

Abstract
The RdxA oxygen insensitive nitroreductase of the human gastric pathogen Helicobacter pylori is responsible for the susceptibility of this organism to the redox active prodrug metronidazole (MTZ). Loss-of-function mutations in rdxA are primarily responsible for resistance to this therapeutic. RdxA exhibits potent NADPH oxidase activity under aerobic conditions and metronidazole reductase activity under strictly anaerobic conditions. Here we report the crystal structure of RdxA, which is a homodimer exhibiting domain swapping and containing two molecules of FMN bound at the dimer interface. We have found a gap between the side chain of Tyr47 and the isoalloxazine ring of FMN that seems appropriate for substrate binding. The structure does not include residues 97–128, which corresponds to a locally unstable part of the NTR from E. coli, and might be involved in cofactor binding. Comparison of H pylori RdxA to other oxidoreductases of known structure suggests RdxA may belong to a new subgroup of oxidoreductases in which a cysteine sidechain close to the FMN cofactor could be involved in the reductive activity. In this respect, mutation of C159 to A or S (C159A/S) has resulted in loss of MTZ reductase activity, but not NADPH oxidase activity. The RdxA structure allows interpretation of the many loss-of-function mutations previously described, including those affecting C159, a residue whose interaction with FMN is required for nitroreduction of MTZ. Our studies provide unique insights into the redox behavior of the flavin in this key enzyme for metronidazole activation, and with potential use in gene therapy.
Keywords: nitroreductase, metronidazole, Helicobacter pylor, prodrug, flavoprotein, antibiotic resistance
INTRODUCTION
Nitroreductases (NTR) catalyze the reduction of nitro groups of structurally diverse aromatic and heterocyclic compounds that includes nitrotoluenes, nitrofurans, and nitroimidazoles [1,2]. These enzymes can be subdivided into two general classes based on one or two electron transfer mechanisms [3,4]. The oxygen sensitive class represented by cytochrome P450 reductase and xanthine dehydrogenase catalyze one-electron transfers (nitro anion) that in the presence of molecular oxygen produces superoxide anions [3]. The oxygen insensitive class, represented by the homodimeric NAD(P)H/FMN NTRs commonly found in bacteria, catalyze a two-step, 4e− reduction of the nitro group, producing DNA damaging and mutagenic hydroxylamine adducts [3–6]. These NTRs, represented by NfsB of Escherichia coli and RdxA of Helicobacter pylori also catalyze the reduction of molecular oxygen in a two electron transfer that produces hydrogen peroxide [7].
In vitro studies indicate that NTRs differ widely in substrate preference with enteric NTRs reducing prodrugs like nitrofurans, but not substrates of much lower redox potential such as metronidazole (MTZ) [1]. Thus, enteric bacteria are generally resistant to the action of metronidazole unless the NTR genes are over expressed. For example, in the Salmonella typhimurium strains used in Ames testing, overexpression of NfsB results in a higher mutation frequency that is attributed to increased reduction of MTZ [8]. In contrast, the RdxA NTR of H. pylori efficiently reduces metronidazole and is the major activating enzyme in this species [5,7]. Direct enzyme assays performed under strictly anaerobic conditions, which simulate the highly anaerobic bacterial cytoplasm, established that the metronidazole reductase activity of RdxA was 60-fold higher than the rate measured for the NfsB of E. coli and for a second nitroreductase of H. pylori FrxA [7]. Expression of RdxA in other bacteria that are not otherwise susceptible to MTZ, renders them susceptible [5,7]. In allelic exchange bacterial genetic studies, plasmid borne rdxA serves as a suicide gene that can be selected against with MTZ (loss of plasmid vector) [9,10]. NTRs have been employed in adenovirus prodrug gene therapy systems to activate prodrug CB1954 in treatment of prostate cancer [11]. Our studies show that RdxA is 10 times more efficient than NfsB in activation of CB1954 [7] and might be amenable to protein engineering to develop a more efficient enzyme.
The differences in nitroreduction capacity of an NTR likely depend on the midpoint reduction potential of the FMN cofactor (Em ~ 190 mV and low range of −380 mV) [12]. Metronidazole (Em -486) [13] is clearly outside this range, so RdxA appears to be unique among NTRs in its capacity to reduce this prodrug. However, in other flavoproteins, such as flavodoxins, hydrophobic or negatively charged amino acids can contribute to even lower redox potentials [14]. Studies by Olekhnovich et al determined that RdxA was also a potent NADPH oxidase that reduced molecular oxygen to hydrogen peroxide [7]. These studies also revealed that trace amounts of oxygen in the enzyme assay mixture was sufficient to inhibit metronidazole reductase activity of RdxA [7]. In the original communication on RdxA [5], we noted that the pI for RdxA was ~8 and that the protein contained 6 cysteine residues; whereas, the NTRs of enteric bacteria generally have one or two cysteine residues and more acidic pI values (pI =5.4 to 5.6). It is reasonable to consider the possibility that amino acid composition and in particular the cysteine residues influence the local redox range of the FMN cofactor in enabling more efficient reduction of metronidazole. Removal of molecular oxygen might allow reduction of cysteine thiols and the accompanying conformational changes required to lower the redox potential of FMN. To address mechanistic questions of aerobic and anaerobic substrate specificity, we have solved the crystal structure of RdxA and used site directed mutagenesis of a uniquely positioned cysteine residue near the FMN to establish its requirement for the anaerobic reduction of MTZ.
RESULTS AND DISCUSSION
Overall structure of RdxA from H. pylori
Two functional homodimers are present in the asymmetric unit of the tetragonal crystals. Each monomer consists of 210 amino acids folded in an α+β motif (Figure 1) containing eight α-helices and five β-strands. The dimer exhibits domain swapping and is organized into two folding domains. The central core of each domain is formed by a five-stranded sheet (β5β1β2β4β3) composed of four antiparallel strands from one monomer (strands β1β2β4β3) plus strand β5 from the other one. Surrounding this sheet there are three small α-helices on one side (α3,α4 and α7) and two larger α-helices on the other (α2 and α6). Helices α2 and α6, together with α1, packed perpendicularly onto the C-terminus of helix α6, form the dimer interface. Helix α1 additionally packs onto helix α2 and α6 of the other monomer. The C-terminal, residues 198–210 of one monomer, containing the small α8 and β5, surround part of the other monomer where they contact α2 and β1. Besides the two protein chains, there are two FMN molecules inserted in the interface of the dimer with their re faces accessible to the solvent. Each FMN is surrounded by β3 strand and the loop connecting helices α1 and α2 from one monomer, plus the loop connecting helix α2 and β1 strand from the other monomer. In the structure, no electronic density could be observed for residues 97–128 (chain A) and 90–133 (chain B).
