

Recent global studies revealed that mRNA alternative polyadenylation (APA) is widespread in eukaryotic transcriptomes and is an important mechanism for gene regulation. APA deregulation has been found in a variety of diseases. This article reviews recent progress in the field and provides a global perspective on the prevalence, mechanisms, and functional impact of APA.
Keywords: alternative polyadenylation, genomics, pre-mRNA processing
Abstract
Recent studies have revealed widespread mRNA alternative polyadenylation (APA) in eukaryotes and its dynamic spatial and temporal regulation. APA not only generates proteomic and functional diversity, but also plays important roles in regulating gene expression. Global deregulation of APA has been demonstrated in a variety of human diseases. Recent exciting advances in the field have been made possible in a large part by high throughput analyses using newly developed experimental tools. Here I review the recent progress in global studies of APA and the insights that have emerged from these and other studies that use more conventional methods.
INTRODUCTION
3′ end processing is an essential step of eukaryotic mRNA maturation, which typically involves a cotranscriptional endonucleolytic cleavage and the subsequent addition of a string of adenosines (Colgan and Manley 1997; Zhao et al. 1999; Danckwardt et al. 2008; Millevoi and Vagner 2009; Chan et al. 2011). The poly(A) site (PAS) is defined by multiple cis-elements. For canonical mammalian PASs, these cis-elements include an AAUAAA hexamer, a U- or GU-rich downstream element (DSE), and other auxiliary sequences including the U-rich upstream stimulatory element (USE). PAS recognition relies on several multisubunit protein complexes. The CPSF (cleavage and polyadenylation specificity factor) complex recognizes the AAUAAA hexamer, while the CstF (cleavage stimulation factor) and CFIm (cleavage factor Im) complexes bind the DSE and UGUA motifs, respectively. These factors in turn recruit CFIIm (cleavage factor IIm) and other proteins to form the 3′ processing complex in which cleavage and polyadenylation take place (Colgan and Manley 1997; Zhao et al. 1999; Shi et al. 2009; Chan et al. 2011). The majority of the core 3′ processing factors are well conserved.
For a large portion of eukaryotic genes, their mRNAs have multiple alternative 3′ ends that are formed by cleavage/polyadenylation at distinct sites, a phenomenon known as alternative polyadenylation (APA) (Di Giammartino et al. 2011; Lutz and Moreira 2011). Various systems and nomenclatures have been used for classification of APA. Here I consider two basic groups based on the distribution of the alternative PASs (Fig. 1). The first group is called same exon APA (SE-APA), in which the alternative PASs are found within the same terminal exon. In the second group, referred to as different exon APA (DE-APA), the alternative PASs are located within distinct exons. DE-APA can be further divided into three subgroups based on the sequence context of the PASs (Fig. 1). In DE-APA3 (nomenclature partially adapted from Zhang et al. 2010), the usage of alternative 3′ splice sites is coupled with cleavage/polyadenylation within mutually exclusive terminal exons. In the second subgroup, called DE-APA5, when an alternative 5′ splice site within an internal exon is skipped, cleavage/polyadenylation occurs at a downstream otherwise intronic PAS. Unlike DE-APA3 and DE-APA5, which are coupled with alternative splicing, the third subgroup involves the usage of a PAS within a constitutive internal exon that results in a truncated mRNA (Yao et al. 2012), and therefore is called DE-APAI (I for internal). The various types of APA described here may have different regulatory mechanisms and functional consequences, which will be further discussed below. It should be pointed out that although most previous APA studies have focused on mRNAs, APA has also been reported for noncoding RNAs (Liu et al. 2010). The classification system proposed here could be applied to the APA of noncoding RNAs as well.
FIGURE 1.

Classification of mRNA alternative polyadenylation. Exons are shown as boxes and introns as lines. The arrows point to the 3′ end of the transcript. For splicing events, the 5′ and 3′ splice sites are connected by dotted lines. Positions of the alternative poly(A) sites (PASs) are marked. Distinct terminal exons are colored in red or green and the composite exons are shown in red and orange.
APA STUDIES: FROM SINGLE GENES TO GLOBAL ANALYSES
In 1980, the earliest examples of APA were discovered in the mRNAs of the IgM and dihydrofolate reductase (DHFR) genes (Alt et al. 1980; Early et al. 1980; Rogers et al. 1980; Setzer et al. 1980). It was shown that the APA (DE-APA, according to the nomenclature proposed in this article) isoforms of IgM encode distinct proteins, the secreted or the membrane-bound forms of IgM (Alt et al. 1980; Early et al. 1980; Rogers et al. 1980). In contrast, the APA (SE-APA) isoforms of DHFR differ in their 3′ untranslated regions (3′ UTRs) (Setzer et al. 1980). These early studies also suggested that APA is developmentally regulated. Since then, evidence for APA had been slowly accumulating in the literature. By 1997 APA was reported for the mRNAs of ∼80 genes (Edwalds-Gilbert et al. 1997). The potential prevalence of APA was first revealed by global characterizations of APA through bioinformatic analyses of the Expressed Sequence Tag (EST) data (Tian et al. 2005; Yan and Marr 2005). These studies suggested that over 50% of human genes undergo APA and many APA events display tissue-specific patterns (Zhang et al. 2005). In 2008 and the following year, the APA field was ushered into the “global analyses era” by several pioneering studies that, using microarray-based approaches, detected widespread and coordinated APA changes during T-cell activation (Sandberg et al. 2008), neuronal activity (Flavell et al. 2008), and development (Ji et al. 2009). These studies were quickly followed by the introduction of high-throughput sequencing-based techniques for global APA analyses that dramatically accelerated the pace of discovery in the field (Jan et al. 2010; Mangone et al. 2010; Ozsolak et al. 2010; Shepard et al. 2011). In the past 3 yr alone, the RNA polyadenylation profiles have been characterized for yeast (Ozsolak et al. 2010), Caenorhabditis elegans (Jan et al. 2010; Mangone et al. 2010; Haenni et al. 2012), Drosophila (Smibert et al. 2012), Arabidopsis (Wu et al. 2011; Sherstnev et al. 2012), and mammals (Ozsolak et al. 2010; Fox-Walsh et al. 2011; Fu et al. 2011; Shepard et al. 2011; Derti et al. 2012; Jenal et al. 2012; Lin et al. 2012). In most of these species, the polyadenylation profiles have been characterized for different tissues and developmental stages. Recent global studies have identified new APA regulators and novel regulatory mechanisms and revealed widespread APA changes associated with human diseases. Here, I review the technologies that propelled the recent advances and new insights that were generated by these global analyses as well as other studies using conventional methods.
TECHNOLOGIES FOR GLOBAL APA ANALYSES
The earliest global experimental studies of APA used various microarray platforms (Flavell et al. 2008; Sandberg et al. 2008; Ji et al. 2009). In these studies, the APA profile of a gene was deduced by the ratio between the average signal intensities of probes targeting the extended region that are found only in the longer APA isoform and that of other probes targeting the same gene. The success of these studies in detecting widespread APA changes demonstrated that microarrays are effective tools for global APA analyses. Additionally, when splicing-sensitive arrays are used, both alternative splicing and APA information can be obtained from the same analysis (Flavell et al. 2008; Sandberg et al. 2008). However, microarrays also have several drawbacks. For example, microarrays cannot directly detect novel PASs or map the PAS locations precisely, and quantification may be challenging for mRNAs with more than two APA isoforms. Finally, the availability of microarrays may be another limiting factor, especially for less-studied species.
The advent of high-throughput sequencing has dramatically transformed biomedical research in recent years. For example, RNA-seq has become the method of choice for gene expression analyses (Ozsolak and Milos 2011). For mRNA analysis, a cDNA library generated from fragmented poly(A)+ RNAs is sequenced and the resulting reads are mapped to the reference genome to obtain information on mRNA sequence and relative abundance. mRNA-seq is highly quantitative and is particularly powerful for analyzing mRNA isoform expression. mRNA 3′ ends can be mapped using reads that contain a string of untemplated adenosines (Nagalakshmi et al. 2008; Wang et al. 2008). Quantification of APA isoforms with mRNA-seq data is achieved by calculating the ratio of read density in the region found only in the longer APA isoform and that in the common region. An mRNA-seq analysis of the human transcriptome, including 10 diverse tissues and five mammary epithelial or breast cancer cell lines, has provided a wealth of information on tissue-specific APA and the coordinated regulation of alternative splicing and APA (Wang et al. 2008). However, similar to microarrays, quantification may be difficult for mRNAs with more than two APA isoforms. More importantly, because only a small portion of the reads can be used to distinguish different APA isoforms, and even less reads map to the poly(A) junctions, greater sequencing depths are required for accurately mapping PASs and reliably quantifying APA isoforms.
