

Background: Trim28 appears up-regulated in many cancers.
Results: In early stage lung tumors high Trim28 correlates with increased overall survival and Trim28 reduces cell proliferation in model lung cancer cell lines through E2F interactions.
Conclusion: Trim28 may have a tumor suppressing role in the early stages of lung cancer.
Significance: These results suggest a complex role for Trim28 in lung cancer.
Keywords: Cancer, Cell Cycle, Cell Growth, E2F Transcription Factor, Lung Cancer
Abstract
Trim28 is a poorly understood transcriptional co-factor with pleiotropic biological activities. Although Trim28 mRNA is found in many studies to be up-regulated in both lung and breast cancer tissues relative to normal adjacent tissue, we found that within a panel of early-stage lung adenocarcinomas high levels of Trim28 protein correlate with better overall survival. This surprising observation suggests that Trim 28 may have anti-proliferative activity within tumors. To test this hypothesis, we used shRNAi to generate Trim28-knockdown breast and lung cancer cell lines and found that Trim28 depletion led to increased cell proliferation. Likewise, overexpression of Trim28 led to decreased cell proliferation. Confocal microscopy indicated co-localization of E2F3 and E2F4 with Trim28 within the cell nucleus, and co-immunoprecipitation assays demonstrated that Trim28 can bind both E2F3 and E2F4. Trim28 overexpression inhibited the transcriptional activity of E2F3 and E2F4, whereas Trim28 deficiency enhanced their activity. Co-immunoprecipitations further indicated that Trim28 bridges an interaction between E2Fs 3 and 4 and HDAC1. Promoter-reporter assays demonstrated that the ability of HDAC1 to repress E2F3 and E2F4-driven transcription is dependent on Trim28. Trim28 depletion increased E2F3 and E2F4 DNA binding activity, as measured by chromatin-immunoprecipitation (ChIP) assays while simultaneously reducing HDAC1 binding. Finally, ChIP-ReChIP experiments demonstrated that Trim/E2F complexes exist on several E2F-regulated promoters. Taken together, these results suggest that Trim28 has anti-proliferative activity in lung cancers via repression of members of the E2F family that are critical for cell proliferation.
Introduction
Tripartite motif containing 28 (Trim28)2 (also known as KAP1, KRIP1, and TIF1β) is one of 60 members of the Trim2 (tripartite motif-containing) family of transcriptional co-factors that regulate chromatin organization (1). Trim28 appears essential for maintaining pluripotency in a stem cell model (2) and Trim28-null mice die prior to gastrulation (3). Trim28 is also necessary for the differentiation of mouse embryonic stem cells (4, 5). Paradoxically, Trim28 has also been implicated in antagonizing erythroid differentiation in adult tissues (7).
Published data suggest an oncogenic role for Trim28 in cancer. With one exception (8), microarray studies in lung cancer that have included a significant number of normal tissue samples for comparison have demonstrated a statistically significant increase in the Trim28 mRNA in tumors relative to normal tissue (9–11). Similar trends have been observed in breast cancer (see Trim28 in the Oncomine Database). The most detailed study of Trim28 mRNA expression was performed on 91 gastric cancer samples as compared with distal normal gastric tissue from the same patient using quantitative polymerase chain reaction (PCR) (12). In that study (12), the authors demonstrated that high mRNA expression correlated with significantly decreased survival and that knockdown of Trim28 in two gastric cancer cell lines resulted in reduced proliferation and sensitivity to anoikis. Interestingly, the study (12) also demonstrated that 1/3 of the gastric tumors had reduced Trim28 mRNA expression; unfortunately, the significance of this observation was not explored.
The Human Protein Atlas database reports on Trim28 protein expression in various tumors relative to adjacent normal tissues. The findings from the database have indicated that Trim28 protein is up-regulated in a variety of cancers (including breast cancer) but that it is down-regulated in other tumors (including lung cancer) relative to adjacent normal tissues. These results suggest that Trim28 may play complex roles in human cancer.
E2F1 is one of 8 members of the E2F family of transcription factors; these factors have overlapping and distinct roles in the regulation of cell proliferation and survival (13–16). Our published work has demonstrated that Trim28 can interact with and inhibit the E2F1 transcription factor (17). This E2F1/Trim28 interaction was shown to block the potent apoptosis-inducing activity of E2F1. The E2F1/Trim28 interaction was independent of members of the Rb family, but did involve the recruitment of HDAC1 to E2F1 and the regulation of E2F1 acetylation.
In the current work, we further explored the interaction of Trim28 with the E2F family and the potential role(s) that these complexes may play in cancer. In particular, we focused on lung adenocarcinomas. We believe that the results described here reconcile the apparently contradictory observations regarding the role of Trim28 in lung cancer. Specifically, we propose that Trim28 is up-regulated at the transcriptional level in lung adenocarcinomas relative to normal in an “attempt” to restrain cell growth. In patients where this attempt is successful, overall survival is longer because the tumor growth is restrained due to the ability of Trim28 to restrain the growth-promoting activity of E2F3 and E2F4.
EXPERIMENTAL PROCEDURES
Mining Public Databases
Two microarray studies using the Affymetrix platform were used to evaluate Trim28 mRNA expression (Affymetrix probe set 200990_at) in non-small cell lung cancer (NSCLC) patients and in normal tissues. The study of Hou et al. (11) included 91 NSCLC patients (45 adenocarcinomas, 27 squamous cell carcinomas, and 19 large cell carcinomas) and 65 paired normal tissues. This database allowed us to determine whether Trim28 has differential expression between tumor and normal tissue, as well as among the three histological types of NSCLC. The study of Shedden et al. (18) encompassed 442 lung adenocarcinoma patients, including 114 with stage IA. This database allowed us to determine if Trim28 mRNA expression within tumors was associated with overall survival.
Tissue Array and AQUA
Our tissue microarray and automated quantitative immunofluorescence analysis (AQUA) procedures have been described previously (19–21). The tissue array included a total of 186 samples (n = 85 for stage IA and n = 101 for stage IB). Immunofluorescence conditions were optimized using control H1299 cells compared with a stable Trim28 knockdown H1299 cell line. Trim28 (green) was detected using anti-Trim28 primary antibody (Bethyl Laboratories) and a goat anti-rabbit Alexa 488 conjugate as secondary antibody. DAPI was used to stain nuclei (blue), and anti-cytokeratin (red, goat anti-rabbit Alexa 455 conjugate) was used to identify epithelial cells and to exclude stromal cells.