FIGURE 1. Helicobacter pylori RdxA structure (PDB code: 3QDL).

A. Ribbon drawing of the content of the asymmetric unit in the RdxA crystal. B. Ribbon drawing of the functional dimer of RdxA. Each monomer and the corresponding FMN molecules are colored in yellow (chain A) and green (chain B)
Using the Dali server [15] the structure of RdxA has been aligned and compared with other homologous structures of nitroreductases with which it shows no more than 29% sequence identity (Figure 2). The alignment shown in figure 2 does not contain residues 97–128 of RdxA, nor the presumably equivalent residues in the other proteins. The homologous reductases belong to two distinct groups: nitroreductases and flavin reductases. The closest structural homologue of RdxA is the NTR from Streptococcus mutans (PDB code: 3GAG). The other enzymes exhibiting high structural homology are also nitroreductases. Nevertheless, the Staphylococcus epidermidis, FMN.FAD reductase presents more similarity in structure than some prokaryotic NTRs, including that of E. coli.
FIGURE 2. A. Structure-based sequence alignment of homologous prokaryotic reductases.

Secondary structure for RdxA was assigned by DSSP server [39] and is shown above its sequence. The parentheses indicate the position of missing P97-S128 residues. B. Alignment of H97-S128 region in RdxA with the corresponding sequences in other NTRs. This alignment was performed with Clustal W [40].
FMN cofactor binding site
The RdxA dimer contains two molecules of FMN. For each FMN, all the residues interacting with the phosphate moiety are located in the same monomer, while the isoalloxazine and the ribityl moieties are bound at the dimer interface and form H-bonds and hydrophobic interactions with residues of both monomers (Figure 3).
FIGURE 3. Interactions at the FMN binding site.
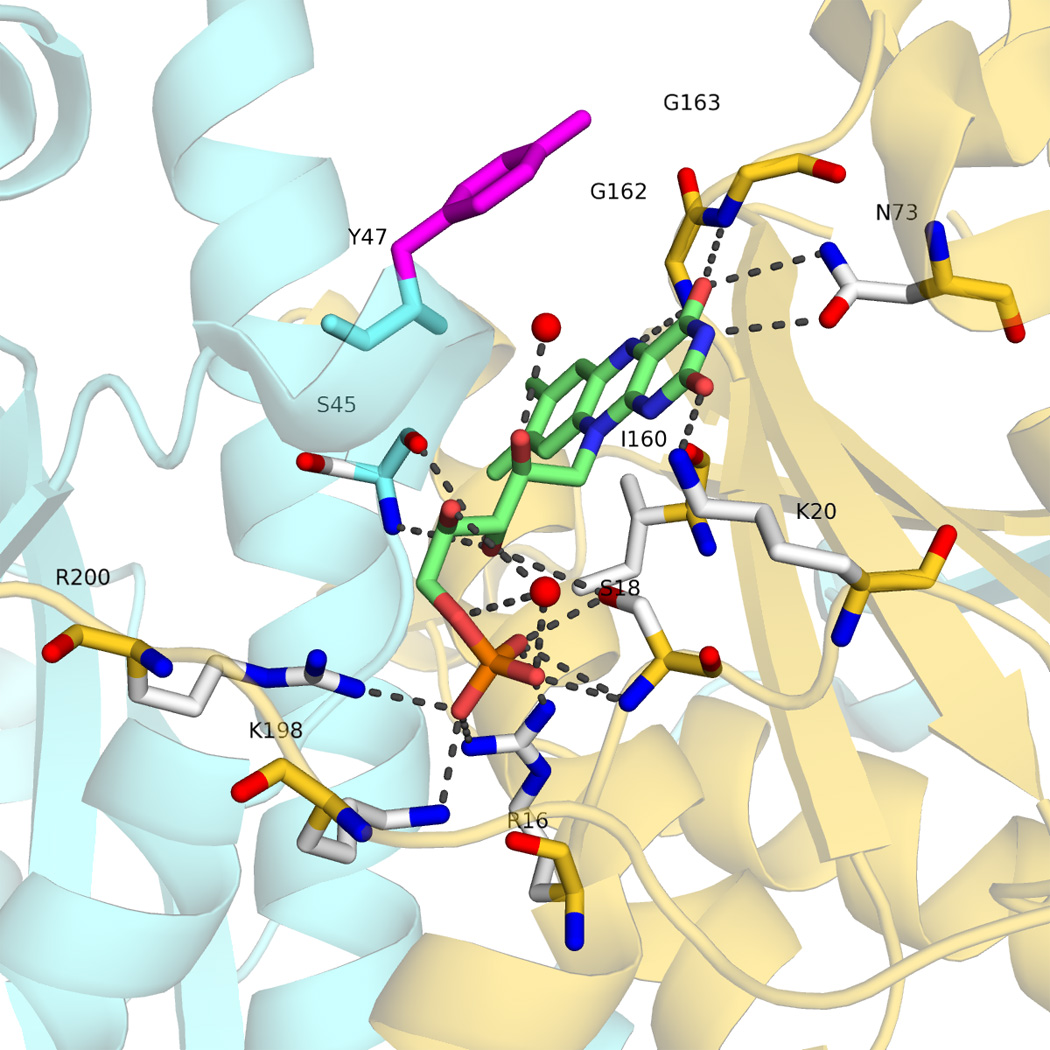
H-bonding network in the FMN binding site in monomer A. Chain A is colored in aquamarine and chain B in gold, Tyr47 belongs to monomer B. The oxygen atoms of the two water molecules involved in the interaction network are shown as red spheres.
At the si face, the Ile160 O and side chain atoms are packed onto the isoalloxazine ring of FMN, with the O atom located at 3.2 Å from N5 and N10. The position equivalent to that of Ile 160 is occupied in most of the homologous reductases by a Pro residue (Figure 2) with the exception of the NTR from S. pneumoniae and of FPase I which bears Ile and Thr residues, respectively. In all cases, these residues form stacking contacts with the isoalloxazine ring. Also noticeable at the si face is the presence of Cys159, whose S atom points toward the flavin and is located at 3.6–5.2 Å of N1 (depending on the chain). At the re face, the side chain of Tyr47 from the other monomer lays parallel on the isoalloxacine ring (closest distance of 6.0 Å) and seems appropriately located to allow the intercalation of substrates, as it has been observed in other flavoenzymes [16,17]. The equivalent position in the aligned NTR (Figure 2) is not conserved.
