

Abstract
Nuclear architecture defects have been shown to correlate with the manifestation of a number of human diseases as well as aging1-4. It is then plausible that diseases whose manifestations correlate with aging might be connected to the appearance of nuclear aberrations over time. We decided to evaluate nuclear organization in the context of aging-associated disorders by focusing on a Leucine Rich Repeat Kinase 2 (LRRK2) dominant mutation (G2019S) shown to associate with familial and sporadic Parkinson’s Disease (PD), as well as impairment of adult neurogenesis in mice5. Here, we report on the generation of PD patient-derived induced pluripotent stem cells (iPSCs) and the implications of LRRK2(G2019S) in human neural stem cell (NSC) populations. Mutant NSCs showed increased susceptibility to proteasomal stress as well as passage-dependent deficiencies in clonal expansion and neuronal differentiation. Disease phenotypes were rescued by targeted correction of the LRRK2(G2019S) mutation with its wild-type counterpart in PD-iPSCs and recapitulated upon targeted knock-in of LRRK2(G2019S) in human embryonic stem cells (hESCs). Analysis of human brain tissue showed nuclear envelope impairment in clinically diagnosed Parkinson’s patients. Altogether, our results identify the nucleus as a previously unknown cellular organelle in Parkinson’s pathology and may help open new avenues for PD diagnoses as well as potential development of therapeutics targeting this fundamental cell structure.
To study the role that the nuclear envelope might have during aging-related neurodegenerative processes we chose to focus our attention on Parkinson’s disease (PD), the second most prevalent neurodegenerative disease among aging individuals. LRRK2 is a large multidomain protein bearing kinase activity10 whose mutations correlate with inherited and sporadic PD10, 11. Specifically, G2019S mutation leads to an increase in LRRK2 kinase activity correlating with the manifestation of disease3,10-13. However, the molecular and cellular implications of LRRK2(G2019S) during PD manifestation and progression remain elusive.
To this end, we first decide to evaluate potential novel interactions. Flag-tagged version of LRRK2(G2019S) and subsequent MudPIT analysis led to the identification of unreported nuclear components in LRRK2(G2019S)-containing protein complexes (Supplementary Fig. 2a and b and Supplementary Table 1). Furthermore, LRRK2(G2019S) overexpression resulted in deformed nuclei with a pedal-like structure (Supplementary Fig. 2c-d).
The fact that LRRK2(G2019S) leads to impaired adult neurogenesis in mice 5, led us to take advantage of iPSC technologies and their potential for directed differentiation into specific human cell lineages (Supplementary Figs. 3 and 4). Differentiation of iPSCs into neural progenitors followed by serial passaging resembling cellular “aging”14 showed progressive deterioration of nuclear architecture in mutant but not in wild-type NSCs (ipsNSCs-LRRK2(G2019S) and ipsNSCs-wt respectively). Nuclear aberrations started to manifest by passage 14 and progressively resulted in compartmentalized pedal-like nuclei (Fig. 1a-f, Supplementary Figs. 5c, 6 and 7). Staining for Lamin B1 in late-passage differentiated as well as LRRK2(G2019S)-overexpressing wild-type NSCs (e.g. passage 19), demonstrated a markedly enlarged nuclear area, accompanied by a significant decrease in nuclear circularity, in ipsNSCs-LRRK2(G2019S) (Fig.1c-e, Supplementary Fig. 5c and Supplementary Fig. 8a). Noticeably, late passage ipsNSCs-LRRK2(G2019S) exhibited local loss of Lamin B1 and B2 at specific folds of the nuclear envelope (Fig. 1c,f and g, and Supplementary Figs 6e and 7) whereas all other examined nuclear components remained correctly localized (Fig. 1f and Supplementary Figs. 6e and 7). Co-immunoprecipitation experiments revealed that ectopically expressed LRRK2(G2019S) associated in protein complexes with both, Lamin B1 and Lamin B2 (Fig. 1h). Furthermore, increased phosphorylation of Lamin B1 and Lamin B2 was observed in late passage ipsNSC-LRRK2(G2019S) (Supplementary Fig. 8b), suggesting that excessive LRRK2 kinase activation may directly or indirectly stimulate phosphorylation of B-type lamins in NSCs. Lastly, analysis of mitochondrial structure, an organelle frequently implicated in PD pathogenesis10, demonstrated the normal expression of the outer mitochondrial membrane receptor TOM20 as well as the mitochondrial heat shock protein HSP60 in both NSCs-wt and NSCs-LRRK2(G2019S) (Supplementary Fig. 9).