Statistical Methods
Three datasets were analyzed: one tissue array and two microarray datasets (11, 18). For the tissue array, a square-root transformation was performed to normalize AQUA-generated Trim28 scores. For the microarray data, the Affymetrix probeset, 200990_at, was used to measure Trim28 expression. One-way ANOVA (with Tukey's method to adjust for post-hoc comparison) was used to test for group differences for categorical variables (such as histology). Spearman correlation analysis was used to test for trends in continuous or ordinal variables (such as tumor size and smoking status). To assess overall survival, Trim28 expression was dichotomized into low and high groups based on the median value of all measurements. Survival curves were generated according to Kaplan-Meier, and the log-rank test was used to test for survival differences between the low- and high-Trim28 groups. Student's t test was used to compare values obtained in laboratory experiments. A two sided p value < 0.05 was considered statistically significant. All statistical analyses were performed using R software.
Cell Lines
Original cell lines were obtained from the ATCC. MDA-MB-231, 293FT, and A549 cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) with 10% fetal bovine serum. H1299 cells were cultured in DMEM supplemented with 5% fetal bovine serum. H1299 cells were used for transient transfection experiments due to their reliable transfection efficiency. To produce Trim28 shRNA lentivirus, 293FT cells were transfected with pCMV-VSV-G, pCMV-Δ8.2, and lentiviral plasmid pLKO.1-scramble or pLKO.1-TRIM28 shRNA (TRCN0000017998). Culture supernatant was collected 48 and 72 h post-transfection and passed through 0.45-μm filters. Aliquots of lentiviral stocks were stored at −80 °C. MDA-MB-231, A549, and H1299 cells were infected with shRNA lentiviruses, and 1 μg/ml puromycin was added 72 h after infection. For the MDA-MB-231 cells, pools of puromycin-resistant cells were utilized, whereas for A549 and H1299 cells, individual clones were derived by colony selection. Specifically, cells were selected with puromycin for 3 weeks, and then individual puromycin-resistant A549 clones were derived and screened for Trim28 expression by Western blot. Control cell lines derived using the empty vector were similarly derived. To generate control and Trim28 overexpressing cell lines, A549 cells were transfected with pcDNA3 or pcDNA-FLAG-HA-TRim28 and selected in G418. Single clones were screened for Trim28 expression.
Growth and Colony Formation Assays
For growth assays, 1000 cells were seeded per well in a 24-well plate in triplicate. Cells were incubated at 37 °C in a humidified incubator for different periods of time. After incubation, cells were washed with PBS and trypsinized and counted. For colony formation assays, 5000 cells were seeded in a p100 plate in triplicate and incubated at 37 °C in a humidified incubator for 7–10 days. After incubation, cells were washed with PBS once and stained with 0.5% crystal violet for 1 min at room temperature. Cells were washed with PBS, and images were scanned using an Epson Expression 1680 scanner. Ten percent acetic acid was added to each plate and incubated with shaking for 20 min. One-half ml of supernatant was diluted 1:4 in water and its absorbance measured at 590 nm.
Plasmid Vectors
The pLKO.1-TRIM28 shRNA set (TRCN0000017998, TRCN0000017999, TRCN0000018000, TRCN0000018001, TRCN0000018002) was purchased from Open Biosystems. pcDNA3-FLAG-HA-TIF1β and pcDNA3.1-Myc-TIF1β (22) were kind gifts from Dr. Muriel Aubry (Université de Montréal). pcDNA3-E2F3 (23), pcDNA3-E2F4 (23), pGL3-p27 (24) and pGL2-E2F1 (25) have been previously described.
The novel expression plasmids pCMV-3xFLAG-E2F3 and pCMV-3xFLAG-E2F4 were generated by PCR using primers: E2F3 forward: 5′-AGCAAGCTTATGAGAAAGGGAATCCAG-3′, E2F3 reverse: 5′- ATAGAATTCTCAACTACACATGAAGTC-3′; E2F4 forward: 5′-ATCAAGCTTATGGCGGAGGCCGGGCCA-3′, E2F4 reverse: 5′-TATGAATTCTCAGAGGTTGAGAACAGG-3′. The products were cleaved with HindIII and EcoRI and cloned into those sites of pCMV-3xFlag-myc-26 vector (E6401, Sigma). pEGFP-E2F4 was constructed by subcloning an E2F4 expression cassette into pEGFP-C2 (Clontech).
Adenovirus
Ad-green fluorescent protein (GFP) (28), Ad-E2F1 (28, 29), Ad-E2F3A, and Ad-E2F4 (29) have been previously described, as referenced. Viruses were propagated in 293FT cells and titrated with the Adeno-X Rapid Titer Kit (BD Biosciences, 631028). Western blots verified absence of E1A expression (to ensure non-recombination).
Luciferase Assays
Cells (50,000/well) were cultured in 24-well plates and transfected with a mixture containing 500 ng of expression plasmid or control vector (pcDNA3), 100 ng of firefly luciferase reporter plasmid (pGL2/pGL3-based vectors, Promega), 10 ng of Renilla luciferase reporter (pRL-TK, Promega), and carrier DNA (sheared salmon sperm DNA) to an equal amount of total DNA in each transfection. Cells were harvested 48 h after transfection, and luciferase assays were performed using the Dual-Luciferase Reporter Assay System following the manufacturer's protocol (Promega). Experiments were done in triplicate. To control for transfection efficiency, firefly luciferase values were normalized to the values for Renilla luciferase.
Analytical Methods
Immunoprecipitation and immunoblot experiments were performed as previously described (30). E2F1 (sc-193), E2F3 (sc-878), E2F4 (sc-866), p107 (sc-318), and GFP (sc-9996) antibodies were purchased from Santa Cruz Biotechnology. Flag (F3165) and β-actin (A5441) antibodies were from Sigma. Myc (#2276) antibody was from Cell Signaling Technology. Trim28 antibody (A300–275A) was from Bethyl Laboratories. Anti-cyclin-D3 (C28620) and anti-p27 (610242) were purchased from BD Transduction Laboratories.
Confocal Microscopy
Cells were seeded in a Lab-TakTM eight-chamber slide (Thermal Scientific) 1 day before experimentation. After they were rinsed twice in PBS, cells were fixed in 4% paraformaldehyde solution followed by permeabilization in 0.1% Triton X-100/PBS and incubation in 10% SDS/PBS. Cells were then sequentially incubated in 10% bovine serum albumin, primary antibodies, and secondary antibodies with DAPI. E2F3 c-18 (Santa Cruz Biotechnology), E2F4 A302–133A (Bethyl Laboratories), and Trim28 ab22553 (Abcam) were used as primary antibodies. Goat anti-mouse IgG-FITC (Santa Cruz Biotechnology) and AlexaFluor 594-conjugated goat anti-rabbit (Invitrogen) were used as secondary antibodies. After incubation in secondary antibodies, cells were washed with PBS three times and covered with Vectasheild mounting media (Vector Labs) and coverslips. Cells were imaged by the Microscopy Core at Moffitt Cancer Center using a Leica SP5 AOBS tandem scanning inverted confocal microscope.