To harness the power of high-throughput sequencing specifically for RNA polyadenylation analysis, a number of methods have been developed for targeted sequencing of poly(A) junctions. The most commonly used methods rely on oligo(dT)-primed reverse transcription–PCR (RT–PCR) for library construction (Mangone et al. 2010; Fox-Walsh et al. 2011; Fu et al. 2011; Shepard et al. 2011; Derti et al. 2012). Different versions of this method have been described with modifications designed to improve library construction and/or to facilitate sequencing (Shepard et al. 2011; Derti et al. 2012; Martin et al. 2012). The obvious advantage of the approach is its simplicity and quantitative nature. The main disadvantage, however, is the possibility of oligo(dT) priming at internal A-rich sequences at the RT step, resulting in potentially higher false discovery rate. To address these limitations, multiple computational approaches have been developed to filter out the majority of sequencing reads generated by internal priming (Mangone et al. 2010; Derti et al. 2012). Recent studies have demonstrated that these methods are highly effective in detecting global APA changes (Fu et al. 2011; Shepard et al. 2011; Jenal et al. 2012; Martin et al. 2012). A more sophisticated and labor-intensive method, called 3P-seq, uses more enzymatic steps for library construction, but avoids oligo(dT) priming and the associated internal priming issue (Jan et al. 2010). Due to its low false-positive rate, 3P-seq is especially suited for accurately mapping the 3′ ends of mRNAs. However, some of the enzymatic steps in 3P-seq, such as RNA adaptor ligation, are known to introduce bias and could affect its quantitative performance (Hafner et al. 2011). cDNA libraries prepared using either the oligo(dT)-primed RT–PCR or the 3P-seq method are most commonly sequenced on the Illumina platform, which offers high yield, low error rate, and multiplexing capability (Ozsolak and Milos 2011). In contrast, a third method, called direct RNA sequencing (DRS), uses the Helicos single-molecule sequencing platform (Ozsolak et al. 2010). In this method, the poly(A)+ RNAs are captured on oligo(dT) coated slides and the RNAs are directly sequenced by synthesis. DRS is believed to be more quantitative, as individual RNAs are sequenced without RT, PCR, or any other enzymatic reactions associated with typical cDNA library construction (Ozsolak and Milos 2011). Additionally, internal priming is rare in DRS, and attomole levels of RNAs are sufficient for DRS analysis (Ozsolak et al. 2010). When compared with the Illumina system, the disadvantages of DRS include higher error rate, shorter read lengths, lower yield (read counts), and the lack of multiplexing capability (Ozsolak and Milos 2011). Although at present no direct comparison of the three methods is available, all have been successfully applied to global analyses of polyadenylation. It should be emphasized, however, that novel PASs identified using these ultra-sensitive deep-sequencing methods must be carefully validated using independent methods to distinguish between genuine and functionally relevant PASs from sequencing noise and the intrinsic heterogeneity of cleavage/polyadenylation sites (Pauws et al. 2001).
Another key technology that is critical for mechanistic studies of APA regulation is the global mapping of protein–RNA interactions by UV cross-linking immunoprecipitation coupled with high-throughput sequencing (HITS–CLIP or CLIP–seq) (Ule et al. 2003; Licatalosi et al. 2008). Modifications of this method, such as PAR–CLIP (Photoactivatable-Ribonucleoside-Enhanced CLIP) and iCLIP (individual-nucleotide resolution CLIP), were subsequently introduced to improve the cross-linking efficiency and/or mapping resolution (Hafner et al. 2010; Konig et al. 2010). Recently, PAR–CLIP has been applied to globally mapping the RNA-binding sites for many core 3′ processing factors (Martin et al. 2012). Integration of transcriptome-wide protein–RNA interaction mapping with global functional analyses allows for construction of information-rich mechanistic models, called “RNA maps” (Licatalosi et al. 2008; Yeo et al. 2009). This approach has proven highly effective in elucidating the functions of RNA-binding factors in the regulation of pre-mRNA processing.
INSIGHTS FROM GLOBAL STUDIES OF APA
Recent global studies have made significant contributions to our understanding of APA. First, recent studies revealed that APA is much more pervasive than previously appreciated. Secondly, global analyses revealed new APA regulatory mechanisms at the transcriptome level. Thirdly, global studies have begun to reveal the functional consequences of APA and its role in a number of human diseases including cancer. These new insights are discussed in detail below.
APA is a pervasive and evolutionarily conserved mechanism for gene regulation in eukaryotes
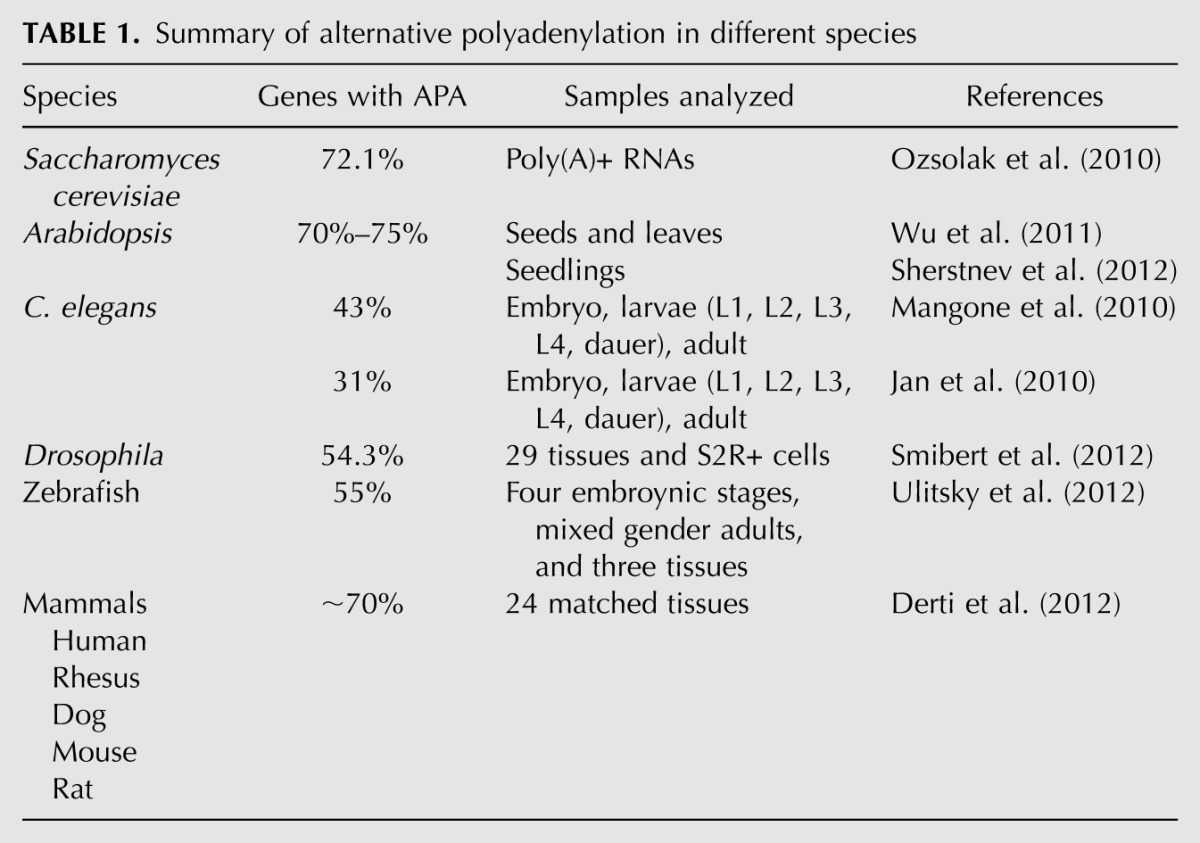
Table 1 summarizes the results of recent global RNA polyadenylation mapping in various eukaryotic species. Thirty to seventy percent of genes from all species studied are estimated to produce alternatively polyadenylated mRNAs. In human, the latest estimate is that ∼70% of genes undergo APA (Derti et al. 2012), which is significantly higher than earlier estimates based on EST data (Tian et al. 2005; Yan and Marr 2005). Perhaps most surprisingly, APA was detected for the mRNAs of ∼70% of genes in the budding yeast Saccharomyces cerevisiae (Ozsolak et al. 2010), a prevalence level comparable to that observed in the plant and human transcriptomes (Wu et al. 2011; Derti et al. 2012; Sherstnev et al. 2012). Although the large number of novel PASs detected in these high-throughput sequencing analyses need to be rigorously validated and further analyzed, recent global studies provided evidence that APA is more widespread than previously thought. The prevalence of APA in all eukaryotes tested is in sharp contrast to alternative splicing, which is extremely rare in yeast but nearly universal in human (Wang et al. 2008) and indicates that APA is an ancient phenomenon.
