could be recapitulated by treatment with hemin, and abrogated by treatment with the HO inhibitor tin protoporphyrin (18). We found that bacteria accumulated in circulating neutrophils, and that these neutrophils were defective in killing S. typhimurium, associated with impairment of their oxidative burst response which is an essential mechanism for killing S. typhimurium (21). This was due to heme-mediated induction of HO-1 in granulocyte precursors in bone marrow, causing neutrophils leaving the bone marrow to have a reduced capacity to mount an effective oxidative burst. It is not known whether bacteria accumulate preferentially in neutrophils in humans with malaria and NTS co-infections, but in the pre-antibiotic era neutrophils and NTS were often found co-localized in abscesses which formed at the site of intramuscular quinine injection in co-infected individuals (9). Although neutrophil function has not been extensively studied in malaria, there are several case reports of patients with severe malarial hemolysis spontaneously developing fungal sepsis (52-54), which is typically associated with neutropenia and neutrophil dysfunction.
The present study was designed to determine whether Gambian children with P. falciparum malaria have evidence of neutrophil dysfunction similar to that observed in mice infected with P. yoelli 17XNL. We hypothesized that the neutrophil oxidative burst would be impaired in children with malaria, and the severity of this impairment would be related to hemolysis and HO-1 induction. However, we also predicted that the impairment of neutrophil function would be relatively mild, because declining malaria transmission in The Gambia has lead to a decrease in the incidence of severe malarial anemia and malaria-NTS co-infection (7). Overall, our results are consistent with our hypothesis: the oxidative burst activity of circulating neutrophils was profoundly abnormal in subjects with acute P. falciparum malaria, and most severely impaired in children with the highest parasite burdens and greatest hemolysis, albeit the magnitude of this impairment did not translate into a clinically significant defect in neutrophil killing of S. typhimurium in vitro. We also found that these abnormalities persisted for at least 56 days, and that bacterial phagocytosis and killing appeared to deteriorate during the early convalescent period (up to 28 days), findings which may be consistent with descriptions of increased susceptibility to NTS bacteremia in children who have recently had malaria (6, 10), and the gradual emergence of dysfunctional neutrophils from bone marrow following HO-1 induction during the acute infection (18). To facilitate comparison with our studies in mice (18), we assessed neutrophil function using PMA as the stimulus for the oxidative burst. Although this is not a physiological stimulus, the advantages of this method are: i) that it produces a strong oxidative burst (55-56) which is clearly distinguished from any low-level activation caused by malaria infection per se, ii) it is not dependent on phagocytosis (which might also be impaired by malaria) (55-56), and iii) variations in the magnitude of the PMA-induced oxidative burst are directly related to the ability of humans to survive infections (57).
Consistent with data from mice and humans (18, 42, 58), we observed induction of HO-1 during acute malaria. Although dissecting the causal and consequential pathways of HO-1 induction is difficult in an observational study, we constructed a conceptual model of likely pathways leading to HO-1 induction based on existing literature (Figure 4) (44), and used this model to guide our statistical analysis. Plasma HO-1 levels correlated more strongly with plasma lactate and CRP concentrations than with plasma heme concentrations, suggesting that plasma HO-1 may be predominantly a response to inflammation and hypoxia and a marker of cell damage. As noted previously, intracellular HO-1 protein expression was not significantly upregulated in acute malaria in circulating neutrophils (42), presumably because they lack CD163 expression, whereas monocytes did show evidence of increased HO-1 protein expression in acute malaria. However there was not a significant univariate association between total plasma heme concentration and HO-1 expression in monocytes, which could be explained statistically by the reduced levels of monocyte surface CD163 expression in acute malaria. This explanation is fully consistent with the subjects in this study having only mild hemolysis (most had hemoglobin values within the normal range for their age (30)), none had severe malarial anemia and only half had low haptoglobin levels) and with the assumption (as hemopexin levels were not depleted) that very little of the total circulating heme represents cell-free heme. In this case, HO-1 induction due to hemolysis would be expected to proceed predominantly through CD163-mediated uptake of haptogobin-hemoglobin complexes, and reduction