

Background: Hepatocarcinoma cancer (HCC) occurs more often in men than in women, and little is known about its underlying molecular mechanisms.
Results: We identify that 17β-estradiol (E2) could suppress tumor growth via regulating the polarization of macrophages.
Conclusion: Estrogen functions as a suppressor for macrophage alternative activation.
Significance: These studies introduce a novel mechanism for suppressing male-predominant HCC.
Keywords: Estrogen, Estrogen Receptor, Macrophages, Tumor Immunology, Tumor Therapy, Alternatively Activated Macrophages, Hepatocellular Carcinoma, Tumor Progression, Tumor-associated Macrophages (TAMs), Alternatively Activated Macrophage (M2)
Abstract
Hepatocarcinoma cancer (HCC), one of the most malignant cancers, occurs significantly more often in men than in women; however, little is known about its underlying molecular mechanisms. Here we identified that 17β-estradiol (E2) could suppress tumor growth via regulating the polarization of macrophages. We showed that E2 re-administration reduced tumor growth in orthotopic and ectopic mice HCC models. E2 functioned as a suppressor for macrophage alternative activation and tumor progression by keeping estrogen receptor β (ERβ) away from interacting with ATP5J (also known as ATPase-coupling factor 6), a part of ATPase, thus inhibiting the JAK1-STAT6 signaling pathway. These studies introduce a novel mechanism for suppressing male-predominant HCC.
Introduction
Hepatocelluar carcinoma (HCC)2 is the most common primary malignancy of the liver in adults and the third most common cause of cancer death worldwide (1). Previous reports have shown that HCC is a male-predominant cancer associated with chronic hepatitis (2). Men are about 3–5 times more likely to develop HCC than women; a similar pronounced gender disparity can be seen in rodent HCC models (3–5). In addition, males have a worse prognosis in comparison with females (6).
Because the liver is a sexually dimorphic organ, sex hormones are potential agents for bringing about the gender disparity seen in HCC progression. Studies showed that 17β-estradiol (E2) administration markedly diminished the inflammation and injury associated with a chemical carcinogen of diethylnitrosamine (7). Administration of E2 to male mice also inhibited the development of chemically diethylnitrosamine-induced HCC (8), although several preliminary clinical trials targeting sex hormone pathways had proved disappointing (9, 10). Meanwhile, some studies have shown that proinflammatory IFNγ cytokines may promote HCC in a male-predominant fashion due to high sensitivity of the masculinized liver to loss of sex-specific transcriptional balance (2), and Naugler et al. proposed that gender disparity in liver cancer was due to sex differences in MyD88-dependent interleukin-6 (IL-6) production by Kupffer cells (7). Furthermore, Naugler et al. (7) discovered that estrogen-mediated inhibition of IL-6 production by Kupffer cells, which reduced liver cancer risk in females, may also be used to prevent HCC in males. These results may introduce a novel mechanism of inflammation-associated carcinogenesis consistent with male-predominant HCC risk (9).
Tumor-associated macrophages (TAMs), macrophages within the tumor microenvironment facilitate tumor angiogenesis, promote extracellular matrix degradation and its remodeling and promote tumor cell motility (11). Recent studies have revealed a direct communication between macrophages and tumor cells that leads to the invasion and egress of tumor cells into blood vessels (12); a high density of these tumor-associated macrophages correlates with poor prognosis in over 80% of studies published (13). Overexpression of colony-stimulating factor 1 (Csf-1) accelerated tumor progression and metastasis in a mouse model of breast cancer, and these effects were induced by expression of the polyoma middle T oncoprotein in mammary epithelial cells (14). In contrast, blocking expression of mouse Csf-1 in a xenograft model (mice engrafted with human tumor cells) reduced the growth and metastatic capacity of the tumor cells. This was associated with reduced invasion by host tumor-associated macrophages (15) and demonstrated that macrophages appeared to be directly involved in tumor progression and metastasis. Indeed, in human breast cancers, there is a positive correlation between poor prognosis and the density of tumor-associated macrophages (16).
Zeisberger et al. (17) reported that clodronate-liposome-mediated depletion of tumor-associated macrophages combined with new antiangiogenic or cytotoxic therapies posed a promising new approach with high clinical potential. Moreover, depletion of tumor-associated macrophages with zoledronic acid and clodrolip enhanced the effect of sorafenib in HCC mice models by antimetastatic and antiangiogenic effects. Zoledronic acid is clinically available and promising when combined with sorafenib for HCC patients. Subsequently, TAMs represent a major component of tumor-inflammatory infiltrate (18), and several lines of evidence suggest that these cells are important targets for cancer therapy (11).
Macrophage origins, lineage, and regulation by growth factors have been recently reviewed (19). The “classically activated” macrophages (M1) are involved in the responses of Th1 cells to pathogens. The “alternatively activated” macrophages (M2) instead respond to Th2-type cytokines, such as IL-4 and IL-13, and are involved in fibrosis, tissue repair, and humoral immunity (20). Mantovani et al. (21) have suggested that macrophages in tumors could convert from M1 to M2. Recent gene profiling experiments on TAMs supported this shift to an immunoregulatory type (22–24), indicating that the functions of macrophages are converted by tumor environment from inhibiting tumor growth to tumor trophic roles.
In this study, we used orthotopic and ectopic mouse models with mouse HCC cell lines to investigate the role of M2 in tumor progression, and we showed a possible link among estrogen, M2, and HCC gender disparity. We demonstrated that estrogen could suppress HCC progression through inhibiting macrophage alternative activation. Additionally, we showed that estrogen exerted its repressive function by inhibiting the Jak-Stat6 signaling pathway through estrogen receptor β (ERβ), not estrogen receptor α (ERα). Taken together, our findings indicated that M2 played a protumor role in HCC and also provided a possible explanation for the disparity between male and female HCC occurrence.
MATERIALS AND METHODS
Reagents
RPMI 1640 medium and DMEM were supplied by Invitrogen. DMEM high glucose medium was purchased from Invitrogen. Except for PPT and DPN (Tocris, Bristol, UK), other chemicals were purchased from Sigma unless otherwise stated.
Mouse IL-4 was purchased from Peprotech (London, UK) and resuspended in PBS before use. Final cytokine concentrations used are described in detail under “Results” (specific for each experiment). CD206 (mouse macrophage mannose receptor) antibody was purchased from AbD serotec (Oxford, UK).
Mice and Surgical Procedure
Male and female BALB/c mice (6 weeks, 20–25 g) were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China) and bred in our animal facilities under specific pathogen-free conditions. Animal welfare and experimental procedures were carried out in strict compliance with the “Guide for the Care and Use of Laboratory Animals” (Ministry of Science and Technology of China, 2006) and the related ethical regulations of Nanjing University. The heterotopic tumor model was set up by injecting 1 × 106 Heps cells under skin of BALB/c mice. Orthotopic liver tumors were induced by introhepatic tumor cell injection of Heps murine hematoma cells. Briefly, mice were anesthetized with 70 mg/kg sodium pentobarbital injected intraperitoneally and then placed in a supine position on the procedure table. The liver was exposed through a small vertical incision made below the xyphoid, and then 1 × 106 Heps cells suspended in 10 μl of PBS were slowly injected into the left lobe of the liver using a microinjector. After injection, a sterile cotton swab was placed on the injection site, and light pressure was applied for 1 min to ensure hemostasis. The pinprick was sealed with NEXABAN liquid topical tissue adhesive (Closure Medical Corp., Raleigh, NC). The abdominal musculature and skin were then closed.