Fig.1. LRRK2(G2019S) mutation results in progressive deterioration of nuclear architecture in ipsNSCs.
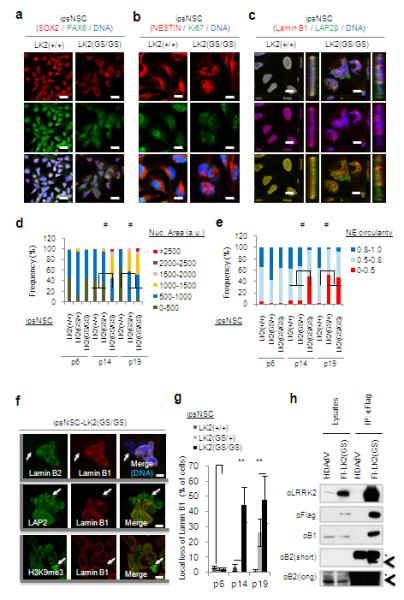
a-b, Immunofluorescence analysis of the indicated neural progenitor markers and proliferation markers Ki67 at passage 14 (p14). Nuclei were stained with Hoechst. Scale bars, 20 μm (a), and 10 μm (b). c, Immunofluorescence of Lamin B1 and LAP2 at passage 14. Nuclei were stained with Hoechst. Projections of y-z axes from representative nuclei are shown on the right of each panel. Scale bars, 10 μm. d-e, Quantifications of nuclear area (d) and nuclear envelope (NE) circularity (e) in differentiated NSCs at passage 6, 14, and 19. a.u., arbitrary units. At least 100 randomly chosen cells were counted. #p<10−15. f, Subcellular distributions of Lamin B1, Lamin B2, LAP2β*** and H3K9me3 were determined at passage 15. Arrows indicate nuclear envelope microdomains deficient for Lamin B types. Scale bars, 5 μm. g, Quantifications of local loss of Lamin B1 in ipsNSCs. Data are shown as mean±s.d. n=3. **p<0.01. h, Extracts from wild-type esNSCs transduced with HDAdV(venus) vector (HDAdV) or HDAdV(venus)-Flag-LRRK2(G2019S)(Fl-LK2(GS)) were immunoprecipitated (IP) with an anti-Flag antibody, followed by immunoblotting analysis for the indicated proteins. Asterisks indicate non-specific bands, and arrowheads indicate Lamin B2.
We next wondered whether LRRK2(G2019S)-elicited nuclear defects contributed to epigenetic alterations such as those observed during cellular aging1, 2, 15. Fluorescence in situ hybridization revealed a dramatic increase of centromeric signals accompanied by reorganization of centromeric, but not telomeric, heterochromatin in passage 15 ipsNSCs-LRRK2(G2019S) (Supplementary Fig. 10a). Genome-wide epigenetic analysis demonstrated passage-dependent differences in global H3K4me3 modifications, an epigenetic mark associated with human neuronal aging16, at the analyzed promoter regions (Fig. 2a and Supplementary Fig. 10b). At passage 15, 437 genes in ipsNSCs-LRRK2(G2019S) displayed ≥5-fold enrichment for the epigenetic mark H3K4me3 as compared to ipsNSCs-wt whereas no significant differences were observed when comparing undifferentiated iPSCs. Interestingly, most of these genes were associated with neurogenesis and neural function (Supplementary Table 2).
Fig. 2. LRRK2(G2019S) mutant ipsNSCs show deficiency in clonal expansion, spontaneous neuronal differentiation, and exhibit enhanced susceptibility to proteasomal stress-induced apoptosis.