in surface CD163 would be expected to limit HO-1 induction (45). In contrast, severe hemolysis would be expected to generate cell free heme, and lead to HO-1 induction and heme catabolism in cells expressing the surface receptor (CD91) for heme-hemopexin complexes, which appears to be invariantly expressed during infection. Indeed, we previously found elevated carboxyhemoglobin levels, an indirect measure of HO activity, only in children with severe malarial anemia suggesting that heme catabolism is constrained in acute malaria and only detectably increased in cases with the most severe hemolysis (59). It is conceivable that by reducing CD163 expression in the context of infection, monocytes are rendered relatively resistant to HO-1 induction by hemolysis, perhaps preventing HO-1-mediated impairment of their normal inflammatory responses (45, 60). However, it is presently unknown whether either CD163 or CD91 expression are required for HO-1 induction in immature myeloid cells and their progenitors in human bone marrow. If HO-1 induction in bone marrow is responsible for the observed neutrophil dysfunction (as it is in mice (18)), it may be either independent of CD163, or CD163 may not be down regulated in the bone marrow to the same extent as in blood monocytes. We did not examine the effect of HMOX1 promoter (GT)n length polymorphisms in this study because the majority of subjects had uncomplicated malaria with mild hemolysis and we expected that under these circumstances genetic polymorphisms would have a relatively small effect and a much larger sample size would have been required (42).
In apparent contradiction of our findings in mice, killing of S. typhimurium by neutrophils was not noticeably impaired on day 0, but deteriorated over the next 4 weeks. This finding is perhaps not surprising because none of the children had severe hemolysis or severe anemia, which are the major risk factors for malaria-NTS coinfection (5-6), suggesting that their bactericidal capacity should not be seriously impaired. A possible explanation is that, despite the accumulation of a rhodamineLo neutrophil population, the oxidative burst of the rhodamineHi neutrophil population was higher on days 0 and 7 than at later time points, and enhanced bactericidal activity among rhodamineHi cells may initially compensate for the lack of killing among the rhodamineLo population, particularly if the rhodamineHi cells showed preferential phagocytosis of the opsonised S. typhimurium. Unfortunately our assays could not simultaneously assess phagocytosis and oxidative burst in the same cells. We predict, however, that bacterial killing would be seriously impaired in children with severe hemolytic anemia. The acquired defect of neutrophil function that we observed in children with malaria in this study might be considered analogous the neutrophil defect observed in female carriers of X-linked chronic granulomatous disease, where around 50% of neutrophils have defective oxidative burst activity due to random inactivation of the X-chromosome, but there is not increased susceptibility to infection (61) and in vitro bactericidal activity may be normal (62). However in some carriers, inactivation of the X-chromosome becomes skewed, and when less than 15% of neutrophils are able to make a normal oxidative burst, susceptibility to infections is markedly increased (20). Consistent with this, in a large European registry of chronic granulomatous disease patients, Salmonella has been reported as by far the most common cause of septicemia (whilst fungi and Staphylococcus aureus are the most common causes of chronic lung and deep tissue infections, due to the persistent defect in oxidative burst activity) (20). We propose that in patients with malaria a threshold proportion of abnormal neutrophils in blood may be required to produce susceptibility to NTS, and when this threshold is exceeded the degree of susceptibility may then also be determined by the magnitude of the impairment of oxidative burst capacity. Factors which may determine whether the proportion of dysfunctional neutrophils exceeds this putative threshold may include: the duration of infection and the severity of the inflammatory response, which may both influence the mobilization of dysfunctional neutrophils from bone marrow (18); the severity of hemolysis, since cell-free heme itself promotes neutrophil mobilization (18); and possibly host factors such as age and genetic background. The strong correlation we observed between impaired neutrophil oxidative burst and total parasite biomass allows us to predict that children with high parasite burden (who are also most likely to have severe hemolysis) would have the most impaired oxidative burst. These children may also have depleted hemopexin levels and accumulate cell free heme, which can itself mobilize neutrophils from bone marrow (18), perhaps increasing the proportion of abnormal neutrophils above a threshold required to induce susceptibility to NTS.
