

SUMMARY
Defects in the availability of heme substrates or the catalytic activity of the terminal enzyme in heme biosynthesis, ferrochelatase (Fech), impair heme synthesis, and thus cause human congenital anemias1,2. The inter-dependent functions of regulators of mitochondrial homeostasis and enzymes responsible for heme synthesis are largely unknown. To uncover this unmet need, we utilized zebrafish genetic screens and cloned mitochondrial ATPase inhibitory factor 1 (atpif1) from a zebrafish mutant with profound anemia, pinotage (pnt tq209). We now report a direct mechanism establishing that Atpif1 regulates the catalytic efficiency of vertebrate Fech to synthesize heme. The loss of Atpif1 impairs hemoglobin synthesis in zebrafish, mouse, and human hematopoietic models as a consequence of diminished Fech activity, and elevated mitochondrial pH. To understand the relationship among mitochondrial pH, redox potential, [2Fe-2S] clusters, and Fech activity, we used (1) genetic complementation studies of Fech constructs with or without [2Fe-2S] clusters in pnt, and (2) pharmacological agents modulating mitochondrial pH and redox potential. The presence of [2Fe-2S] cluster renders vertebrate Fech vulnerable to Atpif1-regulated mitochondrial pH and redox potential perturbations. Therefore, Atpif1 deficiency reduces the efficiency of vertebrate Fech to synthesize heme, resulting in anemia. The novel mechanism of Atpif1 as a regulator of heme synthesis advances the understanding of mitochondrial heme homeostasis and red blood cell development. A deficiency of Atpif1 may contribute to important human diseases, such as congenital sideroblastic anemias and mitochondriopathies.
A deficiency in heme, which is used in a wide variety of metabolic and regulatory pathways in cells3, results in pathological conditions that range from mild anemia to early in utero death4. As an essential component of hemoglobin, the individual enzymes and substrates of heme biosynthesis have been well studied2; however, key gaps remain in our knowledge of genes that regulate iron and heme trafficking and homeostasis. This incomplete understanding prevents researchers from developing targeted therapies for a broad range of disorders, including congenital anemias and porphyrias, as well as metabolic and neurological disorders.
We recovered pnt, a zebrafish non-lethal recessive mutant, from an unbiased ethyl nitrosourea (ENU) mutagenesis screen5 for defects in circulating erythroid cells6. pnt embryos were anemic (Fig. 1a) despite normal expression of erythroid cell markers, β-globin and band-3 (data not shown). Based on red cell indices, the erythrocytes from pnt embryos that survive to adult stage exhibited hypochromic, microcytic anemia (Supplementary Fig. 1a). Histological analysis of adult pnt hematopoietic tissues, showed no gross morphological defects (Supplementary Fig. 1b).
Fig. 1. Disruption of atpif1 in pinotage (pnttq209) produces hypochromic anemia.
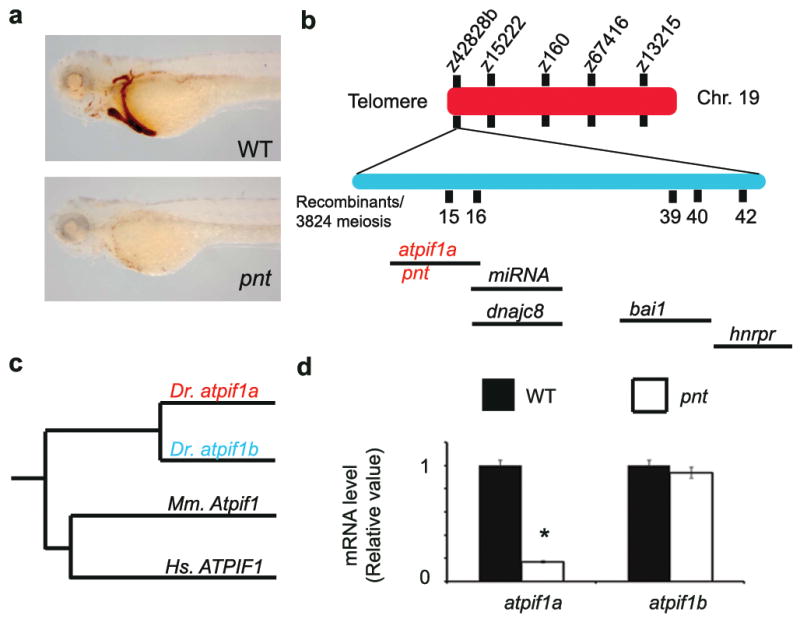
a, pnt embryos are severely anemic. Wild-type (WT) embryo at 72 hpf exhibits o-dianisidine stained (brown) hemoglobinized cells. b, The positional cloning of the pnt locus on zebrafish chromosome (Chr.) 19. A positional cloning effort with 1,912 diploid pnt embryos identified the closest linked genetic marker, z42828b. We initiated a chromosomal walk, at a distance of ~0.01 centimorgan (cM) from the pnt locus. The BAC clone, encompassing the pnt locus, is shown below, along with the annotated genes within the critical physical contig. c, Phylogenetic dendrogram showing the amino acid homology between the various atpif1 genes. D. rerio (Dr) atpif1a aligns with its related paralog, atpif1b, on zebrafish Chr. 17. Both atpif1a and atpif1b are shown clustering with their functional mammalian orthologs from mouse (Mm) and human (Hs). d, qRT-PCR analysis of atpif1a and atpif1b mRNA in pnt and WT embryos, showing reduced atpif1a and normal atpif1b mRNA level in pnt. *p<0.05 (t-test, n=3)