a, Pair-wise comparisons of quantile normalized log2-read counts within +/− 2.5 kb of the Transcription Start Site (TSS) or RefSeq genes. b, passage 16 ipsNSCs-LK2(GS/GS) demonstrated significantly lower colony forming capacity. #1 and #2 represent two independent NSC lines. Data are shown as mean±s.d. n=3. **p<0.01. c, Immunofluorescence analysis of the neuronal marker MAP2 in spontaneous differentiation experiments. Arrows indicate deformed nuclei. Percentages of neuronal differentiation efficiency, indicated by the ratio of MAP2 positive cells to total cell nuclei, are shown in corners. See Supplementary Fig. 11a (left panels) for wide field images. Scale bar, 20 μm. d, 14 days after ipsNSC seeding, ipsNSCs-LK2(GS/GS) at passage 16 demonstrated significantly lower neuronal differentiation capability as compared to wild-type counterparts. Data are shown as mean±s.d. n=3. **p<0.01. e. Cell death assays upon MG132 (10 μM, 20 h)-treatment in ipsNSCs-LK2(+/+) and ipsNSCs-LK2(GS/GS) at passage 15. Representative results from three independent experiments are shown.
These results led us to hypothesize that LRRK2(G2019S) might progressively affect other in vitro NSC functions such as clonal expansion and neural differentiation. Newly generated ipsNSCs-wt and ipsNSCs-LRRK2(G2019S) were similarly capable of both clonal expansion and neural differentiation, whereas only ipsNSCs-wt retained clonogenic and differentiation capacity upon extensive passaging (≥14) (Fig. 2b-d and Supplementary Fig. 8c). Strikingly, passage 16-17 ipsNSCs-LRRK2(G2019S) failed to give rise to MAP2+ (or Tuj1+) cells and resulted in aberrant non-neuronal cellular morphologies upon differentiation (Fig. 2c and d and Supplementary Figs. 8c, 11a and b). Injection of proteasome inhibitors into animal brains has been shown to recapitulate PD phenotypes17. Interestingly, treatment of NSCs with the proteasome inhibitor MG132 resulted in increased apoptosis in ipsNSCs-LRRK2(G2019S) as compared to their wild-type counterparts in a passage-independent manner (Fig. 2e and Supplementary Fig. 11c and d).
To further validate that the observed cellular phenotypes were related to NSCs carrying the LRRK2(G2019S) mutation, we evaluated different somatic cell types as well as generated two different unbiased cellular systems, an isogenic control iPSC line by targeted correction of the LRRK2(G2019S) mutation as well as the reversal by specific knock-in of LRRK2(G2019S) into human Embryonic Stem Cells (hESCs)18-21 (Fig. 3a-c and Supplementary Figs. 12 and 13). Contrary to LRRK2(G2019S) NSCs, long-term in vitro culture of differentiated somatic cell populations, other than NSCs, did not result in misshapen nuclei (Supplementary Fig. 15). LRRK2(G2019S)-corrected iPSC differentiation (c-ipsNSCs-LRRK2(G2019S), resulted in the rescue of aberrant cellular phenotypes as compared to the respective uncorrected controls (Fig. 3d-g). Conversely, LRRK2(G2019S) knock-in in ESCs resulted in the progressive appearance of disrupted nuclear architecture, compromised clonal expansion, impaired neural differentiation and increased susceptibility to proteasomal stress upon differentiation into NSCs [esNSC-LRRK2(G2019S)] (Fig. 3h-k and Supplementary Figs. 13 and 14). Altogether, our results suggested a specific role for LRRK2(G2019S) during the manifestation of passage-dependent NSC abnormal phenotypes and highlighted the nucleus as a potential novel organelle affected in PD.
Fig. 3. Phenotypic analyses of isogenic iPSC and ESC lines in the presence or absence of the LRRK2(G2019S) mutation.