The prolonged duration of abnormal neutrophil oxidative burst activity, extending up to 8 weeks after acute infection in some subjects, was unexpected. This is unlikely to be due to anti-malarial treatment, because artemesinin-based treatments cause mild enhancement of the oxidative burst and suppression of phagocytosis, the opposite of what we observed (63). Two possible explanations are the effect of persisting hemozoin, and fundamental alteration in myelopoiesis. Hemozoin is an insoluble hemin polymer, the end product of hemoglobin digestion inside the parasitised red cell, which is able to induce HO-1 (and impair the oxidative burst in phagocytes) but is not catabolized (13, 64). Prolonged persistence of hemozoin in phagocytic cells in the bone marrow (65) could cause prolonged HO-1 induction and abnormal development of neutrophils (18). Associated with this, or through separate mechanisms, a novel myeloid progenitor phenotype may be generated in P. falciparum malaria, as has been observed in rodent malaria infection (66), which may lead to sustained abnormal granulopoiesis.
The major limitations of our study are that most subjects had relatively mild hemolysis and that we did not have bone marrow samples to confirm HO-1 induction in neutrophil progenitors. To recruit a significant number of subjects with severe malarial hemolysis would require a much larger study, probably conducted in a higher transmission setting. To study prospectively whether the severity of neutrophil dysfunction at recruitment correlated with susceptibility to NTS bacteremia during convalescence would require an even larger and more logistically complex study. To obtain bone marrow aspirates from children with malaria would be difficult to justify ethically unless appropriate sedation and analgesia could be provided without additional risk of complications.
Nevertheless, our findings have a number of important implications. First we show that the oxidative burst capacity of a large proportion of neutrophils is markedly abnormal in children with P. falciparum malaria, supporting the translation of findings in a mouse model (18). Second, neutrophil function recovers only very slowly over the two months after treatment, providing an explanation for the association of susceptibility to NTS with recent malaria (10-11). In the mouse model, hemolysis-induced neutrophil dysfunction could be abrogated by competitive inhibition of HO with tin protoporphyrin (18), but using this treatment in acute malaria would be challenging because HO-1 is also important for tolerance to cytotoxic effects of cell free heme in mouse models (58, 67). Alternative therapeutic strategies would be administration of tin protoporphyrin upon completion of antimalarial treatment, with the aim of restoring neutrophil function during convalescence and preventing the susceptibility to NTS caused by recent malaria, or prioritisation of children at greatest risk of persistent neutrophil dysfunction for prophylactic antibiotic treatment. Third, we propose the down-regulation of the haptoglobin receptor CD163 on the surface of blood monocytes during acute P. falciparum malaria as a novel host-protective homeostatic response to hemolysis and inflammation, which may prevent HO-1 induction from impairing monocyte function. Further experimental studies are needed to confirm the effects of manipulating CD163 expression during infections, but manipulation of this axis would hold promise for both the modulation of inflammation and optimization of iron re-utilization during chronic infections.
Supplementary Material
Acknowledgements
We are grateful to David Conway who helped to set up and manage the recruitment process used in the study, David Jeffries for statistical advice, and to the clinic, laboratory, and administrative staff, field workers, and subjects who participated in the study.
Abbreviations used in this paper
- NTS
non-Typhoid Salmonella
- HO-1
heme oxygenase-1
- MFI
median fluorescent intensity
- CRP
C-reactive protein
Footnotes
This study was supported by a clinical research training fellowship from the Medical Research Council (UK) (G0701427) to A.J.C. and by MRC Institute funding to the MRC Laboratories, The Gambia.