The positional cloning and chromosomal walk identified the most telomeric gene, the mitochondrial ATPase inhibitory factor 1 (atpif1a; zgc:162207), as the most likely candidate for the pnt locus (Fig. 1b). Phylogenetic analysis showed that an ATPase inhibitory factor 1-like protein (atpif1b; zgc: 153321) on Chr. 17 is 71% similar to atpif1a at the amino acid level (Fig. 1d), and is likely the result of gene duplication in teleosts7. Peptide alignments further displayed human (ATPIF1, chromosome 1) and mouse (Atpif1, chromosome 4) homologs for atpif1a and atpif1b (Fig. 1c). Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) showed reduced levels of atpif1a mRNA in pnt embryos (Fig. 1d) and pnt adult kidney marrow compared to respective wild-type (WT) controls (Supplementary Fig. 1c). The levels of atpif1b mRNA were, however, unchanged in pnt embryos (Fig. 1d) and elevated 2 to 3-fold in pnt adult kidney marrow (Supplementary Fig. 1c). Thus, atpif1a is likely the gene disrupted in the pnt locus. Previous studies have shown that mitochondrial Atpif1 regulates the proton motive force via mitochondrial influx of H+ ions, mitochondrial structure, and ATP synthesis, indicating that Atpif1 is required in a wide range of metabolically active tissues8. The broad requirement for Atpif1 is reinforced by the ubiquitous expression of both atpif1a and atpif1b in zebrafish embryos (Supplementary Fig. 1d), and Atpif1 in various mouse adult and fetal organs (Supplementary Fig. 1e).
To verify the loss-of-function phenotype for atpif1a, we injected two atpif1a antisense morpholinos (MO), a splice-blocking (Fig. 2a) and a translational-blocking (data not shown), to knock down atpif1a expression in zebrafish embryos. The atpif1a-silenced embryos (morphants) lacked hemoglobinized cells, as detected by o-dianisidine staining, thereby phenocopying the anemia in pnt embryos (Fig. 2a). The anemic phenotype in the morphant embryos correlates with a reduction of atpif1a mRNA levels, verifying that the splice-blocking MO accurately targeted atpif1a (Fig. 2b, Supplementary Discussion 1, Supplementary Figs. 2a–2d).
Fig. 2. Functional characterization of the atpif1a gene.

a, Splice blocking morpholino (MO) knock down of atpif1a phenocopies the anemia observed in pnt embryos. b, qRT-PCR analysis shows that the anemic phenotype is due to the accurate knockdown of atpif1a. c, Ectopic expression of atpif1a or atpif1b cRNA functionally complements the anemia in pnt embryos at 72 hpf. WT control, pnt, and rescued pnt embryos complemented with atpif1a or atpif1b cRNA are stained with o-dianisidine. The non-functional atpif1a, harboring the E26A mutation, specifically failed to complement the pnt anemia. d, pnt embryos have an AC polymorphism in the 3′ UTR of the atpif1a gene. e, The 3′UTR AC polymorphism co-segregates with the pnt phenotype by SSCP analysis. The SSCP segregation pattern for lanes 1–2 (+/+), lane 3 (+/pnt), and lanes 4–6 (pnt/pnt). f, The mutant AC polymorphism in the 3′UTR of the atpif1a cDNA functionally destabilizes its mRNA. MT construct stably expressed in MEL cells showed reduced atpif1a mRNA levels. *p<0.05 (t-test, n=3)
To further validate that atpif1a is the gene disrupted in pnt, we over-expressed atpif1a cRNA in pnt embryos and subsequently evaluated their hemoglobinization. To assess the specificity of our complementation assay, we also injected pnt embryos with non-functional atpif1a cRNA harboring a mis-sense mutation (E26A) in the regulatory inhibitory domain9. Only functional atpif1a complemented the anemia in pnt embryos (Fig. 2c), thereby confirming that atpif1a is the gene disrupted in pnt embryos. Consistent with 2 to 3-fold increase in atpif1b expression in surviving pnt adults (Supplementary Fig. 1c), the ectopic-expression of atpif1b cRNA also complemented pnt anemia (Fig. 2c), a result of their redundant function. These data suggest that the hypomorphic and viable phenotype of pnt could be attributed to the ability of atpif1b to partially compensate for the loss of function of atpif1a (Supplementary Fig. 1c). This is in contrast to other situations with paralogous genes such as the mitoferrin (mfrn) transporters10.
