

Background: ISCU myopathy is a disease caused by muscle-specific deficiency of the Fe-S cluster scaffold protein ISCU.
Results: MyoD expression enhanced ISCU mRNA mis-splicing, and oxidative stress exacerbated ISCU depletion in patient cells.
Conclusion: ISCU protein deficiency in patients results from muscle-specific mis-splicing as well as oxidative stress.
Significance: Oxidative stress negatively influences the mammalian Fe-S cluster assembly machinery by destabilization of ISCU.
Keywords: Iron, Iron-sulfur protein, Mitochondria, Mitochondrial diseases, Oxidative stress
Abstract
Iron-sulfur (Fe-S) cluster cofactors are formed on the scaffold protein ISCU. ISCU myopathy is a disease caused by an intronic mutation that leads to abnormally spliced ISCU mRNA. We found that two predominant mis-spliced ISCU mRNAs produce a truncated and short-lived ISCU protein product in multiple patient cell types. Expression of the muscle-specific transcription factor MyoD further diminished normal splicing of ISCU mRNA in patient myoblasts, demonstrating that the process of muscle differentiation enhances the loss of normal ISCU mRNA splicing. ISCU protein was nearly undetectable in patient skeletal muscle, but was higher in patient myoblasts, fibroblasts, and lymphoblasts. We next treated patient cells with pro-oxidants to mimic the oxidative stress associated with muscle activity. Brief hydrogen peroxide treatment or incubation in an enriched oxygen atmosphere led to a marked further reduction of ISCU protein levels, which could be prevented by pretreatment with the antioxidant ascorbate. Thus, we conclude that skeletal muscle differentiation of patient cells causes a higher degree of abnormal ISCU splicing and that oxidative stress resulting from skeletal muscle work destabilizes the small amounts of normal ISCU protein generated in patient skeletal muscles.
Introduction
ISCU myopathy, also known as “myopathy with deficiency of succinate dehydrogenase and aconitase,” is a rare autosomal recessive disease found in individuals of northern Swedish descent. The disease is characterized by life-long exercise intolerance in which trivial bouts of exercise can cause tachycardia and palpitations, dyspnea, muscle fatigue, and lactic acidosis (1–3). Many ISCU myopathy patients experience periods of muscle weakness, pain, and swelling associated with rhabdomyolysis and myoglobinuria followed by muscle regeneration and resolution of these symptoms (1, 4). Previous skeletal muscle biopsy analyses revealed a distinctive pattern of biochemical and histological hallmarks in ISCU myopathy, including deficiency of several mitochondrial iron-sulfur (Fe-S) proteins as well as mitochondrial iron overload in affected myofibers (4–7). The most striking deficiencies in Fe-S proteins were observed in aconitase and mitochondrial complex II/succinate dehydrogenase (SDH)2 (5, 7–9), although lesser deficiencies in mitochondrial complex I, III, and the Rieske protein were also reported (5, 8).
The human ISCU protein works in complex with the cysteine desulfurase NFS1 and the accessory proteins ISD11 and frataxin to assemble [2Fe-2S] and [4Fe-4S] clusters (10, 11), which are subsequently transferred to recipient apo-proteins aided by a chaperone complex that includes HSC20 and HSPA9 or a related HSP70 protein (12). ISCU myopathy is caused by a homozygous point mutation (g.7044 G→C) in intron 4 of the ISCU gene that results in retention of a 100-bp fragment of intron sequence between exons 4 and 5 in the ISCU mRNA and inclusion of a premature translational stop codon in the aberrant open reading frame (6, 13). More recently, a more severe progressive myopathy associated with hypertrophic cardiomyopathy, caused by the g.7044 G→C mutation on one allele in combination with a newly characterized heterozygous missense allele (c.149G→A) in ISCU exon 3, was reported (7).
Several recent studies have attempted to identify molecular features that may contribute to the unique tissue specificity and clinical phenotype of ISCU myopathy. A recent study showed that the ratio between normal and mis-spliced ISCU mRNA species in patient muscle biopsies was different from that of patient myoblasts, fibroblasts, and blood samples (14). Consistent with this, researchers reported that skeletal muscle ISCU protein levels were low in patient autopsy material as compared with heart, liver, and kidney (15), whereas a separate study reported the identity of several muscle-specific cellular RNA binding factors that influence the splicing of the mutant ISCU mRNA (16). Importantly, a biopsy of regenerating ISCU myopathy patient muscle taken soon after an episode of rhabdomyolysis showed far higher ISCU protein and SDH activity levels than a biopsy of mature muscle fibers taken 9 years later, which suggested that higher levels of functional ISCU protein might be present in newly regenerated skeletal muscle (4).
Many questions remain regarding the tissue specificity and mechanism of ISCU myopathy. What is the relationship between aberrant ISCU mRNA transcript expression and ISCU protein production in patient tissues? Are there additional factors in skeletal muscle that predispose this tissue to the pathological effects of ISCU depletion? To address these and other questions, we evaluated ISCU mRNA and protein expression in four distinct cell types derived from ISCU myopathy patients. Our findings highlight a unique and multifaceted mechanism of tissue specificity in ISCU myopathy and demonstrate that oxidative stress can directly impact the mitochondrial Fe-S cluster assembly machinery by depleting ISCU protein levels.
EXPERIMENTAL PROCEDURES
Patients, Tissue Biopsies, and Primary Cultures
Three ISCU myopathy patients were included in this study, aged 42, 42, and 66 years, as well as one 43-year-old unaffected heterozygous offspring. Controls consisted of five healthy individuals, ages 22–47 years. Skeletal muscle biopsies from vastus lateralis were obtained and prepared as described previously (6). Written informed consent was given by all individuals involved in the study in accordance with the Institutional Review Boards of the University of Texas Southwestern Medical Center and of Texas Health Presbyterian Hospital of Dallas. All primary cells were maintained in a low oxygen (5% O2) humidified atmosphere at 37 °C and 5% CO2. Primary myoblast cell lines, a generous gift from Dr. Eric Shoubridge, were cultured as described previously (17), except that Lonza SKBM-2 culture medium was used. Primary fibroblasts were cultured in DMEM medium containing 5 mm glucose and 110 mg/liter sodium pyruvate and supplemented with 10% FBS, and lymphoblasts were cultured in RPMI 1640 buffered with 25 mm HEPES and supplemented with 10% FBS. For pro-oxidant exposures with H2O2 and diethylamine-nitric oxide (DEA/NO) (Sigma), the compounds were dissolved in minimal essential medium (Invitrogen) immediately before the start of the experiment and added to cell cultures following two rinses with phosphate-buffered saline.
The lifespan of primary myoblast and fibroblast cultures was extended by infection with retrovirus LXSN16E6E7, which encodes the human papillomavirus E6 and E7 genes and the neomycin resistance cassette (18–20). Culture medium supernatants containing retrovirus were obtained from PA317 LXSN 16E6E7 cells (ATCC), sterile-filtered, and transferred directly to control and patient primary cultures daily for 3 days. All neomycin-resistant cells were harvested and pooled after 6 days of subsequent incubation with 100 μg/ml G418.