One week after tumor implantation, mice were distributed into six groups: male control, female control, surgical castration, surgically ovariectomized (OVX) (surgeries of castration and ovariectomy were performed 2 weeks before tumor implantation), surgically castrated treated with E2, and surgically ovariectomized treated with E2. Mice were injected daily with E2 (50 μg/kg) or vehicle (saline containing 0.1% ethanol) subcutaneously for 3 weeks and then sacrificed. Tumors were weighed after removal of the livers and evaluated by histopathology at the conclusion of this study.
Cell Culture and Estrogen Treatment
Heps cells (Jiangsu Institute of Cancer Research), ANA-1 cells, and Hepa1-6 cells (Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China), were cultured in RPMI 1640 medium and Dulbecco's modified Eagle's medium, respectively, supplemented with 10% fetal bovine serum (PAA, Germany), 2 mm l-glutamine, 100 units/ml penicillin, and 100 units/ml streptomycin in a humidified atmosphere of 5% CO2 at 37 °C.
E2 was dissolved in ethanol and then diluted in medium containing 1% fetal bovine serum to a final ethanol concentration of 0.1%. Control cultures were exposed to equal concentrations of ethanol with no estradiol. Cells were treated daily for 3 days with or without E2.
Histology and Immunohistochemistry
Formalin-fixed paraffin-embedded samples were sectioned and stained with H&E for tumor histology analysis. Sections were blocked for 1 h with 3% BSA and 0.05% Tween 20 diluted in 50 mm Tris-HCl, pH 7.6, containing 150 mm NaCl. Sections were then probed with antibodies against CD206 conjugated to PE (AbD Serotec, Kidlington, UK) and anti-mouse F4/80 antigen-purified (ebioscience, San Diego) followed by anti-rat IgG FITC (Boster, Wuhan, China).
Western Blotting
Cells cultured were harvested and then suspended in whole cell lysis buffer or by a nuclear protein extract kit purchased from Beytotime (Haimen, Jiangsu, China). Protein concentration was determined by BCA reagent from Pierce. The immunoblot experiments were performed as described previously (25). The protein (50 μg/sample) was electrophoresed on 8, 10, 12, or 15% SDS-polyacrylamide gels and then electrotransferred onto polyvinylidene fluoride membrane (Amersham Biosciences). Antibodies against the following proteins were used: p-Stat6, p-Jak1, ERβ (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), ATP5J (Boster), and Gapdh (Kangcheng, Shanghai, China).
Coomassie Brilliant Blue Staining
After electrophoresis, the gels were stained with Brilliant Blue G250. Following electrophoresis, the gel was placed in a solution of 40% methanol, 10% acetic acid, 0.025% Coomassie Brilliant Blue R-250, which had been filtered through Whatman 1 paper. The gel was incubated for 6 h to overnight in the staining solution. The gel was destained with several changes of distilled water until the background was transparent. All steps were done on a rotary shaker with gentle mixing.
Flow Cytometry Analysis
PE-conjugated anti-mouse CD206 (Clone M1) and corresponding isotype controls were purchased from BD Pharmingen (San Diego, CA). 0.05% saponin (Sigma-Aldrich) diluted in phosphate-buffered saline was used as a membrane permeabilization agent for inner membrane staining. Cells were analyzed on a FACSCalibur cytometer using Cellquest software (BD Biosciences). Dead cells were excluded by forward and side scatter characteristics. Statistics presented are based on 10,000 events gated on the population of interest.
Cytokine Assays by Specific Enzyme-linked Immunosorbent Assay (ELISA)
IL-10 and IL-12 in supernatants were quantified using standard sandwich ELISAs according to instruction manuals. IL-10 and IL-12 concentrations are expressed in ng/ml, as calculated from calibration curves from serial dilutions of murine recombinant standards (eBioscience, San Diego) in each assay. The sensitivity of both IL-10 and IL-12 assays was 20 pg/ml.
Immunoprecipitation and LTQ Mass Spectra
Cell proteins were extracted as described above. A sufficient amount of ERβ (Santa Cruz Biotechnology, Inc.) or ATP5J (Boster) antibody was added into 500 μg of proteins and gently rotated at 4 °C overnight. The immunocomplex was captured by adding 20 μl of protein A + G-agarose beads (Beyotime, Jiangsu, China) and gently rotating at 4 °C for 3 h. Then the mixture was centrifuged at 2500 rpm for 5 min at 4 °C, and the supernatant was discarded. The precipitate was washed three times with ice-cold PBS buffer, resuspended in 5× sample buffer, and boiled for 5 min to dissociate the immunocomplex from the beads. The supernatant was collected by centrifugation and subjected to Western blot (10% SDS-PAGE).
Protein bands were excised from SDS-PAGE. The excised bands were then transferred to 1.5-ml tubes loaded with 500 μl of 50% ACN, 25 mm ammonium bicarbonate solution per tube. After being destained for 1 h, gel plugs were dehydrated with 500 μl of 100% ACN for 30 min. The dried gel bands were rehydrated with 30 μl/well trypsin in 25 mm ammonium bicarbonate with an approximately 1:50 enzyme/protein ratio. After staying at 37 °C for 45 min, the tubes were incubated at 37 °C for 8–10 h. After trypsin digestion, the peptide mixtures were extracted with 1% TFA, 5 mm OGP(Octyl β-d-glucopyranoside) at 37 °C for 1 h using 8 μl of the extraction solution per tube. Peptide mixtures were deposited onto an anchor and then were automatically analyzed in an Ultraflex MALDI TOF/TOF mass spectrometer (Bruker Daltonik) with an automated analysis loop using external mass calibration, under the control of FlexControlTM version 2.2 software (Bruker Daltonik GmbH). The protein identification was performed by PMF (peptide mass fingerprinting) and peptide fragment fingerprinting using the database search program MASCOT (MatrixScience).
Macrophage/Tumor Cell Coculture
A PET film 6-hole hanging cell culture chamber (Millipore, Billerica, MA) was used. The ANA-1 macrophages (1 × 105 cells/ml) were put in the wells of the 6-well plate (each well 2 ml); the tumor cells (1 × 105 cells/ml) were put in the filter chamber (each hole 500 μl), allowing diffusion of secreted molecules between the two cell types. The serum concentrations of the two cell types were kept consistent.
Small Interfering RNA (siRNA) Transfection
Cells were plated in 6-well plates 12 h before transfection with siRNA oligonucleotides (Stealth siRNA duplex oligoribonucleotides, Invitrogen). We used LipofectamineTM 2000 reagent (Invitrogen) and Opti-MEM (Invitrogen) according to the manufacturers' recommendations. Treatments were taken 24 h after siRNA transfection.
Cell Invasion Assay
The ability of liver cancer cells to migrate through Matrigel-coated filters was measured using Transwell chambers (Costar, Cambridge, MA) with polycarbonate membranes (8.0-μm pore size) coated with 100 μl of Matrigel (BD Biosciences) on the top side of the membrane. The upper surface of the matrix was challenged with 20,000 Hepa1-6 cells, and cells were kept in serum-free medium containing 0.1% BSA. The lower chamber contained medium supplemented with 10% serum, vehicle, or 10 nm E2. After 6 h, the cells were stained with 0.1% crystal violet solution. Cells and Matrigel on the upper surface of the membrane were removed carefully with a cotton swab.