References
- 1.World malaria report 2011. World Health Organization; Geneva: 2011. [Google Scholar]
- 2.Snow RW, Korenromp EL, Gouws E. Pediatric mortality in Africa: plasmodium falciparum malaria as a cause or risk? Am. J. Trop. Med. Hyg. 2004;71:16–24. [PubMed] [Google Scholar]
- 3.Berkley JA, Bejon P, Mwangi T, Gwer S, Maitland K, Williams TN, Mohammed S, Osier F, Kinyanjui S, Fegan G, Lowe BS, English M, Peshu N, Marsh K, Newton CR. HIV infection, malnutrition, and invasive bacterial infection among children with severe malaria. Clin. Infect. Dis. 2009;49:336–343. doi: 10.1086/600299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scott JA, Berkley JA, Mwangi I, Ochola L, Uyoga S, Macharia A, Ndila C, Lowe BS, Mwarumba S, Bauni E, Marsh K, Williams TN. Relation between falciparum malaria and bacteraemia in Kenyan children: a population-based, case-control study and a longitudinal study. Lancet. 2011;378:1316–1323. doi: 10.1016/S0140-6736(11)60888-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bronzan RN, Taylor TE, Mwenechanya J, Tembo M, Kayira K, Bwanaisa L, Njobvu A, Kondowe W, Chalira C, Walsh AL, Phiri A, Wilson LK, Molyneux ME, Graham SM. Bacteremia in Malawian children with severe malaria: prevalence, etiology, HIV coinfection, and outcome. J. Infect. Dis. 2007;195:895–904. doi: 10.1086/511437. [DOI] [PubMed] [Google Scholar]
- 6.Mabey DC, Brown A, Greenwood BM. Plasmodium falciparum malaria and Salmonella infections in Gambian children. J. Infect. Dis. 1987;155:1319–1321. doi: 10.1093/infdis/155.6.1319. [DOI] [PubMed] [Google Scholar]
- 7.Mackenzie G, Ceesay SJ, Hill PC, Walther M, Bojang KA, Satoguina J, Enwere G, D’Alessandro U, Saha D, Ikumapayi UN, O’Dempsey T, Mabey DC, Corrah T, Conway DJ, Adegbola RA, Greenwood BM. A decline in the incidence of invasive non-typhoidal Salmonella infection in The Gambia temporally associated with a decline in malaria infection. PLoS One. 2010;5:e10568. doi: 10.1371/journal.pone.0010568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayasaka C. Im Verlauf einer Malariakur durch Bacillus enteritidis Gärtner entstandene Meningitis und Sepsis. Tohoku J. Exp. Med. 1933;21:466–504. [Google Scholar]
- 9.Giglioli G. Paratyphoid C and endemic disease of British Guiana: A clinical and pathological outline. B paratyphosum C as a pyogenic organism: case reports. Proc. Roy. Soc. Med. 1929;23:165–167. [PMC free article] [PubMed] [Google Scholar]
- 10.Brent AJ, Oundo JO, Mwangi I, Ochola L, Lowe B, Berkley JA. Salmonella bacteremia in Kenyan children. Pediatr. Infect. Dis. J. 2006;25:230–236. doi: 10.1097/01.inf.0000202066.02212.ff. [DOI] [PubMed] [Google Scholar]
- 11.Nadjm B, Amos B, Mtove G, Ostermann J, Chonya S, Wangai H, Kimera J, Msuya W, Mtei F, Dekker D, Malahiyo R, Olomi R, Crump JA, Whitty CJ, Reyburn H. WHO guidelines for antimicrobial treatment in children admitted to hospital in an area of intense Plasmodium falciparum transmission: prospective study. BMJ. 2010;340:c1350. doi: 10.1136/bmj.c1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urban BC, Roberts DJ. Malaria, monocytes, macrophages and myeloid dendritic cells: sticking of infected erythrocytes switches off host cells. Curr. Opin. Immunol. 2002;14:458–465. doi: 10.1016/s0952-7915(02)00368-0. [DOI] [PubMed] [Google Scholar]