Q-RT-Quantitative Real-time PCR
Total cell RNA was harvested using the RNeasy Mini Kit (Qiagen) following the manufacturer's instructions. Reverse transcription reactions were carried out using iScript cDNA Synthesis Kit (Bio-Rad). Real-time PCR was performed using Bio-Rad iQ SYBR Green Supermix on a MyiQ Single Color real-time PCR detection system.
The following primers were used: CDC25A forward: 5′-GAGGTGAAGAACAGTAATC-3′, reverse: 5′-TGGTCAAGAGAATCAGAATGG-3′; CCNA2 forward: 5′-TGGGAGAATTAAGTTTGATAGATG-3′, reverse: 5′-GAGGTCGGTCTGGTGAAGG-3′; CCND3 forward: 5′-GCTGGAGGTATGTGAGGAG-3′, reverse: 5′-CGTGGTCGGTGTAGATGC-3′; p27 forward: 5′-CAGGAGGACCAGGATGTC-3′, reverse: 5′-TAGAAGAATCGTCGGTTGC-3′; DHFR forward: 5′-TCTTGCTATAACTAAGTGCTTCTC-3′, reverse: 5′-TGTGATGGGTGTGAAATGG-3′; p21 forward: 5′-GGGATGAGTTGGGAGGAG-3′, reverse: 5′-GGTACAAGACAGTGACAGG-3′; GAPDH forward: 5′-GAGTCAACGGATTTGGTCGT-3′; reverse: 5′-TTGATTTTGGAGGGATCTCG-3′.
Chromatin Immunoprecipitation (ChIP) Assay and ChIP-ReChIP
ChIP assays were performed as previously described (30). Briefly, H1299 cells were treated with formaldehyde to create protein-DNA cross-links, and the cross-linked chromatin extracted and sheared by sonication. Protein A beads were precleared and blocked with 1% salmon sperm DNA and 1% bovine serum albumin. Total sheared chromatin corresponding to 4.5 × 106 cells was used for immunoprecipitation with either 5 μg of normal rabbit IgG (12–370, Millipore), 5 μg of anti-E2F3 (C-18, Santa Cruz Biotechnology), 5 μg of anti-E2F4 (C-20, Santa Cruz Biotechnology), 5 μg of anti-TRIM28 (A300–275A, Bethyl Laboratories), 3 μg of anti-acetyl-histone H3K9 (07–352, Millipore), or 5 μg of anti-HDAC1 (ab7028, Abcam). The immunoprecipitates were washed five times, pelleted by centrifugation, and then heated at 65 °C for 4 h to de-crosslink. For ChIP-reChIP, DNA-protein complexes were washed five times and eluted with 20 mm DTT at 37 °C for 30 min and re-subjected to immunoprecipitation. The immunoprecipitates were washed, eluted, and de-crosslinked. After proteins and RNA were degraded by treatment with proteinase K and RNase A, DNA was purified using QIAquick PCR Purification Kit (Qiagen). Real-time PCR was performed using Bio-Rad iQ SYBR Green Supermix on a MyiQ Single Color real-time PCR detection system.
The following PCR primers were used in real-time PCR: CDC25A forward: 5′-AGAAGTTGCTTACTGATTGG-3′, CDC25A reverse: 5′-CGACCTACACCTCTTACC-3′; CDC25A control forward: 5′- TCTACCTCCTTCAGGGCTCA-3′, CDC25A control reverse: 5′-TTGCTGAAGAGTTTGGCCTT-3′; RBL1 forward: 5′-GGAGGCATCTCACTACGC-3′, RBL1 reverse: 5′-TTCCTGGTTAGGCTCTTGG-3′; RBL1 control forward: 5′-GAGGGAGAGGTTGCAGTGAG-3′, RBL1 control reverse: 5′-TTTTGTCCTCCAGCTTTGCT-3′; CCNE2 forward: 5′-ACTTCCCAGCCACCCTATG-3′, CCNE2 reverse: 5′-CATCTCCCGCCAGTTTGC-3′; CCNE2 control forward: 5′-CCACCATCCGTCTATCATCC-3′, CCNE2 control reverse: 5′-TATTTCCAACTCGACCTCGG-3′. Human myoglobin exon 2 primers used as control were purchased from Diagenode (pp-1006–500).
RESULTS
High Tumor Trim28 Correlates with Increased Overall Survival in Early-stage Adenocarcinoma
We examined the expression of Trim28 mRNA using a publically available database (GSE19188), which included 91 NSCLC tumors and 65 adjacent (paired) normal tissues (11). Results in Fig. 1A reveal that Trim28 mRNA is significantly up-regulated in adenocarcinomas, squamous cell carcinomas, and large cell carcinomas relative to normal tissue (p = 0.0003, <0.0001, and <0.0001, respectively). Furthermore, large cell carcinomas had significantly higher levels of Trim28 mRNA than either adenocarcinomas or squamous cell carcinomas (p < 0.0001 and 0.002, respectively).
FIGURE 1.