Arginase Activity Assay
Arginase activity was measured in 104 cell lysates, which were lysed with 25 μl of 0.5% Triton X-100, containing 1 mm phenylmethylsulfonyl fluoride (PMSF) and 2 μg/ml aprotinin, leupeptin, and pepstatin (lysis buffer), respectively. We took 20 μl of this lysate and added 25 mm Tris-HCl, pH 7.5, and 5 μl of 10 mm MnCl2, and the enzyme was activated by heating for 3 min at 56 °C. After arginine hydrolyzes, the urea concentration is measured. One unit of enzyme activity is defined as the amount of enzyme that catalyzes the formation of 1 μmol of urea/min.
Gene Expression Analysis
TRIzol reagent (Invitrogen) was used to prepare total RNA from macrophages or tissues. Total RNA (1.5 μg) was reverse transcribed using a first strand cDNA synthesis kit (Bioteke, Beijing, China). Socs1, Socs3, Shp1, and Shp3 were analyzed by a PCR kit (Bioteke). Arginase-1 (Arg1), found in inflammatory zone protein (Fizz1) and CD206 were analyzed by Q-PCR. Q-PCR assays were carried out on the CFX96 real-time PCR detection system (Bio-Rad), using the Q-PCR kit (Bio-Rad). The comparative threshold method for relative quantification was used, and results are expressed as -fold change. The primers were synthesized by Invitrogen. The entire list of primers can be found in supplemental Fig. 4.
Statistical Analysis
Data were expressed as the mean ± S.E. Statistical analysis was performed by Students' t test when only two value sets were compared and by one-way analysis of variance followed by Dunnett's test when the data involved three or more groups. p < 0.05, p < 0.01, or p < 0.001 was considered statistically significant and indicated by one, two, or three asterisks, respectively.
RESULTS
Estrogen Inhibits Macrophage Alternative Activation in Mouse Liver Tumor Models
Based on our preliminary results, we hypothesized that estrogen played an important role in the gender bias of HCC progression. To test this hypothesis, we examined six groups of mice (described under “Materials and Methods”) in orthotopic liver tumors animal models (Fig. 1 and supplemental Fig. 1) and ectopic mouse liver tumor models (supplemental Fig. 5). The tumor weight in OVX female mice was higher than that in female control; E2-treated OVX female mice demonstrated a reduced tumor weight as compared with OVX female mice. However, there was no significant difference between the weight of tumors that developed in the castrated male mice compared with male control (Fig. 1B), suggesting that androgen did not play a significant role in HCC progression in our model. More importantly, after E2 supplement, tumor weight in castrated male mice was significantly reduced (Fig. 1A and supplemental Fig. 5). These results indicated that estrogen rather than androgen contributed to the gender disparity in HCC progression.
FIGURE 1.

Estrogen inhibits macrophage alternative activation in orthotopic mouse liver tumor models. A, tumors developed in the left hepatic lobe of BALB/c mice 6 weeks after intrahepatic inoculation of 106 Heps cells in each group. a–c, females. a, female control; b, females that underwent ovariotomy; c, E2 (50 μg/kg) administration after ovariotomy. d–f, males. d, male control; e, males that underwent castration; f, E2 (50 μg/kg) administration after castration (n = 8–9). In the OVX female mice, intrahepatic metastasis of liver cancer existed. B, average weights of tumors from each group. Tumors were stripped and weighed. The data are presented as mean ± S.E. (n = 8–9). In OVX mice, the liver tumor size was larger than in the intact mice. After administration of E2, tumor size for OVX mice was small compared with growth in OVX mice without E2 treatment. In male mice, we also found that estrogen administration after castration can result in relatively smaller liver tumors than in the castrated group. C, the macrophage cell numbers in tumor tissue were enumerated directly. Columns show the mean of three different experiments. Error bars, S.E. *, p < 0.05; **, p < 0.01. D, the ratio of CD206 and CD68 was used to demonstrate the relative cell numbers of CD206+ macrophage. E2 supplement decreased the CD206+ macrophage proportion compared with the surgical groups.
Then macrophages in the tumor sections were counted by immunofluorescence staining for CD68 (cluster of differentiation 68) (FITC), CD206 (PE), and nucleus (DAPI) (supplemental Fig. 2). We found that macrophages mainly exist in the form of CD206+. CD206 high expression is an important characterization of M2 macrophages. The CD206+ density in OVX female mice (∼72 cells/field of vision) was about 2-fold more than that in the female control (∼42 cells/field of vision). However, after the administration of exogenous E2, the number of CD206+ cells in E2-treated OVX female mice was almost restored to the level of female control, from 72 to 26 per field of vision (supplemental Fig. 2, a–c). In male mice, there was no change between male control and castration in CD206+ number. However, E2 treatment reduced the number of CD206+, from 65 to 32 per field of vision. The relative value of CD68 and CD206 in each group was shown in Fig. 1, C and D.
In conclusion, the macrophages inside tumor tissue in our model primarily existed in the form of CD206+, and E2 pretreatment reduced the number of CD206+ macrophages. These findings represented a novel characterization of the molecular events underlying gender disparity in HCC progression and may shed light on the pathological mechanisms of estrogen in alternative activation of macrophages.
Estrogen Inhibits Alternative Activation of Tumor-associated Macrophage in Vitro
According to the results above, we found that there was gender disparity in macrophage alternative activation in tumors and hypothesized that estrogen played an important role in suppressing tumor growth through inhibiting macrophage alternative activation. To investigate this hypothesis, we carried out experiments where macrophages were incubated with E2 (10 nm) prior to stimulus of IL-4 (10 ng/ml) or cocultured with Hepa1-6 liver cancer cells (Fig. 2). After incubation, we observed that E2 could strikingly reduce IL-4-induced arginase activity (Fig. 2, A (from 14.83 mm urea to 10.41 mm urea) and B) and mannose receptor CD206 (Fig. 2G). Similar results were obtained in the coculture experiments, where incubation with E2 resulted in a dose-dependent decrease of the mannose receptor CD206 (Fig. 2C). Cytokines in the supernatants released by macrophages in coculture experiments were then quantified by ELISA (Fig. 2, D and E), showing that E2 could inhibit IL-10 in a dose-dependent manner but had no effects on IL-12. Furthermore, we calculated the ratio of IL-10 to IL-12 (Fig. 2F) and found that it was reduced 3-fold in E2 10 nm treatment compared with the control group. In conclusion, E2 treatment significantly inhibited the special cytokine release of alternatively activated macrophages.
FIGURE 2.