The O2N3O4 edge of the isoalloxazine is stabilized by side chain H-bonds between Lys20 and O2, and between Asn73 and N3 and O4. In other reductase sequences, the equivalent position is occupied by Asn or Gln.. The FMN O4 atom additionally forms an H-bond with Gly163, while N5 is H-bound to Gly162. Gly163 is highly conserved in NTRs and flavin reductases (Figure 2) where its small volume has been proposed to allow the entrance of substrate in the space over the re face of the flavin [18]. At the other edge of the isoalloxazine ring, Ile142 contacts one of the methyl groups.
The FMN ribityl forms H bonds with OG of Ser18 of the same monomer, with the main chain of Ser45 of the other monomer and with two water molecules Finally, the phosphoryl group is tightly bound to the structure through contact with the side chains of several residues of the same monomer (Arg16, Ser18 Lys198 and Arg200), plus H-bonds with the N atom of Ser18 and a water molecule.
The missing region of RdxA – The absence of electron density for residues 97–128 (chain A) and 90–133 (chain B) is very likely due to proteolysis during purification. Mass spectrometry data corresponding to dissolved RdxA crystals show a main peak at 12174 kDa, with two additional components at 12370 and 12530 kDa. Possible H+2 peaks appear at 6086 and 6182 kDa. The main peak thus corresponds to approximately half the mass of the RdxA protein (24067 kDa according to the sequence). This indicates that proteolysis of the protein sample has taken place. However, a sample of the same protein preparation that was incubated with NADP+, and did not crystallize in the same conditions, showed by SDS-PAGE to have a mass of 26313 ± 10 Da, close to the theoretical mass of the protein. The missing segments are occupied by helices F and G in the search model, the NTR of E.coli [19]. In this NTR, helix F is part of a solvent-exposed channel at the dimer interface where FMN lies and helix G is supposed to convey substrate specificity. Helices F and G exhibit high B values in the structure, and it has been proposed that mobility of helix F might be important for optimal binding and catalysis [20]. We have recently developed a method to identify locally unstable regions of proteins using computed polarity and packing density profiles of protein buried interfaces. The method successfully identifies the unstable segments of proteins for which structural information of partly unfolded conformations has been reported [21]. The method is based in analysis of PDB files and therefore cannot be used directly to characterize the missing region in RdxA since no coordinates are available for this region. However, it can provide useful insight on this missing region from the analysis of the corresponding region of the search model. Thus, the polarity and packing density profiles of the search model are displayed in Figure 4. The more unstable region of E. coli NTR, characterized by its high content of buried polar area, encompasses residues A89-M139, corresponding to residues P91 and I136 of H pylori RdxA, which roughly mimics the missing segment of the structure. This suggests that the missing region in the structure of RdxA will also be locally unstable and more exposed to proteolysis. It seems that the proposal that helix F of E. coli NTR may exhibit high functional mobility may be extended to the equivalent helix of RdxA and also to helix G. Our observation that NADP+ protects RdxA against proteolytic removal of the missing segment agrees with the role assigned to helix F of E. coli NTR in binding this cofactor [20].
FIGURE 4. Detection of conformationally unstable segments in E. coli NTR by interfacial polarity analysis.

The profile is built by assigning to each residue the ratio of polar/apolar buried surface area associated with the interaction of an 8-residue flanking peptide with the rest of the protein. The sequence segment encompassing the peaks of highest ratios over the 0.5 polarity ratio baseline is indicated by the blue bar. The packing of this segment with the rest of the protein is predicted to be unstable. The full details are described in [21]. The sequence shadowed in grey represents the E. coli NTR segment structurally equivalent to the missing helices in the structure of RdxA.
Classification of metronidazole resistance mutations in RdxA based in their potential impact in RdxA stability, dimerization, FMN binding or enzyme activity
Metronidazole resistance strongly correlates with mutations in RdxA [5,22]. The structure of RdxA allows us to assess and classify the impact of those mutations at the protein level. Mendz and Megraud provided a compilation of mutations in RdxA that includes those resulting in loss of function reported by many laboratories [22] that we have grouped into five classes as follows. Class I mutations are those expected to reduce the affinity of the apoprotein for the FMN cofactor. As explained above, RdxA binds FMN by a combination of H bonds, electrostatic and hydrophobic interactions. Thermodynamic analysis of the energetics of FMN binding in a model flavoprotein [23] indicates that point mutations of FMN binding side chains can significantly decrease the strength of the binding. In RdxA, mutations such as those affecting residues R16, S18, K20, N73, I142, G162 and K200 involved in FMN binding (see above) are expected to decrease the affinity of the apoprotein for FMN, which may be detrimental for function.
Class II mutations are those expected to destabilize dimer formation. RdxA is a tight dimer and the monomer is expected to be conformationally unstable. Thus, mutation of residues that appear at the dimer interface such as L42, S43, R41, Q50, V55, M56, I142, G145, K202, L209, are expected to destabilize the protein. Class III includes mutations of residues not expected to be crucial for FMN binding or for dimerization, as judged from the structure, but potentially associated to the redox function of the enzyme, given their location near the FMN redox centre and their chemical properties. Examples include: C19, Y47, and C159. Class IV is formed by mutations expected to cause protein destabilization not related to dimerization. Obvious examples are the mutations of the buried G149, or those of H17, a residue that is hydrogen bonded with the C terminus of alpha helix 1 [24]. Finally, there are mutations associated to metronidazole resistance, such as those at A143 or V192, for which we find no obvious explanation. They constitute class V. A review of the original citations used by Mendz and Megraud for their composite [22] revealed that RdxA alleles from most MTZ resistant strains also contained multiple mutations, many of which probably do not contribute to loss of enzyme activity. To our knowledge, there has been no systematic directed mutational analysis of RdxA.