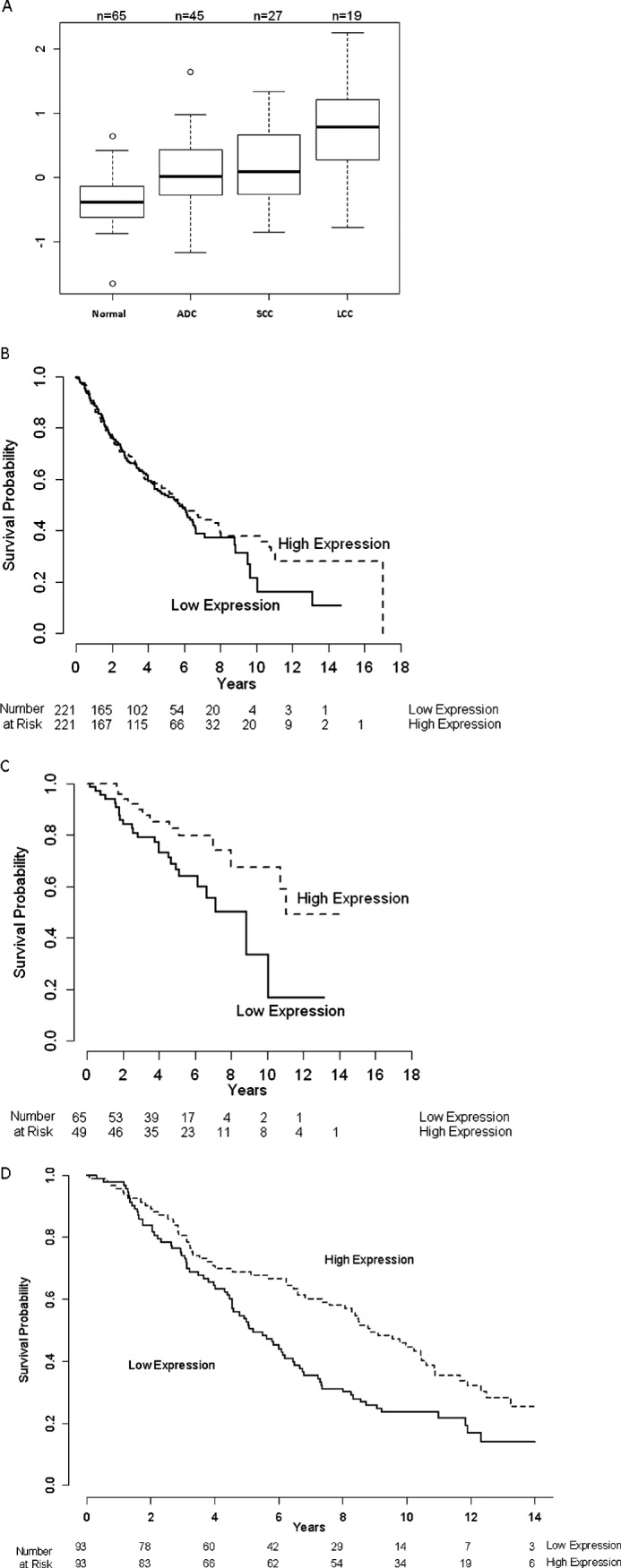
Trim28 levels increase in NSCLC tumors relative to normal tissue, but high Trim28 in early-stage tumors increases overall survival. A, data from (11) were examined. Results indicate that all three NSCLC tumor types have elevated Trim28 mRNA relative to adjacent normal tissue and that large cell carcinoma (LCC) has higher Trim28 than adenocarcinoma (ADC) and squamous cell carcinoma (SCC). p values are as follows: normal-ADC (p = 0.0003), normal-SCC (p < 0.0001), normal-LCC (p < 0.0001), ADC-LCC (p < 0.0001), SSC-LCC (p = 0.002), and ADC-SCC (p = 0.77). B, examination of microarray data from (18), which included a mixture of adenocarcinoma stages, reveals nonsignificant association between Trim28 mRNA expression with overall survival (p = 0.42). C, survival in Stage 1A tumors with high Trim28 is increased (p = 0.02). D, Trim28 protein levels were measured using AQUA in an early stage lung cancer TMA (see “Experimental Procedures”). Results show significant positive association between overall survival and Trim28 levels (p = 0.003).
Next, we examined the correlation between Trim28 expression and overall survival in a published dataset (18). When the entire database was considered, we did not see a significant correlation between Trim28 mRNA expression and overall survival (p = 0.42; Fig. 1B). However, when the data were separated into stages, we discovered that, within stage 1A tumors, high Trim28 correlated with increased overall survival (p = 0.02; Fig. 1C), suggesting that Trim28 might have a tumor suppressor role in the early stages of adenocarcinoma development.
To test the possibility that Trim28 expression might play a tumor suppressor role in early-stage adenocarcinomas, we utilized a previously described tissue microarray (TMA) corresponding to early -stage adenocarcinoma patients (19–21) to test the hypothesis that Trim28 expression might play a tumor suppressor role in these early stage tumors. This array consists of 85 stage IA adenocarcinomas and 102 stage 2B tumors. Immunofluorescence and AQUA analyses were used to assess expression levels of Trim28, as previously described for other markers (21, 31). Our statistical analysis of the TMA data (Fig. 1D) indicated that patients with high Trim28 protein have a significantly longer overall survival (p = 0.003) than patients with lower levels. Our subgroup analysis also showed similar positive correlations between overall survival and Trim28 levels (p = 0.03 for stage IA and p = 0.04 for stage IB). However, we found no statistically significant correlations between Trim28 protein levels and tumor histology, pathological stage, smoking status, tumor size, or number of pack-years of smoking (all p values >0.05).
Trim28 Slows Cell Proliferation
The observation that high Trim28 levels correlated with increased patient survival suggested that Trim28 might have anti-proliferative activity. To test this hypothesis, we depleted Trim28 from two fast-growing cell lines (MDA-MB-231 breast cancer cells and A549 lung cancer cells) using shRNAi. Control cells were derived alongside the Trim28 knockdown lines using a non-silencing shRNAi vector as control. RT-PCR assays were used to measure the levels of Trim28 mRNA in control (empty shRNAi vector) and Trim28 shRNAi-derived cells. As shown in Fig. 2A, our data clearly demonstrated that Trim28 mRNA was efficiently reduced in the knockdown cell lines. Likewise, Western blot analysis (Fig. 2B) demonstrated a very efficient down-regulation of Trim28 protein. As a first estimation of cell growth rate, we plated at low density and monitored growth by counting viable cells on a daily basis for 6 days. Fig. 2C demonstrates that Trim28-deficient cells grew significantly faster that control cell lines. As a second measure of cell proliferation, we plated at very low density and measured the ability to form colonies after 10 days. Resulting colonies were stained with 0.5% crystal violet and counted/quantified. Fig. 2D clearly demonstrates that the Trim28-deficient cells formed colonies much more efficiently than controls. To determine if Trim28 overexpression could reduce cell growth, a Trim28-overexpressing A549 cell line was generated. As evidenced by Fig. 2E, Trim28 overexpression significantly slowed cell proliferation in both growth and colony-formation assays.
FIGURE 2.

Trim28 knockdown in breast and lung cancer cells accelerates cell growth. A and B, two cell lines (breast MDA-MB-231 and lung A549) were depleted of Trim28 using shRNAi. Protein and RNA extracts of control and Trim28 knockdown cells were subjected to real-time PCR (A) and Western blotting (B). C, growth rates of control cells and Trim28 knockdown cells were determined. One thousand cells per well were plated in 24-well plates in 18 wells on day 0. Three wells were harvested by trypsin, collected, and counted each day. Plotted numbers represent the average. Cells approached ∼80% confluency by day 6. Error bars represent the S.D. of three independent experiments. D, colony formation assays were conducted on control and Trim28 knockdown cell lines. Photographs are of representative plates. For quantification, colonies were stained with crystal violet and then washed with 10% acetic acid. The absorbance was measured at 590 nm. Error bars represent the S.D. of three independent experiments, and p values relative to controls are indicated. E, pcDNA3 and Trim28 stable A549 cells were subjected to Western blotting. Growth and colony formation assays were conducted on these two stable cells. Asterisks represent significant p values: *: p < 0.05.