Estrogen inhibits alternative activation of tumor-associated macrophage in vitro. A, E2 inhibits IL-4-induced arginase activity of ANA-1 cells. Values are expressed as means ± S.E. (n = 3 from three separate experiments; **, p < 0.01). B, E2 inhibits cocultured macrophage arginase activity. ANA-1 cells were pretreated with E2 for 2 days before coculture with Hepa1-6 cells. C, E2 inhibits cocultured macrophage mannose receptor expression in a dose-dependent manner. ANA-1 cells were pretreated with E2 for 2 days before coculture with Hepa1-6 cells. Anti-mouse CD206 (PE-conjugated; red) was used to label alternatively activated macrophage, and the percentage was calculated by flow cytometry analysis. Values are expressed as means ± S.E. (n = 3 from three separate experiments; *, p < 0.05). D–F, inflammatory cytokine protein secreted into culture medium. Values were expressed as means ± S.E. (n = 3 from three separate experiments; *, p < 0.05; **, p < 0.01; ***, p < 0.001). D, IL-10; E, IL-12p70; F, ratio of IL-10 to IL-12. G, E2 treatment could significantly inhibit IL-4-induced CD206 expression, as measured by flow cytometry analysis. H, Hepa1-6 cancer cell mobility is inhibited by E2-treated alternative macrophage. Mobility assays were carried out in 24-well Transwell units (cells per treatment condition in triplicate). Hepa1-6 cells were cocultured with no cells (a), control ANA-1 (b), alternatively activated ANA-1 (c), and E2-pretreated alternatively activated ANA-1 (d). After a 6-h incubation period, the moved cells that had passed through the membrane were stained and photographed (magnification, ×100). Error bars, S.E.; * and **, statistically significant p values <0.05 and <0.01, respectively.
Previous studies have indicated that, in the tumor microenvironment, infiltrated inflammatory cells could promote tumor invasion and migration (26). To test whether alternatively activated macrophages could affect tumor invasion in our model, we performed cell invasion assays with Hepa1-6 cells. Analysis of cell invasive assays showed that E2-treated alternatively activated ANA-1 significantly reduced Hepa1-6 cell invasion compared with alternatively activated ANA-1 alone (Fig. 2H, c and d). This suggested that in tumor microenvironment inhibiting macrophage alternative activation may contribute to suppressing cancer cell invasion. Altogether, these results demonstrated that estrogen could suppress macrophage alternative activation and prevent tumor progression and invasion.
Estrogen Inhibits the Alternative Activation of Macrophage through the Jak1-Stat6 Pathway
Based on the findings, we indicated that the antitumor effect of estrogen was mediated through its inhibitory effect on macrophage alternative activation in the tumor microenvironment. It is known that alternatively activated macrophage responded to Th2-type cytokines, such as IL-4. Recent studies have also identified IL-4 as a major regulator of the phenotypes of TAMs (27, 28). IL-4 signaling is initiated when this cytokine binds its cell surface receptor, activating receptor-associated Jaks, followed by recruitment and phosphorylation of Stat6. Once phosphorylated, Stat6 promoted the expression of IL-4-responsive genes, such as CD206, Arg1, and Fizz1. Hence, we tested whether E2 prior incubation could reverse IL-4-induced macrophage alternative activation. We first examined IL-4 receptor α (IL-4Rα); the results indicated that IL-4Rα expression had no changes before or after coculture (data not shown). Interestingly, we found that E2 could significantly reduce the phosphorylation of Stat6 and Jak1 (Fig. 3, A and B) by Western blotting. E2 inhibited phosphorylation of Jak1 in a time-dependent manner in macrophages by IL-4 treatment (Fig. 3B) and phosphorylation of Jak1 and Stat6 in a dose-dependent manner in macrophages (Fig. 3A). We also examined the downstream genes of Jak1-Stat6 (Fig. 4), confirming the inhibitory effects of E2 on macrophage alternative activation.
FIGURE 3.
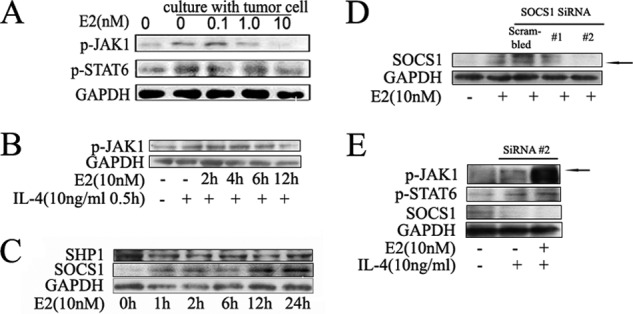
Estrogen inhibits the alternative activation of macrophage through the Jak1-Stat6 pathway. A, estrogen inhibits phosphorylation of Jak1-Stat6 in a dose-dependent manner in macrophages. ANA-1 macrophage cell lines were cocultured with murine hepatocarcinoma cell line Hepa1-6, and different estrogen concentrations (0.1, 1.0, 5.0, and 10 nm) were used to treat cells. Blots were probed with antibodies against p-Jak1 and p-Stat6. Blots were then stripped and reprobed with antibody against Gapdh as an internal control for equal loading. B, estrogen inhibits phosphorylation of Jak1 in a time-dependent manner in macrophages by IL-4 treatment. The cytoplasm protein was extracted, and the protein level was detected by Western blot. Results shown are representative of three independent experiments. C, Western blot analysis of the protein level of Jak1-Stat6 signaling pathway negative regulators after estrogen administration. Results shown are representative of three independent experiments. D and E, ANA-1 cells were transfected with scrambled siRNA (20 pm) and Socs1 siRNA (20 pm), and then treated with E2 (10 nm). The black arrow points to the position of the Socs1 band. D, Western blot analysis of total extracts (E2 36-h treatment after transfection). E, p-Jak1, p-Stat6, and Socs1 from extracts (E2 36-h treatment after transfection and then IL-4 24-h treatment) were analyzed by Western blot. The black arrow points to the position of the p-Jak1 band.
FIGURE 4.

Estrogen inhibits the downstream genes of the Jak1-Stat6 pathway. Total mRNA was extracted from ANA-1 cells treated with IL-4 and/or E2, and downstream genes Arg1 (A), Fizz1 (B), and CD206 (C) were analyzed by quantitative PCR. Error bars, S.E.
On the basis of Fig. 3, A and B, we observed that these E2 inhibitory effects on p-Jak1 occur after 12 h of E2 pretreatment. To examine whether some regulators influence the Jak1-Stat6 pathway, many negative factors, such as suppressor of cytokine signaling (SOCS) and SH2-containing phosphatase (SHP), were measured in RNA isolated from macrophages treated with E2. E2 treatment did not modify the expression of Socs3, Shp1, and Shp3. However, the expression of Socs1 was strikingly increased in macrophages treated with E2 (supplemental Fig. 3).
We also investigated this result in protein levels (Fig. 3C). This result was in line with those obtained in mRNA levels. Furthermore, Socs1-specific RNA interference experiment was carried out (Fig. 3, D and E), and the result confirmed these observations. Taken together, these results demonstrated that E2 inhibited macrophage alternative activation by way of dampening the phosphorylation of Jak1 and Stat6, through inducing Jak1-Stat6 pathway negative regulator Socs1.