TABLE 1.
Summary of alternative polyadenylation in different species
Recent global studies also provided new evidence that the mechanisms for mRNA 3′ processing and APA regulation are evolutionarily conserved. First, the PAS region has conserved sequence characteristics. In all eukaryotic species studied, the overall PAS regions are A/U-rich with alternating U- and A-rich elements (Fig. 2A; Jan et al. 2010; Ozsolak et al. 2010; Martin et al. 2012; Sherstnev et al. 2012; Ulitsky et al. 2012). These include the U-rich USEs (>30 nt upstream of the cleavage site), an A-rich region (from −10 to −30 nt), and U-rich elements both upstream of and downstream from the cleavage sites. For all metazoans and Arabidopsis, AAUAAA is the most common hexamer found in the A-rich regions and its close variants are also detected in a significant portion of PASs (Jan et al. 2010; Ozsolak et al. 2010; Martin et al. 2012; Sherstnev et al. 2012; Ulitsky et al. 2012). Therefore, the nucleotide composition, key cis-elements, and their spatial organization seem well conserved in metazoans and plant. Interestingly, it has been suggested that the alternating U-rich and A-rich sequences could serve double duties in neighboring PASs or on the opposite strands (Jan et al. 2010; Sherstnev et al. 2012). For example, the USE of a downstream PAS could function as the DSE for an adjacent upstream PAS in different registers. The palindromic arrangement of PAS cis-elements also makes it possible for the same genomic region to encode for partially overlapping PAS on different strands for convergent genes (Jan et al. 2010). Similar to the classic examples found in viruses (Fitzgerald and Shenk 1981), a significant number of eukaryotic genes (for example, 14% of yeast genes) are convergently transcribed with overlapping 3′ ends (Nagalakshmi et al. 2008; Ozsolak et al. 2010), and the bidirectional PASs provide a mechanism for genome compaction (Jan et al. 2010; Sherstnev et al. 2012). Therefore, the conserved PAS sequence features may have important implications for the evolution of the genomes (Jan et al. 2010). Secondly, a general pattern for alternative PAS arrangement has been observed for many genes with APA. Overall, the most distal PASs (relative to the 5′ end of the mRNA) tend to have strong consensus features of canonical PAS, including the AAUAAA hexamer and strong DSEs, and are generally well conserved. In contrast, proximal PAS tend to diverge from the canonical sequence features and are generally less well conserved (Fig. 2B). This pattern has been observed in all metazoans studied (Beaudoing et al. 2000; Jan et al. 2010; Shepard et al. 2011; Martin et al. 2012; Smibert et al. 2012; Ulitsky et al. 2012). Interestingly, a similar arrangement was also suggested for alternative PASs in several yeast genes (Sparks and Dieckmann 1998). These observations indicate that the conserved distal PASs may serve as the default sites, while the proximal PASs may mainly play regulatory roles. Finally, the temporal and spatial regulation of APA appears highly conserved. For example, mRNAs in the brain or neuronal cells tend to use the most distal PAS and therefore have the longest 3′ UTRs, while mRNAs in the testis or ovaries tend to favor proximal PAS to generate mRNAs with the shortest 3′ UTRs (Smibert et al. 2012; Ulitsky et al. 2012). In mammals, the APA profiles are more similar among the same tissues in different species than among different tissues in the same species (Derti et al. 2012). Together, these observations strongly suggest that APA is a pervasive and conserved mechanism for eukaryotic gene regulation.
FIGURE 2.

Conserved poly(A) site sequence features and arrangement. (A) Conserved poly(A) site sequence features. U-rich sequences are shown in blue, A-rich sequences in red, UA-rich sequences in pink. Nucleotide positions relative to the cleavage site are labeled. (USE) Upstream sequence element; (ARE) A-rich element; (DSE) downstream sequence element. (B) Conserved arrangement of poly(A) sites. The arrows point to the 3′ end of the transcript. The most distal PAS has strong canonical features and is shown as a blue and red box, similar to A. The proximal PAS tend to diverge from canonical PAS features and are shown as light blue boxes.
Mechanisms for APA regulation: The “polyadenylation code”
APA is a dynamic process and recent global analyses have revealed widespread APA changes in many physiological processes as well as in human diseases. One of the most important observations from these studies is that APA, especially SE-APA, is intimately connected with the cellular proliferation and differentiation status (Sandberg et al. 2008; Ji et al. 2009; Shepard et al. 2011). In proliferating cells, proximal PASs tend to be used to generate mRNA isoforms with shorter 3′ UTRs. In contrast, in resting or differentiated cells distal PASs are favored to generate mRNAs with longer 3′ UTRs. This type of systematic and coordinated APA regulation has been observed upon activation of neurons and T cells (Flavell et al. 2008; Sandberg et al. 2008) and in development (Ji et al. 2009), stem cell differentiation (Shepard et al. 2011), and the generation of induced pluripotency (iPS) cells (Ji and Tian 2009). In some cases, there is evidence suggesting that APA regulation has significant functional consequences. For example, 3′ UTR shortening in the mRNAs of proto-oncogenes have been shown to activate these genes and lead to higher transformation (Mayr and Bartel 2009). However, the mechanisms underlying these APA changes remain poorly understood.
Global studies of RNA polyadenylation have provided a rich resource for deciphering the rules that govern PAS choice, which I refer to as the “polyadenylation code.” To understand the mechanisms for PAS selection, it is helpful to examine the chemical reaction of mRNA 3′ processing itself (Fig. 3A). In this reaction, the essential components include CPSF, CstF, CFIm, CFIIm, PAP, and the PAS-containing pre-mRNA. Two molecules of CstF and CFIm are required as they likely function as dimers (for review, see Chan et al. 2011). The overall reaction rate is a function of the concentrations of all reactants (Fig. 3A). Each alternative PAS has its unique rate constant (k) based on how efficiently it is recognized by the 3′ processing machinery and regulators. In addition, proximal PAS have an intrinsic advantage over the distal ones, as they are transcribed earlier and have more time to be recognized. In other words, the concentration of the proximal PASs is higher than that of the distal PASs during transcription. Therefore, both the sequence and the relative position of the alternative PASs are important factors that ultimately determine APA outcome. Below I present a few specific models of APA regulation that have emerged from previous studies (Fig. 3B–E).
FIGURE 3.

Regulatory mechanisms for poly(A) site selection. (A) Depiction of the mRNA 3′ processing reaction and the reaction rate formula. k is the reaction constant. [X]: Concentration of X. PAS is the poly(A) site-containing pre-mRNA. See text for details. (B–E) Regulatory mechanisms for poly(A) site selection. Poly(A) sites (PAS) are shown as rectangular boxes. Core 3′ processing factors are shown as orange boxes. The directionality of the mRNAs is marked with arrows. In B and E, the strengths of the alternative PAS are labeled. Strong PAS are shown as dark blue boxes and weak PAS as light blue boxes.
The “survival of the fittest” model
PASs with higher affinities for the 3′ processing machinery most likely have greater rate constants and therefore tend to be favored in APA. In addition to the intrinsic strengths of alternative PASs, however, PAS selection can be modulated by the levels of the core 3′ processing factors. The Manley group provided the first evidence for such a mechanism (Takagaki et al. 1996). They observed that during B-cell activation elevated protein levels of the core 3′ processing factor CstF64 promoted the usage of a proximal PAS in the IgM mRNAs that contains a weak CstF64 binding site. However, in resting B cells, which have much lower levels of CstF64 (limited resource), the distal PAS with a strong CstF64 binding site was preferentially recognized. I will refer to this mechanism as the “survival of the fittest” model (Fig. 3B). A similar mechanism was proposed to explain the APA changes of the NF-ATc mRNAs during T-cell activation (Chuvpilo et al. 1999). This mechanism may underlie, at least in part, the global APA differences between proliferating and differentiated cells (Di Giammartino et al. 2011). As mentioned earlier, proximal PASs tend to have limited consensus PAS features (likely weaker sites) and are favored in proliferating cells. In differentiated cells, polyadenylation shifts to the distal PASs that have canonical cis-elements (likely stronger sites). Interestingly, both microarray and sequencing analyses suggest that most core 3′ processing factors are expressed at higher levels in proliferating cells than in differentiated or resting cells (Ji et al. 2009; Elkon et al. 2012). Therefore, higher levels of 3′ processing factors in proliferating cells may promote the recognition of weaker proximal PAS, while the limited amounts of 3′ processing factors in differentiated cells may be preferentially recruited to the stronger distal PASs. The expression of the core 3′ processing factors can be regulated transcriptionally, for example, by the transcription factor E2F (Elkon et al. 2012) or by signaling pathways, including the p38 MAP kinase pathway (Danckwardt et al. 2011). In addition to the molecular abundance of core 3′ processing factors, post-translational modifications or interactions with other factors can modulate their activities or availability in the nucleus (Millevoi and Vagner 2009), which may contribute to APA changes as well.