Animals
All procedures using C57BL/6 mice were approved by the NICHD Animal Care and Use Committee through the National Institutes of Health. Six-month-old animals were deeply anesthetized by injection of an isotonic pentobarbital solution containing sodium heparin, and tissues were exsanguinated by cardiac perfusion with phosphate-buffered saline (PBS), harvested, and frozen in liquid N2 immediately.
siRNA Knockdown of ISCU
A pool of siRNA oligonucleotides directed against ISCU was purchased from Thermo Scientific/Dharmacon (part number L-012837-01-0005); a pool of nontargeting oligonucleotides served as the control. Primary myoblasts were transfected with 100 nm siRNA using the NeonTM transfection system (Invitrogen), with settings of 1400 V, 20-ms pulse width, and two pulses per transfection. The cells were transfected every other day for 6 days and were harvested 2 days after the last transfection.
RNA Preparation, Northern Blots, and qRT-PCR Analysis
RNA from biopsies and cell pellets was prepared using the mirVanaTM RNA isolation kit (Ambion) according to the manufacturer's instructions. qRT-PCR was performed using SYBR® Green (Applied Biosystems) according to the manufacturer's instructions, following reverse transcription of total RNA into cDNA (Applied Biosystems). qRT-PCR primer sequences are listed in Table 1. Relative transcript abundance was calculated using the 2−ΔΔCT method (21), with GAPDH as the internal control. Correct qRT-PCR product size was verified by agarose gel electrophoresis, and automated melting curves were generated and assessed after every experiment. Northern blots were performed with 1% agarose-formaldehyde gels as reported previously (22). DNA probes were produced by random priming of template sequences generated by RT-PCR and the Klenow DNA polymerase fragment (GE Healthcare Megaprime kit), incorporating [α-32P]deoxycytidine triphosphate (6000 Ci/mmol; PerkinElmer Life Sciences). Antisense RNA probes against ISCU mRNA sequences were synthesized using the MAXIScript T7 in vitro transcription kit (Ambion) incorporating [α-32P]cytidine triphosphate (3000 Ci/mmol; PerkinElmer Life Sciences). The probe templates with T7 promoters were generated by PCR using an ISCU patient cDNA library or plasmids encoding the ISCU open reading frame. Primers used to make these probes are listed in Table 1.
TABLE 1.
PCR primers used in this study
| ISCU exon 2–4 DNA probe template | Forward | AAAATCCTAGAAACGTGGGG |
| ISCU exon 2–4 DNA probe template | Reverse | TGCAGTTTCACGGGAGGAAG |
| ISCU exon 4A–intron 4 DNA probe template | Forward | TCCAGCAGAGGAGAAAACTCAGC |
| ISCU exon 4A–intron 4 DNA probe template | Reverse | CCAGGCTTCCTGAAGAATGAGTT |
| ISCU exon 5 DNA probe template | Forward | CTGGCTGAAGATGCAATCAA |
| ISCU exon 5 DNA probe template | Reverse | CAGACCAAGGCATTCTACTG |
| ISCU exon 2–4 T7 RNA probe template | Forward | AAAATCCTAGAAACGTGGGG |
| ISCU exon 2–4 T7 RNA probe template | Reverse | TAATACGACTCACTATAGGGAGATGCAGTTTCACGGGAGGA |
| ISCU exon 4A–intron 4 T7 RNA probe template | Forward | AATCTGTGCTGTTTCCAGC |
| ISCU exon 4A–intron 4 T7 RNA probe template | Reverse | TAATACGACTCACTATAGGGAGACCAGGCTTCCTGAAGAATGAGTT |
| ISCU exon 2–3 qPCR primer | Forward | GGCTCCAGCATGTGGTGACGTA |
| ISCU exon 2–3 qPCR primer | Reverse | GCGGAACCACAGCCAAATGT |
| ISCU exon 4A qPCR primer | Forward | TCCAGCAGAGGAGAAAACTCAGC |
| ISCU exon 4A qPCR primer | Reverse | CCGACAGGAATACCATCCAGACA |
| ISCU exon 5 qPCR primer | Forward | AGCAGGCCACACCAGCTGTT |
| ISCU exon 5 qPCR primer | Reverse | GGAAGCGGCTTCTGAACATCTA |
| ISCU intron 4 qPCR primer | Forward | TAAGTCCCCACACTATCCTGGC |
| ISCU intron 4 qPCR primer | Reverse | GTTCCTTAGCCTCTTTGACCCT |
| MYOD1 qPCR primer | Forward | CGACGGCATGATGGACTACA |
| MYOD1 qPCR primer | Reverse | TGGGCGCCTCGTTGTAGTAG |
| MYH1 qPCR primer | Forward | CTAAAACTCCTGGTGCCATGGA |
| MYH1 qPCR primer | Reverse | TTCCTGCAGATGCGGATGC |
| ISCU1 ORF cloning primer | Forward | TTAAGCTTCCACCATGGTTCTCATTGACATGAGTG |
| ISCU2 ORF cloning primer | Forward | ATTAAGCTTCCACCATGGCGGCGGCTGGGGCTTTC |
| ISCU universal ORF cloning and Myc tagging primer | Reverse | CGCCTCGAGCTACAGATCTTCTTCAGAAATAAGTTTTTGTTCTTTCTTCTCTGCCTCTCCTTTTTTGGG |
Western Blots, Metabolic Labeling, and Immunoprecipitation
SDS-PAGE and Western blotting were performed as described previously (23) using 1.5-mm 4–12% precast Bis-Tris gels (Invitrogen). Mouse anti-MyoD1 and anti-SOD1 were from Abcam. Rabbit anti-citrate synthase was from Sigma-Aldrich. Mouse anti-SDH-B was from MitoSciences. Rabbit polyclonal antisera against ISCU, ISCS, IRP1, IRP2, and m-aconitase were produced from synthetic peptides as described previously (24, 25). Rabbit polyclonal anti-ferrochelatase serum was a kind gift from Dr. Harry Dailey. Metabolic labeling and immunoprecipitation were carried out essentially as described (22).
Gene Expression Constructs
The human MyoD1 ORF in vector pSPORT6 was purchased from Invitrogen, amplified in Escherichia coli, digested with EcoRI and XhoI, and subcloned into the pEBTetD mammalian episomal, tetracycline inducible expression vector (26). ISCU1 and ISCU2 ORF sequences were Myc-tagged and amplified using PCR and reported ISCU ORF plasmid templates (24), restriction-digested, and ligated into the pCEP4 episomal mammalian expression vector. Primary myoblast cell lines were transfected using the NeonTM transfection system (Invitrogen), with settings of 1400 V, 20-ms pulse width, and two pulses per transfection. On the following day, cells were permanently maintained in culture medium containing either 3 μg/ml puromycin (pEBTetD) or 150 μg/ml hygromycin (pCEP4). We found that heterologous gene expression from both plasmids in our primary cells was greatly enhanced by inclusion of 3 mm sodium butyrate in the culture medium.