Estrogen Exerts Its Inhibitory Effects via ERβ but Not ERα
Having confirmed the role of estrogen in HCC progression, we needed to clearly define how estrogen exerted its functions. The biological actions of estrogen are mediated by binding to one of two specific ERs, ERα and ERβ, which belong to the nuclear receptor superfamily, a family of ligand-regulated transcription factors. After binding to the ligand-binding domain of the ER, estrogen initiates a series of molecular events culminating in the activation or repression of target genes. To study the specific role of these two types of ERs in the negative regulation signaling pathway of Jak1-Stat6, we chose the selective agonist PPT of ERα and selective agonist DPN of ERβ to stimulate macrophages, respectively, and then analyzed the macrophage alternative activation state under these two agonists. As shown in Fig. 5, A and B, taking arginase activity as the detection index, we found that DPN exhibited inhibitory effects on the alternative activation of macrophages similar to E2, but this effect was not obtained by PPT. Furthermore, a substantial increase of Socs1 at the protein level (Fig. 5C) was observed when macrophages were treated with E2 and DPN. These results suggested that ERβ was responsible for interfering with the Jak1-Stat6 signaling pathway. To confirm this conclusion, ERβ-siRNA interference experiments were performed (Fig. 5D) independently. The results showed that ERβ-siRNA almost completely abolished the inhibitory effects initiated by E2 on IL-4-induced macrophage alternative activation, similar to the effects of ER-specific antagonist ICI,182780 on macrophage alternative activation. These results confirmed that E2 inhibited macrophage alternative activation mainly through ERβ. Because ERβ-selective agonist DPN could enhance Socs1 protein level, DPN was thought to affect the phosphorylation status of Stat6 by regulating Socs1. These data suggested that E2 supressed macrophage alternative activation and the Jak1-Stat6 pathway via ERβ, not ERα.
FIGURE 5.

Estrogen exerts its inhibitory effects via ERβ but not ERα. The effects of estrogen receptor agonists on macrophage arginase activity were analyzed. Cells were precultured with E2 (nonspecific ER agonist), PPT (ERα-specific agonist) and DPN (ERβ-specific agonist) at the indicated concentration for 48 h and stimulated with IL-4 for another 24 h (A) or cocultured with Hepa1-6 (B). Values were expressed as means ± S.E. (error bars) (n = 6 from three separate experiments; *, p < 0.05 versus IL-4 treated group). C, Western blot analysis of total extracts from macrophages that were exposed to E2 (10 nm), ERα-specific agonist PPT (10 nm), and ERβ-specific agonist DPN (100 nm) for 24 h. Socs1 level was analyzed by immunoblotting after treatment with E2, DPN, and PPT. D, extracellular surface receptor CD206 of ANA-1 macrophage was detected by flow cytometry. Macrophages were pretreated with Estrogen receptor antagonist (ICI182,780) or siRNA to knock down ERβ (siESR2). The treatment in each group was independent from the others. siESR2 could abolish the inhibitory effects of estrogen.
Estrogen Damages the Interaction of ERβ and ATPase in IL-4-stimulated Cells
To determine how ERβ-mediated inhibition of macrophage alternative activation is linked to the Jak1-Stat6 pathway, we performed immunoprecipitation experiments with anti-ERβ under different treatment conditions and used Coomassie Brilliant Blue staining. We detected a weak protein band (black arrow) only in the IL-4-treated group immunoprecipitants. Obviously, this weak band disappeared after pretreatment with E2, restored to a state similar with the control group (Fig. 6A). Then, by the mass spectrometry analysis, we identified this protein as mitochondrial membrane protein ATPase, which catalyzes ATP synthesis and is composed of two linked multisubunit complexes, including ATP5J. Therefore, we chose the subunit membrane protein ATP5J randomly to stand for ATPase. To further define the interaction between ERβ and ATPase in the absence and presence of E2, we continued to perform immunoprecipitation with anti-ERβ and anti-ATP5J (Fig. 6B). The results suggested that interaction between ERβ and ATP5J was impaired after E2 treatment.
FIGURE 6.
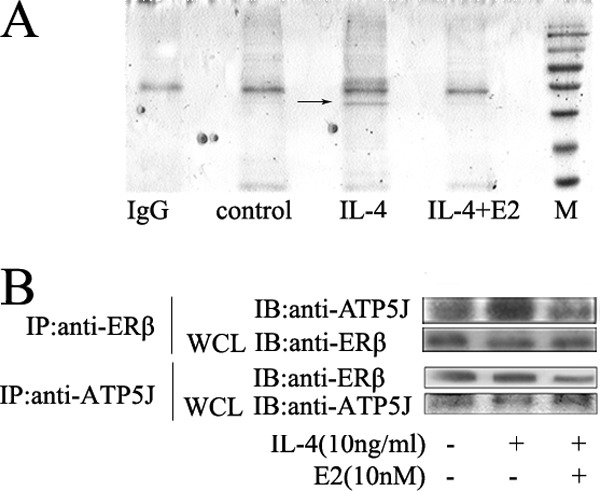
Estrogen damages the interaction of ERβ and ATPase in IL-4-stimulated cells. A, whole cell lysates (WCL) were prepared for immunoprecipitation with anti-ERβ. ANA-1 macrophages were pretreated with E2 for 48 h and stimulated by IL-14 for another 12 h. After SDS-PAGE, Coomassie G250/silver staining was carried out. A band (black arrow) was found with IL-4 treatment, but it disappeared when pretreated with E2. B, coimmunoprecipitation (IP) of ATP5J and ERβ. IB, immunoblot.
Based on our collective results, we propose a cascade of molecular events (Fig. 7) initiated by E2 as a mechanism to explain its suppressive effects on tumor-associated alternatively activated macrophage cells. ERβ mainly located in the mitochondria (29), and it combined with the ATPase under the stimulus of IL-4. Therefore, ERβ was involved in the regulation of ATP production and mitochondrial oxidative phosphorylation in macrophages after IL-4 treatment. However, after E2 activated ERβ, the interaction between ERβ and ATPase (ATP5J) weakened, and ERβ separated from ATPase. ERβ relocation may induce a series of consequences, such as up-regulating the expression of Jak1-Stat6 signaling pathway negative regulator SOCS1, thereby attenuating the macrophage alternative activation and HCC tumor growth.
FIGURE 7.

The speculative model of macrophage alternative activation regulation by estrogen. When estrogen interacts with ERβ, it can keep ERβ away from ATPase. Consequently, this process may enhance Socs1 expression, a negative regulator of the Jak1-Stat6 signaling pathway, to suppress the phosphorylation of Stat6 and inhibit the macrophage alternative phenotype.
DISCUSSION
HCC is a male-predominant cancer, and the rates of HCC in men are typically 2–4 times higher than in women (5, 30). Recent studies using mouse models show that sex hormones play a key role in hepatocarcinogenesis. However, the effects of sex hormones on HCC progression have no consistent conclusion to date. Therefore, we focused on the gender disparity in HCC progression rather than on HCC carcinogenesis. Our results showed that E2 functioned as a suppressor for macrophage alternative activation and tumor progression by keeping estrogen receptor β (ERβ) away from interacting with ATPase-coupling factor 6 (ATP5J), a part of ATPase, thus inhibiting the Jak1-Stat6 signaling pathway. These studies introduce a novel mechanism for suppressing male-predominant HCC.
The Role of Estrogen in HCC Progression
Previous studies on gender disparity in HCC have highlighted the linkage between estrogen and inflammation-induced carcinogenesis (2, 7). Our previous studies have shown that estrogen rather than androgen may contribute to gender disparity in HCC progression (31), and we obtained the same results here.
In the current study, we explored the effects of estrogen on the biological behavior of TAMs and their fate in different hormone environments.