To identify a genetic mutation in the pnt locus, we first sequenced the open reading frame (ORF) and splice junctions of the atpif1a gene and found no mutation. Instead, a polymorphism in the 3′ untranslated region (UTR) of the atpif1a gene was discovered (Fig. 2d). This mutation was linked to the pnt locus following the distinct segregation of WT and mutant (MT) conformations on single-strand conformational polymorphism (SSCP) (Fig. 2e). To establish the functional consequences of the 3′UTR polymorphism on the stability of atpif1a mRNA, we designed constructs containing the zebrafish atpif1a ORF fused with either the WT or polymorphic 3′UTR sequences, and transfected mouse erythroleukemia (MEL) cells with each construct individually for stable selection (Supplementary Fig. 2e). qRT-PCR analysis showed a decrease in steady state atpif1a mRNA levels in cells harboring polymorphic 3′UTR constructs as compared to the WT construct (Fig. 2f, Supplementary Discussion 2). These data functionally demonstrate that the polymorphism in the 3′UTR of the atpif1a gene destabilizes its steady state mRNA. The observed decrease in the atpif1a mRNA level in pnt embryos (Fig. 1d) is consistent with the ascribed function for the 3′UTR polymorphism, which destabilizes atpif1a steady state mRNA and leads to its degradation 11.
In mammalian cells, Atpif1 is located in the inner mitochondrial membrane and primarily regulates the function of the F1F0-ATP synthase12. There is, however, no direct evidence suggesting a role for Atpif1 in heme synthesis or erythropoiesis. To ascertain the role of Atpif1 in mammalian heme synthesis, we utilized short hairpin RNAs (shRNAs) to stably silence Atpif1 in differentiating mammalian erythroid cells: human primary CD34+ (hCD34+) cells, mouse primary fetal liver (MPFL), and MEL cells. The reduced number of hemoglobinized cells in Atpif1-silenced hCD34+ and MEL cells (Fig. 3a, Supplementary Fig. 3a) and the reduced hemoglobin content in MPFL cells transduced with Atpif1-shRNAs (Supplementary Figs. 3b–3c), demonstrate conserved heme-specific function of Atpif1. Consistent with a high turnover of heme in developing erythroblasts13, we observed that the hemoglobinization defect was specific to Atpif1-silenced differentiating erythroid cells. Similarly, we observed that the expression of Atpif1 protein increases along with other proteins required for heme synthesis14, such as Fech and mitoferrin1 (Mfrn1, Slc25a37), during terminal erythroid maturation (Supplementary Fig. 3d). Analogous Atpif1-silencing experiments performed in non-erythroid (NIH3T3) and undifferentiated MEL cells showed no measurable heme defects (Supplementary Fig. 4). This indicates that Atpif1 is essential for terminal erythroid maturation and is not critical for heme synthesis in non-erythroid cells and early erythroid progenitors.
Fig. 3. Loss of Atpif1 produces a hemoglobinization defect in mammalian cells.

a, Silencing of Atpif1 in human CD34+ (left) and MEL (right) cells with shRNAs (Human: H1sh & H2sh; Mouse: M1sh) results in a hemoglobinization defect. b, Western analysis in Atpif1-shRNA (Mouse: M1sh) silenced MEL cells. Atpif1 protein level is reduced in Atpif1-shRNA (M1sh)-silenced cells. However, the mitochondrial structural proteins, AtpB, CoxIV, Vdac1, and Hsp60, are not affected. c, Silencing of Atpif1 elevates the mitochondrial membrane potential (ΔΨm), analyzed using a TMRE fluorescence probe in the presence of verapamil and FCCP. d, Silencing of Atpif1 elevates the import of 59Fe in the mitochondria, consistent with the increased mitochondrial ΔΨm. e, Silencing of Atpif1 does not influence the formation of protoporphyrin IX (PPIX), indicating PPIX and iron are not limited for heme synthesis. f, The level of 59Fe incorporated in heme is greatly reduced in Atpif1-silenced MEL cells, recapitulating the heme synthesis defect in pnt. *p<0.05 (t-test, n=3)
Under physiological conditions, the F1F0-ATP synthase generates a proton motive force and ATP by transporting H+ ions into the mitochondria8, 15. Under oxidative stress, the F1F0-ATP synthase reverses the direction of H+ transport, thus reducing ATP and proton motive force8, 15. Atpif1 inhibits the reversal of F1F0-ATP synthase to reduce ATP hydrolysis, thereby maintaining the mitochondrial membrane potential and proton motive force8. To determine the causal association between the roles of Atpif1 in mitochondrial homeostasis and mammalian heme synthesis, we used stable Atpif1-shRNA-silenced MEL cells. Consistent with the silencing of Atpif1, the Atpif1 protein levels were reduced (Fig. 3b). While excluding the possibility of global mitochondrial structural dysfunction: (1) We found normal protein expression of integral mitochondrial proteins, such as the β-subunit of ATP synthase (AtpB), complex IV (Cox IV), voltage-dependent anionic-selective channel protein 1 (Vdac1), and heat shock protein 60 (Hsp60) (Fig. 3b). (2) Electron microscopic and biochemical analysis revealed normal integrity of the mitochondria; indicating that the gross structural architecture of mitochondria in Atpif1-silenced MEL cells was preserved (Fig. 3b, Supplementary Fig. 5). We, however, found that the loss of Atpif1 in differentiating MEL cells caused a depletion of cellular ATP levels (Supplementary Figs. 6a–6b), an increase in mitochondrial membrane potential (ΔΨm) (Fig. 3c, Supplementary Fig. 6c), and an alkalinization of the mitochondria (Supplementary Fig. 6d).