A brief comparison of Hp RdxA with Hp FrxA and with structurally homologous reductases
The structure of FrxA, a second nitroreductase present in Helicobacter pylori, is available in the PDB (2H0U). While this enzyme alone (regardless of its level of expression) does not contribute to MTZ susceptibility in H. pylori strains mutant in rdxA, double mutants are 2-fold more resistant to MTZ indicating that FrxA is contributing to MTZ susceptibility [25,26]. The FrxA sequence is ~25% identical to that of RdxA, and the two proteins are structurally related. Superposition of the FrxA and RdxA structures shows a RMSD of 1.15 Å for 125 Cα (Figure 5). Interestingly, the missing region of RdxA (residues 97–128) is seen in FrxA to fold into two long α helices (112–124 and 93–103). Conversely, residues 57–76, missing in the FrxA structure, are seen in RdxA to be folded into two helices (80–67 and 73–79) one of them contributing to FMN binding. Overall, the binding of the FMN cofactor is similar in the two nitroreductases, although more water molecules appear bound to the FMN in FrxA. However, within the very conserved segment approaching the re face of the isoalloxacine ring, Tyr47 of RdxA is replaced by an Ile residue in FrxA. The important Cys159 in RdxA (see mutational analysis below) is also present in FrxA (Cys161), similarly positioned close to the FMN N1 atom, so it is possible that it also plays a redox role, which should be mutationally tested. In contrast to cysteine-rich RdxA, where no pair of Cys residues appear appropriately located to form a disulfide bond (see structural analysis below), the sulfur atoms of the only two Cys residues in FrxA: Cys161 and Cys193, are at 5.3 Å, and might form a disulfide bond at some stage of the catalytic cycle. Thus the possible involvement of a disulfide bond in the catalytic cycle of FrxA is worth testing. If found to form, Cys159 in RdxA and Cys 161 in FrxA, although structurally equivalent, would serve a different role in catalysis, which might contribute to the different specificity observed for the two H pylori oxidoreductases. Further differences between the two enzymes that may contribute to tailor their redox potentials include the presence of three aromatic residues close the FMN in RdxA (the putative substrate binding Tyr47, Tyr141 at 4.5 Å from FMN C7M, and Phe72 at 3.8 Å from FMN N3) and the presence of Glu164 at 3.3 Å of FMN C7M in FrxA.
FIGURE 5. Structural comparison of the RdxA and FrxA structures.

A. The RdxA monomer is in blue and the FrxA monomer in gold. The α-helices which are only visible in one of the structures are highlighted in darker colours. B. The two chains o the RdxA dimer are depicted in cyan and green. The two subunits of the biologically relevant FrxA dimer are in red and yellow. The arrows point to the two helices in RdxA not seen in FrxA and to the two helices in FxrA not seen in RdxA
On the other hand, there are more than 30 structures of FMN containing nitroreductases deposited in the PDB (3R6W, 2WQF, 2WZV, 3N2S, 3BM1, 1YKI, 1ZCH, 1KQB, 1F5V, 1YWQ, 2B67, 2I7H, 2R01, 3BEM, 3E10, 3E39, 3EK3, 3EO7, 3EO8, 3GAG, 3GE5, 3GE6, 3GFA, 3GR3, 3H4O, 3HJ9, 3HOI, 3K6H, 3KWK, 3M5K, 3OF4, 3PXV, 4DN2), many of which have been deposited by the Joint Center for Structural Genomics (JCSG)and not further analyzed in an accompanying publication. The superfamily server [27] identifies RdxA as a member of the NADH oxidase/flavin reductase family, of which the server includes 13 members with known 3D structure (1BKJ, 1F5V, 1KQB, 1NEC, 1NOX, 1V5Y, 1VFR, 1YWG, 1ZCH, 2B67, 2FRE, 2IFA, 1DS7). The overall structural fold is relatively well preserved among all these structures, but the primary sequence is not. Based on various insertions and deletions around the FMN binding region, the NADH oxidase/flavin reductase family has been divided into three subclasses [28] According to our crystallographic data, RdxA can now be included into the NOX subclass. Superposition of NOX and RdxA shows that all residues interacting with FMN are conserved except for Cys159, which in NOX corresponds to a valine residue (Figure 6a). Interestingly, this valine is highly conserved among other NADH oxidase/flavin reductase structures (Figure 6b). Thus, RdxA is the first protein of this family that shows a cysteine close to the FMN.
FIGURE 6. FMN cofactor and surrounding residues in RdxA and several other oxidoreductases.

A, FMN environment of RdxA and NOX. RdxA FMN is shown in sticks with RdxA and FrxA carbons in white and light blue respectively. The overall binding mode of FMN is conserved in these enzymes. RdxA main difference with NOX corresponds to the presence of Cys159. B. Residues at the equivalent position to C159 in representative structures (1NOX, 2BKJ, 3GFD) of the three subclasses of the NADH oxidase/flavin reductase superfamily. C. Structural superimposition of 9 structures homologous to RdxA and displaying an FMN close cysteine residue structurally homologous to Cys159 in RdxA. The alignment was performed using the Dali server.
By using the Dali server we have found more than 200 structures that exhibit a rsmd below 3.0Å with RdxA. After aligning the first 200 structures we found that only 9 (2HAY, 3OF4, 2WZV, 3N2S, 2R01, 3BEM, 3EK3, 3EO8, 3GR3), present a Cys residue structurally homologous to Cys159 (Figure 7c). Seven of these structures have been solved by JCSG with no publication associated. The only two publications related to these Cys residues containing nitroreductases, NfrA1from Bacilus subtilis (PDB: 3N2S) [29] and NfnB from Mycobacterium smegmatis (PDB: 2WZV) [30] do not attribute a role to the cysteine at the FMN binding site, perhaps because it is not conserved in most FMN binding enzymes. Based on these findings we propose that RdxA could be included as the representative member of a new subgroup of oxidoreductases that might use the cysteine close to the FMN cofactor to perform its reductive activity. In order to test this hypothesis, it will be interesting to perform activity experiments with the cysteine mutants of the nitroreductases that contain this cysteine. We have begun this task by mutating Cys159, present in RdxA, and analyzing the consequences in the activity of the enzyme (see below).
FIGURE 7. Relative positions of the FMN and six cysteine residues in the RdxA monomer.