Immunofluorescence Microscopy
Double immunofluorescent stainings of primary human myoblasts transfected with two different ISCU expression clones were performed following a general protocol (27) with some modifications. Briefly, equal numbers of cells were attached onto presterilized 12-mm coverslips. After washing, cells were fixed for 30 min with 4% paraformaldehyde in PBS at room temperature. Cells were washed and incubated with 0.4% Triton X-100, 4% normal goat serum, and 2% ovalbumin in PBS for blocking nonspecific binding and cell membrane permeabilization for 30 min. Then cells were treated with the endogenous biotin-blocking kit (Invitrogen) following the manufacturer's protocol. Mouse anti-Myc antibody (1/1000, Covance) mixed with either rabbit anti-SOD1 (1/300, Research Diagnostics Inc.) or rabbit anti-Tom20 (1/300, Santa Cruz Biotechnology) was applied to cells and incubated for 30 min. After washing, cells were incubated with mixture of secondary antibodies (1/400, biotin-conjugated goat anti-mouse/Alexa Fluor 488-conjugated goat anti-rabbit) for 30 min. Anti-Myc signal was detected using Cy3-streptavidin. Finally, cells were mounted with anti-fading medium containing DAPI (Vector Laboratories) and analyzed for ISCU expression in different subcellular compartments using a confocal microscope (Olympus FluoView).
Aconitase In-gel Assay and Electrophoretic Mobility Shift Assay (EMSA)
Aconitase was assayed using a coupled assay following native PAGE separation, as described previously (28). IRP-IRE binding activity was determined by EMSA using a 32P-labeled ferritin IRE probe, as described previously (28).
RESULTS
Altered ISCU mRNA Expression in ISCU Myopathy Patient Muscle Biopsies
To evaluate ISCU mRNA expression in control and ISCU myopathy patient samples, we developed DNA and RNA probes complementary to several regions of the ISCU mRNA transcript, including sequences covering exons 2–4 or exon 5, and the region encompassing the patient-specific exon 4A and adjacent 3′ intronic sequences (Fig. 1A). Northern blots demonstrated that one mRNA species predominated in control RNA extracts (transcript I), whereas two additional patient-specific transcripts denoted as transcripts II and III were present in mRNA from patient muscle biopsies (Fig. 1B). RT-PCR and sequencing of three distinct isolated clones representing these bands confirmed that transcript I represented the correctly spliced ISCU mRNA transcript, whereas transcript II corresponded to an mRNA species in which the 100 bp previously denoted as exon 4A (6) was included, and transcript III was composed of exon 4A as well as the remaining 1073-bp 3′ intronic sequence (Fig. 1C). Quantitative RT-PCR further confirmed that exon 4A, as well as downstream regions of intron 4, were highly expressed in patient muscle biopsies (Fig. 1D). Using high affinity 32P-labeled RNA probes, we observed several additional high molecular weight patient-specific mRNA species in fibroblasts, likely indicating the existence of partially spliced ISCU mRNA intermediates that were not observed in the patient muscle biopsies (Fig. 1, E and F, denoted by asterisks). Finally, we compared the pattern of ISCU mRNA expression in patient muscle biopsies directly with that of cultured patient-derived myotubes and fibroblasts and found that the residual amount of normal-sized ISCU mRNA (transcript I) was clearly lowest in the patient muscle biopsy (Fig. 1G). Further Northern blot analysis demonstrated that total ISCU mRNA expression was decreased in all three patient cell types relative to respective controls, whereas expression of the ISCU myopathy-specific ISCU mRNA transcripts II and III was abundant in all tested patient cell types (data not shown).
FIGURE 1.

Altered ISCU mRNA expression patterns in ISCU myopathy patients. A, a schematic representation of ISCU mRNA showing the single intronic point mutation within the fourth intron, canonical, and aberrant splicing patterns and regions of Northern blot probe complementarity. mito, mitochondrial; cyto, cytosolic; TRUNC., truncated. B, hybridization of ISCU-specific 32P-labeled DNA probes with control and patient vastus lateralis biopsy RNA demonstrated normal (I) and additional patient-specific (II and III) ISCU mRNA bands. C, RT-PCR and sequencing using primers flanking ISCU exons 4 and 5 confirmed the identity of the three major ISCU bands (I–III), with transcript II containing 100 bp and transcript III containing 1173 bp of intronic sequence. D, qRT-PCR analysis of three ISCU myopathy patients and seven control biopsies using primer sets for ISCU exon 4A and downstream intron 4 sequences. E and F, Northern blot analysis of ISCU mRNA expression in control (Ctl) and patient (Pat) muscle biopsies and fibroblasts using high specific activity RNA probes revealed several larger ISCU-specific mRNA bands in ISCU myopathy patient fibroblasts. Methylene blue-stained 18 S ribosomal RNA served as a loading control. G, Northern blot analysis of control and patient muscle biopsies alongside myotubes and fibroblasts demonstrates a decreased residual level of normally spliced ISCU mRNA in the patient muscle biopsy.
MyoD-induced Muscle Lineage Differentiation Caused Decreased Levels of Normally Spliced ISCU mRNA in Patient Myoblasts
The data here and in other studies (14, 15) suggest that greater amounts of normally spliced ISCU mRNA exist in other patient tissues such as heart, liver, fibroblasts, or myoblasts as compared with skeletal muscle. To further explore the tissue specificity of abnormal splicing, we generated stably transfected patient myoblasts with a plasmid harboring the muscle-specific MyoD1 transcription factor driven by a tetracycline-inducible promoter. In these cells, doxycycline treatment resulted in an ∼50-fold induction of MyoD1 mRNA expression and a concomitant ∼6-fold induction of myosin heavy chain 1 (MYH1) mRNA relative to uninduced cells, indicating that the muscle differentiation program was initiated (Fig. 2A). Western blots confirmed strong induction of MyoD1 protein expression in transfected control and patient cell lines (Fig. 2B). RT-PCR of ISCU mRNA using primers flanking the ISCU exon 4–5 boundary demonstrated a shift in the relative abundance of normal versus abnormally spliced ISCU mRNA in ISCU myopathy patient cells (Fig. 2C). The amount of normally spliced ISCU mRNA (transcript I) decreased, whereas the amount of patient-specific ISCU mRNA containing the 100-bp exon 4A (transcript II) increased with MyoD induction (Fig. 2C). We followed ISCU mRNA expression during induction of MyoD1 in these cells for 0, 12, 24, or 48 h, and we observed that expression of the abnormal transcript II increased as the length of MyoD induction times increased, whereas expression of the normal transcript I decreased, as assessed by Northern blot (Fig. 2D) and RT-PCR (Fig. 2E).
FIGURE 2.