a, Schematic representation of HDAdV-based correction of the G2019S mutation in the LRRK2 gene locus. Half arrows indicate the primer sites for PCR (P1, P2, P3 and P4). The probes for Southern analysis are shown as black bars (a, 5′ probe; b, neo probe; c, 3′ probe). Red X indicates the mutation site in exon 41. Blue triangles indicate the FLPo recognition target (FRT) site. P indicates the PflFI sites. b, Sequencing results of the G2019S mutation site in exon 41 of the LRRK2 gene in LRRK2(G2019S) heterozygous mutant iPSCs before (left; LK2(GS/+)) and after (right; C-LK2(GS/+) ) gene correction. c, Southern blot analysis of LRRK2(G2019S) heterozygous mutant iPSCs (LK2(GS/+)) and their gene-corrected counterparts bearing neo cassette (C-LK2(neo/+)). d, Subcellular distributions of Lamin B1 and Lamin B2 in representative ipsNSCs-LK2(GS/+) and ipsNSCs-C-LK2(GS/+) at passage 19. e, cell death assays upon MG132 treatment in corrected and uncorrected ipsNSCs. f-g, Colony formation (f) and neuronal differentiation (g) assay of ipsNSCs at passage 19. h, Immunofluorescence analysis for Lamin B1 and H3K9me3 in esNSCs-H9 and esNSCs-H9-LK2(GS/+) at passage 14. i, cell death assays upon MG132 treatment in knock-in differentiated ESCs. j, Colony formation assay on esNSCs-H9 and esNSCs-H9-LK2(GS/+) at passage 14. k, Spontaneous neuronal differentiation assay of esNSCs-H9 and esNSCs-H9-LK2(GS/+) at passage 14. For e-g and i-k, data are shown as mean±s.d. n=3.** p<0.01. For d and h, scale bars, 10 μm. Nuclei were stained with Hoechst. Arrowheads denote nuclear microdomains deficient for Lamin B1.
Since LRRK2(G2019S) displays enhanced Ser/Thr protein kinase activation that correlates with its pathogenic role in PD10, 11, 22-24, we investigated whether kinase activity correlated with nuclear degeneration (Supplementary Fig. 16). Our results suggested that cell passaging further enhanced LRRK2(G2019S)-mediated phosphorylation of a number of different substrates including 4E-BP1, a known LRRK2 target 24 (Fig. 4a and Supplementary Fig. 16a-b). Interestingly, Ser935 phosphorylation, an indicator for LRRK2 activation12, 22, 25, 26, was increased in passage 15 ipsNSCs-LRRK2(G2019S) relative to total LRRK2 levels (Fig. 4a and Supplementary Fig. 16c).
Fig. 4. Inhibition of LRRK2 kinase activity rescues LRRK2(G2019S)-associated phenotypic defects in NSCs and morphological analysis of nuclear envelope in PD brain slices.

a, Immunoblotting analysis of passage 15 ipsNSCs-LK2(+/+) and ipsNSCs-LK2(GS/GS) Data are shown as mean±s.d. n=3, **p<0.01. b, Immunoblotting analysis of indicated phosphorylated protein substrates in ipsNSCs. Data are shown as mean±s.d. n=3, **p<0.01. c, Nuclear morphology analysis in ipsNSCs-LK2(GS/GS) at passage 15 treated with In-1 for 4 days, compared to untreated controls. Nuclei were stained with Hoechst. Arrows indicate nuclear microdomains deficient for Lamin B1. Scale bars, 5 μm. d-e, Quantification of nuclear area (d) and nuclear envelope (NE) circularity (e) in ipsNSCs at passage 15 treated and untreated with In-1 for 4 days. a.u., arbitrary units. #p<10−15. f, ipsNSCs-LK2(GS/GS) at passage 17 were treated with or without In-1 for 8 days, followed by treatment with MG132 for 20 h. Data are shown as mean±s.d. n=3. **p<0.01. g, Hierarchical clustering of gene expression profiles in esNSCs-H9, esNSCs-H9-LK2(GS/+) and esNSCs-H9-LK2(GS/+) treated with 3 μM In-1 for 5 days at passage 14. h, Immunofluorescence analysis in PD patients bearing the LRRK2(G2019S) mutation (Right panel) and age-matched healthy individuals (Left panel). Data are shown as mean±s.e.m. n=5. Scale bars, 25 μm.