Heme synthesis requires the incorporation of ferrous ion (Fe2+) into protoporphyrin IX (PPIX), which is catalyzed by Fech on the matrix side of the inner mitochondrial membrane16. Since the loss of Atpif1 results primarily in a defect of heme synthesis in differentiating erythroid cells, we analyzed the mitochondrial levels of heme substrates, heme, and their enzymes. We have previously shown that Mfrn1, an inner mitochondrial membrane transporter, facilitates the import of Fe into erythroid mitochondria10; which is dependent upon the mitochondrial ΔΨm17,18. Consistent with the increased mitochondrial ΔΨm in Atpif1-silenced MEL cells (Fig. 3c, Supplementary Fig. 6c), the uptake of radio-labeled iron (59Fe) into the mitochondria was increased (Fig. 3d), indicating that the loss of Atpif1 increases mitochondrial iron load. Iron transported into the mitochondrial matrix is either: 1) utilized to make [2Fe-2S] clusters, or 2) incorporated into PPIX by Fech to generate heme1, 2, 3, 13. Atpif1-silenced MEL cells had normal levels of mitochondrial PPIX (Fig. 3e), suggesting that enzymes and substrates necessary to make this heme precursor are not rate-limiting. The incorporation of 59Fe into PPIX to generate heme, however, was reduced (Fig. 3f), despite having sufficient levels of Fech substrates, iron and PPIX. We predicted that the catalytic efficiency of Fech might have been compromised, and thus, we measured the protein expression levels of Fech and Fech activity. Although normal levels of Fech protein were present, Fech activity was reduced (Fig. 4a), suggesting that the defect in heme synthesis in pnt could be attributed to a reduction in Fech activity. We found that the basal mitochondrial pH of control cells was pH 7.4, and for Atpif1-silenced MEL cells was pH 8.6 (Fig. 4b, Supplementary Fig. 6d). Since Atpif1-silenced MEL cells have higher basal mitochondrial pH, we predicted that (1) elevated mitochondrial pH might influence the efficiency of Fech to make heme, and (2) the Fech isolated from these cells is already harmfully influenced by the elevated pH. To experimentally support our hypothesis, we pre-incubated the purified human Fech at pH 7.4, 8.0, 8.5, and 9.0 for 1 hr, and then run Fech assay at pH 7.4. We found that pre-incubation at elevated pH have harmful impact on human Fech, and thus reduces its capacity to make heme (Supplementary Fig. 6e). In addition, we found that increasing mitochondrial pH to 8.5 and pH 9.0 would further lower Fech activity in both control and Atpif1-silenced MEL cells (Fig. 4c), indicating the Fech activity is a function of mitochondrial pH (Supplementary Discussion 3). Altogether, these data suggest that the ability of Fech to generate heme was compromised due to the increase in mitochondrial pH that occurs in Atpif1-silenced differentiating MEL cells.
Fig. 4. Atpif1 regulates heme synthesis by modulating Fech activity.