MyoD-induced differentiation in patient myoblasts leads to decreased amounts of the normally spliced ISCU mRNA isoform. A, pooled clones of control and patient primary myoblasts stably harboring the episomal plasmid pEBTetD-MyoD (see “Experimental Procedures”) were incubated in growth medium or differentiation medium (Diff. Medium) in the presence or absence of 1 μg/ml doxycycline (DOX) for 48 h. MyoD1 and MYH1 mRNA expression was measured by qRT-PCR. Two-tailed t tests were used to evaluate statistical significance (**, p < 0.01) B, inducible MyoD protein expression in control and patient myoblasts was verified by Western blot. C, the pattern of ISCU mRNA expression in patient myoblasts in the presence or absence of MyoD overexpression was assessed by qualitative RT-PCR using primers flanking ISCU exons 4 and 5. The control is undifferentiated myoblast mRNA from a healthy individual. PCR bands I–III correspond to the bands identified in Fig. 1C. D, the time course of ISCU mRNA expression in differentiating myoblasts was followed by Northern blot using a 32P-labeled ISCU RNA probe. E, RT-PCR using primers flanking ISCU exons 4 and 5 was performed on cDNA generated from the same RNA as in panel D.
Decreased Expression of Full-length ISCU Protein Led to Low ISCU Levels in Cultured ISCU Patient Primary Cells
To investigate the relationship between altered ISCU mRNA expression and aberrant ISCU protein expression in ISCU myopathy patient tissues, we conducted metabolic labeling experiments with cultured myoblast, fibroblast, and lymphoblast cells derived from patients and controls. Newly synthesized ISCU protein was extracted and immunoprecipitated from radiolabeled cells, revealing a band of ∼13 kDa that corresponded with the predicted size of the truncated patient-specific ISCU protein following import into the mitochondrion and cleavage of the mitochondrial transit peptide (Fig. 3, A–C). We also found decreased amounts of the full-sized ∼15-kDa mitochondrial ISCU band in the myoblasts and fibroblasts, relative to respective controls (Fig. 3, A–C). A pulse-chase experiment in patient myoblasts demonstrated that the truncated ISCU protein had an extremely short half-life (<30 min), whereas the full-sized mature mitochondrial ISCU isoform was relatively stable throughout the 2-h experiment (Fig. 3D). Finally, Western blotting demonstrated that total ISCU protein levels were decreased substantially in myoblasts and fibroblasts and minimally in lymphoblasts relative to controls (Fig. 3E). These data demonstrated that aberrantly spliced ISCU mRNA led to synthesis of the unstable truncated ISCU protein in all patient cell types that were tested, but levels of the normal mitochondrial ISCU protein varied substantially between different cell types.
FIGURE 3.
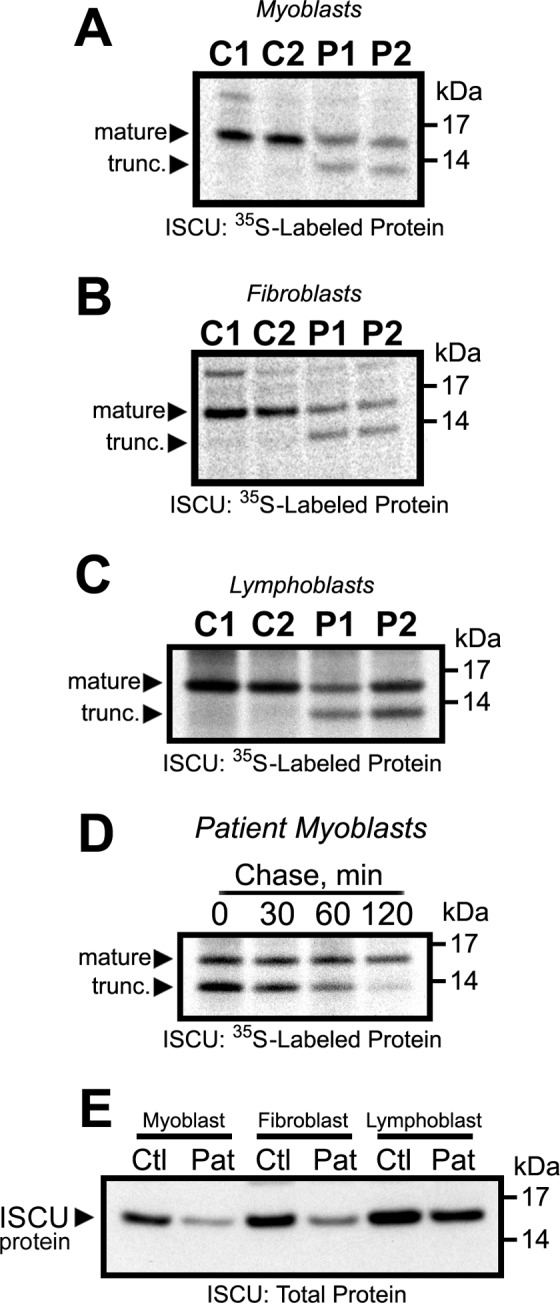
An unstable truncated patient-specific ISCU protein is produced in ISCU myopathy patient myoblasts. A–C, metabolic labeling of control (C1 and C2) and patient (P1 and P2) primary cells with [35S]cysteine and [35S]methionine for 30 min followed by immunoprecipitation and SDS-PAGE revealed the presence of a truncated (trunc.) mitochondrial ISCU band as well as decreased synthesis of the normal-sized mitochondrial ISCU in patient myoblasts (A), fibroblasts (B), and lymphoblasts (C). D, a pulse-chase experiment was performed to follow the fate of the normal-sized and truncated ISCU protein products over time (see “Experimental Procedures”). E, Western blots using an antibody against ISCU demonstrated decreased total ISCU protein levels in patient (Pat) primary cells. Ctl, control.
ISCU and NFS1 Protein Levels Were Comparatively Low in Skeletal Muscle Tissue
To compare the relative level of ISCU depletion in patient skeletal muscle to other patient cell types, we performed Western blots using control and patient skeletal muscle biopsy material. Biopsies from vastus lateralis of either an ISCU myopathy patient or a healthy control showed markedly lower ISCU protein levels in comparison with cultured human myoblasts, fibroblasts, and lymphoblasts (Fig. 4A), although ISCU mRNA in the control muscle biopsy was not low relative to the other cell lines (Fig. 4B). The ISCU binding partner, NFS1, which provides inorganic sulfide for formation of Fe-S clusters on ISCU, was also relatively low in the control biopsy and was undetectable in the patient biopsy (Fig. 4A). Notably, NFS1 was also mildly decreased in patient myoblasts and fibroblasts as compared with controls (Fig. 4A). Western blots for the other Fe-S assembly core machinery members in the muscle biopsies revealed mild depletion of the obligate NFS1 binding partner, ISD11 (11, 29, 30), in three ISCU myopathy patients, whereas expression of frataxin (FXN) increased slightly, an interesting result because reduced expression of FXN causes Friedreich ataxia, and little is known about how frataxin expression is regulated (31, 32) (Fig. 4C). We also checked expression of the Fe-S-containing enzymes aconitase and succinate dehydrogenase subunit B (SDH-B), which rely on the core Fe-S assembly machinery to synthesize and donate their Fe-S clusters. Mitochondrial aconitase and SDH-B proteins were abundantly expressed in the control muscle biopsy, whereas SDH-B was virtually undetectable in the ISCU myopathy patient biopsy (Fig. 4D) despite abundant expression of the mitochondrial matrix enzyme citrate synthase, which has been used as a proxy for cellular mitochondrial volume (5).
FIGURE 4.