Although the “survival of the fittest” model can explain many APA events, global studies suggest that the relationship between the levels of 3′ processing factors and PAS selection is more complicated. For example, it has been reported recently that CFIm68 promotes the usage of distal PASs in many mRNAs (Martin et al. 2012). Depletion of CFIm68 by RNAi results in a widespread APA shift toward proximal PASs, which, in general, are poorer binders for CFIm68. The observed APA changes are consistent with earlier studies of CFIm-mediated APA regulation (Kubo et al. 2006; Kim et al. 2010). These studies suggest that the protein levels of different core 3′ processing factors may influence the global APA profile differently. Refinement of this model will require deeper understanding of the interactions between the core 3′ processing factors and the PAS sequence and more comprehensive studies on APA regulation mediated by the core 3′ processing factors.
The agonist/antagonist model
Recognition of alternative PASs by the core 3′ processing factors can be modulated, either positively or negatively, by regulatory factors (Fig. 3C). The best-known examples of APA regulators are RNA-binding proteins that function through two mechanisms. First, some APA regulators directly compete with the core 3′ processing factors for binding to the PAS region. For example, it was shown that the mammalian PTB and Drosophila sex lethal proteins can modulate PAS recognition by competing with CstF64 for binding to the DSEs (Castelo-Branco et al. 2004; Gawande et al. 2006). Similarly, recent studies demonstrated that the RNA-binding protein HuR regulates the APA of its own transcripts by competing with CstF64 for binding to one of the alternative PASs, thereby suppressing its usage (Dai et al. 2012; Mansfield and Keene 2012). Global analyses revealed that when Nova2, a brain-specific RNA-binding protein, binds to RNAs within the PAS regions, it tends to inhibit the usage of that PAS, possibly by a steric hindrance mechanism (Licatalosi et al. 2008). The epithelial specific RNA-binding proteins ESRP1/2 may regulate APA through a similar mechanism as they bind to GU-rich motifs, and thus may directly compete with CstF64 for the DSEs (Dittmar et al. 2012). Another example of this type of regulators is PABPN1. In addition to its well-characterized functions in polyadenylation and poly(A) tail length control (Eckmann et al. 2011), PABPN1 has recently been shown to suppress the usage of proximal PAS globally (Jenal et al. 2012). As mentioned earlier, proximal PASs tend to lack consensus PAS sequences, such as the AAUAAA hexamer. It was proposed that PABPN1 competes with CPSF for binding to the A-rich regions at proximal PASs, thereby inhibiting their usage. When PABPN1 is depleted, however, CPSF can recognize these PASs and promote their usage (Jenal et al. 2012).
The second type of APA regulators binds to mRNA sequences outside of the PAS region to regulate their usage. U1 snRNP is one of the best-characterized examples. In vitro studies demonstrated that U1 snRNP bound to a 5′ splice site suppresses cleavage/polyadenylation at downstream PAS, which is mediated by U1-70K and U1A inhibition of PAP (Gunderson et al. 1994). Recent global studies showed that U1 globally suppresses premature cleavage/polyadenylation within introns and that U1 depletion leads to the activation of intronic PASs and global APA changes in a concentration-dependent manner (Kaida et al. 2010; Vorlova et al. 2011; Berg et al. 2012). Although overall U1 levels are not known to fluctuate, global transcriptional activation has been suggested to cause a transient U1 shortage, which in turn results in changes in PAS selection (Berg et al. 2012). In addition to negative regulators such as U1, some RNA-binding factors can promote PAS recognition. FCA and FPA are RNA-binding proteins that play important roles in flowering time control in Arabidopsis (Simpson et al. 2003; Hornyik et al. 2010). Both proteins regulate the APA of their own transcripts by binding to RNAs near the proximal PASs to promote their usage. For FCA, its effect on PAS selection relies on its interaction with the FY protein, a highly conserved core 3′ processing factor (Simpson et al. 2003).
The “poisedness” model
It is now appreciated that all the steps of mRNA biogenesis are tightly coupled and one step could affect another (Hirose and Manley 2000; Maniatis and Reed 2002; Bentley 2005; Moore and Proudfoot 2009). For example, most core 3′ processing factors have been shown to associate with the transcription machinery and these interactions play an important role for efficient 3′ processing (Chan et al. 2011). Accumulating evidence suggests that transcription factors can regulate the efficiency of cotranscriptional recruitment of 3′ processing factors, which in turn modulates APA. For example, ELL2, a transcription elongation factor, has been suggested to promote the loading of CstF onto the transcription machinery so that CstF is more poised to engage with PAS, thereby promoting the usage of proximal PASs (Martincic et al. 2009). I refer to this mechanism as the “poisedness” model (Fig. 3D). A recent global study provided further evidence for this mechanism by demonstrating that the promoters and transcriptional activities have a significant impact on APA (Ji et al. 2011). Reporter assays and bioinformatic analyses of RNA-seq data from human and mouse tissues suggested a correlation between higher transcription activity and higher usage of the proximal PASs. Additionally, more RNA polymerase II (RNAP II) pausing and differential nucleosome occupancy and histone modification patterns were observed at the proximal PASs for highly expressed genes. These results support a model in which transcriptional activators promote efficient recruitment of the mRNA 3′ processing machinery, and thus promote the recognition of the proximal PASs. The PAF complex may play an important role in this mechanism given its functions in coordinating transcription, histone modifications, and mRNA 3′ processing (Rozenblatt-Rosen et al. 2009). It was shown in vitro that transcriptional activators stimulate cotranscriptional mRNA 3′ processing by directly recruiting the PAF complex, which in turn recruits CPSF and CstF (Nagaike et al. 2011). It will be of interest to test the impact of the PAF complex on global APA profiles. The Mediator, a transcriptional coactivator complex, has also been shown to influence the global APA profile by modulating the occupancy of HnRNP L, a Mediator-associated protein and a regulator of mRNA alternative processing, on its target transcripts (Huang et al. 2012). These studies provided evidence that the efficiency of cotranscriptional recruitment of the 3′ processing machinery and regulatory factors is an important determinant of the APA outcome.
The “first come, first served” model
As mentioned before, proximal PAS have an intrinsic advantage over the distal ones as they are transcribed earlier (“first come, first served”) (Fig. 3E). Conceptually, such an advantage should correlate with the distance between alternative PASs and the transcription elongation rate. Indeed, it has recently been shown that APA is modulated by the transcription elongation rate. Using the Drosophila polo mRNAs as a model, Pinto et al. (2011) showed that a RNAP II mutant with slower elongation rate kinetically favors the recognition of the proximal PAS. In contrast, in cells expressing the wild-type RNAP II, the intrinsic advantage of the proximal PAS becomes less significant, and as a result, the relative usage of the distal PAS increases (Pinto et al. 2011). A similar mechanism has been previously shown to regulate alternative splicing (de la Mata et al. 2003). Although it remains poorly understood how transcription elongation rate is regulated, accumulating evidence suggests that chromatin and chromatin-associated factors may play an important role (Brown et al. 2012). For example, CTCF is a DNA-binding factor that insulates active genomic regions from inactive ones and promotes long-range interactions in the genome (Phillips and Corces 2009). Recently, it has been shown that CTCF, when bound to DNA in the coding regions, causes local RNAP II pausing, which in turn promotes the recognition of upstream splice sites (Shukla et al. 2011). Furthermore, CTCF–DNA interactions are inhibited by DNA methylation, thereby linking DNA methylation to alternative splicing regulation (Shukla et al. 2011). It is likely that similar mechanisms could influence APA as well. In fact, it has been reported that genomic imprinting regulates the APA of the mouse H13 and Herc3/Nap1l5 transcripts (Wood et al. 2008; Cowley et al. 2012). In both cases, the genomic sequences encoding alternative PASs are separated by a CpG island, which is differentially methylated on the paternal and maternal alleles. Transcripts expressed from the unmethylated allele are polyadenylated at the PAS upstream of the CpG island to produce a truncated mRNA, while those expressed from the methylated allele favor the downstream PAS. Similar to the mechanism for CTCF-mediated alternative splicing regulation, a methylation-sensitive DNA-binding factor may bind to the CpG island on the unmethylated allele, cause RNAP II pausing, and promote the usage of the upstream PAS. This model needs to be tested in future studies.
It is important to note that the transcription activities of a gene are not directly correlated with its transcription elongation rate. In fact, as discussed above, high transcription activity promotes the usage of proximal PASs through the “poisedness” model, while higher elongation rate leads to enhanced recognition of the distal PASs (the “first come, first served” model).