- 13.Schwarzer E, Turrini F, Ulliers D, Giribaldi G, Ginsburg H, Arese P. Impairment of macrophage functions after ingestion of Plasmodium falciparum-infected erythrocytes or isolated malarial pigment. J. Exp. Med. 1992;176:1033–1041. doi: 10.1084/jem.176.4.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roux CM, Butler BP, Chau JY, Paixao TA, Cheung KW, Santos RL, Luckhart S, Tsolis RM. Both hemolytic anemia and malaria parasite-specific factors increase susceptibility to Nontyphoidal Salmonella enterica serovar typhimurium infection in mice. Infect. Immun. 2010;78:1520–1527. doi: 10.1128/IAI.00887-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaye D, Hook EW. The Influence of Hemolysis or Blood Loss on Susceptibility to Infection. J. Immunol. 1963;91:65–75. [PubMed] [Google Scholar]
- 16.Kaye D, Hook EW. The Influence of Hemolysis on Susceptibility to Salmonella Infection: Additional Observations. J. Immunol. 1963;91:518–527. [PubMed] [Google Scholar]
- 17.Kaye D, Merselis JG, Jr., Hook EW. Influence of Plasmodium berghei infection on susceptibility to salmonella infection. Proc. Soc. Exp. Biol. Med. 1965;120:810–813. doi: 10.3181/00379727-120-30661. [DOI] [PubMed] [Google Scholar]
- 18.Cunnington AJ, de Souza JB, Walther M, Riley EM. Malaria impairs resistance to Salmonella through heme- and heme oxygenase-dependent dysfunctional granulocyte mobilization. Nat. Med. 2011;18:120–127. doi: 10.1038/nm.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kappas A, Drummond GS, Manola T, Petmezaki S, Valaes T. Sn-protoporphyrin use in the management of hyperbilirubinemia in term newborns with direct Coombs-positive ABO incompatibility. Pediatrics. 1988;81:485–497. [PubMed] [Google Scholar]
- 20.van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, Espanol T, Fischer A, Kurenko-Deptuch M, Mouy R, Petropoulou T, Roesler J, Seger R, Stasia MJ, Valerius NH, Weening RS, Wolach B, Roos D, Kuijpers TW. Chronic granulomatous disease: the European experience. PLoS One. 2009;4:e5234. doi: 10.1371/journal.pone.0005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mastroeni P, Vazquez-Torres A, Fang FC, Xu Y, Khan S, Hormaeche CE, Dougan G. Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. II. Effects on microbial proliferation and host survival in vivo. J. Exp. Med. 2000;192:237–248. doi: 10.1084/jem.192.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gondwe EN, Molyneux ME, Goodall M, Graham SM, Mastroeni P, Drayson MT, MacLennan CA. Importance of antibody and complement for oxidative burst and killing of invasive nontyphoidal Salmonella by blood cells in Africans. Proc. Natl. Acad. Sci. USA. 2010;107:3070–3075. doi: 10.1073/pnas.0910497107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walther M, Jeffries D, Finney OC, Njie M, Ebonyi A, Deininger S, Lawrence E, Ngwa-Amambua A, Jayasooriya S, Cheeseman IH, Gomez-Escobar N, Okebe J, Conway DJ, Riley EM. Distinct roles for FOXP3 and FOXP3 CD4 T cells in regulating cellular immunity to uncomplicated and severe Plasmodium falciparum malaria. PLoS Pathog. 2009;5:e1000364. doi: 10.1371/journal.ppat.1000364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.WHO Severe falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 2000;94 S1/1-S1/18. [Google Scholar]
- 25.Ceesay SJ, Casals-Pascual C, Nwakanma DC, Walther M, Gomez-Escobar N, Fulford AJ, Takem EN, Nogaro S, Bojang KA, Corrah T, Jaye MC, Taal MA, Sonko AA, Conway DJ. Continued decline of malaria in The Gambia with implications for elimination. PLoS One. 2010;5:e12242. doi: 10.1371/journal.pone.0012242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richardson MP, Ayliffe MJ, Helbert M, Davies EG. A simple flow cytometry assay using dihydrorhodamine for the measurement of the neutrophil respiratory burst in whole blood: comparison with the quantitative nitrobluetetrazolium test. J. Immunol. Methods. 1998;219:187–193. doi: 10.1016/s0022-1759(98)00136-7. [DOI] [PubMed] [Google Scholar]