Steady-state ISCU and NFS1 protein levels are inherently low in muscle tissue. Protein levels in lysates from control (Ctl) and patient (Pat) vastus lateralis biopsies and primary cells were assessed by Western blots. A, Fe-S assembly proteins ISCU and ISCS. Short and long film exposures are shown for completeness. B, ISCU mRNA levels were assessed by Northern blot in a healthy control vastus lateralis biopsy and in primary cells derived from a healthy individual. Methylene blue-stained 18 S and 28 S ribosomal RNA bands served as loading control. C, Western blots assessed levels of Fe-S assembly cofactors ISD11 (LYRM4), Fxn, and NFS1 in control and patient muscle biopsies. D, Fe-S recipient proteins succinate dehydrogenase subunit B and mitochondrial aconitase (mACO). All proteins above were probed on the same filter, and citrate synthase served as a loading control for mitochondrial protein. E, expression of the Fe-S assembly proteins ISCU and NFS1, as well as the Fe-S recipient proteins succinate dehydrogenase subunit B and mitochondrial aconitase in multiple mouse tissues was evaluated by Western blotting. F, siRNA-mediated knockdown of ISCU protein in control patient myoblasts led to decreased NFS1 and ISD11 protein levels, whereas FXN levels increased. Citrate synthase and mitochondrial complex IV, subunit IV (CIV-IV) proteins served as loading controls.
In tissues taken from a healthy control mouse, ISCU and NFS1 protein levels were also lowest in skeletal muscle, whereas ISCU levels were highest in heart (Fig. 4E) and were much higher in the liver, kidney, and brain. Again, SDH-B and mitochondrial aconitase were abundant in both mouse heart and skeletal muscle (Fig. 4E). Finally, similar alterations in expression of the core Fe-S assembly machinery were observed in control primary myoblasts depleted of ISCU by siRNA; NFS1 and ISD11 proteins were less abundant, whereas FXN protein expression was increased by ISCU depletion (Fig. 4F). Together, these data demonstrate that protein levels of critical components of the mitochondrial Fe-S assembly machinery are markedly lower in skeletal muscle as compared with other cell types, and the abundance of other members of the core mitochondrial Fe-S assembly machinery, which consists of ISCU, NFS1, ISD11, and FXN, are affected by alterations in ISCU expression.
Mitochondrial ISCU Restored Fe-S Protein Activities and Iron Homeostasis in ISCU Myopathy Patient Myoblasts
The ISCU protein is found in two distinct isoforms in human cells, namely a low abundance cytosolic isoform known as ISCU1 and a far more abundant mitochondrial isoform known as ISCU2 (24, 28). We stably expressed ISCU1 and ISCU2 in patient-derived myoblast cell lines to test whether these proteins could rescue defects in Fe-S cluster proteins and cellular iron metabolism in these cells. Using confocal microscopy, we verified that the cytosolic and mitochondrial forms of our ISCU1 and ISCU2 constructs localized as predicted (Fig. 5, A and B). Mitochondrial and cytosolic aconitase activities were enhanced by expression of mitochondrial ISCU (ISCU2) in patient cells, whereas IRE binding activity of IRP1 decreased (Fig. 5C), indicating that more IRP1 was in the Fe-S-containing aconitase form. Furthermore, IRP2 protein levels decreased in ISCU2-expressing cells, and more subtly in ISCU1-expressing cells, whereas H-ferritin protein levels increased upon expression of ISCU2, consistent with correction of cytosolic iron depletion that frequently occurs in conjunction with disturbed mitochondrial Fe-S synthesis (28, 30, 33). Together, these data demonstrate that mitochondrial and cytosolic Fe-S enzymes and iron homeostasis in ISCU myopathy patient myoblasts can be restored by increasing mitochondrial ISCU levels.
FIGURE 5.

Expression of mitochondrial ISCU restores Fe-S protein activities and iron homeostasis in ISCU myopathy patient-derived myoblasts. A and B, primary patient myoblasts were stably transfected with a plasmid encoding Myc-tagged cytosolic ISCU (ISCU1) or mitochondrial ISCU (ISCU2). Proper subcellular localization was verified by confocal microscopy, with double-immunostaining with anti-Myc and SOD1 (cytosol) or TOM20 (mitochondrion); nuclei were stained with DAPI. C, ISCU, IRP2, and ferritin protein levels were assessed by Western blot, and mitochondrial aconitase (mACO) and cytosolic aconitase (cACO) activity levels and IRP-IRE binding activity were assessed as described under “Experimental Procedures.”
Impaired Synthesis of ISCU Protein Rendered Patient Myoblasts and Fibroblasts More Sensitive to Oxidative Stress
Skeletal muscle has long been known to be the source of significant reactive oxygen species during both rest and exercise (34–36). Fe-S proteins are a well known target of reactive oxygen species (37), and intact Fe-S cluster assembly machinery is needed for regeneration of Fe-S enzyme activities following oxidative damage (28). The propensity for working skeletal muscles to produce reactive oxygen species led us to ask whether ISCU protein deficiency rendered patient primary myoblasts more sensitive to oxidative stress. We exposed control and patient myoblasts to a 1-h H2O2 pulse followed by incubation/recovery in normal medium for 4 or 8 h. Recovery of mitochondrial and cytosolic aconitase activities was impaired in the patient myoblasts as compared with control cells, and activation of IRP-IRE binding activity was greater and more sustained in patient cells (Fig. 6A). Unexpectedly, ISCU protein levels were also significantly diminished by H2O2 treatment, with ISCU levels in the patient cells falling to nearly undetectable levels (Fig. 6A). We also explored the effects of NO on the myoblasts. Although ISCU protein levels were not measurably decreased following treatment with the NO donor DEA/NO, impaired recovery of aconitase activity and elevated IRP-IRE binding activity were again observed in the patient cells (Fig. 6B). Because ISCU protein in patient cells appeared to be readily decreased by H2O2, we tested the ability of antioxidants to prevent this effect. Patient myoblasts were pretreated with either 0.2 mm or 0.5 mm sodium ascorbate or with 0.5 mm reduced glutathione for 12 h, washed twice with normal medium, and exposed to 1 mm H2O2 for 1 h. We found that ascorbate, but not glutathione, prevented the rapid H2O2-mediated decrease in ISCU protein levels (Fig. 6C).
FIGURE 6.

Impaired synthesis of ISCU protein renders patient myoblasts and fibroblasts more sensitive to oxidative stress. A, control and patient myoblasts were pulsed with H2O2 for 1 h followed by a chase in normal culture medium for 4 or 8 h. ISCU protein levels, mitochondrial aconitase (mACO) and cytosolic aconitase (cACO) activity levels, and IRP-IRE binding activity were measured. B, control and patient myoblasts were also tested for their ability to recover from a 1-h pulse with 1 mm DEA/NO. C, preincubation of primary patient myoblasts with sodium ascorbate (vitamin C) prevented H2O2-mediated depletion of ISCU protein. Preincubation with reduced glutathione (GSH) had no effect on ISCU depletion. D, control and patient primary fibroblasts were tested for their ability to recover aconitase activity and IRP-IRE binding activity in a 5% O2 atmosphere following a 16-h challenge in a 95% O2/5% CO2 atmosphere. E, control (Ctl) and patient (Pat) myoblasts expressing either mitochondrial or cytosolic ISCU were grown in a 95% O2 atmosphere for 16 h followed by recovery at 6% O2 for 24 h.