Results revealed that in comparison with the female mice, the male mice exhibited larger tumor weight (Fig. 1, A and B, and supplemental Fig. S1), and the tumor weight in OVX female mice significantly increased compared with female control mice; however, castration had no impact on the HCC tumor volume among tumor-bearing male mice. Meanwhile, E2 supplement reduced tumor volume in E2-treated OVX and E2-treated castrated mice with preestablished liver cancer (Fig. 1B). These results demonstrated that gender disparity existed in HCC progression and that E2 played a critical role in this process. However, androgen has no effects on HCC progression.
To fully appreciate the important role of sex hormones on HCC progression and mimic the growth of human HCC comprehensively, we developed orthotopic models by injecting Heps hepatocellular carcinoma cells directly into the liver parenchyma of BALB/c mice. Prior to ours, several orthotopic transplantation models using human liver cancer cell lines and athymic nude mice have been described (32, 33). However, one obvious disadvantage of these models is that they do not permit the study of complex interactions between tumor cells and the host immune system, which have been shown to significantly influence tumor progression and metastatic potential (34). Our mouse models contended with this problem and improved on this basis, providing us with a useful, reproducible, and consistent model wherein we could manipulate hormone levels to examine their effect on HCC tumor progression.
The Alteration of Macrophage Activation Status in HCC Progression
Solid tumors consist of neoplastic cells, non-malignant stromal cells, and migratory hematopoietic cells. Complex interaction between the cell types in this microenvironment regulates tumor growth, progression, angiogenesis, and metastasis (35). There is strong evidence that this microenvironment is inflammatory and that activation of the innate immune system plays a role in the progression of cancer. One such inflammatory cell that has the potential to promote cancer progression is the macrophage. There is a growing body of preclinical and clinical evidence associating abundance of TAM with poor prognosis. According to Condeelis and Pollard (11), TAMs are obligate partners for malignant cell migration, invasion, and metastases in many different cancers. In addition, these TAMs also enhance tumor progression by supporting tumor-associated angiogenesis and suppression of antitumor immune responses, such as impairing cytotoxic CD8+T cell immune responses (26, 36).
In our study, we found that the number of TAMs in male control mouse models was significantly greater than that in female control mice (Fig. 1, C and D, and supplemental Fig. 2). More importantly, TAMs have been shown to acquire the hallmark properties of “alternatively activated” macrophages, such as the ability to tune inflammatory and immune responses and to promote angiogenesis and tissue remodeling (20). Consistent with this notion, our murine results showed that TAMs in mouse model tumors existed mainly in the form of CD206+ status (Fig. 1, C and D, and supplemental Fig. S2). CD206 is an important characterization of M2 macrophages. We also found that there was a great gender disparity of CD206+ macrophages of TAMs between male control and female control mice in the organization level in both orthotopic and ectopic mouse HCC models, and E2 administration resulted in a reduction of CD206+ macrophages in both E2-treated castration and E2-treated OVX. Interestingly, similar findings were reported by Ding et al. (37). In their study, they demonstrated that high macrophage infiltration predicts poor prognosis in patients with HCC. When macrophages infiltrated, tumor-derived factors in the HCC microenvironment might divert macrophage functions toward an immunosuppressive and protumoral phenotype, which was defined as TAMs (37). Our work in vitro also supported this notion. E2 treatment significantly reduced the arginase activity and the expression of CD206. Collectively, these results delineated the connections between E2, TAMs, and HCC cells and identified tumor-associated alternatively activated macrophages as a key factor in HCC progression.
As mentioned above, our in vitro and in vivo data revealed that E2 was able to suppress macrophage alternative activation and thus attenuate HCC progression significantly, indicating that with liver cancer deteriorating, estrogen may play a role in inhibiting alternative activation of macrophage and anti-inflammatory factors, such as IL-10. However, Karin and co-workers (7) found that in early stages of hepatocarcinogenesis, estrogen seemed to be able to suppress MyD88-dependent IL-6 production pathway in Kupffer cells to inhibit liver carcinogenesis. Thus, our results may be a novel mechanism for the tumor-suppressing function of estrogen during the progression of HCC, which seems not to exhibit an anti-inflammation function but to change the activation patterns of macrophage.
Estrogen Inhibits Macrophage Alternative Activation Mainly through ERβ
Previous studies indicated a critical role for the Jak1-Stat6 signaling pathway in maturation of anti-inflammatory alternatively activated macrophages. Moreover, Ohmori and Hamilton (38) confirmed that Stat6 was necessary for the IL-4-mediated inflammatory suppression. Meanwhile, the IL-4 signaling pathways are subject to negative regulation by several mechanisms, such as SHP proteins (39, 40) and the Socs family (41). In addition, in recent years, IL-4 has also been identified to be a major regulator of the phenotypes of TAMs (27, 28). We reported here that E2 pretreatment could inhibit the phosphorylation of Jak1 and Stat6 in a dose- and time-dependent manner in vitro (Fig. 3, A and B).
A previous study (42) reported that E2-liganded ER-β could directly modulate Socs1 expression by binding to the Socs1 promoter. These authors proposed that ER-β regulated three classes of genes (42). Socs1 genes belonged to the class II genes not regulated by unliganded ERβ but regulated by E2-bound ERβ. In our study, the effects of E2 on Socs1 expression are consistent with theirs. The PCR (supplemental Fig. 3) and Western blot results showed that the expression of Socs1 was strikingly increased in macrophages pretreated with E2 (Fig. 3C), and Socs1-specific RNA interferences confirmed this observation. Taken together, the findings presented here demonstrated that E2 promoted the expression of negative regulator Socs1 of the Jak1-Stat6 signaling pathway.
The biological functions of estrogen are mainly mediated by estrogen binding to one of the two specific ERs, ERα and ERβ (43). And our data demonstrated that E2 suppressed macrophage alternative activation mainly via ERβ.
In addition, our work raised an intriguing question as to how ERβ exerts its function to inhibit the phosphorylation of Stat6. In the presence of IL-4, we found that ERβ interacted with membrane protein ATPase (Fig. 6A), which catalyzed ATP synthesis from ADP and inorganic phosphate (Pi) by using the electrochemical potential of protons (or sodium ions in some bacteria) across the membrane (44), whereas after pretreating with E2, the interaction was impaired (Fig. 6B). ERβ was separated from ATPase and turned to interacting with other mitochondrial proteins or was translocated into the cytoplasm from the chondriosome to affect some other factors such as Socs1, exerting its inhibitory effect on macrophage alternative activation. However, after immunoprecipitation with ATP5J, ERβ protein expression was not significantly different among the groups. This may be due to the fact that ATP5J was only one part of the ATPase; ERβ could interact with other parts. These results suggested that ERβ was involved in the regulation of ATP production and mitochondrial oxidative phosphorylation in macrophages. This hypothesis was supported by the literature indicating that ERβ is involved in mitochondrial membrane potential maintenance and mitochondrial vulnerability (29). Importantly, relocation of the ERβ after separating from ATPase requires further study.