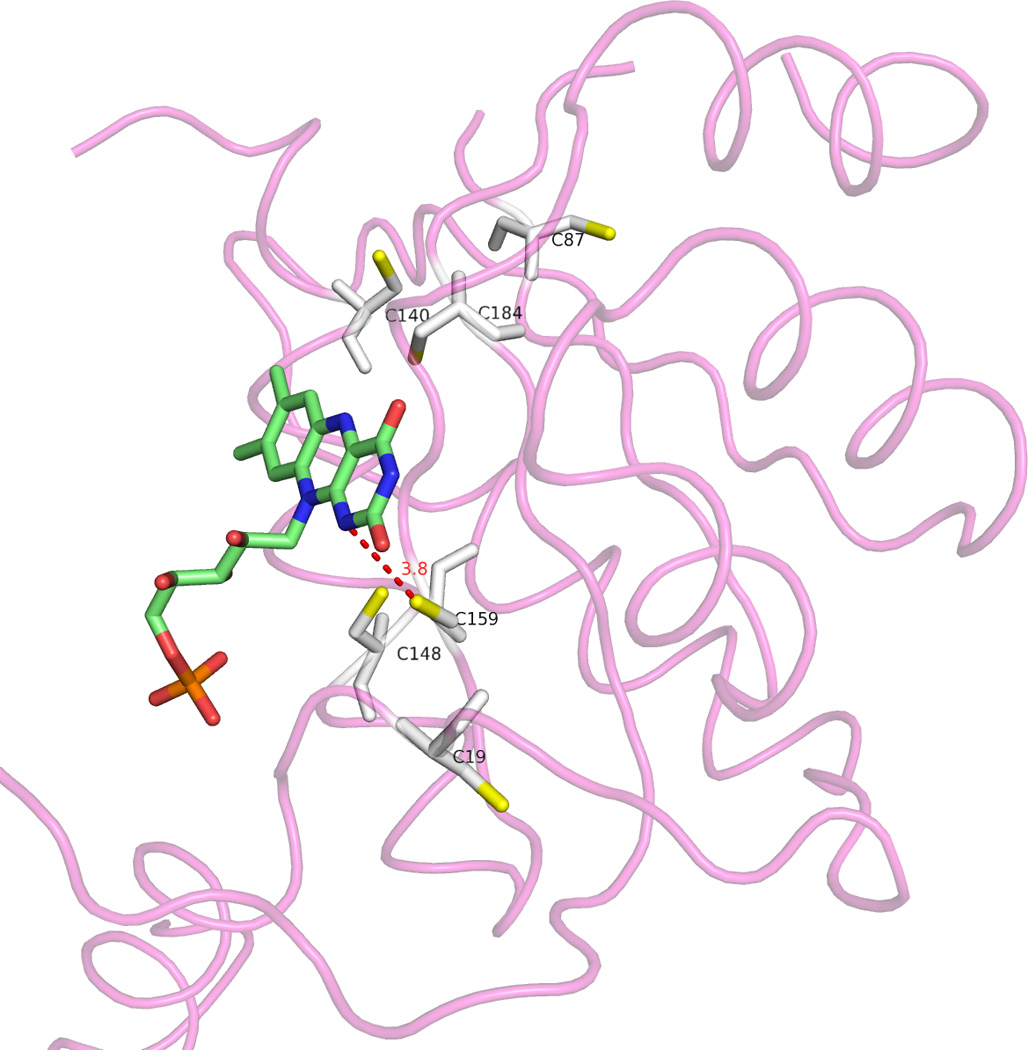
We show the complete chain D in tube representation and the cysteine residues (S atoms in yellow) and FMN cofactor in sticks representation. We also depict the short distance (red dotted line and number) between the SG atom of Cys 159 and the N1 atom of the isoalloxazine ring.
Structural and mutational insight into a possible role of cysteine residues in metronidazole reduction
RdxA contains 6 cysteine residues per subunit (Figure 7), while other NTRs average about 1 cysteine (sequences shown in Figure 2). There is a possibility that the reported distinct redox behavior of RdxA (efficient reduction of low potential metronidazole plus high NADPH oxidase activity) is related to some of those cysteines. Inspection of Figure 2 indicates that none of the six cysteines is conserved among nitroreductases from other bacteria. Two of them, Cys87 and Cys159, appear in three of the other NTRs, but never simultaneously. Since sequence comparison of structurally homologous NTRs offers no information on the potential significance of the RdxA cysteines, we have aligned the sequences of four RdxAs belonging to H. pylori and related species (Figure 8) to see which ones are conserved. In addition, we included the FrxA nitroreductase, which is more closely related to NfsB, but has been implicated in MTZ susceptibility and resistance [25]. Of the six cysteine residues in H. pylori RdxA, Cys 19, 140, 159 and 184 are conserved in the other Helicobacter species, and two of them, Cys 184 and 159 appear close to the isoalloxazine ring in the H. pylori RdxA structure ( Figure 7). However, only Cys159 points to the ring, its S atom being close to the N1 FMN atom (at 3.6 Å in chain A). The proximity of RdxA Cys159 to FMN raises the possibility that this residue plays a role in metronidazole reduction. In this respect we propose that, since the N1 atom of FMN can become protonated in its 2e− reduced hydroquinone form, Cys159 might donate a proton to N1 to facilitate the formation of the neutral hydroquinone. If that were the case, the resulting thiolate ion could destabilize the hydroquinone thus decreasing the FMN redox potential, which seems required for RdxA to reduce metronidazole. A similar key role in providing a proton to the N5 atom of FMN has been proposed for the single cysteine residue of the light receptor phototropin [31], although in that case the thiolate reacts to form a covalent adduct with FMN.
FIGURE 8. Sequence alignment of RdxA belonging to H. pylori and related species.

Alignments were assembled using ClustalW of the published protein sequences for RdxA proteins. H. bizzozeronii, H. felis, H. pylori strain 26695, H. acinonychis and FrxA of H. pylori 26695. The conservation of cysteine residues in all RdxA proteins from H. pylori strains available is identical to those of the HP26695 strain. The cysteine residues are bolded and the conserved cysteines are numbered.
To test our proposal, we changed Cys159 to either alanine or serine. The mutations were confirmed by DNA sequencing and the purified proteins were analyzed for enzymatic activity. As seen in Table 2, RdxA C159S and RdxA C159A both exhibited increased NADPH oxidase activity (40 and 70%, respectively) when compared with WT RdxA. This higher NADPH oxidase activity might reflect a greater access of molecular oxygen to the active site in the mutants. On the other hand, there were no differences in nitroreductase activity with either nitrofurazone or the prodrug CB1954 as substrates under aerobic conditions. However, under strictly anaerobic conditions required for MTZ reductase activity, the RdxAC159S and the RdxAC159A mutants showed 62 and 72% decreased MTZ reductase activity, respectively (see Table 2) compared with WT enzyme activity. It is clear that Ala residues cannot donate protons and Ser resides are very unlikely to do so except in extreme pH conditions or very special protein environments. The diminished MTZ reductase activity displayed by the Ser and Ala mutants, compared to Cys containing WT is in agreement with our proposed mechanism. We would like to note that the six cysteines present in each RdxA monomer are clustered on a plane roughly parallel to the FMN si face (Figure 7). No disulfide bridge is formed in the structure. The two closest cysteine residues are Cys19 and Cys159, with their sulfur atoms at 7.1–7.5 Å. This large distance, together with the results of our mutational analysis on Cys159 make unlikely that a disulfide bond between those two residues participate in RdxA catalysis. Finally, in light of the different MTZ activity of RdxA and NfsB nitroreductase, future studies might explore the addition to NfsB nitroreductase of a cysteine equivalent to Cys159 to evaluate the thiol effect on redox potential and gain of MTZ reductase activity.