PAS selection is most likely under combinatorial control of multiple mechanisms. For example, tissue- or developmental stage-specific APA profiles may be generated by several possible mechanisms. The core 3′ processing factors or APA regulators may be expressed at different levels in different tissues or at different times during development (Ji et al. 2009). For instance, CstF64τ, a CstF64 paralog, is highly expressed in testis (Wallace et al. 1999; Dass et al. 2007). As described earlier, the APA regulators, Nova2 and ESRP1/2, are specifically expressed in the brain and epithelial cells, respectively (Ule et al. 2005; Warzecha et al. 2009). Thus, these factors may contribute to tissue-specific PAS selection. Similarly, multiple mechanisms have been proposed to explain the 3′ UTR lengthening during differentiation, including changes in 3′ processing factor abundances and U1 snRNP availability (Ji et al. 2009; Berg et al. 2012). Therefore, a major challenge in deciphering the “polyadenylation code” is to integrate all of the different mechanisms in a quantitative manner.
Functional impact of APA
A major contribution of the recent global studies is the realization that APA is a widespread mechanism for eukaryotic gene regulation. APA could impact gene expression through several mechanisms. First, different APA isoforms, especially DE-APA isoforms, may encode distinct proteins. A comprehensive human transcriptome analysis using RNA-seq found that over 5000 human genes produce APA isoforms that have differences in their coding regions, and half of these APA events showed tissue-specific profiles (Wang et al. 2008). Therefore, like alternative splicing, APA significantly expands the proteome diversity. The proteins encoded by APA isoforms may have distinct functional properties. As mentioned earlier, the IgM protein expressed from the shorter APA isoform is secreted, while the product of the longer isoform is membrane bound (Alt et al. 1980; Early et al. 1980; Rogers et al. 1980). In a recently described example, polyadenylation within a constitutively spliced internal exon of the EPRS mRNA converts a Tyr codon into a stop codon (Yao et al. 2012). The shorter EPRS protein isoform expressed from the truncated mRNAs attenuates the function of the full-length protein in translation inhibition. Furthermore, some of the APA isoforms are poorly translated or the expressed proteins are unstable. Therefore, the generation of nonfunctional APA isoforms can be viewed as a mechanism to turn off gene expression post-transcriptionally. For example, both CstF77 and HuR have been shown to autoregulate their own expression by stimulating the usage of an alternative PAS that leads to the production of truncated or unstable mRNAs (Juge et al. 2000; Dai et al. 2012; Mansfield and Keene 2012).
Secondly, APA isoforms differ in their 3′ UTRs, which can modulate the localization of the mRNAs. Many mRNAs are known to localize to specific subcellular loci from yeast to human, and the sequences required for such localization are often found in the 3′ UTRs (Kislauskis and Singer 1992). This mechanism is particularly prevalent in polarized cells, including neurons (Jung et al. 2012). Indeed, we have recently observed widespread 3′ UTR lengthening during the differentiation of mouse embryonic stem cells to neurons (Shepard et al. 2011). Two recent global analyses of APA in Drosophila and zebrafish showed that the mRNAs in the brain have the longest average 3′ UTRs (Smibert et al. 2012; Ulitsky et al. 2012). These studies indicate that mRNAs acquire their localization signals through APA changes during neurodevelopment. However, no global analyses of the localization patterns for APA isoforms have been reported.
Thirdly, APA isoforms may have different stabilities and/or translation efficiencies. It is well known that many RNA sequence elements in the 3′ UTR, such as the AU-rich and GU-rich elements (AREs and GREs) and microRNA target sites, can negatively regulate mRNA stability (Di Giammartino et al. 2011). Therefore, it has been suggested that shorter APA isoforms are generally more stable and produce more proteins (Sandberg et al. 2008; Mayr and Bartel 2009). More than half of the putative microRNA target sites are located downstream from the first PASs (Legendre et al. 2006), indicating widespread cross-talk between microRNA- and APA-mediated gene regulation. An EST-based global analysis showed that miR-1 or miR-124-targeted APA isoforms are down-regulated in tissues where these two microRNAs are expressed (Legendre et al. 2006). A number of examples have been reported in which APA changes lead to production of mRNAs with shorter 3′ UTRs, thereby evading microRNA-mediated inhibition (Wirsing et al. 2011; Boutet et al. 2012). Other than these sequence-specific mechanisms, the sheer length of the 3′ UTR may influence the stability of the mRNA through the nonsense-mediated mRNA decay (NMD) pathway. NMD is an RNA quality control mechanism that selectively degrades mRNAs harboring premature stop codons (Hwang and Maquat 2011). Although it is still not fully understood what RNA characteristics define NMD targets (Rebbapragada and Lykke-Andersen 2009), the 3′ UTR lengths have been suggested to be an important determinant of NMD from yeast to human (Amrani et al. 2004; Hogg and Goff 2010). According to this model, the NMD factor UPF1 associates with 3′ UTRs to promote NMD, and the level of UPF1 recruitment is proportional to the 3′ UTR length (Hogg and Goff 2010). Therefore, the APA isoforms with longer 3′ UTRs are expected to be more susceptible to NMD. Consistent with this model, it has recently been shown by microarray analyses that physiological NMD target mRNAs have greater median 3′ UTR lengths than nontarget mRNAs (Yepiskoposyan et al. 2011). Because the activity of the NMD pathway is regulated during development (Bruno et al. 2011), it is possible that changes in NMD activities could lead to changes in the relative abundance of APA isoforms.
Finally, the functional significance of APA at the organism level has been highlighted by a recent study. Pinto et al. (2011) showed that the mRNAs of the polo gene are alternatively polyadenylated, and the isoform with longer 3′ UTR is translated more efficiently. Interestingly, when the distal PAS is genetically disrupted so that only the short APA isoform is produced, the transgenic flies die at the pupa stage due to proliferation defects in the precursor cells of the abdomen (Pinto et al. 2011). Together, these studies highlight the functional impact of APA regulation on the development and functioning of eukaryotic species.
Global analyses of APA regulation in human diseases
Aberrant mRNA 3′ processing has long been implicated in human diseases (Danckwardt et al. 2008). Genetic mutations or single-nucleotide-polymorphisms (SNPs) can lead to loss or gain of PASs in functionally important genes. The resultant alterations in the expression of affected genes have been shown to cause or contribute to a number of human diseases, including thalassemia (Higgs et al. 1983; Orkin et al. 1985), Fabry disease (Bishop et al. 1988), metachromatic leukodystrophy (Gieselmann et al. 1989), amyotrophic lateral sclerosis (ALS) (Lin et al. 1998), IPEX (immune dysfunction, polyendocrinopathy, enteropathy, X-linked) (Bennett et al. 2001), and facioscapulohumeral muscular dystrophy (FSHD) (Lemmers et al. 2010). Even subtle changes to a PAS may have pathological consequences. For example, in many highly proliferative mantle cell lymphoma (MCL) tumors, mutations at a proximal PAS of the proto-oncogene cyclin D1 lead to higher levels of its mRNAs with shorter 3′ UTRs, which is more stable and produces more proteins. This shift in cyclin D1 APA profile is associated with an inferior survival rate in MCL patients (Wiestner et al. 2007).
Recent studies have begun to analyze global APA changes in a number of human diseases, including cancer. Mayr and Bartel (2009) observed APA-mediated 3′ UTR shortening for the mRNAs of a number of proto-oncogenes in cancer cell lines compared with the corresponding normal tissues and immortalized nontransformed cell lines. The APA isoforms of these proto-oncogenes with shorter 3′ UTRs were shown to be more stable, generate greater amounts of proteins, and most importantly, lead to higher neoplastic transformation rates (Mayr and Bartel 2009). Based on these observations, it was proposed that 3′ UTR shortening through APA plays an important role in oncogene activation in the absence of genetic mutations. Several recent studies suggest that 3′ UTR shortening in cancers is not limited to proto-oncogenes. For example, a comparison between the immortalized but nontransformed mammary epithelial cell line MCF10A and the breast cancer cell line MCF7 detected a global APA shift toward proximal PASs in MCF7 cells (Fu et al. 2011). However, a similar shift was not observed in a different breast cancer cell line MB231 (Fu et al. 2011). A comparison of five different cancers and their corresponding normal tissues revealed a widespread increase in the relative levels of the shorter APA isoforms (Lin et al. 2012). Although APA changes seem to occur in many cancers, it remains unclear what role APA regulation plays in cancer development. A recent study suggests that 3′ UTR shortening is more strongly associated with proliferation than with transformation induced by oncogene activation (Elkon et al. 2012). This may indicate that, among all of the hallmarks of cancer (Hanahan and Weinberg 2011), APA regulation mainly contributes to sustaining proliferation.