a, Ferrochelatase (Fech) protein levels are normal (top right); however, the Fech activity is reduced in Atpif1-silenced MEL cells. b, Silencing of Atpif1 increases the mitochondrial matrix pH to 8.6. The histogram summarizes values at resting conditions and after challenge with oligomycin that normalizes the initial difference. c, Analysis of Fech activity as a function of pH. The elevation of mitochondria pH to 8.5 & 9.0 markedly reduced the Fech activity. d, Drugs lowering mitochondrial membrane potential reverses the anemic phenotype due to loss of Atpif1. Treatment of FCCP and 2, 4-DNP complements the anemic phenotype of Atpif1-silenced MEL cells. e, Aconitase activity, a marker for mitochondrial [2Fe-2S] cluster synthesis, is normal in Atpif1-silenced MEL cells. f, The presence of [2Fe-2S] cluster makes Fech susceptible to mitochondrial pH alteration in the absence of Atpif1. Yeast Fech, lacking [2Fe-2S], complements pnt anemia, indicating a resistance to pH changes in the absence of atpif1. Zebrafish Fech, containing [2Fe-2S], does not complement pnt anemia, indicating its susceptibility to mitochondrial alkalinization. g, The [2Fe-2S] of Fech is sensitive to the reduction in redox potential. The treatment of DTN reduces human Fech activity, and does not affect yeast Fech activity. h, Reactive oxygen species (ROS) are not responsible for pnt anemia. Treatment of pnt embryos with N-acetyl cysteine (NAC) does not reverse pnt anemia. i, A proposed mechanistic model of Atpif1 function in the maintenance of mitochondrial pH and regulation of Fech activity for heme synthesis. Mitochondrial heme synthesis requires Fech to incorporate iron into PPIX at physiological pH. Atpif1 normally preserves the mitochondrial pH. Loss of Atpif1 alkalinizes mitochondrial pH, the presence of [2Fe-2S] cluster makes Fech susceptible to mitochondrial pH and redox potential perturbations, and consequently reduces its catalytic efficiency for the production of heme. *p<0.05 (t-test, n=3); †p<0.05 vs. control cells at pH 8.5 (t-test, n=3)
We further found that the lowering mitochondrial membrane potential and thus mitochondrial pH, using pharmacological agents carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP; 1 mM; 1hr) and 2, 4-dintro-phenol (2,4-DNP; 500 μM; 1hr)19, could rescue anemic phenotype in Atpif1-silencd MEL cells (Fig. 4d). These data reinforce our observation that Atpif1 regulates heme synthesis via its modulation of the mitochondrial Ψm and mitochondrial pH.
Atpif1-silenced MEL cells have normal cytosolic and mitochondrial [2Fe-2S] cluster biogenesis as they have normal activities of proteins dependent on [2Fe-2S] cluster synthesis, such as cytosolic xanthine oxidase (Supplementary Fig. 7a), and mitochondrial aconitase (Fig. 4e). The post-translational stability of Fech apoprotein depends on iron availability and an intact [2Fe-2S] cluster assembly machinery20, 21. However, the functional role of the [2Fe-2S] cluster in Fech activity remains unknown (Supplementary Discussion 4). All vertebrate Fech enzymes possess [2Fe-2S] clusters, whereas Fech from the yeast Saccharomyces cerevisiae does not have a cluster16, 21. Based on these data and our current observations, we hypothesized that the [2Fe-2S] cluster of Fech could influence Fech activity in situ by rendering Fech responsive to changes in the mitochondrial proton motive force, and thus mitochondrial pH and redox potential.
To experimentally test whether [2Fe-2S] cluster-bound Fech responded to the capacity of Atpif1 to regulate mitochondrial proton motive force and thus mitochondrial pH, we designed constructs containing either zebrafish Fech (with [2Fe-2S]) or yeast Fech (without [2Fe-2S]) with the vertebrate mitochondrial targeting pro-peptide for cRNA injections. Both constructs, irrespective of the presence of [2Fe-2S] clusters, functionally complemented a genetic deficiency for zebrafish Fech in freixenet6, 22 (frxtu271, Supplementary Fig. 7b). When either zebrafish Fech or yeast Fech cRNA was injected into pnt embryos, the yeast Fech rescued pnt anemia, while zebrafish Fech did not (Fig. 4f). These data suggest that it is the [2Fe-2S] cluster of the zebrafish Fech that makes this enzyme vulnerable to pH perturbations of the mitochondrial matrix in atpif1-deficient pnt embryos. Therefore, Atpif1 plays an essential role in stabilizing the activity of [2Fe-2S] cluster-containing Fech by modulating changes in mitochondrial matrix pH. Based upon our results, we infer that yeast Fech, lacking the [2Fe-2S] cluster, is resistant to mitochondrial pH changes in pnt embryos. Consistent with our observation that the activity of yeast Fech would be unaffected by mitochondrial pH (Fig. 4f), we found that the deficiency of Inh1 did not produce a defect in heme synthesis (data not shown). These data show that Atpif1 regulates heme synthesis in higher vertebrate erythroid tissues, while its counterpart in baker’s yeast, Inh1, does not regulate heme synthesis.