Table 2.
Effect of mutation of residue Cys159 on the RdxA NADPH oxidase and nitroreductase activities
| Electron acceptor | Conditionsa | Activityb |
||
|---|---|---|---|---|
| RdxA WT | C159S | C159A | ||
| Oxygen | aerobic | 2.8 +/− 0.1 | 3.8 +/− 0.1 | 4.7 +/− 0.1 |
| Nitrofurazone | aerobic | 7.8 +/− 0.2 | 8.9 +/− 0.3 | 7.9 +/− 0.2 |
| CB1954 | aerobic | 2.6 +/− 0.2 | 2.1 +/− 0.1 | 2.1 +/− 0.1 |
| Metronidazole | anaerobic | 0.21 +/− 0.02 | 0.08 +/− 0.01 | 0.06 +/− 0.01 |
Conditions: 23°C; 10 mM Tris/HCl pH 7.5; 150 µM NADPH for aerobic assays and 30 µM for anaerobic assay with MTZ. The activities presented are averages of three determinations per substrate. For substrates other than oxygen, the reported activities are corrected for NADPH oxidase activity.
Activity given as: µM substrate s−1 µM protein−1
EXPERIMENTAL PROCEDURES
Protein purification and enzyme assays
Purification of the native and mutant RdxA proteins and enzyme assays were performed as described previously (7). Specific activity measurements were performed in 1-cm-path-length quartz cuvettes in buffer A [10 mM Tris-HCl pH 7.5 containing NAD(P)H (150 µM) and appropriate substrate (50–150 µM)] as previous described [7]. The reduction of MTZ was determined under anaerobic conditions by either allowing the NADPH-oxidase activity of RdxA to render the contents of the cuvette anaerobic or by employing a glucose oxidase/catalase system as previously described [7]. Specific activities are presented as µM substrate s−1 µM protein−1.
Site-directed mutagenesis
The RdxA C159A and C159S mutants were generated by PCR using the following primers: RdxA C159A: 5’-TGGGATTGGATAGTGCCATTATTGGAGGCT;
RdxAC159S 5’-TGGGATTGGATAGTAGCATTATTGGAGGCT;
5RdxA_NdeI 5'-GGGAATTCCATATGGAATTTTTGGATCAAG;
3RdxA_BamHI 5'-CGCGGATCCTCACAACCAAGTAATCGCATC. Mutated rdxA genes were cloned into NdeI – BamHI sites of the pET29B vector (Novagen). All DNA inserts were verified by automated DNA sequencing at the Genewiz, Inc DNA Sequencing Service (www.genewiz.com).
Crystallization and Data Collection
Crystals of H. pylori RdxA were grown by the sitting drop method at 21°C. The reservoir solution contained 25% PEG 3350, 0.1 M Bis-Tris pH 6.5 and 0.2 M magnesium chloride. The drops were setup with 1 µl of this solution and 1 µl of protein dissolved in 20 mM Hepes, pH 7.5 at 9 mg/ml. After 3 days at 20°C, square shaped crystals appeared belonging to the P41 tetragonal space group with unit cell parameters of: a = 49.4 Å, b = 49.4 Å, c = 304.5 Å. The crystals had a twin fraction of 0.458, therefore, they apparently belonged to a higher symmetry space group P41212. Matthews’s coefficient [32] calculations for the P41212 space group suggested the presence of a dimer of dimers (Figure 1A) in the asymmetric unit with Vm=1.94 Å3 Da−1 and a solvent content of approximately 37%. The crystals were cryoprotected by soaking in the mother liquor supplied with 20% glycerol and then flash-cooled under the nitrogen stream at 100 K. Two data sets at 2.0 Å and 3.4 Å resolution were collected at ESRF beam lines ID23-2 and BM-14, respectively. Images were collected with 0.1 degrees of rotation given the length of the c axis of the unit cell. Data reduction was carried with MOSFLM [33] and SCALA [34] programs (Table 1).
Table 1.
Data collection and structure refinement parameters
| P41 (High-resolution) | P41 (Low-resolution) | |
|---|---|---|
| Data collection | ||
| Wavelength | 0.8726 | 0.97625 |
| Images | 1300 | 1800 |
| Unit cell parameters | a = 49.44 Å b = 49.44 Å c = 304.48 Å |
a = 49.25 Å b = 49.25 Å c = 304.07 Å |
| Resolution | 41.46–2.0 (2.11–2.10) | 60.81–3.43 (3.62–3.43) |
| Rmerge | 0.145 (0.320) | 0.077 (0.10) |
| Completeness | 99.7 (100) | 100 (100) |
| Multiplicity | 5.5 (5.3) | 7.6 (7.2) |
| I/σ | 8.1 (4.3) | 20.4 (17.1) |
| Alpha_twin | 0.458 | 0.48 |
| No.Reflections (observed) | 266704 | |
| No.Reflections (unique) | 48807 | |
| Structure refinement | ||
| R-factor (%) | 18.8 | |
| R-free (%) | 22.0 | |
| No Reflections (R-Free) | 2511 | |
| No. water molecules | 170 | |
| No. glycerol molecules | 2 | |
| Rmsd values | ||
| Bond lengths (A°) | 0.006 | |
| Bond angles (deg.) | 0.917 |
Phases calculation and model building
The low-resolution dataset processed in P422 space group and the RdxA sequence of H. pylori were submitted to BALBES [35] server for molecular replacement (MR) in the 8 possible space groups. Using RdxA sequence, BALBES found 21 structures above 15% identity but only the top six were used to generate 15 models for the MR search. The best solution was found in P41212 using a Escherichia coli nitroreductase domain (PDB CODE: 1ICR) as a template. The selection of this domain, in which 2 helices were deleted, turned out to be critical since crystal formation is driven by a proteolyzed form of RdxA, evidenced only after solving the structure. The solution found with BALBES was used as a search-model for MR with PHASER using the high-resolution data set processed in P41212. Then, after several cycles of refinement with REFMAC 36] combined with automatic building using BUCCANER [37], the RdxA model resulted in a Rfact/Rfree of 0.33/0.38. Next, the model was manually completed with COOT [38] and refined in REFMAC [36] with an Rfact/Rfree of 0.26/0.33 respectively. The model was used later to perform MR with PHASER using the high-resolution dataset processed in P41. Finally, after applying the twin operator during refinement in REFMAC [36] the Rfact and Rfree reached acceptable values of 0.18 and 0.22 respectively (Table 1). The final model has been deposited in the Protein Data Bank (PDB code: 3QDL).