Chemical inhibition has been previously shown to protect against LRRK2 associated toxicity in vitro and in vivo13, 22. Accordingly, treating late-passage ipsNSCs-LRRK2(G2019S) with LRRK2-In-112, 22 resulted in reduced phosphorylation of LRRK2 downstream targets and significantly rescued the aberrant cellular parameters observed in late-passage ipsNSCs-LRRK2(G2019S), (Fig. 4b-f, Supplementary Fig. 6a-c, and Supplementary Movie 1). Microarray analysis demonstrated that 5-day incubation of passage 14 esNSCs-LRRK2(G2019S) with LRRK2-In-1 was sufficient to restore a gene expression signature similar to that of wild-type esNSCs and significantly different from untreated controls (Fig. 4g; Supplementary Table 3). Further studies on cross-talking signaling pathways suggested that LRRK2-In-1 was capable of restoring both nuclear morphology and colony-like growth in late passage ipsNSCs-LRRK2(G2019S), whereas, LY294002, a PI3K inhibitor, partially rescued nuclear morphology (Supplementary Fig. 16c and 17).
Lastly, we investigated nuclear morphology in post-mortem human brain samples. Lamin B1 immunostaining demonstrated a high proportion of cells displaying altered nuclear morphologies within the hippocampal dentate gyrus, but not in non-neurogenic areas such as cortex regions, in all the analyzed LRRK2(G2019S)-PD patients’ samples as compared to their respective age-matched controls (78.82%±6.28 and 16.49%±4.26 respectively) (Fig. 4h, Supplementary Table 5 and Supplementary Fig. 18). Interestingly, nuclear aberrations were not only present in LRRK2(G2019S) but also in several idiopathic PD human brain samples (47.28%±4.217). Of note, control samples also displayed nuclear disruption to a certain extent, which might be potentially due to the described association of physiological aging and the nuclear envelope27 (Fig. 4h and Supplementary Fig. 18). Whereas due to the reduced number of samples (n=5) we cannot conclude that the above mentioned phenotypes have a 100% penetrance in all PD patients, these results indicate the possibility that dysfunctional NSC pools, and/or their respective neuronal derivatives at early stages of maturation, might contribute to the hippocampal and subventricular zone-related age-dependent non-motor symptoms, including depression, anxiety and hyposmia, associated with LRRK2(G2019S)-bearing individuals and PD5, 29.
Altogether, LRRK2(G2019S)-elicited aberrant phenotypic manifestations seemed particularly prominent in human NSC populations and were more noticeable at late passages. Of most relevance, our studies further indicate the suitability of iPSC-based models of disease and aging not only for the in vitro recapitulation of known disease phenotypes, as extensively described, but also as a platform for discovery and study of otherwise novel and elusive cell parameters and populations30 (Supplementary Fig. 1). Furthermore, through the analysis of human brain samples, our study demonstrates that these cellular aberrations can also be found in vivo. The fact that this validation was only possible in human samples once a reliable iPSC in vitro system was employed further highlights the potential of patient-specific iPSCs, not only for disease modeling but, most importantly, for the advancement of disease pathology knowledge.
Method summary
iPSC generation
Parkinson’s disease fibroblasts were obtained from Telethon Genetic Biobank Network and brain patient samples from the Biobank of hospital clinic (IDIBAPS) in Barcelona. Normal fibroblasts were purchased from Coriell Cell Repository. Fibroblasts were reprogrammed with retroviruses expressing OCT4, SOX2, KLF4. The generated iPSC lines were maintained in hESC medium on iMEF feeder cells or in mTeSR on Matrigel 1, 18.
Targeted gene correction in iPSCs-LRRK2(G2019S) and generation of isogenic hESC line with a LRRK2(G2019S) mutation
Gene targeting in iPSCs-LRRK2(G2019S) and H9 ESCs were performed using an HDAdV-based method 18-20.