Mammalian aconitase is known to readily lose enzymatic activity upon exposure to an enriched O2 atmosphere (38). We found that mitochondrial aconitase activity in patient fibroblasts was minimally affected by exposure to high oxygen levels in control cells, whereas cytosolic aconitase activity was profoundly diminished. In patient cells, both mitochondrial and cytosolic aconitase activities were very diminished by exposure to high oxygen levels. Unexpectedly, ISCU protein levels were also reduced by high oxygen levels in control and patient fibroblasts (Fig. 6D). Similar results were observed in patient myoblasts, where oxygen exposure resulted in diminished activities of both aconitases and greatly elevated IRP-IRE binding activity in patient cells (Fig. 6E). Patient myoblasts heterologously expressing mitochondrial ISCU showed significantly improved recovery of aconitase activities and reduction of IRE binding activities after a high oxygen pulse, whereas cells expressing cytosolic ISCU did not recover well (Fig. 6E), indicating that the cytosolic ISCU isoform alone cannot rescue the mitochondrial Fe-S synthesis and cytosolic iron homeostasis disruptions in ISCU-deficient cells.
DISCUSSION
Since the identification of the single intronic point mutation in the ISCU gene that underlies the pathophysiology of ISCU myopathy, several groups have suggested that tissue-specific splicing explains the muscle-specific disease phenotype (4, 14–16). In this study, we have extended these findings mechanistically by demonstrating that progress toward terminal skeletal muscle differentiation, induced by MyoD expression, leads to decreased residual levels of normally spliced ISCU mRNA expression in patient cells (Fig. 7A). This further compromise of ISCU expression in patient skeletal muscle leads to impaired responsiveness to the inherent stresses of oxidative metabolism, in which the Fe-S assembly machinery is needed for regeneration of Fe-S proteins damaged by oxidative stress (Fig. 7B).
FIGURE 7.

The impact of aberrant ISCU mRNA expression and oxidative stress on ISCU protein synthesis and degradation in ISCU myopathy patient muscle mitochondria. A, an intronic point mutation (g.7044 G→C) leads to decreased expression of the normally spliced ISCU mRNA and the appearance of two predominant patient-specific ISCU mRNA transcripts in all tested patient tissues, both of which encode a truncated (TRUNC.) ISCU protein that is rapidly degraded after synthesis. MyoD expression and maturation of skeletal muscle myofibers lead to further decreases in the expression of normally spliced ISCU mRNA, eventually leading to pathologically low ISCU protein levels in mature skeletal muscles. B, generation of reactive oxygen species such as hydrogen peroxide and superoxide can exacerbate Fe-S cluster deficiency in enzymes and respiratory chain complexes of patient skeletal muscles by causing further depletion of ISCU protein, which cannot be rapidly resynthesized due to the ISCU mRNA splicing defect. TCA cycle, tricarboxylic acid cycle; mACO, mitochondrial aconitase.
Functional ISCU mRNA and Protein Levels Are Diminished during Skeletal Muscle Differentiation in ISCU Myopathy Patients
Our results indicate that there is a continuum in the spectrum of abnormal ISCU mRNA splicing in patient tissues as we found evidence for abnormal splicing in several different patient cell types. All tested patient cells abundantly expressed an ISCU mRNA containing the entire intronic sequence downstream of the patient-specific exon 4A (transcript III; see Fig. 1C). This longer mRNA transcript might be more stable than the canonical patient-specific transcript (transcript II) because the 56 bp of sequence between the premature stop codon and the exon 5 junction make transcript II a good candidate for nonsense-mediated decay (39). The altered pattern of ISCU mRNA splicing that we observed in patient skeletal muscle and in MyoD-expressing cells is likely a result of altered expression of one or more RNA-binding proteins and splicing factors. Such factors may include IGF2BP1, RBM39, and PTBP1, which were recently found to influence the retention of exon 4A during splicing of the mutant ISCU mRNA (16).
In comparison with patient skeletal muscle tissue, the relatively high level of ISCU protein expression that we observed in patient myoblasts (Fig. 4) offers insight into the nonprogressive nature of ISCU myopathy. Residual ISCU protein levels introduced to regenerating patient muscle tissue by the fusion of satellite cells would allow a window of time in which ISCU expression was sufficiently high to allow for more normalized levels of Fe-S protein expression. This idea is consistent with a recent report on an ISCU myopathy patient in which significantly higher levels of ISCU protein expression and succinate dehydrogenase activity were detected in the regenerating muscle fibers of a patient recovering from an acute attack of rhabdomyolysis (4).
Muscles utilize several different metabolic pathways to produce ATP depending on the myofiber type and level of contractile activity, ranging from almost exclusive dependence on fatty acid oxidation during times of rest to predominant utilization of glucose/glycogen during periods of maximal exertion (40). Cellular respiration requires many Fe-S-containing proteins, and therefore the need for Fe-S cluster synthesis and repair is likely to be great in skeletal muscle. Although we do not know why ISCU and NFS1 are expressed at low levels in skeletal muscle relative to other tissues, these inherently low levels of Fe-S cluster assembly proteins in normal skeletal muscle likely make skeletal muscle particularly vulnerable to the effects of further ISCU depletion.
Oxidative Stress Exacerbates the Phenotype of Impaired Fe-S Cluster Assembly in ISCU-deficient Patient Cells
Skeletal muscle is unique in that it generates large transient bursts of reactive oxygen species during both low intensity and high intensity exercise (34, 35, 41, 42) and even during prolonged inactivity (36). Furthermore, mitochondrial respiratory chain defects such as those that accompany depletion of Fe-S-containing enzymes in ISCU myopathy may increase mitochondrial production of reactive oxygen species (43). A surprising observation in our experiments was the rapid decrease in ISCU protein levels following H2O2 or high O2 treatments. We hypothesize that nascent [2Fe-2S] clusters on ISCU (44) may be particularly vulnerable to damage by reactive oxygen species as previous work has shown that H2O2 can readily oxidize [2Fe-2S] clusters on E. coli IscU, resulting in release of iron and sulfide (45).
A recent study demonstrated that apo-ISCU (Isu) in yeast cells is rapidly degraded by the Lon-type AAA+ protease Pim1 (46). If human ISCU is similarly degraded when it lacks an Fe-S cluster, then oxidant-mediated cluster dissolution would lead to a decreased protein half-life, an effect seen in several other mammalian Fe-S proteins (22, 47). Alternatively, ISCU degradation might be stimulated by oxidant-induced modification of amino acid residues of human ISCU, such as intramolecular disulfide bond formation or irreversible oxidation of active site cysteine thiols to sulfinic acid (S-O2H).