The Significance of Our Results May Suggest a New Insight for HCC Therapy
Taken together, the findings presented here supported the notion that estrogen could inhibit macrophage alternative activation, resulting in attenuated HCC progression. Because HCC therapy has failed to improve overall survival in patients with advanced HCC, efforts to develop new drug treatments have shifted from systemic chemotherapy to targeted treatment against the tumor-stromal interaction. Therefore, targeting components of the tumor microenvironment will be a better auxiliary way to suppress HCC progression. For example, sorafenib, an oral multikinase inhibitor, is the most successful medication of this kind. It inhibits Vegfr-2/-3 (vascular endothelial growth factor receptor-2/3) and Pdgfr (PDGF receptor) as well as Raf kinase, disrupting tumor-stromal interactions and resulting in decreased cell proliferation and angiogenesis. In addition, because tumor-associated macrophages accumulation correlates with tumor progression, it is feasible to convert tumor-associated macrophage polarization in order to inhibit tumor growth. In T241 tumors, by skewing TAM polarization away from the M2 macrophage to a tumor-inhibiting M1-like phenotype, Hrg (histidine-rich glycoprotein) promotes antitumor immune responses and vessel normalization, effects known to decrease tumor growth and metastasis and to enhance chemotherapy (45). Therefore, considering the important role of tumor-associated M2 in the tumor microenvironment, estrogen therapy may be feasible in treating HCC, especially for males. However, despite the disadvantages of hormone replacement therapy, estrogen analogues may contribute as a complementary therapy in HCC treatment and may also be a possible adjuvant therapy for controlling HCC. Our research has proposed that estrogen-attenuated HCC progression, through inhibiting tumor-associated macrophage alternative activation, therefore selectively targeting macrophages in combination with targeting the cancer cells, may be a promising trend for tumor cure.
Supplementary Material
Acknowledgments
We thank Chris T. Chen (Laboratory for Translational Research, Harvard Medical School) and Nina Y. Xue (University of California San Diego, La Jolla, CA) for critical comments and grammatical correction of the manuscript. We thank Dr. Ning Su (School of Basic Medical Sciences, Southeast University, China) for excellent technical assistance.
This work was supported by National Natural Science Foundation of China Projects 31070764, 91013015, and 81121062 and National Key Basic Research Program of China Project 2010CB912203.

This article contains supplemental Figs. 1–5.
- HCC
- hepatocellular carcinoma
- ER
- estrogen receptor
- E2
- 17β-estradiol
- OVX
- ovariectomized
- Shp
- SH2-containing phosphatase
- Socs
- suppressor of cytokine signaling
- M1
- classically activated macrophage(s)
- M2
- alternatively activated macrophages(s)
- TAM
- tumor-associated macrophage
- OVX
- ovariectomized
- PE
- phycoerythrin
- p-Jak1 and p-Stat6
- phosphorylated Jak1 and Stat6, respectively
- DPN
- 2,3-bis(4-hydroxyphenyl)-propionitrile
- PPT
- 4,4′,4″-(4-propyl-c-pyrazole-1,3,5-triyl)tris-phenol.
REFERENCES
- 1. Thomas M. B., Jaffe D., Choti M. M., Belghiti J., Curley S., Fong Y., Gores G., Kerlan R., Merle P., O'Neil B., Poon R., Schwartz L., Tepper J., Yao F., Haller D., Mooney M., Venook A. (2010) Hepatocellular carcinoma. Consensus recommendations of the National Cancer Institute Clinical Trials Planning Meeting. J. Clin. Oncol. 28, 3994–4005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rogers A. B., Theve E. J., Feng Y., Fry R. C., Taghizadeh K., Clapp K. M., Boussahmain C., Cormier K. S., Fox J. G. (2007) Hepatocellular carcinoma associated with liver-gender disruption in male mice. Cancer Res. 67, 11536–11546 [DOI] [PubMed] [Google Scholar]
- 3. Ghebranious N., Sell S. (1998) Hepatitis B injury, male gender, aflatoxin, and p53 expression each contribute to hepatocarcinogenesis in transgenic mice. Hepatology 27, 383–391 [DOI] [PubMed] [Google Scholar]
- 4. Maeda S., Kamata H., Luo J. L., Leffert H., Karin M. (2005) IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 121, 977–990 [DOI] [PubMed] [Google Scholar]
- 5. Bosch F. X., Ribes J., Díaz M., Cléries R. (2004) Primary liver cancer. Worldwide incidence and trends. Gastroenterology 127, S5–S16 [DOI] [PubMed] [Google Scholar]
- 6. El-Serag H. B., Mason A. C., Key C. (2001) Trends in survival of patients with hepatocellular carcinoma between 1977 and 1996 in the United States. Hepatology 33, 62–65 [DOI] [PubMed] [Google Scholar]
- 7. Naugler W. E., Sakurai T., Kim S., Maeda S., Kim K., Elsharkawy A. M., Karin M. (2007) Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 317, 121–124 [DOI] [PubMed] [Google Scholar]
- 8. Nakatani T., Roy G., Fujimoto N., Asahara T., Ito A. (2001) Sex hormone dependency of diethylnitrosamine-induced liver tumors in mice and chemoprevention by leuprorelin. Jpn. J. Cancer Res. 92, 249–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. CLIP Group (Cancer of the Liver Italian Programme) (1998) Tamoxifen in treatment of hepatocellular carcinoma. A randomized control trial. Lancet 352, 17–20 [PubMed] [Google Scholar]
- 10. Liu C. L., Fan S. T., Ng I. O., Lo C. M., Poon R. T., Wong J. (2000) Treatment of advanced hepatocellular carcinoma with tamoxifen and the correlation with expression of hormone receptors. A prospective randomized study. Am. J. Gastroenterol. 95, 218–222 [DOI] [PubMed] [Google Scholar]
- 11. Condeelis J., Pollard J. W. (2006) Macrophages. Obligate partners for tumor cell migration, invasion, and metastasis. Cell 124, 263–266 [DOI] [PubMed] [Google Scholar]
- 12. Goswami S., Sahai E., Wyckoff J. B., Cammer M., Cox D., Pixley F. J., Stanley E. R., Segall J. E., Condeelis J. S. (2005) Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 65, 5278–5283 [DOI] [PubMed] [Google Scholar]
- 13. Bingle L., Brown N. J., Lewis C. E. (2002) The role of tumor-associated macrophages in tumor progression. Implications for new anticancer therapies. J. Pathol. 196, 254–265 [DOI] [PubMed] [Google Scholar]
- 14. Lin E. Y., Nguyen A. V., Russell R. G., Pollard J. W. (2001) Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 193, 727–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pollard J. W. (2004) Tumor-educated macrophages promote tumor progression and metastasis. Nat. Rev. Cancer 4, 71–78 [DOI] [PubMed] [Google Scholar]
- 16. Lin E. Y., Gouon-Evans V., Nguyen A. V., Pollard J. W. (2002) The macrophage growth factor CSF-1 in mammary gland development and tumor progression. J. Mammary Gland Biol. Neoplasia 7, 147–162 [DOI] [PubMed] [Google Scholar]