Statistical analysis
Results are presented as mean±s.d. or mean±s.e.m. for at least three independent biological replicates. Comparisons were performed with student’s t-test. Distributions of nuclear area and nuclear envelope (NE) circularity were analyzed with the Kolmogorov-Smirnov test.
Supplementary Material
Acknowledgements
We would like to thank K. Mitani, P. Ng, A. Lieber, Y. Imai, M. A. Miyawaki, Filocamo, S. Goldwurm, Telethon Genetic Biobank Network and Neurological Tissue Bank of the Biobank-Hospital Clínic-IDIBAPS for kindly providing human brain tissue, F. Gage, M. Hetzer, J. Yao, Y. Mu, D. Yu, E. Gelpí, G. Bai, and Z.J. Liu for helpful discussions, M. Joens and J. Fitzpatrick of the Waitt Advanced Biophotonics Core Facility for performing TEM analysis, M. Marti and C. Gomez for teratoma and karyotyping analysis, F. Osakada for statistics analysis, and M. Schwarz, P. Schwarz, and L. Laricchia-Robbio for administrative help. G.H.L. was supported by “Thousand Young Talents” program of China, National Laboratory of Biomacromolecules, “Strategic Priority Research Program” of the Chinese Academy of Sciences. J.Q. was partially supported by an AFAR/Ellison Medical Foundation postdoctoral fellowship. K.S. was partially supported by a Uehara Memorial Foundation research fellowship. E.N. was partially supported by an F.M. Kirby Foundation postdoctoral fellowship. B.R. was supported by NIH (ES017166) and LICR. J.Y. was supported by a NIH grant (P41 RR011823). J.C.I.B. was supported by grants from the Glenn Foundation, G. Harold and Leila Y. Mathers Charitable Foundation, Sanofi, The California Institute of Regenerative Medicine, Ellison Medical Foundation, Helmsley Charitable Trust, MINECO, and Fundacion Cellex.
Footnotes
Author’s contribution GHL, JQ, KS prepared the figures, designed and performed all in vitro experiments. EN and NM designed and performed in vivo experiments. AG, JK, RPS, XX, WZ, YL and CRE provided technical assistance. ID performed teratoma studies. FY and ML performed mRNA analysis. SR performed and provided cell cycle analysis expertise. BR, UW and AK performed and analyzed epigenetic studies. JT and JYIII provided reagents and designed aging studies. GHL, JQ, KS, EN, ISM and JCIB wrote the manuscript. Reprints, permissions, correspondence and requests for materials should be addressed to the attention of GHL and JCIB.
References
- 1.Liu GH, et al. Recapitulation of premature ageing with iPSCs from Hutchinson-Gilford progeria syndrome. Nature. 2011;472:221–225. doi: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dechat T, et al. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22:832–853. doi: 10.1101/gad.1652708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kudlow BA, Kennedy BK, Monnat RJ., Jr. Werner and Hutchinson-Gilford progeria syndromes: mechanistic basis of human progeroid diseases. Nat Rev Mol Cell Biol. 2007;8:394–404. doi: 10.1038/nrm2161. [DOI] [PubMed] [Google Scholar]
- 4.Worman HJ, Ostlund C, Wang Y. Diseases of the nuclear envelope. Cold Spring Harb Perspect Biol. 2010;2:a000760. doi: 10.1101/cshperspect.a000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winner B, et al. Adult neurogenesis and neurite outgrowth are impaired in LRRK2 G2019S mice. Neurobiol Dis. 2011;41:706–716. doi: 10.1016/j.nbd.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang KH, et al. Nuclear envelope dispersion triggered by deregulated Cdk5 precedes neuronal death. Mol Biol Cell. 2011;22:1452–1462. doi: 10.1091/mbc.E10-07-0654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tran D, Chalhoub A, Schooley A, Zhang W, Ngsee JK. Amyotrophic Lateral Sclerosis Mutant VAPB Causes a Nuclear Envelope Defect. J Cell Sci. 2012 doi: 10.1242/jcs.102111. [DOI] [PubMed] [Google Scholar]
- 8.Padiath QS, et al. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet. 2006;38:1114–1123. doi: 10.1038/ng1872. [DOI] [PubMed] [Google Scholar]
- 9.Woulfe JM. Abnormalities of the nucleus and nuclear inclusions in neurodegenerative disease: a work in progress. Neuropathol Appl Neurobiol. 2007;33:2–42. doi: 10.1111/j.1365-2990.2006.00819.x. [DOI] [PubMed] [Google Scholar]