Mass spectrometry analyses
Drops containing the crystals were dissolved (4 µL crystal sample plus 2 µL SA [Sinapinic acid, 70:30 ACN:TFA 0.1 %] matrix elution), over a ground Steel Massive 384 target plate (Bruker). The sample was then desalted using the ZipTip C4 desalting method. As a control, an RdxA sample containing NADP+ was acidified adding 1% TFA (trifluoroacetic acid) and protein was concentrated and desalted by passing it through ZipTip C18 columns (Millipore) following the manufacturer’s instructions. Protein was eluted with 50% ACN/0,1% TFA/H2O followed by 70% ACN/0,1% TFA/H2O. MALDI-TOF MS was performed using a 4800plus MALDI-TOFTOF (Applied Biosystems) in the linear mode with accelerating voltage of 20 kV, mass range of 6000 to 80000 Da, 1000 shots/spectrum and laser intensity of 4800. Spectra were calibrated externally using a standard protein mixture (ProteoMass Protein MALDI-MS Calibration Kit MSCAL3, Sigma)
ACKNOWLEDGMENTS
This work was supported by NIH grant R01DK073823 to P.S.H and grants BFU2010-16297 (Ministerio de Ciencia e Innovación: MICINN, Spain) and Grupo Protein Targets B89 (Diputación General de Aragón, Spain) to JS. VEA was funded by Banco Santander Central Hispano, Fundación Carolina and Universidad de Zaragoza and is now recipient of a doctoral fellowship awarded by Consejo Superior de Investigaciones Científicas, JAE program. We thank the staff of the European Synchrotron Radiation Facility (Grenoble, France) for assistance with X-ray data collection under the Block Allocation Group MX1130/MX1282.
Abbreviations
- NTR
nitroreductase
- IPTG
isopropyl-β-D-thiogalactopyranoside
- PEG
Polyethylene glycol
- Bis-Tris
Bis(2-hydroxyethyl)amino-tris(hydroxymethyl)methane
- ACN
acetonitrile
- TFA
trifluoroacetic acid
- CB 1954
5-(aziridin-1-yl)-2,4-dinitrobenzamide
- MTZ
2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethanol
Footnotes
Database: Structural data have been deposited in the Protein Data Bank under accession number 3QDL
REFERENCES
- 1.Bryant C, DeLuca M. Purification and characterization of an oxygen-insensitive NAD(P)H nitroreductase from Enterobacter cloacae. J Biol Chem. 1991;266:4119–4125. [PubMed] [Google Scholar]
- 2.Race PR, Lovering AL, White SA, Grove JI, Searle PF, Wrighton CW, Hyde EI. Kinetic and structural characterisation of Escherichia coli nitroreductase mutants showing improved efficacy for the prodrug substrate CB1954. J Mol Biol. 2007;368:481–492. doi: 10.1016/j.jmb.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 3.Koder RL, Haynes CA, Rodgers ME, Rodgers DW, Miller AF. Flavin thermodynamics explain the oxygen insensitivity of enteric nitroreductases. Biochemistry. 2002;41:14197–14205. doi: 10.1021/bi025805t. [DOI] [PubMed] [Google Scholar]
- 4.Nivinskas H, Koder RL, Anusevicius Z, Sarlauskas J, Miller AF, Cenas N. Two-electron reduction of nitroaromatic compounds by Enterobacter cloacae NAD(P)H nitroreductase: description of quantitative structure-activity relationships. Acta Biochim Pol. 2000;47:941–949. [PubMed] [Google Scholar]
- 5.Goodwin A, Kersulyte D, Sisson G, Veldhuyzen van Zanten SJ, Berg DE, Hoffman PS. Metronidazole resistance in Helicobacter pylori is due to null mutations in a gene (rdxA) that encodes an oxygen-insensitive NADPH nitroreductase. Mol Microbiol. 1998;28:383–393. doi: 10.1046/j.1365-2958.1998.00806.x. [DOI] [PubMed] [Google Scholar]
- 6.Sisson G, et al. Metronidazole activation is mutagenic and causes DNA fragmentation in Helicobacter pylori and in Escherichia coli containing a cloned H pylori RdxA(+) (Nitroreductase) gene. J Bacteriol. 2000;182:5091–5096. doi: 10.1128/jb.182.18.5091-5096.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olekhnovich IN, Goodwin A, Hoffman PS. Characterization of the NAD(P)H oxidase and metronidazole reductase activities of the RdxA nitroreductase of Helicobacter pylori. Febs J. 2009;276:3354–3364. doi: 10.1111/j.1742-4658.2009.07060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carroll CC, Warnakulasuriyarachchi D, Nokhbeh MR, Lambert IB. Salmonella typhimurium mutagenicity tester strains that overexpress oxygen-insensitive nitroreductases nfsA and nfsB. Mutat Res. 2002;501:79–98. doi: 10.1016/s0027-5107(02)00018-0. [DOI] [PubMed] [Google Scholar]
- 9.Morash MG, Brassinga AK, Warthan M, Gourabathini P, Garduño RA, Goodman SD, Hoffman PS. Reciprocal expression of integration host factor and HU in the developmental cycle and infectivity of Legionella pneumophila. Appl Environ Microbiol. 2009:1826–1837. doi: 10.1128/AEM.02756-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LeBlanc JJ, Brassinga AK, Ewann F, Davidson RJ, Hoffman PS. An ortholog of OxyR in Legionella pneumophila is expressed postexponentially and negatively regulates the alkyl hydroperoxide reductase (ahpC2D) operon. J Bacteriol. 2008;190:3444–3455. doi: 10.1128/JB.00141-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel P, Young JG, Mautner V, Ashdown D, Bonney S, Pineda RG, Collins SI, Searle PF, Hull D, Peers E, Chester J, Wallace DM, Doherty A, Leung H, Young LS, James ND. A phase I/II clinical trial in localized prostate cancer of an adenovirus expressing nitroreductase with CB1954. Mol Ther. 2009;17:1292–1299. doi: 10.1038/mt.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nivinskas H, Koder RL, Anusevicius Z, Sarlauskas J, Miller AF, Cenas N. Quantitative structure-activity relationships in two-electron reduction of nitroaromatic compounds by Enterobacter cloacae NAD(P)H:nitroreductase. Arch Biochem Biophys. 2001;385:170–178. doi: 10.1006/abbi.2000.2127. [DOI] [PubMed] [Google Scholar]