- 27.Hirai H, Kubo H, Yamaya M, Nakayama K, Numasaki M, Kobayashi S, Suzuki S, Shibahara S, Sasaki H. Microsatellite polymorphism in heme oxygenase-1 gene promoter is associated with susceptibility to oxidant-induced apoptosis in lymphoblastoid cell lines. Blood. 2003;102:1619–1621. doi: 10.1182/blood-2002-12-3733. [DOI] [PubMed] [Google Scholar]
- 28.Dondorp AM, Desakorn V, Pongtavornpinyo W, Sahassananda D, Silamut K, Chotivanich K, Newton PN, Pitisuttithum P, Smithyman AM, White NJ, Day NP. Estimation of the total parasite biomass in acute falciparum malaria from plasma PfHRP2. PLoS Med. 2005;2:e204. doi: 10.1371/journal.pmed.0020204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raes A, Van Aken S, Craen M, Donckerwolcke R, Vande Walle J. A reference frame for blood volume in children and adolescents. BMC Pediatr. 2006;6:3. doi: 10.1186/1471-2431-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adetifa IM, Hill PC, Jeffries DJ, Jackson-Sillah D, Ibanga HB, Bah G, Donkor S, Corrah T, Adegbola RA. Haematological values from a Gambian cohort--possible reference range for a West African population. Int. J. Lab. Hematol. 2009;31:615–622. doi: 10.1111/j.1751-553X.2008.01087.x. [DOI] [PubMed] [Google Scholar]
- 31.Soldin SJ, Brugnara C, Wong EC. Pediatric Reference Intervals. AACC Press; 2007. [Google Scholar]
- 32.de Haas M, Kerst JM, van der Schoot CE, Calafat J, Hack CE, Nuijens JH, Roos D, van Oers RH, von dem Borne AE. Granulocyte colony-stimulating factor administration to healthy volunteers: analysis of the immediate activating effects on circulating neutrophils. Blood. 1994;84:3885–3894. [PubMed] [Google Scholar]
- 33.Sheppard FR, Kelher MR, Moore EE, McLaughlin NJ, Banerjee A, Silliman CC. Structural organization of the neutrophil NADPH oxidase: phosphorylation and translocation during priming and activation. J. Leukoc. Biol. 2005;78:1025–1042. doi: 10.1189/jlb.0804442. [DOI] [PubMed] [Google Scholar]
- 34.Fendel R, Brandts C, Rudat A, Kreidenweiss A, Steur C, Appelmann I, Ruehe B, Schroder P, Berdel WE, Kremsner PG, Mordmuller B. Hemolysis is associated with low reticulocyte production index and predicts blood transfusion in severe malarial anemia. PLoS One. 2010;5:e10038. doi: 10.1371/journal.pone.0010038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferreira A, Balla J, Jeney V, Balla G, Soares MP. A central role for free heme in the pathogenesis of severe malaria: the missing link? J. Mol. Med. (Berl.) 2008;86:1097–1111. doi: 10.1007/s00109-008-0368-5. [DOI] [PubMed] [Google Scholar]
- 36.Muller-Eberhard U, Javid J, Liem HH, Hanstein A, Hanna M. Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood. 1968;32:811–815. [PubMed] [Google Scholar]
- 37.Nielsen MJ, Moestrup SK. Receptor targeting of hemoglobin mediated by the haptoglobins: roles beyond heme scavenging. Blood. 2009;114:764–771. doi: 10.1182/blood-2009-01-198309. [DOI] [PubMed] [Google Scholar]
- 38.O’Donnell A, Fowkes FJ, Allen SJ, Imrie H, Alpers MP, Weatherall DJ, Day KP. The acute phase response in children with mild and severe malaria in Papua New Guinea. Trans. R. Soc. Trop. Med. Hyg. 2009;103:679–686. doi: 10.1016/j.trstmh.2009.03.023. [DOI] [PubMed] [Google Scholar]