- 10.Cookson MR. The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat Rev Neurosci. 2010;11:791–797. doi: 10.1038/nrn2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cookson MR, Bandmann O. Parkinson’s disease: insights from pathways. Hum Mol Genet. 2010;19:R21–27. doi: 10.1093/hmg/ddq167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng X, et al. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat Chem Biol. 2011;7:203–205. doi: 10.1038/nchembio.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee BD, et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat Med. 2010;16:998–1000. doi: 10.1038/nm.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li W, et al. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc Natl Acad Sci U S A. 2011;108:8299–8304. doi: 10.1073/pnas.1014041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krishnan V, et al. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc Natl Acad Sci U S A. 2011;108:12325–12330. doi: 10.1073/pnas.1102789108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheung I, et al. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc Natl Acad Sci U S A. 2010;107:8824–8829. doi: 10.1073/pnas.1001702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie W, et al. Proteasome inhibition modeling nigral neuron degeneration in Parkinson’s disease. J Neurochem. 2010;115:188–199. doi: 10.1111/j.1471-4159.2010.06914.x. [DOI] [PubMed] [Google Scholar]
- 18.Liu GH, et al. Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell. 2011;8:688–694. doi: 10.1016/j.stem.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suzuki K, et al. Highly efficient transient gene expression and gene targeting in primate embryonic stem cells with helper-dependent adenoviral vectors. Proc Natl Acad Sci U S A. 2008;105:13781–13786. doi: 10.1073/pnas.0806976105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li M, et al. Efficient correction of hemoglobinopathy-causing mutations by homologous recombination in integration-free patient iPSCs. Cell Res. 2011 doi: 10.1038/cr.2011.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aizawa E, et al. Efficient and accurate homologous recombination in hESCs and hiPSCs using helper-dependent adenoviral vectors. Mol Ther. 2012;20:424–431. doi: 10.1038/mt.2011.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudenko IN, Chia R, Cookson MR. Is inhibition of kinase activity the only therapeutic strategy for LRRK2-associated Parkinson’s disease? BMC Med. 2012;10:20. doi: 10.1186/1741-7015-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanao T, et al. Activation of FoxO by LRRK2 induces expression of proapoptotic proteins and alters survival of postmitotic dopaminergic neuron in Drosophila. Hum Mol Genet. 2010;19:3747–3758. doi: 10.1093/hmg/ddq289. [DOI] [PubMed] [Google Scholar]
- 24.Gehrke S, Imai Y, Sokol N, Lu B. Pathogenic LRRK2 negatively regulates microRNA-mediated translational repression. Nature. 2010;466:637–641. doi: 10.1038/nature09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nichols RJ, et al. 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem J. 2010;430:393–404. doi: 10.1042/BJ20100483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dzamko N, et al. Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem J. 2010;430:405–413. doi: 10.1042/BJ20100784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–1063. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coffinier C, et al. Deficiencies in lamin B1 and lamin B2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Mol Biol Cell. 2011;22:4683–4693. doi: 10.1091/mbc.E11-06-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poulopoulos M, et al. Clinical and Pathological Characteristics of LRRK2 G2019S Patients with PD. J Mol Neurosci. 2012;47:139–143. doi: 10.1007/s12031-011-9696-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tiscornia G, Vivas EL, Belmonte JC. Diseases in a dish: modeling human genetic disorders using induced pluripotent cells. Nat Med. 2011;17:1570–1576. doi: 10.1038/nm.2504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.