Trim28 Interacts with E2F3 and E2F4
Previous work has demonstrated that Trim28 can interact with E2F1 and repress its ability to drive apoptosis independent of Rb (17). However, E2F1 is only one member of a family of E2Fs, and numerous experiments have suggested that E2F1 is not essential for proliferation but rather plays a specialized role in linking the E2F pathway to apoptosis (32, 33). For these reasons, an interaction with E2F1 would not likely explain Trim28's anti-proliferative activity. In contrast, interactions with E2F3 would be more likely to explain a tumor suppressor activity for Trim28. In particular, E2F3 is considered the only member of the E2F gene family that is individually essential for proliferation (34, 35). For these reasons, we sought to test the hypothesis that Trim28 might regulate cellular proliferation via interaction with E2F3.
E2F1 and E2F3 have for some time been placed in of a group of E2Fs considered the “activating E2Fs”, whereas E2F4 is a member of a group of E2Fs considered the “repressing E2Fs” (36). Although these distinctions are somewhat artificial, they provide a basic framework for the general classification of the E2F family. By way of comparison with E2F3, we also examined Trim28 interaction with E2F4 as a representative member of the repressive E2Fs. Because of limitations of reagents, we did not investigate Trim28 interaction with E2Fs 5 through 8.
As a first step in this analysis, H1299 cells were transfected with expression vectors for a Myc epitope-tagged expression vector for Trim28 (Myc-Trim28) with and without co-transfection with expression vectors for E2Fs 3, specifically E2F3A (37–39), and 4. Extracts of transfected cells were then subjected to immunoprecipitation (IP). Fig. 3, A and B demonstrate that Myc-Trim28 can co-immunoprecipitate both E2F3 and E2F4 and that antibodies against E2F3 and E2F4 can co-immunoprecipitate Trim28. Fig. 3C demonstrates that endogenous Trim28 interacts with both E2F3 and E2F4.
FIGURE 3.

Trim28 forms complexes with E2F3 and E2F4. A and B, H1299 cells were transiently transfected with expression plasmids for E2F3, E2F4, or Myc-Trim28 (Myc-epitope tagged), as indicated. Whole cell lysates (WCL) of transfected cells were subjected to immunoprecipitation (IP) and then immunoblotting (IB). C, H1299 cells were transfected with expression plasmids for E2F3 or E2F4 at different amount (+: 2 μg; +++: 6 μg) or pcDNA3 (−) as indicated. WCLs of transfected cells were subjected to IP and then IB. D, A549 cells were stained (as described under “Experimental Procedures”) and examined using confocal immunofluorescence microscopy, as follows: E2F3 (red, top row), E2F4 (red, bottom row), Trim28 (green), and DAPI (blue; nuclei). Merged images are in the last column, yellow represents co-localization of E2Fs and Trim28.
We next sought to determine if Trim28 and E2Fs 3 and 4 interact in intact whole cells in absence of overexpression or cell lysis, either of which might allow unnatural associations. For this, we used confocal immunofluorescent microscopy in which whole cells were simultaneously stained with Trim28 antibody, an E2F antibody and DAPI (to identify the cell nucleus). Fig. 3D highlights the individual images for each stain as well as the merged image in which yellow indicates E2F/Trim 28 co-localization. E2F3 and Trim28 are both exclusively nuclear, whereas a significant fraction of E2F4 is found in the cytoplasm, as reported (40). The merged confocal image clearly indicates a very strong colocalization of Trim28 with E2F3 and significant colocalization with nuclear E2F4.
Trim28 Represses E2F-driven Transcription
Trim28 is generally considered to be a transcriptional co-repressor via the recruitment of histone-modifying enzymes, including histone acetylases and histone methyltransferases (SETDB1) (1, 42–46). However, under certain circumstances, Trim28 functions as a co-activator (6, 47, 48). To address whether Trim28 interaction with E2Fs increases or decreases their transcriptional activity, we examined the activity of three E2F family members (expressed using adenovirus) in control H1299 cells and in H1299 cells stably depleted of Trim28 by virtue of a shRNA. Fig. 4A demonstrates that H1299 control cells express significant Trim28, whereas Trim28 is essentially undetectable in the cells expressing the Trim28 shRNAi. Fig. 4A also demonstrates that the individual viruses expressed the expected E2F protein and did not affect the expression of Trim28 (present in one cell line and absent in the other) or p107 (a member of the Rb family used here simply as a control). Extracts of these same cells were next subjected to quantitative real-time PCR to measure the levels of six well-characterized E2F-regulated transcripts. Fig. 4B demonstrates that Trim28 deficiency almost universally enhanced the transcriptional activity of each E2F family on these model promoters. One exception is the p21 promoter, which was down-regulated in the Trim28-deficient cells. This observation suggests that Trim28 may down-regulate the p21 promoter in an E2F-independent manner.
FIGURE 4.

Trim28 and HDAC1 cooperate to repress E2F3- and E2F4-driven transcription. A, control and Trim28 knockdown H1299 cells were transfected in triplicate with the indicated promoter-reporter, pRL-TK reporter, and protein expression plasmids. Cells were harvested 48 h after transfection for determination of luciferase levels. B, H1299 cells were transfected in triplicate with the indicated promoter-reporter, pRL-TK reporter, and protein expression plasmids. Cells were harvested 48 h after transfection for determination of luciferase levels. C and D, control and Trim28 knockdown cells were infected with the indicated adenoviruses. The expression of individual proteins was verified by Western blotting with E2F-specific antibodies (minor cross-reactivity is evident in C). The expression of target genes was determined by real-time PCR (D). Asterisks represent significant p values, as follows: *: p < 0.05, **: p < 0.01, and ***: p < 0.005.
To test whether the changes in mRNA levels observed in Fig. 4B were promoter-driven, plasmids expressing E2F3 and E2F4 were co-transfected into these two H1299 cell lines along with a p27 (24) or cyclin D3 promoter-reporter vector, which we have previously demonstrated to be E2F regulated (26) and which also showed strong E2F regulation in Fig. 4B. Fig. 4C demonstrates that transfection of either E2F3 or E2F4 can significantly activate both of these promoters and that this activation is greater in cells lacking Trim28.
Our previous work suggested that HDAC1 may play a role in E2F1-Trim28-mediated transcriptional repression (17). To test this with regard to E2F3 and E2F4, H1299 cells were co-transfected with E2F promoter reporters together with various combinations of E2F, HDAC, and Trim28 expression vectors. Fig. 4D reveals that either Trim28 or HDAC1 can individually inhibit E2F3- or E2F4-driven transcription slightly. However, Trim28 and HDAC1 together are more efficient than either individual repressor alone.
Trim28 Promotes the Interaction between HDAC1 and E2Fs 3 and 4
Next, we wanted to ask whether the ability of HDAC1 to repress E2F3- and 4-driven transcription was dependent on Trim28. To answer this question, we used the Trim28-deficient H1299 cell line. Fig. 5A reveals that depletion of Trim28 completely abolished the interactions between HDAC1 and both E2F3 and -4. Likewise, Fig. 5B reveals that increased expression of Trim28 dramatically increased the total amount of HDAC1 associated with either E2F3 or -4.