Global deregulation of APA was also detected in cardiac hypertrophy (the enlargement of the heart), which could occur under both physiological and pathological conditions (Park et al. 2011). A microarray-based analysis detected higher usage of the proximal PAS and overall 3′ UTR shortening in hypertrophy compared with normal hearts (Park et al. 2011). Such APA changes are accompanied by the up-regulation of many microRNA target mRNAs, suggesting coupling between APA- and microRNA-mediated gene regulation (Park et al. 2011). 3′ UTR shortening was also observed in oculopharyngeal muscular dystrophy (OPMD) (de Klerk et al. 2012; Jenal et al. 2012). This neuromuscular disease is caused by “prion”-like mutant PABPN1 proteins that have an expanded polyalanine region due to a trinucleotide (GCN) expansion in the PABPN1 gene. Recent studies suggested that the mutant PABPN1 protein, called trePABPN1, acts as a dominant-negative factor by trapping the wild-type protein in insoluble nuclear inclusion bodies (Jenal et al. 2012). Global mapping of RNA polyadenylation in trePABPN1-expressing cells and using an OPMD mouse model revealed a systematic shift of the APA profile toward proximal PASs, similar to the effect of PABPN1 loss-of-function (de Klerk et al. 2012; Jenal et al. 2012). It will be important to understand how the observed aberrant APA pattern contributes to the pathology of OPMD and why only certain muscle cells are affected by this defect.
CONCLUSIONS AND FUTURE PERSPECTIVES
Thanks to the recent wave of global studies, APA has now been recognized as a major mechanism for eukaryotic gene regulation. Regulatory mechanisms and functional impacts of APA are being revealed at an unprecedented pace. But a number of important issues remain to be addressed by future studies. First, major efforts are needed to systematically characterize the APA regulatory mechanisms and to compile a “polyadenylation code.” For example, it remains difficult to accurately identify PASs based on the sequence information and to predict the “strength” of known PASs. For splicing, the strength of a splice site can be estimated by its complementarity with the snRNAs (Yeo and Burge 2004). Although the basic cis-elements for PASs are known, quantification of PAS strength using sequence information has not been adequately accomplished. Additionally, future studies are needed to systematically identify regulatory cis-elements, such as “enhancers” and “silencers” of polyadenylation and trans-acting regulatory factors that recognize them. A key challenge will be to integrate the various mechanisms to achieve an understanding of APA regulation at the systems level. Secondly, although widespread APA changes have been observed under a wide variety of physiological and pathological conditions, relatively little is known about the specific functional consequences of these changes. In most cases, the cause and effect relationship between APA changes and the involved biological processes remains to be determined, and the functionally important APA changes remain to be identified and characterized. Finally, as aberrant APA associates with many human diseases, it will be important to explore whether APA can be specifically and effectively manipulated in vivo to help treat these diseases. Antisense oligo-based methods have been successfully used to modulate alternative splicing of many human genes and are being tested for clinical purposes (Kole et al. 2012). A similar approach has recently been used to modulate APA (Vorlova et al. 2011). It will be of great interest to test whether this method can be applied to treating diseases caused by aberrant mRNA polyadenylation.
ACKNOWLEDGMENTS
I thank Dr. Russ Carstens, Klemens Hertel, Bin Tian, Yi Xing, and members of the Shi lab for discussions and critically reading the manuscript. Research in my laboratory is supported by NIH (R01GM090056), ACS (RSG-12-186), and start-up funds from UC Irvine.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.035899.112.
REFERENCES
- Alt FW, Bothwell AL, Knapp M, Siden E, Mather E, Koshland M, Baltimore D 1980. Synthesis of secreted and membrane-bound immunoglobulin μ heavy chains is directed by mRNAs that differ at their 3′ ends. Cell 20: 293–301 [DOI] [PubMed] [Google Scholar]
- Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A 2004. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 432: 112–118 [DOI] [PubMed] [Google Scholar]
- Beaudoing E, Freier S, Wyatt JR, Claverie JM, Gautheret D 2000. Patterns of variant polyadenylation signal usage in human genes. Genome Res 10: 1001–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CL, Brunkow ME, Ramsdell F, O'Briant KC, Zhu Q, Fuleihan RL, Shigeoka AO, Ochs HD, Chance PF 2001. A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA→AAUGAA) leads to the IPEX syndrome. Immunogenetics 53: 435–439 [DOI] [PubMed] [Google Scholar]
- Bentley DL 2005. Rules of engagement: Co-transcriptional recruitment of pre-mRNA processing factors. Curr Opin Cell Biol 17: 251–256 [DOI] [PubMed] [Google Scholar]
- Berg MG, Singh LN, Younis I, Liu Q, Pinto AM, Kaida D, Zhang Z, Cho S, Sherrill-Mix S, Wan L, et al. 2012. U1 snRNP determines mRNA length and regulates isoform expression. Cell 150: 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop DF, Kornreich R, Desnick RJ 1988. Structural organization of the human α-galactosidase A gene: Further evidence for the absence of a 3′ untranslated region. Proc Natl Acad Sci 85: 3903–3907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutet SC, Cheung TH, Quach NL, Liu L, Prescott SL, Edalati A, Iori K, Rando TA 2012. Alternative polyadenylation mediates microRNA regulation of muscle stem cell function. Cell Stem Cell 10: 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SJ, Stoilov P, Xing Y 2012. Chromatin and epigenetic regulation of pre-mRNA processing. Hum Mol Genet doi: 10.1093/hmg/dds353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno IG, Karam R, Huang L, Bhardwaj A, Lou CH, Shum EY, Song HW, Corbett MA, Gifford WD, Gecz J, et al. 2011. Identification of a microRNA that activates gene expression by repressing nonsense-mediated RNA decay. Mol Cell 42: 500–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelo-Branco P, Furger A, Wollerton M, Smith C, Moreira A, Proudfoot N 2004. Polypyrimidine tract binding protein modulates efficiency of polyadenylation. Mol Cell Biol 24: 4174–4183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S, Choi EA, Shi Y 2011. Pre-mRNA 3′-end processing complex assembly and function. Wiley Interdiscip Rev RNA 2: 321–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuvpilo S, Zimmer M, Kerstan A, Glockner J, Avots A, Escher C, Fischer C, Inashkina I, Jankevics E, Berberich-Siebelt F, et al. 1999. Alternative polyadenylation events contribute to the induction of NF-ATc in effector T cells. Immunity 10: 261–269 [DOI] [PubMed] [Google Scholar]
- Colgan DF, Manley JL 1997. Mechanism and regulation of mRNA polyadenylation. Genes Dev 11: 2755–2766 [DOI] [PubMed] [Google Scholar]
- Cowley M, Wood AJ, Bohm S, Schulz R, Oakey RJ 2012. Epigenetic control of alternative mRNA processing at the imprinted Herc3/Nap1l5 locus. Nucleic Acids Res doi: 10.1093/nar/gks654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W, Zhang G, Makeyev EV 2012. RNA-binding protein HuR autoregulates its expression by promoting alternative polyadenylation site usage. Nucleic Acids Res 40: 787–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danckwardt S, Hentze MW, Kulozik AE 2008. 3′ end mRNA processing: Molecular mechanisms and implications for health and disease. EMBO J 27: 482–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danckwardt S, Gantzert AS, Macher-Goeppinger S, Probst HC, Gentzel M, Wilm M, Grone HJ, Schirmacher P, Hentze MW, Kulozik AE 2011. p38 MAPK controls prothrombin expression by regulated RNA 3′ end processing. Mol Cell 41: 298–310 [DOI] [PubMed] [Google Scholar]
- Dass B, Tardif S, Park JY, Tian B, Weitlauf HM, Hess RA, Carnes K, Griswold MD, Small CL, Macdonald CC 2007. Loss of polyadenylation protein τCstF-64 causes spermatogenic defects and male infertility. Proc Natl Acad Sci 104: 20374–20379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Klerk E, Venema A, Anvar SY, Goeman JJ, Hu O, Trollet C, Dickson G, den Dunnen JT, van der Maarel SM, Raz V, et al. 2012. Poly(A) binding protein nuclear 1 levels affect alternative polyadenylation. Nucleic Acids Res doi: 10.1093/nar/gks655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, Kornblihtt AR 2003. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell 12: 525–532 [DOI] [PubMed] [Google Scholar]