- 39.Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immunol. 2010;31:318–324. doi: 10.1016/j.it.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delanghe JR, Langlois MR. Hemopexin: a review of biological aspects and the role in laboratory medicine. Clin. Chim. Acta. 2001;312:13–23. doi: 10.1016/s0009-8981(01)00586-1. [DOI] [PubMed] [Google Scholar]
- 41.Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood. 2005;106:2572–2579. doi: 10.1182/blood-2005-03-1185. [DOI] [PubMed] [Google Scholar]
- 42.Walther M, De Caul A, Aka P, Njie M, Amambua-Ngwa A, Walther B, Predazzi IM, Cunnington A, Deininger S, Takem EN, Ebonyi A, Weis S, Walton R, Rowland-Jones S, Sirugo G, Williams SM, Conway DJ. HMOX1 gene promoter alleles and high HO-1 levels are associated with severe malaria in Gambian children. PLoS Pathog. 2012;8:e1002579. doi: 10.1371/journal.ppat.1002579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mackintosh CL, Beeson JG, Marsh K. Clinical features and pathogenesis of severe malaria. Trends Parasitol. 2004;20:597–603. doi: 10.1016/j.pt.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 44.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol. Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 45.Van Gorp H, Delputte PL, Nauwynck HJ. Scavenger receptor CD163, a Jack-of-all-trades and potential target for cell-directed therapy. Mol. Immunol. 2010;47:1650–1660. doi: 10.1016/j.molimm.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 46.Kusi KA, Gyan BA, Goka BQ, Dodoo D, Obeng-Adjei G, Troye-Blomberg M, Akanmori BD, Adjimani JP. Levels of soluble CD163 and severity of malaria in children in Ghana. Clin. Vaccine Immunol. 2008;15:1456–1460. doi: 10.1128/CVI.00506-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kowanko IC, Ferrante A, Clemente G, Kumaratilake LM. Tumor necrosis factor primes neutrophils to kill Staphylococcus aureus by an oxygen-dependent mechanism and Plasmodium falciparum by an oxygen-independent mechanism. Infect. Immun. 1996;64:3435–3437. doi: 10.1128/iai.64.8.3435-3437.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wright J, Thomas P, Serjeant GR. Septicemia caused by Salmonella infection: an overlooked complication of sickle cell disease. J. Pediatr. 1997;130:394–399. doi: 10.1016/s0022-3476(97)70201-4. [DOI] [PubMed] [Google Scholar]
- 49.Bennet IL, Hook EW. Infectious Diseases (Some Aspects of Salmonellosis) Annual Review of Medicine. 1959;10:1–20. [Google Scholar]
- 50.Berkley JA, Lowe BS, Mwangi I, Williams T, Bauni E, Mwarumba S, Ngetsa C, Slack MP, Njenga S, Hart CA, Maitland K, English M, Marsh K, Scott JA. Bacteremia among children admitted to a rural hospital in Kenya. N. Engl. J. Med. 2005;352:39–47. doi: 10.1056/NEJMoa040275. [DOI] [PubMed] [Google Scholar]
- 51.Morpeth SC, Ramadhani HO, Crump JA. Invasive non-Typhi Salmonella disease in Africa. Clin. Infect. Dis. 2009;49:606–611. doi: 10.1086/603553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soesan M, Kager PA, Leverstein-van Hall MA, van Lieshout JJ. Coincidental severe Plasmodium falciparum infection and disseminated candidiasis. Trans. R. Soc. Trop. Med. Hyg. 1993;87:288–289. doi: 10.1016/0035-9203(93)90131-9. [DOI] [PubMed] [Google Scholar]
- 53.Eckerle I, Ebinger D, Gotthardt D, Eberhardt R, Schnabel PA, Stremmel W, Junghanss T, Eisenbach C. Invasive Aspergillus fumigatus infection after Plasmodium falciparum malaria in an immuno-competent host: case report and review of literature. Malar. J. 2009;8:167. doi: 10.1186/1475-2875-8-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hocqueloux L, Bruneel F, Pages CL, Vachon F. Fatal invasive aspergillosis complicating severe Plasmodium falciparum malaria. Clin. Infect. Dis. 2000;30:940–942. doi: 10.1086/313814. [DOI] [PubMed] [Google Scholar]