The redox potential of mitochondrial matrix at pH 7.4 is -280 mV, and it is reduced to -370 mV at pH 8.423. Since the mitochondrial pH is inversely related to mitochondrial redox potential23, we further analyzed the susceptibility of the [2Fe-2S] of Fech to reduction in mitochondrial redox potential. When we treated human Fech (with [2Fe-2S]) and yeast Fech (without [2Fe-2S]) with a strong reducing agent, sodium dithionate (DTN, 10 mM), we found that the DTN treatment could only reduce the activity of human Fech and not the activity of yeast Fech (Fig. 4g). This further suggests that [2Fe-2S] of vertebrate Fech is sensitive to the reduction in the redox potential. Thus, the Fech activity is reduced in Atpif1-silenced MEL cells as its [2Fe-2S] cluster is sensitive to an elevation of pH (Fig. 4b, 4c, 4d, 4f) and a reduction in redox potential (Fig. 4g), and not due to a defect in [2Fe-2S] cluster biosynthetic assembly (Fig. 4e, Supplementary Fig. 7a).
To exclude an anemic phenotype in pnt as a causal effect of reactive oxygen species (ROS) induced heme degradation, we treated pnt mutant embryos with N-acetyl cysteine (NAC, 10 μM, 48 hr), a potent antioxidant employed for ROS studies in zebrafish embryos24, 25, and found that NAC could not rescue pnt anemia (Fig. 4h). This demonstrates that anemic phenotype in pnt is not due to increased ROS generation. The heme-oxygenase-1 enzyme (Hmox1) is a ROS-responsive, cellular enzyme responsible for heme degradation. We found normal transcript levels of Hmox1 in control and Atpif1-silenced MEL cells (Supplementary Fig. 7c). Consistent with our model, it has been demonstrated that 1) only overexpression of Atpif1, not loss of Atpif1 activity, induces ROS production, independent of the Hif1α pathway26, and 2) the overexpression of Atpif1 did not affect cellular hydrogen peroxide levels, indicating that the Atpif1-mediated superoxide signal is of mild-intensity and localized in mitochondria26. Altogether, these data suggest that ROS and heme degradation are not responsible for the heme defect we observed in erythroid cells.
In summary, Atpif1 modulates the mitochondrial pH and redox potential, and thus allows Fech to efficiently catalyze the incorporation of iron into PPIX to produce heme. Loss of Atpif1 allows the mitochondrial pH and, consequently, the redox potential to change to a level that reduces [2Fe-2S] cluster-containing Fech activity, thereby reducing heme synthesis, resulting in hypochromic anemia in pnt (Fig. 4i). Previous human studies have identified ALAS2, FXN, ABCB7, and SLC25A38 as the genetic causes of congenital sideroblastic anemias27, 28. Data presented herein demonstrate that Atpif1 should now be added to the list of proteins whose native function is required for normal erythropoiesis. This makes Atpif1 a candidate gene for the understanding and treatment of human sideroblastic anemias and mitochondrial disorders.
METHODS SUMMARY
All procedures were approved by the Animal Care and Use Committee of Boston Children’s Hospital. We performed complete blood count and Wright-Geimsa analyses on peripheral blood recovered from adult pnt. Genetic mapping and positional cloning were utilized to identify zgc: 162207 (atpif1a) as the candidate gene for the pnt locus on zebrafish Chr. 19. We employed qRT-PCR using TaqMan gene expression assays (Applied Biosystems, Carlsbad, CA) to measure levels of atpif1a and atpif1b mRNA. Morpholinos (Gene Tools, Philomath, OR) against splice-site of atpif1a and atpif1b were designed and injected in WT embryos to verify loss-of-function phenotype. The cRNA for atpif1a, atpif1a-E26A, and atpif1b were injected in pnt embryos for complementation. The cDNA prepared from WT and pnt embryos was sequenced, and the polymorphism in the 3′UTR of the atpif1a sequence was verified using SSCP gels.
We silenced Atpif1 in hCD34+, MPFL and MEL using shRNAs. The Atpif1-silenced, differentiated hCD34+ and MEL cells were stained with o-dianisidine to measure hemoglobinized cells, while MPFL cells were treated with Drabkin’s reagent to measure relative hemoglobin content. The loss of Atpif1 protein and the state of mitochondrial structural proteins in MEL cells were verified using western and electron microscopic analyses. We analyzed fluorescent intensities of TMRE as a function of mitochondrial membrane potential, Mg green as a function of ATP levels, and ratio of carboxy SNARF-1 to Mitotracker green as a function of the mitochondrial matrix pH8, 12.