Recently, the E. coli Isc system was observed to be inactivated by low levels of H2O2, likely by disruption of Fe-S cluster assembly on IscU or prevention of cluster transfer from IscU (48). In this case, the suf Fe-S assembly/repair operon was activated to restore Fe-S enzyme activities. As mammalian cells are not known to possess homologs of the suf genes, it appears that mammalian cells may be very compromised by difficulties with synthesizing and repairing Fe-S clusters during periods of stress. Importantly, ascorbic acid (vitamin C) protected ISCU from H2O2-mediated degradation in our patient cells, suggesting that targeted antioxidant therapy might be of benefit to ISCU myopathy patients.
In summary, we suggest that the tissue specificity in ISCU myopathy patients is attributable to several factors, including tissue and differentiation-specific abnormal mRNA splicing, impaired ability of patient muscle tissue to recover from oxidative stress because of ISCU deficiency, and further direct effects of oxidative stress on the stability of the remaining ISCU protein (Fig. 7). These factors synergize to lead to significant and pathological deficiency of ISCU in mature muscle cells, whereas other cell types expressing greater residual levels of ISCU protein are spared.
Acknowledgments
We thank Dr. Manu Hegde for helpful scientific discussions and Dr. Eric Shoubridge for providing us with the primary myoblast cells. We would also thank Michele Allen and Dr. Danielle Springer for expert technical assistance with the mouse work. Drs. Deliang Zheng and Gennadiy Kovtunovych provided ideas and suggestions that greatly improved the quality of this work.
This work was supported, in whole or in part, by the National Institutes of Health through the NICHD Intramural Research Program (to T. A. R.) and NIAMS Grant R01 AR050597 (to R. G. H.). This work was also supported by a Veterans Affairs Merit Review (to R. G. H.).
- SDH
- succinate dehydrogenase
- SDH-B
- SDH subunit B
- DEA/NO
- diethylamine-nitric oxide
- IRE
- iron response element
- IRP
- iron response protein
- FXN
- frataxin
- qRT-PCR
- quantitative RT-PCR
- qPCR
- quantitative PCR
- Bis-Tris
- 2-(bis(2-hydroxyethyl)amino)-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Larsson L. E., Linderholm H., Mueller R., Ringqvist T., Soernaes R. (1964) Hereditary metabolic myopathy with paroxysmal myoglobinuria due to abnormal glycolysis. J. Neurol. Neurosurg. Psychiatry 27, 361–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Drugge U., Holmberg M., Holmgren G., Almay B. G., Linderholm H. (1995) Hereditary myopathy with lactic acidosis, succinate dehydrogenase, and aconitase deficiency in northern Sweden: a genealogical study. J. Med. Genet. 32, 344–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Linderholm H., Müller R., Ringqvist T., Sörnäs R. (1969) Hereditary abnormal muscle metabolism with hyperkinetic circulation during exercise. Acta Med. Scand. 185, 153–166 [DOI] [PubMed] [Google Scholar]
- 4. Kollberg G., Melberg A., Holme E., Oldfors A. (2011) Transient restoration of succinate dehydrogenase activity after rhabdomyolysis in iron-sulphur cluster deficiency myopathy. Neuromuscul. Disord. 21, 115–120 [DOI] [PubMed] [Google Scholar]
- 5. Haller R. G., Henriksson K. G., Jorfeldt L., Hultman E., Wibom R., Sahlin K., Areskog N. H., Gunder M., Ayyad K., Blomqvist C. G., et al. (1991) Deficiency of skeletal muscle succinate dehydrogenase and aconitase. Pathophysiology of exercise in a novel human muscle oxidative defect. J. Clin. Invest. 88, 1197–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mochel F., Knight M. A., Tong W. H., Hernandez D., Ayyad K., Taivassalo T., Andersen P. M., Singleton A., Rouault T. A., Fischbeck K. H., Haller R. G. (2008) Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am. J. Hum. Genet. 82, 652–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kollberg G., Tulinius M., Melberg A., Darin N., Andersen O., Holmgren D., Oldfors A., Holme E. (2009) Clinical manifestation and a new ISCU mutation in iron-sulphur cluster deficiency myopathy. Brain 132, 2170–2179 [DOI] [PubMed] [Google Scholar]
- 8. Hall R. E., Henriksson K. G., Lewis S. F., Haller R. G., Kennaway N. G. (1993) Mitochondrial myopathy with succinate dehydrogenase and aconitase deficiency. Abnormalities of several iron-sulfur proteins. J. Clin. Invest. 92, 2660–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Linderholm H., Essén-Gustavsson B., Thornell L. E. (1990) Low succinate dehydrogenase (SDH) activity in a patient with a hereditary myopathy with paroxysmal myoglobinuria. J. Intern. Med. 228, 43–52 [DOI] [PubMed] [Google Scholar]
- 10. Tsai C. L., Barondeau D. P. (2010) Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry 49, 9132–9139 [DOI] [PubMed] [Google Scholar]
- 11. Schmucker S., Martelli A., Colin F., Page A., Wattenhofer-Donzé M., Reutenauer L., Puccio H. (2011) Mammalian frataxin: an essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron-sulfur assembly complex. PloS One 6, e16199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Uhrigshardt H., Singh A., Kovtunovych G., Ghosh M., Rouault T. A. (2010) Characterization of the human HSC20, an unusual DnaJ type III protein, involved in iron-sulfur cluster biogenesis. Hum. Mol. Genet. 19, 3816–3834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Olsson A., Lind L., Thornell L. E., Holmberg M. (2008) Myopathy with lactic acidosis is linked to chromosome 12q23.3–24.11 and caused by an intron mutation in the ISCU gene resulting in a splicing defect. Hum. Mol. Genet. 17, 1666–1672 [DOI] [PubMed] [Google Scholar]
- 14. Sanaker P. S., Toompuu M., Hogan V. E., He L., Tzoulis C., Chrzanowska-Lightowlers Z. M., Taylor R. W., Bindoff L. A. (2010) Differences in RNA processing underlie the tissue specific phenotype of ISCU myopathy. Biochim. Biophys. Acta 1802, 539–544 [DOI] [PubMed] [Google Scholar]
- 15. Nordin A., Larsson E., Thornell L. E., Holmberg M. (2011) Tissue-specific splicing of ISCU results in a skeletal muscle phenotype in myopathy with lactic acidosis, while complete loss of ISCU results in early embryonic death in mice. Hum. Genet. 129, 371–378 [DOI] [PubMed] [Google Scholar]
- 16. Nordin A., Larsson E., Holmberg M. (2012) The defective splicing caused by the ISCU intron mutation in patients with myopathy with lactic acidosis is repressed by PTBP1 but can be derepressed by IGF2BP1. Hum. Mutat. 33, 467–470 [DOI] [PubMed] [Google Scholar]
- 17. Shoubridge E. A., Johns T., Boulet L. (1996) Use of myoblast cultures to study mitochondrial myopathies. Methods Enzymol. 264, 465–475 [DOI] [PubMed] [Google Scholar]