- Derti A, Garrett-Engele P, MacIsaac KD, Stevens RC, Sriram S, Chen R, Rohl CA, Johnson JM, Babak T 2012. A quantitative atlas of polyadenylation in five mammals. Genome Res 22: 1173–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giammartino DC, Nishida K, Manley JL 2011. Mechanisms and consequences of alternative polyadenylation. Mol Cell 43: 853–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmar KA, Jiang P, Park JW, Amirikian K, Wan J, Shen S, Xing Y, Carstens RP 2012. Genome-wide determination of a broad ESRP-regulated posttranscriptional network by high-throughput sequencing. Mol Cell Biol 32: 1468–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Early P, Rogers J, Davis M, Calame K, Bond M, Wall R, Hood L 1980. Two mRNAs can be produced from a single immunoglobulin μ gene by alternative RNA processing pathways. Cell 20: 313–319 [DOI] [PubMed] [Google Scholar]
- Eckmann CR, Rammelt C, Wahle E 2011. Control of poly(A) tail length. Wiley Interdiscip Rev RNA 2: 348–361 [DOI] [PubMed] [Google Scholar]
- Edwalds-Gilbert G, Veraldi KL, Milcarek C 1997. Alternative poly(A) site selection in complex transcription units: Means to an end? Nucleic Acids Res 25: 2547–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkon R, Drost J, van Haaften G, Jenal M, Schrier M, Oude Vrielink JA, Agami R 2012. E2F mediates enhanced alternative polyadenylation in proliferation. Genome Biol 13: R59 doi: 10.1186/gb-2012-13-7-r59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald M, Shenk T 1981. The sequence 5′-AAUAAA-3′ forms parts of the recognition site for polyadenylation of late SV40 mRNAs. Cell 24: 251–260 [DOI] [PubMed] [Google Scholar]
- Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, Markenscoff-Papadimitriou E, Bear DM, Greenberg ME 2008. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron 60: 1022–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox-Walsh K, Davis-Turak J, Zhou Y, Li H, Fu XD 2011. A multiplex RNA-seq strategy to profile poly(A+) RNA: Application to analysis of transcription response and 3′ end formation. Genomics 98: 266–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sun Y, Li Y, Li J, Rao X, Chen C, Xu A 2011. Differential genome-wide profiling of tandem 3′ UTRs among human breast cancer and normal cells by high-throughput sequencing. Genome Res 21: 741–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawande B, Robida MD, Rahn A, Singh R 2006. Drosophila Sex-lethal protein mediates polyadenylation switching in the female germline. EMBO J 25: 1263–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieselmann V, Polten A, Kreysing J, von Figura K 1989. Arylsulfatase A pseudodeficiency: Loss of a polyadenylylation signal and N-glycosylation site. Proc Natl Acad Sci 86: 9436–9440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson SI, Beyer K, Martin G, Keller W, Boelens WC, Mattaj LW 1994. The human U1A snRNP protein regulates polyadenylation via a direct interaction with poly(A) polymerase. Cell 76: 531–541 [DOI] [PubMed] [Google Scholar]
- Haenni S, Ji Z, Hoque M, Rust N, Sharpe H, Eberhard R, Browne C, Hengartner MO, Mellor J, Tian B, et al. 2012. Analysis of C. elegans intestinal gene expression and polyadenylation by fluorescence-activated nuclei sorting and 3′-end-seq. Nucleic Acids Res 40: 6304–6318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Renwick N, Brown M, Mihailovic A, Holoch D, Lin C, Pena JT, Nusbaum JD, Morozov P, Ludwig J, et al. 2011. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA 17: 1697–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA 2011. Hallmarks of cancer: The next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Higgs DR, Goodbourn SE, Lamb J, Clegg JB, Weatherall DJ, Proudfoot NJ 1983. Alpha-thalassaemia caused by a polyadenylation signal mutation. Nature 306: 398–400 [DOI] [PubMed] [Google Scholar]
- Hirose Y, Manley JL 2000. RNA polymerase II and the integration of nuclear events. Genes Dev 14: 1415–1429 [PubMed] [Google Scholar]
- Hogg JR, Goff SP 2010. Upf1 senses 3′UTR length to potentiate mRNA decay. Cell 143: 379–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornyik C, Terzi LC, Simpson GG 2010. The spen family protein FPA controls alternative cleavage and polyadenylation of RNA. Dev Cell 18: 203–213 [DOI] [PubMed] [Google Scholar]
- Huang Y, Li W, Yao X, Lin QJ, Yin JW, Liang Y, Heiner M, Tian B, Hui J, Wang G 2012. Mediator complex regulates alternative mRNA processing via the MED23 subunit. Mol Cell 45: 459–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, Maquat LE 2011. Nonsense-mediated mRNA decay (NMD) in animal embryogenesis: To die or not to die, that is the question. Curr Opin Genet Dev 21: 422–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan CH, Friedman RC, Ruby JG, Bartel DP 2010. Formation, regulation and evolution of Caenorhabditis elegans 3′UTRs. Nature 469: 97–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenal M, Elkon R, Loayza-Puch F, van Haaften G, Kuhn U, Menzies FM, Oude Vrielink JA, Bos AJ, Drost J, Rooijers K, et al. 2012. The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell 149: 538–553 [DOI] [PubMed] [Google Scholar]
- Ji Z, Tian B 2009. Reprogramming of 3′ untranslated regions of mRNAs by alternative polyadenylation in generation of pluripotent stem cells from different cell types. PLoS ONE 4: e8419 doi: 10.1371/journal.pone.0008419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Lee JY, Pan Z, Jiang B, Tian B 2009. Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc Natl Acad Sci 106: 7028–7033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Luo W, Li W, Hoque M, Pan Z, Zhao Y, Tian B 2011. Transcriptional activity regulates alternative cleavage and polyadenylation. Mol Syst Biol 7: 534 doi: 10.1038/msb.2011.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juge F, Audibert A, Benoit B, Simonelig M 2000. Tissue-specific autoregulation of Drosophila suppressor of forked by alternative poly(A) site utilization leads to accumulation of the suppressor of forked protein in mitotically active cells. RNA 6: 1529–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Yoon BC, Holt CE 2012. Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat Rev Neurosci 13: 308–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, Dreyfuss G 2010. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 468: 664–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Yamamoto J, Chen Y, Aida M, Wada T, Handa H, Yamaguchi Y 2010. Evidence that cleavage factor Im is a heterotetrameric protein complex controlling alternative polyadenylation. Genes Cells 15: 1003–1013 [DOI] [PubMed] [Google Scholar]
- Kislauskis EH, Singer RH 1992. Determinants of mRNA localization. Curr Opin Cell Biol 4: 975–978 [DOI] [PubMed] [Google Scholar]
- Kole R, Krainer AR, Altman S 2012. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov 11: 125–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J 2010. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17: 909–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo T, Wada T, Yamaguchi Y, Shimizu A, Handa H 2006. Knock-down of 25 kDa subunit of cleavage factor Im in Hela cells alters alternative polyadenylation within 3′-UTRs. Nucleic Acids Res 34: 6264–6271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre M, Ritchie W, Lopez F, Gautheret D 2006. Differential repression of alternative transcripts: A screen for miRNA targets. PLoS Comput Biol 2: e43 doi: 10.1371/journal.pcbi.0020043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camano P, Dauwerse JG, Snider L, Straasheijm KR, van Ommen GJ, Padberg GW, et al. 2010. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 329: 1650–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, et al. 2008. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456: 464–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CL, Bristol LA, Jin L, Dykes-Hoberg M, Crawford T, Clawson L, Rothstein JD 1998. Aberrant RNA processing in a neurodegenerative disease: The cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 20: 589–602 [DOI] [PubMed] [Google Scholar]
- Lin Y, Li Z, Ozsolak F, Kim SW, Arango-Argoty G, Liu TT, Tenenbaum SA, Bailey T, Monaghan AP, Milos PM, et al. 2012. An in-depth map of polyadenylation sites in cancer. Nucleic Acids Res doi: 10.1093/nar/gks637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Marquardt S, Lister C, Swiezewski S, Dean C 2010. Targeted 3′ processing of antisense transcripts triggers Arabidopsis FLC chromatin silencing. Science 327: 94–97 [DOI] [PubMed] [Google Scholar]
- Lutz CS, Moreira A 2011. Alternative mRNA polyadenylation in eukaryotes: An effective regulator of gene expression. Wiley Interdiscip Rev RNA 2: 22–31 [DOI] [PubMed] [Google Scholar]
- Mangone M, Manoharan AP, Thierry-Mieg D, Thierry-Mieg J, Han T, Mackowiak SD, Mis E, Zegar C, Gutwein MR, Khivansara V, et al. 2010. The landscape of C. elegans 3′UTRs. Science 329: 432–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, Reed R 2002. An extensive network of coupling among gene expression machines. Nature 416: 499–506 [DOI] [PubMed] [Google Scholar]
- Mansfield KD, Keene JD 2012. Neuron-specific ELAV/Hu proteins suppress HuR mRNA during neuronal differentiation by alternative polyadenylation. Nucleic Acids Res 40: 2734–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Gruber AR, Keller W, Zavolan M 2012. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep 1: 753–763 [DOI] [PubMed] [Google Scholar]