We prepared 59Fe-saturated transferrin, and measured 59Fe incorporated in mitochondria and complexed in heme using a gamma counter. We examined PPIX levels and the catalytic efficiency of Fech in MEL cells using spectrophotometric analysis. The MEL cells were treated with FCCP and 2,4-DNP, followed by analysis for hemoglobinized cells. Human and yeast Fech were treated with DTN, and subsequently their catalytic efficiency were measured. Aconitase activity was determined as a measure of [2Fe-2S] cluster levels29. The cRNA for zebrafish Fech or yeast Fech was injected in pnt embryos, and their efficiency to rescue the anemia in pnt was measured using o-dianisidine staining and verified by using SSCP analysis10. Statistical analyses were performed by paired or un-paired t-test. Significance was set at p<0.05.
Supplementary Material
Acknowledgments
We thank members of our lab (Michelle Cassim, Adam Kaplan, Gordon Hildick-Smith, and Heidi Anderson) and colleagues (Seth L. Alper, Karen Pepper, and Nikolaus S. Trede) for critical review of the manuscript, Terrence C. Law for pnt adult blood characterization, Howard Mulhern for help with the electron microscopy, Christopher Lawrence and his team for the zebrafish husbandry. This research was supported in part by the Cooley’s Anemia Foundation (D.I.S., C.C.), the March of Dimes Foundation (B.H.P.), the American Heart Association (J.D.C., A.E.M.), the Dutch National Science Fund (I.J.S.), the Fondation Soldati pour la Recherche en Cancerologie (G.V.), the Burroughs Welcome Fund (N.N.D.), the NIDDK (D.I.S, B.H.P., A.N., J.K., H.A.D., S.M.H.), and the NHLBI (D.I.S., B.H.P).
Footnotes
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
GenBank accession numbers. Sequences are available in GenBank as follows: Zebrafish atpif1a (NM_001089521.1), zebrafish atpif1b (NM_001044859), mouse Atpif1 (NC_000070.5), and human ATPIF1 (NC_005104.2). Reprints and permissions information is available at npg.nature.com/reprints and permissions.
Author Contributions. D.I.S. and B.H.P. originally conceived the project, designed and performed the experiments, analyzed data, and wrote the manuscript. N.T.M., A.S., L.L., D.M.W., and J.K. measured 59Fe uptake in mitochondria and complexed in heme, PPIX levels, Fech activity, xanthine oxidase and aconitase activities, and heme levels in yeast knock-out for Inh1, and participated in scientific discussions. J.D.C., I.J.S., E.L.P., and N.L.B. did microinjections in zebrafish embryos. S.K.H., G.V., C.C., and W.C. helped with zebrafish colony maintenance and protein experiments. Y.Z. helped with high resolution meiotic mapping. A.N., S.N.H., and B.L.E. helped with silencing ATPIF1 in human primary CD34+ cells. S.M.H. helped with silencing Atpif1 in MFPL cells. A.E.M., T.A.D., and H.A.D. created Fech constructs for injection, measured Fech activity as a function of pH, Fech activity, analyzed Fech structure for [2Fe-2S] clusters, and participated in scientific discussions. D.F., J.M.W., M.C. and N.N.D. helped to design and measure mitochondrial physiological parameters. C.B. analyzed pnt adult blood parameters. D.C. and M.D.F. helped with electron microscopic analysis of mitochondrial structure and disease.
Author InformationThe authors declare no competing financial interests
References
- 1.Schultz IJ, Chen C, Paw BH, Hamza I. Iron and porphyrin trafficking in heme biogenesis. J Biol Chem. 2010;285:26753–9. doi: 10.1074/jbc.R110.119503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iolascon A, De Falco L, Beaumont C. Molecular basis of inherited microcytic anemia due to defects in iron acquisition or heme synthesis. Haematologica. 2009;94:395–408. doi: 10.3324/haematol.13619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Severance S, Hamza I. Trafficking of heme and porphyrins in metazoa. Chem Rev. 2009;109:4596–616. doi: 10.1021/cr9001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson KE, Sassa S, Bishop DF, Desnick RJ. In: The Online Metabolic & Molecular Basis of Inherited Disease. Bishop DF, editor. The McGraw-Hill Press; New York, NY: 2011. pp. 1–153. [Google Scholar]
- 5.Shin JT, Fishman MC. From Zebrafish to human: modular medical models. Annu Rev Genomics Hum Genet. 2002;3:311–40. doi: 10.1146/annurev.genom.3.031402.131506. [DOI] [PubMed] [Google Scholar]
- 6.Ransom DG, et al. Characterization of zebrafish mutants with defects in embryonic hematopoiesis. Development. 1996;123:311–9. doi: 10.1242/dev.123.1.311. [DOI] [PubMed] [Google Scholar]
- 7.Postlethwait JH, et al. Vertebrate genome evolution and the zebrafish gene map. Nat Genet. 1998;18:345–9. doi: 10.1038/ng0498-345. [DOI] [PubMed] [Google Scholar]