FIGURE 5.

Trim28 promotes HDAC1/E2F3 and HDAC1/E2F4 interactions. A, control and Trim28 knockdown cells were transfected with the indicated expression plasmids. Whole cell lysates (WCL) of transfected cells were subjected to immunoprecipitation (IP) and then immunoblotting (IB). B, H1299 cells were transfected with the indicated expression plasmids. WCLs of transfected cells were subjected to IP and then IB. C, control and Trim28 knockdown H1299 cells were transfected in triplicate with the indicated promoter-reporter, pRL-TK reporter, and protein expression plasmids. Cells were harvested 48 h after transfection for determination of luciferase levels. Asterisks represent significant p values, as follows: *: p < 0.05, **: p < 0.01, and ***: p < 0.005.
To determine the functional consequences of the HDAC/Trim28/E2F interactions observed in Fig. 5, A and B, we asked whether HDAC1's ability to repress E2F-driven transcription is dependent on Trim28. Cells proficient (empty shRNAi) and deficient in Trim28 expression (Trim28 shRNAi) were transfected with an E2F-regulated promoter reporter (see Fig. 5C) and combinations of HDAC1 and E2F expression vectors. Fig. 5C reveals that E2Fs dramatically up-regulated both the p27 and cyclin D3 promoter independent of Trim28 expression. In Trim28-expressing cells, HDAC1 significantly reduced the ability of E2F to activate transcription. In contrast, in cells with reduced Trim28, HDAC1-mediated repression was minimal (p27 reporter) or non-existent (cyclin D3 reporter) in cells with reduced Trim28.).
Trim28 Depletion Increases the Association of E2F3 and -4 with DNA in Vivo, while Decreasing the HDAC Interaction
Next, we sought to establish the potential functional consequences of the E2F/HDAC interaction. We considered two alternative mechanisms (although these two mechanisms are not necessary mutually exclusive). First, it is possible that HDAC is recruited to promoters by E2F/Trim28 to deacetylate histones, thereby repressing transcription by an active process. Alternatively, HDAC1 may inhibit E2F-driven transcription by deacetylating the E2F, thereby decreasing its affinity for DNA (49) and reducing E2F-driven transcriptional activation.
ChIP assays were used to determine whether Trim28 affected the association of E2F3 and E2F4 with E2F-regulated promoters. Promoters for this analysis were chosen based on previous genome-wide screens that identified these as both E2F (50) and Trim28-associated (41). Fig. 6A highlights results of ChIP assays using antibodies against E2F3 and -4 and PCR primers for the CDC25A, RBL1, and cyclin E2 promoters in Trim28-proficient and -deficient cells. Clearly Trim 28 deficiency increases the association of both E2F3 and E2F4 with these promoters. We also included primers that target 2 kb upstream of the E2F binding site on these promoters as controls; E2F3 and E2F4 showed only limited binding to these regions.
FIGURE 6.

Trim28 influences E2F3, E2F4, and HDAC1 DNA occupancy in vivo. A, C, and D, ChIP assays were conducted in control and Trim28 knockdown cell lines. Quantitative PCR was performed to examine the occupancy of IgG, E2F3, E2F4, Ac-H3K9, HDAC1, and Trim28 proteins on E2F binding sites of CDC25A, RBL1, and CCNE2 promoters as well as 2 kb upstream of E2F binding sites on CDC25A, RBL1, and CCNE2 promoters. Additionally, in this experiment, results were normalized to relative occupancy over the occupancy of the myoglobin intron 2. B, ChIP-reChIP was conducted in H1299 cells. DNA-protein complexes were immunoprecipitated with E2F3 and E2F4 antibodies followed by immunoprecipitating with Trim28 antibody. Quantitative PCR was performed as described above. Asterisks represent significant p values, as follows: *: p < 0.05, **: p < 0.01, and ***: p < 0.005.
To confirm that E2Fs and Trim28 are simultaneously recruited to promoters, we performed a ChIP-reChIP experiment in which DNA-protein complexes are first ChIPed with E2F antibodies, washed and then reChIPed with Trim28 antibody. As shown in Fig. 6B, Trim28 and E2F3/4 reside on the same regions on the CDC25A, RBL1, and CCNE2 promoters, whereas no interaction is detected when distal upstream primers are used for DNA detection.
We next used the ChIP assay to determine whether Trim28 affects the recruitment of HDAC1 to the CDC25A, RBL1, and CCNE2 promoters, as would be predicted from the results shown in Fig. 5. Fig. 6C reveals that HDAC1 association with these promoters is indeed dramatically reduced in the absence of Trim28. Finally, we asked whether the reduction of HDAC1 association with the E2F/Trim28 regulated promoter would result in their increased histone acetylation. Indeed, Fig. 6D demonstrates a significant increase in histone H3 acetylation in the absence of Trim28. Because histone H3 acetylation generally coincides with increased promoter activity, this observation is consistent with the model that Trim28 represses E2F-driven transcription by recruiting HDAC1, which in turn deacetylates histone H3 reducing promoter activity.
DISCUSSION
Herein, we demonstrate that early-stage adenocarcinoma patients with high levels of Trim28 have a longer overall survival than patients with low Trim28. This observation comes in the midst of numerous studies that demonstrate that the Trim28 message is in fact elevated in tumor tissue relative to normal tissues. We believe this report reconciles these seemingly contradictory observations and adds significant clarity to the mechanisms by which Trim28 participates in cell cycle control and tumorigenesis, at least with respect to lung cancer. We believe our results show that Trim28 is up-regulated in early tumors in an “attempt” to restrain cell growth. We propose that patients in whom this attempt is successful in the early stages of disease have better overall survival because the tumor cells have reduced proliferative capacity.
The up-regulation of Trim28 in tumor tissue is likely transcriptional, but we have not yet investigated the mechanism. An obvious hypothesis would be that Trim28 might be E2F-regulated, as is the cdk inhibitor p27 (24), and functions as a negative feedback loop in circumstances in which the E2F pathway is regulated. However, Trim28 does not appear to be cell cycle regulated and does not appear to be altered or influenced significantly by E2F or Rb depletion (unpublished observations). Future work will address the mechanism of Trim28 up-regulation in tumors.