- Martincic K, Alkan SA, Cheatle A, Borghesi L, Milcarek C 2009. Transcription elongation factor ELL2 directs immunoglobulin secretion in plasma cells by stimulating altered RNA processing. Nat Immunol 10: 1102–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Bartel DP 2009. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138: 673–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millevoi S, Vagner S 2009. Molecular mechanisms of eukaryotic pre-mRNA 3′ end processing regulation. Nucleic Acids Res 38: 2757–2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Proudfoot NJ 2009. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 136: 688–700 [DOI] [PubMed] [Google Scholar]
- Nagaike T, Logan C, Hotta I, Rozenblatt-Rosen O, Meyerson M, Manley JL 2011. Transcriptional activators enhance polyadenylation of mRNA precursors. Mol Cell 41: 409–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin SH, Cheng TC, Antonarakis SE, Kazazian HH Jr 1985. Thalassemia due to a mutation in the cleavage-polyadenylation signal of the human β-globin gene. EMBO J 4: 453–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Milos PM 2011. RNA sequencing: Advances, challenges and opportunities. Nat Rev Genet 12: 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Kapranov P, Foissac S, Kim SW, Fishilevich E, Monaghan AP, John B, Milos PM 2010. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell 143: 1018–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JY, Li W, Zheng D, Zhai P, Zhao Y, Matsuda T, Vatner SF, Sadoshima J, Tian B 2011. Comparative analysis of mRNA isoform expression in cardiac hypertrophy and development reveals multiple post-transcriptional regulatory modules. PLoS ONE 6: e22391 doi: 10.1371/journal.pone.0022391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauws E, van Kampen AH, van de Graaf SA, de Vijlder JJ, Ris-Stalpers C 2001. Heterogeneity in polyadenylation cleavage sites in mammalian mRNA sequences: Implications for SAGE analysis. Nucleic Acids Res 29: 1690–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JE, Corces VG 2009. CTCF: Master weaver of the genome. Cell 137: 1194–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto PA, Henriques T, Freitas MO, Martins T, Domingues RG, Wyrzykowska PS, Coelho PA, Carmo AM, Sunkel CE, Proudfoot NJ, et al. 2011. RNA polymerase II kinetics in polo polyadenylation signal selection. EMBO J 30: 2431–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbapragada I, Lykke-Andersen J 2009. Execution of nonsense-mediated mRNA decay: What defines a substrate? Curr Opin Cell Biol 21: 394–402 [DOI] [PubMed] [Google Scholar]
- Rogers J, Early P, Carter C, Calame K, Bond M, Hood L, Wall R 1980. Two mRNAs with different 3′ ends encode membrane-bound and secreted forms of immunoglobulin μ chain. Cell 20: 303–312 [DOI] [PubMed] [Google Scholar]
- Rozenblatt-Rosen O, Nagaike T, Francis JM, Kaneko S, Glatt KA, Hughes CM, LaFramboise T, Manley JL, Meyerson M 2009. The tumor suppressor Cdc73 functionally associates with CPSF and CstF 3′ mRNA processing factors. Proc Natl Acad Sci 106: 755–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB 2008. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science 320: 1643–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setzer DR, McGrogan M, Nunberg JH, Schimke RT 1980. Size heterogeneity in the 3′ end of dihydrofolate reductase messenger RNAs in mouse cells. Cell 22: 361–370 [DOI] [PubMed] [Google Scholar]
- Shepard PJ, Choi E, Lu J, Flanagan LA, Hertel KJ, Shi Y 2011. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA 17: 761–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherstnev A, Duc C, Cole C, Zacharaki V, Hornyik C, Ozsolak F, Milos PM, Barton GJ, Simpson GG 2012. Direct sequencing of Arabidopsis thaliana RNA reveals patterns of cleavage and polyadenylation. Nat Struct Mol Biol 19: 845–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR 3rd, Frank J, Manley JL 2009. Molecular architecture of the human pre-mRNA 3′ processing complex. Mol Cell 33: 365–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S 2011. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479: 74–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson GG, Dijkwel PP, Quesada V, Henderson I, Dean C 2003. FY is an RNA 3′ end-processing factor that interacts with FCA to control the Arabidopsis floral transition. Cell 113: 777–787 [DOI] [PubMed] [Google Scholar]
- Smibert P, Miura P, Westholm JO, Shenker S, May G, Duff MO, Zhang D, Eads BD, Carlson J, Brown JB, et al. 2012. Global patterns of tissue-specific alternative polyadenylation in Drosophila. Cell Rep 1: 277–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks KA, Dieckmann CL 1998. Regulation of poly(A) site choice of several yeast mRNAs. Nucleic Acids Res 26: 4676–4687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagaki Y, Seipelt RL, Peterson ML, Manley JL 1996. The polyadenylation factor CstF-64 regulates alternative processing of IgM heavy chain pre-mRNA during B cell differentiation. Cell 87: 941–952 [DOI] [PubMed] [Google Scholar]
- Tian B, Hu J, Zhang H, Lutz CS 2005. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res 33: 201–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB 2003. CLIP identifies Nova-regulated RNA networks in the brain. Science 302: 1212–1215 [DOI] [PubMed] [Google Scholar]
- Ule J, Ule A, Spencer J, Williams A, Hu JS, Cline M, Wang H, Clark T, Fraser C, Ruggiu M, et al. 2005. Nova regulates brain-specific splicing to shape the synapse. Nat Genet 37: 844–852 [DOI] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Subtelny AO, Koppstein D, Bell GW, Sive H, Bartel DP 2012. Extensive alternative polyadenylation during zebrafish development. Genome Res 22: 2054–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorlova S, Rocco G, Lefave CV, Jodelka FM, Hess K, Hastings ML, Henke E, Cartegni L 2011. Induction of antagonistic soluble decoy receptor tyrosine kinases by intronic polyA activation. Mol Cell 43: 927–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace AM, Dass B, Ravnik SE, Tonk V, Jenkins NA, Gilbert DJ, Copeland NG, MacDonald CC 1999. Two distinct forms of the 64,000 Mr protein of the cleavage stimulation factor are expressed in mouse male germ cells. Proc Natl Acad Sci 96: 6763–6768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP 2009. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell 33: 591–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiestner A, Tehrani M, Chiorazzi M, Wright G, Gibellini F, Nakayama K, Liu H, Rosenwald A, Muller-Hermelink HK, Ott G, et al. 2007. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood 109: 4599–4606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirsing A, Senkel S, Klein-Hitpass L, Ryffel GU 2011. A systematic analysis of the 3′UTR of HNF4A mRNA reveals an interplay of regulatory elements including miRNA target sites. PLoS ONE 6: e27438 doi: 10.1371/journal.pone0027438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AJ, Schulz R, Woodfine K, Koltowska K, Beechey CV, Peters J, Bourc'his D, Oakey RJ 2008. Regulation of alternative polyadenylation by genomic imprinting. Genes Dev 22: 1141–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Liu M, Downie B, Liang C, Ji G, Li QQ, Hunt AG 2011. Genome-wide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proc Natl Acad Sci 108: 12533–12538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Marr TG 2005. Computational analysis of 3′-ends of ESTs shows four classes of alternative polyadenylation in human, mouse, and rat. Genome Res 15: 369–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao P, Potdar AA, Arif A, Ray PS, Mukhopadhyay R, Willard B, Xu Y, Yan J, Saidel GM, Fox PL 2012. Coding region polyadenylation generates a truncated tRNA synthetase that counters translation repression. Cell 149: 88–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Burge CB 2004. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11: 377–394 [DOI] [PubMed] [Google Scholar]
- Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH 2009. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol 16: 130–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yepiskoposyan H, Aeschimann F, Nilsson D, Okoniewski M, Muhlemann O 2011. Autoregulation of the nonsense-mediated mRNA decay pathway in human cells. RNA 17: 2108–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Lee JY, Tian B 2005. Biased alternative polyadenylation in human tissues. Genome Biol 6: R100 doi: 10.1186/gb-2005-6-12-r100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Frias MA, Mele A, Ruggiu M, Eom T, Marney CB, Wang H, Licatalosi DD, Fak JJ, Darnell RB 2010. Integrative modeling defines the Nova splicing-regulatory network and its combinatorial controls. Science 329: 439–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Hyman L, Moore C 1999. Formation of mRNA 3′ ends in eukaryotes: Mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev 63: 405–445 [DOI] [PMC free article] [PubMed] [Google Scholar]