- 18. Halbert C. L., Demers G. W., Galloway D. A. (1991) The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J. Virol. 65, 473–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu X., Dakic A., Chen R., Disbrow G. L., Zhang Y., Dai Y., Schlegel R. (2008) Cell-restricted immortalization by human papillomavirus correlates with telomerase activation and engagement of the hTERT promoter by Myc. J. Virol. 82, 11568–11576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lochmüller H., Johns T., Shoubridge E. A. (1999) Expression of the E6 and E7 genes of human papillomavirus (HPV16) extends the life span of human myoblasts. Exp. Cell Res. 248, 186–193 [DOI] [PubMed] [Google Scholar]
- 21. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 22. Crooks D. R., Ghosh M. C., Haller R. G., Tong W. H., Rouault T. A. (2010) Posttranslational stability of the heme biosynthetic enzyme ferrochelatase is dependent on iron availability and intact iron-sulfur cluster assembly machinery. Blood 115, 860–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Crooks D. R., Ghosh M. C., Braun-Sommargren M., Rouault T. A., Smith D. R. (2007) Manganese targets m-aconitase and activates iron regulatory protein 2 in AF5 GABAergic cells. J. Neurosci. Res. 85, 1797–1809 [DOI] [PubMed] [Google Scholar]
- 24. Tong W. H., Rouault T. (2000) Distinct iron-sulfur cluster assembly complexes exist in the cytosol and mitochondria of human cells. EMBO J. 19, 5692–5700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Land T., Rouault T. A. (1998) Targeting of a human iron-sulfur cluster assembly enzyme, nifs, to different subcellular compartments is regulated through alternative AUG utilization. Mol. Cell 2, 807–815 [DOI] [PubMed] [Google Scholar]
- 26. Bach M., Grigat S., Pawlik B., Fork C., Utermöhlen O., Pal S., Banczyk D., Lazar A., Schömig E., Gründemann D. (2007) Fast set-up of doxycycline-inducible protein expression in human cell lines with a single plasmid based on Epstein-Barr virus replication and the simple tetracycline repressor. FEBS J. 274, 783–790 [DOI] [PubMed] [Google Scholar]
- 27. Jeong S. Y., David S. (2003) Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J. Biol. Chem. 278, 27144–27148 [DOI] [PubMed] [Google Scholar]
- 28. Tong W. H., Rouault T. A. (2006) Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab. 3, 199–210 [DOI] [PubMed] [Google Scholar]
- 29. Wiedemann N., Urzica E., Guiard B., Müller H., Lohaus C., Meyer H. E., Ryan M. T., Meisinger C., Mühlenhoff U., Lill R., Pfanner N. (2006) Essential role of Isd11 in mitochondrial iron-sulfur cluster synthesis on Isu scaffold proteins. EMBO J. 25, 184–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shi Y., Ghosh M. C., Tong W. H., Rouault T. A. (2009) Human ISD11 is essential for both iron-sulfur cluster assembly and maintenance of normal cellular iron homeostasis. Hum. Mol. Genet. 18, 3014–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rouault T. A. (2012) Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis. Model. Mech. 5, 155–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martelli A., Napierala M., Puccio H. (2012) Understanding the genetic and molecular pathogenesis of Friedreich's ataxia through animal and cellular models. Dis. Model. Mech. 5, 165–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li K., Besse E. K., Ha D., Kovtunovych G., Rouault T. A. (2008) Iron-dependent regulation of frataxin expression: implications for treatment of Friedreich ataxia. Hum. Mol. Genet. 17, 2265–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Alessio H. M., Goldfarb A. H., Cutler R. G. (1988) MDA content increases in fast- and slow-twitch skeletal muscle with intensity of exercise in a rat. Am. J. Physiol. 255, C874–C877 [DOI] [PubMed] [Google Scholar]
- 35. Sen C. K. (1995) Oxidants and antioxidants in exercise. J. Appl. Physiol. 79, 675–686 [DOI] [PubMed] [Google Scholar]
- 36. Kavazis A. N., Talbert E. E., Smuder A. J., Hudson M. B., Nelson W. B., Powers S. K. (2009) Mechanical ventilation induces diaphragmatic mitochondrial dysfunction and increased oxidant production. Free Radic. Biol. Med. 46, 842–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gardner P. R., Fridovich I. (1991) Superoxide sensitivity of the Escherichia coli aconitase. J. Biol. Chem. 266, 19328–19333 [PubMed] [Google Scholar]
- 38. Gardner P. R., Nguyen D. D., White C. W. (1994) Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc. Natl. Acad. Sci. U.S.A. 91, 12248–12252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nagy E., Maquat L. E. (1998) A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem. Sci. 23, 198–199 [DOI] [PubMed] [Google Scholar]
- 40. Holloszy J. O., Kohrt W. M. (1996) Regulation of carbohydrate and fat metabolism during and after exercise. Annu. Rev. Nutr. 16, 121–138 [DOI] [PubMed] [Google Scholar]
- 41. Meydani M., Evans W., Handelman G., Fielding R. A., Meydani S. N., Fiatarone M. A., Blumberg J. B., Cannon J. G. (1992) Antioxidant response to exercise-induced oxidative stress and protection by vitamin E. Ann. N.Y. Acad. Sci. 669, 363–364 [DOI] [PubMed] [Google Scholar]
- 42. Meydani M., Evans W. J., Handelman G., Biddle L., Fielding R. A., Meydani S. N., Burrill J., Fiatarone M. A., Blumberg J. B., Cannon J. G. (1993) Protective effect of vitamin E on exercise-induced oxidative damage in young and older adults. Am. J. Physiol. 264, R992–R998 [DOI] [PubMed] [Google Scholar]
- 43. Kirkinezos I. G., Moraes C. T. (2001) Reactive oxygen species and mitochondrial diseases. Semin. Cell Dev. Biol. 12, 449–457 [DOI] [PubMed] [Google Scholar]
- 44. Marinoni E. N., de Oliveira J. S., Nicolet Y., Raulfs E. C., Amara P., Dean D. R., Fontecilla-Camps J. C. (2012) (IscS-IscU)2 complex structures provide insights into Fe2S2 biogenesis and transfer. Angew. Chem. Int. Ed. Engl. 51, 5439–5442 [DOI] [PubMed] [Google Scholar]
- 45. Bitoun J. P., Wu G., Ding H. (2008) Escherichia coli FtnA acts as an iron buffer for re-assembly of iron-sulfur clusters in response to hydrogen peroxide stress. Biometals 21, 693–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Song J. Y., Marszalek J., Craig E. A. (2012) Cysteine desulfurase Nfs1 and Pim1 protease control levels of Isu, the Fe-S cluster biogenesis scaffold. Proc. Natl. Acad. Sci. U.S.A. 109, 10370–10375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guillon B., Bulteau A. L., Wattenhofer-Donzé M., Schmucker S., Friguet B., Puccio H., Drapier J. C., Bouton C. (2009) Frataxin deficiency causes upregulation of mitochondrial Lon and ClpP proteases and severe loss of mitochondrial Fe-S proteins. FEBS J. 276, 1036–1047 [DOI] [PubMed] [Google Scholar]
- 48. Jang S., Imlay J. A. (2010) Hydrogen peroxide inactivates the Escherichia coli Isc iron-sulphur assembly system, and OxyR induces the Suf system to compensate. Mol. Microbiol. 78, 1448–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]