This report also expands our previous analysis (17) of the interaction between Trim28, HDAC1, and the E2F family. We demonstrate that Trim28 can bind both E2F3 and E2F4 and inhibit their transcriptional activity on numerous endogenous E2F-regulated promoters and in the context of promoter/reporter assays. Co-immunoprecipitation assays indicated that Trim28 bridges an interaction between E2Fs 3 and 4 and HDAC1 since Trim28 deficiency dramatically reduced the amount of HDAC1 associated with E2Fs 3 and 4. Promoter/reporter assays demonstrated that the HDAC1 ability to repress E2F3 and E2F4-driven transcription is dependent on Trim28. Trim28 depletion increased E2F3 and 4 DNA binding activity as measured by ChIP assays while simultaneously reducing HDAC1 binding. Recruitment of HDAC1 to E2F-regulated promoters is likely responsible, at least in part, for the promoter down-regulation. This conclusion is supported by the observation that Trim28 deficiency reduced the amount of HDAC1 associated with E2F-regulated promoters, leading to significantly increased histone H3 acetylation, which generally correlates with increased transcriptional activity. Taken together, these results suggest that Trim28 has anti-proliferative activity in lung cancers via repression of members of the E2F family that are critical for cell proliferation.
This work was supported, in whole or in part, by NCI, National Institutes of Health Grants CA119997, CA129343, CA163068, and CA118809 and the Moffitt Cancer Center (to W. D. C. and L. C.).
- Trim28
- tripartite motif containing 28
- AQUA
- automated quantitative immunofluorescence analysis
- NSCLC
- non-small cell lung cancer.
REFERENCES
- 1. Iyengar S., Farnham P. J. (2011) KAP1 protein: an enigmatic master regulator of the genome. J. Biol. Chem. 286, 26267–26276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hu G., Kim J., Xu Q., Leng Y., Orkin S. H., Elledge S. J. (2009) A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 23, 837–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cammas F., Mark M., Dollé P., Dierich A., Chambon P., Losson R. (2000) Mice lacking the transcriptional corepressor TIF1β are defective in early postimplantation development. Development 127, 2955–2963 [DOI] [PubMed] [Google Scholar]
- 4. Cammas F., Oulad-Abdelghani M., Vonesch J. L., Huss-Garcia Y., Chambon P., Losson R. (2002) Cell differentiation induces TIF1β association with centromeric heterochromatin via an HP1 interaction. J. Cell Sci. 115, 3439–3448 [DOI] [PubMed] [Google Scholar]
- 5. Cammas F., Herzog M., Lerouge T., Chambon P., Losson R. (2004) Association of the transcriptional corepressor TIF1beta with heterochromatin protein 1 (HP1): an essential role for progression through differentiation. Genes Dev. 18, 2147–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deleted in proof
- 7. Nakamura Y., Yamagata T., Maki K., Sasaki K., Kitabayashi I., Mitani K. (2006) TEL/ETV6 binds to corepressor KAP1 via the HLH domain. Int. J. Hematol. 84, 377–380 [DOI] [PubMed] [Google Scholar]
- 8. Su L. J., Chang C. W., Wu Y. C., Chen K. C., Lin C. J., Liang S. C., Lin C. H., Whang-Peng J., Hsu S. L., Chen C. H., Huang C. Y. (2007) Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics 8, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beer D. G., Kardia S. L., Huang C. C., Giordano T. J., Levin A. M., Misek D. E., Lin L., Chen G., Gharib T. G., Thomas D. G., Lizyness M. L., Kuick R., Hayasaka S., Taylor J. M., Iannettoni M. D., Orringer M. B., Hanash S. (2002) Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat. Med. 8, 816–824 [DOI] [PubMed] [Google Scholar]
- 10. Landi M. T., Dracheva T., Rotunno M., Figueroa J. D., Liu H., Dasgupta A., Mann F. E., Fukuoka J., Hames M., Bergen A. W., Murphy S. E., Yang P., Pesatori A. C., Consonni D., Bertazzi P. A., Wacholder S., Shih J. H., Caporaso N. E., Jen J. (2008) Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS ONE 3, e1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hou J., Aerts J., den Hamer B., van Ijcken W., den Bakker M., Riegman P., van der Leest C., van der Spek P., Foekens J. A., Hoogsteden H. C., Grosveld F., Philipsen S. (2010) Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS ONE 5, e10312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yokoe T., Toiyama Y., Okugawa Y., Tanaka K., Ohi M., Inoue Y., Mohri Y., Miki C., Kusunoki M. (2009) Ann. Surg. Oncol. 17, 821–828 [DOI] [PubMed] [Google Scholar]
- 13. DeGregori J., Johnson D. G. (2006) Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Curr. Mol. Med. 6, 739–748 [DOI] [PubMed] [Google Scholar]
- 14. Johnson D. G., Degregori J. (2006) Putting the oncogenic and tumor suppressive activities of E2F into context. Curr. Mol. Med. 6, 731–738 [DOI] [PubMed] [Google Scholar]
- 15. Chen H. Z., Tsai S. Y., Leone G. (2009) Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat. Rev. Cancer 9, 785–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lammens T., Li J., Leone G., De Veylder L. (2009) Atypical E2Fs: new players in the E2F transcription factor family. Trends Cell Biol. 19, 111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang C., Rauscher F. J., 3rd, Cress W. D., Chen J. (2007) Regulation of E2F1 function by the nuclear corepressor KAP1. J. Biol. Chem. 282, 29902–29909 [DOI] [PubMed] [Google Scholar]
- 18. Shedden K., Taylor J. M., Enkemann S. A., Tsao M. S., Yeatman T. J., Gerald W. L., Eschrich S., Jurisica I., Giordano T. J., Misek D. E., Chang A. C., Zhu C. Q., Strumpf D., Hanash S., Shepherd F. A., Ding K., Seymour L., Naoki K., Pennell N., Weir B., Verhaak R., Ladd-Acosta C., Golub T., Gruidl M., Sharma A., Szoke J., Zakowski M., Rusch V., Kris M., Viale A., Motoi N., Travis W., Conley B., Seshan V. E., Meyerson M., Kuick R., Dobbin K. K., Lively T., Jacobson J. W., Beer D. G. (2008) Gene expression-based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat. Med. 14, 822–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Haura E. B., Zheng Z., Song L., Cantor A., Bepler G. (2005) Activated epidermal growth factor receptor-Stat-3 signaling promotes tumor survival in vivo in non-small cell lung cancer. Clin. Cancer Res. 11, 8288–8294 [DOI] [PubMed] [Google Scholar]
- 20. Zheng Z., Bepler G., Cantor A., Haura E. B. (2005) Small tumor size and limited smoking history predicts activated epidermal growth factor receptor in early-stage non-small cell lung cancer. Chest 128, 308–316 [DOI] [PubMed] [Google Scholar]