- 8.Campanella M, Parker N, Tan CH, Hall AM, Duchen MR. IF(1): setting the pace of the F(1)F(o)-ATP synthase. Trends Biochem Sci. 2009;34:343–50. doi: 10.1016/j.tibs.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 9.Ando C, Ichikawa N. Glutamic acid in the inhibitory site of mitochondrial ATPase inhibitor, IF(1), participates in pH sensing in both mammals and yeast. J Biochem. 2008;144:547–53. doi: 10.1093/jb/mvn100. [DOI] [PubMed] [Google Scholar]
- 10.Shaw GC, et al. Mitoferrin is essential for erythroid iron assimilation. Nature. 2006;440:96–100. doi: 10.1038/nature04512. [DOI] [PubMed] [Google Scholar]
- 11.Houseley J, Tollervey D. The many pathways of RNA degradation. Cell. 2009;136:763–76. doi: 10.1016/j.cell.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 12.Campanella M, et al. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab. 2008;8:13–25. doi: 10.1016/j.cmet.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 13.Richardson DR, et al. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci U SA. 2010;107:10775–82. doi: 10.1073/pnas.0912925107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nilsson R, et al. Discovery of genes essential for heme biosynthesis through large-scale gene expression analysis. Cell Metab. 2009;10:119–30. doi: 10.1016/j.cmet.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walker JE. The regulation of catalysis in ATP synthase. Curr Opin Struct Biol. 1994;4:912–8. doi: 10.1016/0959-440x(94)90274-7. [DOI] [PubMed] [Google Scholar]
- 16.Lanzilotta WN, Dailey HA. In: Human ferrochelatase in Handbook of Metalloproteins. Messerschmidt A, editor. 4&5. John Wiley & Sons, Ltd; Chichester, UK: 2011. pp. 138–146. [Google Scholar]
- 17.Lange H, Kispal G, Lill R. Mechanism of iron transport to the site of heme synthesis inside yeast mitochondria. J Biol Chem. 1999;274:18989–96. doi: 10.1074/jbc.274.27.18989. [DOI] [PubMed] [Google Scholar]
- 18.Froschauer EM, Schweyen RJ, Wiesenberger G. The yeast mitochondrial carrier proteins Mrs3p/Mrs4p mediate iron transport across the inner mitochondrial membrane. Biochim Biophys Acta. 2009;1788:1044–50. doi: 10.1016/j.bbamem.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 19.Park D, et al. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature. 2011;477:220–4. doi: 10.1038/nature10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crooks DR, Ghosh MC, Haller RG, Tong WH, Rouault TA. Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron-sulfur cluster assembly machinery. Blood. 2010;115:860–9. doi: 10.1182/blood-2009-09-243105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Medlock AE, Dailey HA. Examination of the activity of carboxyl-terminal chimeric constructs of human and yeast ferrochelatases. Biochemistry. 2000;39:7461–7. doi: 10.1021/bi000134p. [DOI] [PubMed] [Google Scholar]
- 22.Childs S, et al. Zebrafish dracula encodes ferrochelatase and its mutation provides a model for erythropoietic protoporphyria. Curr Biol. 2000;10:1001–4. doi: 10.1016/s0960-9822(00)00653-9. [DOI] [PubMed] [Google Scholar]
- 23.Hu J, Dong L, Outten CE. The redox environment in the mitochondrial intermembrane space is maintained separately from the cytosol and matrix. J Biol Chem. 2008;283:29126–34. doi: 10.1074/jbc.M803028200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu D, et al. miR-451 protects against erythroid oxidant stress by repressing 14-3-3zeta. Genes Dev. 2010;24:1620–33. doi: 10.1101/gad.1942110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.North TE, et al. PGE2-regulated wnt signaling and N-acetylcysteine are synergistically hepatoprotective in zebrafish acetaminophen injury. Proc Natl Acad Sci U S A. 2010;107:17315–20. doi: 10.1073/pnas.1008209107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Formentini L, Sanchez-Arago M, Sanchez-Cenizo L, Cuezva JM. The Mitochondrial ATPase Inhibitory Factor 1 Triggers a ROS-Mediated Retrograde Prosurvival and Proliferative Response. Mol Cell. 2012;45:731–42. doi: 10.1016/j.molcel.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 27.Sheftel AD, Richardson DR, Prchal J, Ponka P. Mitochondrial iron metabolism and sideroblastic anemia. Acta Haematol. 2009;122:120–33. doi: 10.1159/000243796. [DOI] [PubMed] [Google Scholar]
- 28.Camaschella C. Hereditary sideroblastic anemias: pathophysiology, diagnosis, and treatment. Semin Hematol. 2009;46:371–7. doi: 10.1053/j.seminhematol.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 29.Li L, Kaplan J. A mitochondrial-vacuolar signaling pathway in yeast that affects iron and copper metabolism. J Biol Chem. 2004;279:33653–61. doi: 10.1074/jbc.M403146200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.