- 17. Zeisberger S. M., Odermatt B., Marty C., Zehnder-Fjällman A. H., Ballmer-Hofer K., Schwendener R. A. (2006) Clodronate-liposome-mediated depletion of tumor-associated macrophages. A new and highly effective antiangiogenic therapy approach. Br. J. Cancer 95, 272–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mantovani A., Porta C., Rubino L., Allavena P., Sica A. (2006) Tumor-associated macrophages (TAMs) as new target in anticancer therapy. Drug Discov. Today 3, 361–366 [Google Scholar]
- 19. Pollard J. W. (2009) Trophic macrophages in development and disease. Nat. Rev. Immunol. 9, 259–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mantovani A., Sozzani S., Locati M., Allavena P., Sica A. (2002) Macrophage polarization. Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23, 549–555 [DOI] [PubMed] [Google Scholar]
- 21. Mantovani A., Sica A. (2010) Macrophages, innate immunity, and cancer. Balance, tolerance, and diversity. Curr. Opin. Immunol. 22, 231–237 [DOI] [PubMed] [Google Scholar]
- 22. Biswas S. K., Gangi L., Paul S., Schioppa T., Saccani A., Sironi M., Bottazzi B., Doni A., Vincenzo B., Pasqualini F., Vago L., Nebuloni M., Mantovani A., Sica A. (2006) A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-κB and enhanced IRF-3/STAT1 activation). Blood 107, 2112–2122 [DOI] [PubMed] [Google Scholar]
- 23. Pucci F., Venneri M. A., Biziato D., Nonis A., Moi D., Sica A., Di Serio C., Naldini L., De Palma M. (2009) A distinguishing gene signature shared by tumor-infiltrating Tie2-expressing monocytes, blood “resident” monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood 114, 901–914 [DOI] [PubMed] [Google Scholar]
- 24. Ojalvo L. S., King W., Cox D., Pollard J. W. (2009) High density gene expression analysis of tumor-associated macrophages from mouse mammary tumors. Am. J. Pathol. 174, 1048–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bao X., Cui J., Wu Y., Han X., Gao C., Hua Z., Shen P. (2007) The roles of endogenous reactive oxygen species and nitric oxide in triptolide-induced apoptotic cell death in macrophages. J. Mol. Med. 85, 85–98 [DOI] [PubMed] [Google Scholar]
- 26. Qian B. Z., Pollard J. W. (2010) Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. DeNardo D. G., Barreto J. B., Andreu P., Vasquez L., Tawfik D., Kolhatkar N., Coussens L. M. (2009) CD4+ T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16, 91–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gocheva V., Wang H. W., Gadea B. B., Shree T., Hunter K. E., Garfall A. L., Berman T., Joyce J. A. (2010) IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 24, 241–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang S. H., Sarkar S. N., Liu R., Perez E. J., Wang X., Wen Y., Yan L. J., Simpkins J. W. (2009) Estrogen receptor β as a mitochondrial vulnerability factor. J. Biol. Chem. 284, 9540–9548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hoenerhoff M. J., Pandiri A. R., Lahousse S. A., Hong H. H., Ton T. V., Masinde T., Auerbach S. S., Gerrish K., Bushel P. R., Shockley K. R., Peddada S. D., Sills R. C. (2011) Global gene profiling of spontaneous hepatocellular carcinoma in B6C3F1 mice. Similarities in the molecular landscape with human liver cancer. Toxicol. Pathol. 39, 678–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xu H., Wei Y., Zhang Y., Xu Y., Li F., Liu J., Zhang W., Han X., Tan R., Shen P. (2012) Estrogen attenuates tumour progression in hepatocellular carcinoma. J Pathol. 228, 216–229 [DOI] [PubMed] [Google Scholar]
- 32. Yao X., Hu J. F., Daniels M., Yien H., Lu H., Sharan H., Zhou X., Zeng Z., Li T., Yang Y., Hoffman A. R. (2003) A novel orthotopic tumor model to study growth factors and oncogenes in hepatocarcinogenesis. Clin. Cancer Res. 9, 2719–2726 [PubMed] [Google Scholar]
- 33. Lu Y. S., Kashida Y., Kulp S. K., Wang Y. C., Wang D., Hung J. H., Tang M., Lin Z. Z., Chen T. J., Cheng A. L., Chen C. S. (2007) Efficacy of a novel histone deacetylase inhibitor in murine models of hepatocellular carcinoma. Hepatology 46, 1119–1130 [DOI] [PubMed] [Google Scholar]
- 34. de Visser K. E., Eichten A., Coussens L. M. (2006) Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 6, 24–37 [DOI] [PubMed] [Google Scholar]
- 35. Albini A., Magnani E., Noonan D. M. (2010) The tumor microenvironment. Biology of a complex cellular and tissue society. Q. J. Nucl. Med. Mol. Imaging 54, 244–248 [PubMed] [Google Scholar]
- 36. Barajas M., Mazzolini G., Genové G., Bilbao R., Narvaiza I., Schmitz V., Sangro B., Melero I., Qian C., Prieto J. (2001) Gene therapy of orthotopic hepatocellular carcinoma in rats using adenovirus coding for interleukin 12. Hepatology 33, 52–61 [DOI] [PubMed] [Google Scholar]
- 37. Ding T., Xu J., Wang F., Shi M., Zhang Y., Li S. P., Zheng L. (2009) High tumor-infiltrating macrophage density predicts poor prognosis in patients with primary hepatocellular carcinoma after resection. Hum. Pathol. 40, 381–389 [DOI] [PubMed] [Google Scholar]
- 38. Ohmori Y., Hamilton T. A. (1998) STAT6 is required for the anti-inflammatory activity of interleukin-4 in mouse peritoneal macrophages. J. Biol. Chem. 273, 29202–29209 [DOI] [PubMed] [Google Scholar]
- 39. Lu X., Malumbres R., Shields B., Jiang X., Sarosiek K. A., Natkunam Y., Tiganis T., Lossos I. S. (2008) PTP1B is a negative regulator of interleukin 4-induced STAT6 signaling. Blood 112, 4098–4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Myers M. P., Andersen J. N., Cheng A., Tremblay M. L., Horvath C. M., Parisien J. P., Salmeen A., Barford D., Tonks N. K. (2001) TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J. Biol. Chem. 276, 47771–47774 [DOI] [PubMed] [Google Scholar]
- 41. Dickensheets H., Vazquez N., Sheikh F., Gingras S., Murray P. J., Ryan J. J., Donnelly R. P. (2007) Suppressor of cytokine signaling-1 is an IL-4-inducible gene in macrophages and feedback inhibits IL-4 signaling. Genes Immun. 8, 21–27 [DOI] [PubMed] [Google Scholar]
- 42. Vivar O. I., Zhao X., Saunier E. F., Griffin C., Mayba O. S., Tagliaferri M., Cohen I., Speed T. P., Leitman D. C. (2010) Estrogen receptor β binds to and regulates three distinct classes of target genes. J. Biol. Chem. 285, 22059–22066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pettersson K., Gustafsson J. A. (2001) Role of estrogen receptor β in estrogen action. Annu. Rev. Physiol. 63, 165–192 [DOI] [PubMed] [Google Scholar]
- 44. Okuno D., Iino R., Noji H. (2011) Rotation and structure of F0F1-ATP synthase. J. Biochem. 149, 655–664 [DOI] [PubMed] [Google Scholar]
- 45. Rolny C., Mazzone M., Tugues S., Laoui D., Johansson I., Coulon C., Squadrito M. L., Segura I., Li X., Knevels E., Costa S., Vinckier S., Dresselaer T., Åkerud P., De Mol M., Sälomaki H., Phillipson M., Wyns S., Larsson E., Buysschaert I., Botling J., Himmelreich U., Van Ginderachter J. A., De Palma M., Dewerchin M., Claesson-Welsh L., Carmeliet P. (2011) HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through down-regulation of PlGF. Cancer Cell 19, 31–44 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.