- 21. Zheng Z., Chen T., Li X., Haura E., Sharma A., Bepler G. (2007) DNA synthesis and repair genes RRM1 and ERCC1 in lung cancer. N. Engl. J. Med. 356, 800–808 [DOI] [PubMed] [Google Scholar]
- 22. Mascle X. H., Germain-Desprez D., Huynh P., Estephan P., Aubry M. (2007) Sumoylation of the transcriptional intermediary factor 1β (TIF1β), the co-repressor of the KRAB Multifinger proteins, is required for its transcriptional activity and is modulated by the KRAB domain. J. Biol. Chem. 282, 10190–10202 [DOI] [PubMed] [Google Scholar]
- 23. Flores A. M., Kassatly R. F., Cress W. D. (1998) E2F-3 accumulation is regulated by polypeptide stability. Oncogene 16, 1289–1298 [DOI] [PubMed] [Google Scholar]
- 24. Wang C., Hou X., Mohapatra S., Ma Y., Cress W. D., Pledger W. J., Chen J. (2005) Activation of p27Kip1 expression by E2F1. A negative feedback mechanism. J. Biol. Chem. 280, 12339–12343 [DOI] [PubMed] [Google Scholar]
- 25. Johnson D. G., Ohtani K., Nevins J. R. (1994) Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev. 8, 1514–1525 [DOI] [PubMed] [Google Scholar]
- 26. Ma Y., Yuan J., Huang M., Jove R., Cress W. D. (2003) Regulation of the cyclin D3 promoter by E2F1. J. Biol. Chem. 278, 16770–16776 [DOI] [PubMed] [Google Scholar]
- 27.Deleted in proof
- 28. Freeman S. N., Ma Y., Cress W. D. (2008) RhoBTB2 (DBC2) is a mitotic E2F1 target gene with a novel role in apoptosis. J. Biol. Chem. 283, 2353–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kowalik T. F., DeGregori J., Schwarz J. K., Nevins J. R. (1995) E2F1 overexpression in quiescent fibroblasts leads to induction of cellular DNA synthesis and apoptosis. J. Virol. 69, 2491–2500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ma Y., Chen L., Wright G. M., Pillai S. R., Chellappan S. P., Cress W. D. (2010) CDKN1C negatively regulates RNA polymerase II C-terminal domain phosphorylation in an E2F1-dependent manner. J. Biol. Chem. 285, 9813–9822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Camp R. L., Chung G. G., Rimm D. L. (2002) Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat. Med. 8, 1323–1327 [DOI] [PubMed] [Google Scholar]
- 32. Weinberg R. A. (1996) E2F and cell proliferation: a world turned upside down. Cell 85, 457–459 [DOI] [PubMed] [Google Scholar]
- 33. Leone G., Sears R., Huang E., Rempel R., Nuckolls F., Park C. H., Giangrande P., Wu L., Saavedra H. I., Field S. J., Thompson M. A., Yang H., Fujiwara Y., Greenberg M. E., Orkin S., Smith C., Nevins J. R. (2001) Myc requires distinct E2F activities to induce S phase and apoptosis. Mol. Cell 8, 105–113 [DOI] [PubMed] [Google Scholar]
- 34. Leone G., DeGregori J., Yan Z., Jakoi L., Ishida S., Williams R. S., Nevins J. R. (1998) E2F3 activity is regulated during the cell cycle and is required for the induction of S phase. Genes Dev. 12, 2120–2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chong J. L., Tsai S. Y., Sharma N., Opavsky R., Price R., Wu L., Fernandez S. A., Leone G. (2009) E2f3a and E2f3b contribute to the control of cell proliferation and mouse development. Mol. Cell. Biol. 29, 414–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Trimarchi J. M., Lees J. A. (2002) Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 3, 11–20 [DOI] [PubMed] [Google Scholar]
- 37. He Y., Armanious M. K., Thomas M. J., Cress W. D. (2000) Identification of E2F-3B, an alternative form of E2F-3 lacking a conserved N-terminal region. Oncogene 19, 3422–3433 [DOI] [PubMed] [Google Scholar]
- 38. Leone G., Nuckolls F., Ishida S., Adams M., Sears R., Jakoi L., Miron A., Nevins J. R. (2000) Identification of a novel E2F3 product suggests a mechanism for determining specificity of repression by Rb proteins. Mol. Cell. Biol. 20, 3626–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adams M. R., Sears R., Nuckolls F., Leone G., Nevins J. R. (2000) Complex transcriptional regulatory mechanisms control expression of the E2F3 locus. Mol. Cell. Biol. 20, 3633–3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lindeman G. J., Gaubatz S., Livingston D. M., Ginsberg D. (1997) The subcellular localization of E2F-4 is cell-cycle dependent. Proc. Natl. Acad. Sci. U.S.A. 94, 5095–5100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. O'Geen H., Squazzo S. L., Iyengar S., Blahnik K., Rinn J. L., Chang H. Y., Green R., Farnham P. J. (2007) Genome-wide analysis of KAP1 binding suggests autoregulation of KRAB-ZNFs. PLoS Genetics 3, e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sripathy S. P., Stevens J., Schultz D. C. (2006) The KAP1 corepressor functions to coordinate the assembly of de novo HP1-demarcated microenvironments of heterochromatin required for KRAB zinc finger protein-mediated transcriptional repression. Mol. Cell. Biol. 26, 8623–8638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Groner A. C., Meylan S., Ciuffi A., Zangger N., Ambrosini G., Denervaud N., Bucher P., Trono D. (2010) PLoS Genetics 6, e1000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schultz D. C., Ayyanathan K., Negorev D., Maul G. G., Rauscher F. J., 3rd. (2002) SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 16, 919–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schultz D. C., Friedman J. R., Rauscher F. J., 3rd. (2001) Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2α subunit of NuRD. Genes Dev. 15, 428–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ayyanathan K., Lechner M. S., Bell P., Maul G. G., Schultz D. C., Yamada Y., Tanaka K., Torigoe K., Rauscher F. J., 3rd. (2003) Regulated recruitment of HP1 to a euchromatic gene induces mitotically heritable, epigenetic gene silencing: a mammalian cell culture model of gene variegation. Genes Dev. 17, 1855–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chang C. J., Chen Y. L., Lee S. C. (1998) Coactivator TIF1β interacts with transcription factor C/EBPβ and glucocorticoid receptor to induce α1-acid glycoprotein gene expression. Mol. Cell. Biol. 18, 5880–5887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rambaud J., Desroches J., Balsalobre A., Drouin J. (2009) TIF1β/KAP-1 is a coactivator of the orphan nuclear receptor NGFI-B/Nur77. J. Biol. Chem. 284, 14147–14156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marzio G., Wagener C., Gutierrez M. I., Cartwright P., Helin K., Giacca M. (2000) E2F family members are differentially regulated by reversible acetylation. J. Biol. Chem. 275, 10887–10892 [DOI] [PubMed] [Google Scholar]
- 50. Bieda M., Xu X., Singer M. A., Green R., Farnham P. J. (2006) Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 16, 595–605 [DOI] [PMC free article] [PubMed] [Google Scholar]