- 55.Avendano A, Sales-Pardo I, Marin L, Marin P, Petriz J. Oxidative burst assessment and neutrophil-platelet complexes in unlysed whole blood. J. Immunol. Methods. 2008;339:124–131. doi: 10.1016/j.jim.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 56.Lun A, Schmitt M, Renz H. Phagocytosis and oxidative burst: reference values for flow cytometric assays independent of age. Clin. Chem. 2000;46:1836–1839. [PubMed] [Google Scholar]
- 57.Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, Uzel G, DeRavin SS, Priel DA, Soule BP, Zarember KA, Malech HL, Holland SM, Gallin JI. Residual NADPH oxidase and survival in chronic granulomatous disease. N. Engl. J. Med. 2010;363:2600–2610. doi: 10.1056/NEJMoa1007097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seixas E, Gozzelino R, Chora A, Ferreira A, Silva G, Larsen R, Rebelo S, Penido C, Smith NR, Coutinho A, Soares MP. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc. Natl. Acad. Sci. U S A. 2009;106:15837–15842. doi: 10.1073/pnas.0903419106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cunnington AJ, Kendrick SF, Wamola B, Lowe B, Newton CR. Carboxyhemoglobin levels in Kenyan children with Plasmodium falciparum malaria. Am. J. Trop. Med. Hyg. 2004;71:43–47. [PubMed] [Google Scholar]
- 60.Rushworth SA, MacEwan DJ, O’Connell MA. Lipopolysaccharide-induced expression of NAD(P)H:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J. Immunol. 2008;181:6730–6737. doi: 10.4049/jimmunol.181.10.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wolach B, Scharf Y, Gavrieli R, de Boer M, Roos D. Unusual late presentation of X-linked chronic granulomatous disease in an adult female with a somatic mosaic for a novel mutation in CYBB. Blood. 2005;105:61–66. doi: 10.1182/blood-2004-02-0675. [DOI] [PubMed] [Google Scholar]
- 62.Repine JE, Clawson CC, White JG, Holmes B. Spectrum of function of neutrophils from carriers of sex-linked chronic granulomatous disease. J. Pediatr. 1975;87:901–907. doi: 10.1016/s0022-3476(75)80902-4. [DOI] [PubMed] [Google Scholar]
- 63.Wenisch C, Parschalk B, Zedwitz-Liebenstein K, Wernsdorfer W, Graninger W. The effect of artemisinin on granulocyte function assessed by flow cytometry. J. Antimicrob. Chemother. 1997;39:99–101. doi: 10.1093/jac/39.1.99. [DOI] [PubMed] [Google Scholar]
- 64.Schwarzer E, De Matteis F, Giribaldi G, Ulliers D, Valente E, Arese P. Hemozoin stability and dormant induction of heme oxygenase in hemozoin-fed human monocytes. Mol. Biochem. Parasitol. 1999;100:61–72. doi: 10.1016/s0166-6851(99)00031-6. [DOI] [PubMed] [Google Scholar]
- 65.Hanscheid T, Egan TJ, Grobusch MP. Haemozoin: from melatonin pigment to drug target, diagnostic tool, and immune modulator. Lancet Infect. Dis. 2007;7:675–685. doi: 10.1016/S1473-3099(07)70238-4. [DOI] [PubMed] [Google Scholar]
- 66.Belyaev NN, Brown DE, Diaz AI, Rae A, Jarra W, Thompson J, Langhorne J, Potocnik AJ. Induction of an IL7-R(+)c-Kit(hi) myelolymphoid progenitor critically dependent on IFN-gamma signaling during acute malaria. Nat. Immunol. 2010;11:477–485. doi: 10.1038/ni.1869. [DOI] [PubMed] [Google Scholar]
- 67.Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, Chora A, Rodrigues CD, Gregoire IP, Cunha-Rodrigues M, Portugal S, Soares MP, Mota MM. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat. Med. 2007;13:703–710. doi: 10.1038/nm1586. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.