- 13.Yarlett N, Yarlett NC, Lloyd D. Ferredoxin-dependent reduction of nitroimidazole derivatives in drug-resistant and susceptible strains of trichomonas vaginalis. Biochemical Pharmacology. 1986;35:1703–1708. doi: 10.1016/0006-2952(86)90327-8. [DOI] [PubMed] [Google Scholar]
- 14.Ghisla S, Massey V. Mechanisms of flavoprotein-catalyzed reactions. Eur J Biochem. 1989;181:1–17. doi: 10.1111/j.1432-1033.1989.tb14688.x. [DOI] [PubMed] [Google Scholar]
- 15.Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biterova EI, Turanov AA, Gladyshev VN, Barycki JJ. Crystal structures of oxidized and reduced mitochondrial thioredoxin reductase provide molecular details of the reaction mechanism. Proc Natl Acad Sci U S A. 2005;102:15018–15023. doi: 10.1073/pnas.0504218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roldan MD, Perez-Reinado E, Castillo F, Moreno-Vivian C. Reduction of polynitroaromatic compounds: the bacterial nitroreductases. FEMS Microbiol Rev. 2008;32:474–500. doi: 10.1111/j.1574-6976.2008.00107.x. [DOI] [PubMed] [Google Scholar]
- 18.Haynes CA, Koder RL, Miller AF, Rodgers DW. Structures of nitroreductase in three states: effects of inhibitor binding and reduction. J Biol Chem. 2002;277:11513–11520. doi: 10.1074/jbc.M111334200. [DOI] [PubMed] [Google Scholar]
- 19.Parkinson GN, Skelly JV, Neidle S. Crystal structure of FMN-dependent nitroreductase from Escherichia coli B: a prodrug-activating enzyme. J Med Chem. 2000;43:3624–3631. doi: 10.1021/jm000159m. [DOI] [PubMed] [Google Scholar]
- 20.Lovering AL, Hyde EI, Searle PF, White SA. The structure of Escherichia coli nitroreductase complexed with nicotinic acid: three crystal forms at 1.7 A, 1.8 A and 2.4 A resolution. J Mol Biol. 2001;309:203–213. doi: 10.1006/jmbi.2001.4653. [DOI] [PubMed] [Google Scholar]
- 21.Angarica VE, Sancho J. Protein dynamics governed by interfaces of high polarity and low packing density. PLoS One. 2012 doi: 10.1371/journal.pone.0048212. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mendz GL, Megraud F. Is the molecular basis of metronidazole resistance in microaerophilic organisms understood? Trends Microbiol. 2002;10:370–375. doi: 10.1016/s0966-842x(02)02405-8. [DOI] [PubMed] [Google Scholar]
- 23.Lostao A, El Harrous M, Daoudi F, Romero A, Parody-Morreale A, Sancho J. Dissecting the energetics of the apoflavodoxin-FMN complex. J Biol Chem. 2000;275:9518–9526. doi: 10.1074/jbc.275.13.9518. [DOI] [PubMed] [Google Scholar]
- 24.Sancho J, Serrano L, Fersht AR. Histidine residues at the N- and C-termini of alpha-helices: perturbed pKas and protein stability. Biochemistry. 1992;31:2253–2258. doi: 10.1021/bi00123a006. [DOI] [PubMed] [Google Scholar]
- 25.Jeong JY, Mukhopadhyay AK, Dailidiene D, Wang Y, Velapatino B, Gilman RH, Parkinson AJ, Nair GB, Wong BC, Lam SK, Mistry R, Segal I, Yuan Y, Gao H, Alarcon T, Brea ML, Ito Y, Kersulyte D, Lee HK, Gong Y, Goodwin A, Hoffman PS, Berg DE. Sequential inactivation of rdxA (HP0954) and frxA (HP0642) nitroreductase genes causes moderate and high-level metronidazole resistance in Helicobacter pylori. J. Bacteriol. 2000;182:5082–5090. doi: 10.1128/jb.182.18.5082-5090.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeong JY, Mukhopadhyay AK, Akada JK, Dailidiene D, Hoffman PS, Berg DE. Roles of FrxA and RdxA nitroreductases of Helicobacter pylori in susceptibility and resistance to metronidazole. J Bacteriol. 2001;183:5155–5162. doi: 10.1128/JB.183.17.5155-5162.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gough J, Karplus K, Hughey R, Chothia C. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol. 2001 Nov 2;313(4):903–919. doi: 10.1006/jmbi.2001.5080. [DOI] [PubMed] [Google Scholar]
- 28.Koike H, Sasaki H, Kobori T, Zenno S, Saigo K, Murphy ME, Adman ET, Tanokura M. 1.8 A crystal structure of the major NAD(P)H:FMN oxidoreductase of a bioluminescent bacterium, Vibrio fischeri: overall structure, cofactor and substrate-analog binding, and comparison with related flavoproteins. J. Mol. Biol. 1998;280:259–273. doi: 10.1006/jmbi.1998.1871. [DOI] [PubMed] [Google Scholar]
- 29.Cortial S, et al. NADH oxidase activity of Bacillus subtilis nitroreductase NfrA1: insight into its biological role. FEBS Lett. 2010;584(18):3916–3922. doi: 10.1016/j.febslet.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 30.Manina G, et al. Biological and structural characterization of the Mycobacterium smegmatis nitroreductase NfnB, and its role in benzothiazinone resistance. Molecular Microbiology. 2010;77(5):1172–1185. doi: 10.1111/j.1365-2958.2010.07277.x. [DOI] [PubMed] [Google Scholar]
- 31.Crosson S, Moffat K. Structure of a flavin-binding plant photoreceptor domain: insights into light-mediated signal transduction. Proc Natl Acad Sci U S A. 2001;98:2995–3000. doi: 10.1073/pnas.051520298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 33.Leslie AGW. Joint CCP4 and ESF-EAMCB Newsletter on Protein Crystallography. Daresbury, Warrington, UK: SERC Laboratory; 1992. Recent changes to the MOSFLM package for processing film and image plate data. [Google Scholar]
- 34.Evans PR. Scaling of MAD data. In: Wilson KS, Davies G, Ashton AW, Bailey S, editors. Proceedings of CCP4 Study Weekend on Recent Advances in Phasing. Warrington, UK: CCLRC Daresbury Laboratory; 1997. pp. 97–102. [Google Scholar]
- 35.Long F, Vagin AA, Young P, Murshudov GN. BALBES: a molecular-replacement pipeline. Acta Crystallogr D Biol Crystallogr. 2008;64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 37.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 38.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]