

Background: Preventing unnecessary cell death is essential for DNA-damaged cells to carry out the DNA repair process.
Results: Cdc7 inhibits the Cul4-DDB1Cdt2-dependent Tob degradation.
Conclusion: Cdc7 enables mild DNA-damaged cells to keep their viability by competing with the Tob degradation system.
Significance: Cells deal with moderate DNA damage not only by cessation of the cell cycle but also through direct mediated pro-survival signaling.
Keywords: Apoptosis, DNA Damage, Protein Degradation, Signal Transduction, Tumor Suppressor Gene, Cdc7, Tob, Cell Survival
Abstract
Cells respond to DNA damage by activating alternate signaling pathways that induce proliferation arrest or apoptosis. The correct balance between these two pathways is important for maintaining genomic integrity and preventing unnecessary cell death. The mechanism by which DNA-damaged cells escape from apoptosis during DNA repair is poorly understood. We show that the DNA replication-initiating kinase Cdc7 actively prevents unnecessary death in DNA-damaged cells. In response to mild DNA damage, Tob levels increase through both a transcriptional mechanism and protein stabilization, resulting in inhibition of pro-apoptotic signaling. Cells lacking Cdc7 expression undergo apoptosis after mild DNA damage, where Cul4-DDB1Cdt2 induces Tob ubiquitination and subsequent degradation. Cdc7 phosphorylates and interacts with Tob to inhibit the Cul4-DDB1Cdt2-dependent Tob degradation. Thus, Cdc7 defines an essential pro-survival signaling pathway by contributing to stabilization of Tob, thereby the viability of DNA-damaged cells being maintained.
Introduction
The integrity of genomic DNA is constantly under threat in every organism. DNA damage on the genome is introduced by reactive oxygen species, stochastic errors in replication or recombination, or environmental genotoxins. Two cellular strategies have evolved for coping with DNA damage: DNA damage is repaired or DNA-damaged cells are removed by apoptosis. Apparently, organisms respond differently to DNA damage depending on the type and severity of the damage (1, 2). Although activation of the cell cycle checkpoint in response to mild DNA damage provides an opportunity to repair the damage, apoptosis should be suppressed to prevent unnecessary cell destruction. One of the important unresolved problems is how DNA-damaged cells undergoing DNA repair escape from apoptosis.
Cdc7 is an evolutionarily conserved kinase that promotes DNA replication origin firing (3, 4). Aberrant DNA replication and DNA damage immediately lead to activation of the S-phase checkpoint, which suppresses late origin firing to prevent further DNA replication and stabilizes stalled replication forks to ensure proper fork restart following removal of the replication error or DNA damage. Whereas some studies have suggested that Cdc7 activity is inhibited through the dissociation of the active Cdc7 complex following DNA damage (5, 6), recent reports have indicated that Cdc7 plays an active role in regulating the S-phase checkpoint and DNA damage checkpoint responses. Upon DNA damage, the integrity of the Cdc7 kinase complex and its activity is unaffected (7–9), although inhibition of Cdc7 in the presence of genotoxic agents increases cell death (8). Moreover, Cdc7 is involved in down-regulating the S-phase checkpoint signaling (9). These studies suggest that Cdc7 contributes to cell survival during genotoxic stress response and triggers DNA replication re-initiation. However, the precise molecular mechanism by which Cdc7 suppresses cell death and a role of Cdc7 activity before removal of DNA damage have not been elucidated.
Tob is a member of the Tob/BTG anti-proliferative protein family. The family consists of six members in mammals (10) and some of them are important for cell cycle regulation and tumorigenesis (11–14). The interaction between this family of proteins and transcription factors is relevant to their ability of regulating cell growth (13, 15). Intriguingly, the Tob/BTG family proteins are also involved in cellular responses to DNA damage. For instance, upon DNA damage, expression of several tob/btg family genes is induced in a p53-dependent manner (15, 16). Furthermore, proteasomal degradation of Tob, which is independent of p53, is closely associated with DNA damage-induced apoptosis (17). It is important to identify molecules that regulate the Tob expression level to deepen our understanding of the mechanism for cell fate decision in response to DNA damage. In this study, we show that Cdc7 and Tob cooperate to prevent cells from undergoing DNA damage-induced apoptosis by competing with Cul4-DDB1Cdt2-dependent proteasome activity, suggesting that the Cdc7-Tob axis actively maintains survival of DNA-damaged cells to provide an opportunity to repair the damage.
EXPERIMENTAL PROCEDURES
Cells and Reagents
U2OS and NUGC3 cells were grown in RPMI supplemented with 10% fetal bovine serum (FBS). HeLa, COS7, and HT1080 cells were grown in Dulbecco's modified Eagle's medium with 10% FBS. Actinomycin D and etoposide were purchased from Sigma and used at 2.5 μg/ml and 10 μm, respectively. The following compounds were purchased from Calbiochem and used: SP600125 (20 μm), CGK733 (20 μm), wortmannin (1 μm), U0126 (10 μm), SB203580 (20 μm), Z-VAD-fmk2 (30 μm), and Cdc7 inhibitor, PHA-767491 (18) (1 μm).
Antibodies
A mouse Tob monoclonal antibody was raised against a recombinant Autographa californica multiple nuclear polyhedrosis virus displaying the fusion protein containing amino acids 263–345 of Tob as previously described (19). Antibodies recognizing poly(ADP-ribose) polymerase (PARP) (Zymed Laboratories Inc.), DDB1 (Bethyl Laboratories), α-tubulin (DM1A; Sigma), MCM2 (N-19; Santa Cruz), phospho-MCM2 (Bethyl Laboratories), and FLAG (M2; Sigma) were also used. Antibodies recognizing cleaved caspase 3 and phosphor-Chk1 were purchased from Cell Signaling Technology. Anti-Cdc7, anti-ataxia telangiectasia mutated (ATM), anti-ATM-Rad3-related (ATR), anti-Chk1, and anti-Chk2 were from MBL. The anti-high mobility group B1 (HMGB1) monoclonal antibody (HAP46.5), anti-HMGB1 polyclonal antibody, and anti-Cdc7 antibody (DCS-341) were from Abcam. The anti-Cdt2 polyclonal antibody was as described previously (20).
Expression Vector Construction and Transient Transfection
Enhanced green fluorescent protein (EGFP) expression vector was constructed by inserting the cDNA of EGFP into pcDNA3.1 vector (Invitrogen). Tob point mutants (in pME18S vector or pET26b vector) were generated by polymerase chain reaction. We used TransIT-LT1 (MirusBio) when introducing expression vectors into COS7 cells.
Retroviral Infection and Small Interfering RNA (siRNA) Transfection
Amphotropic retrovirus packaging cells (platA cells) were transfected with the retroviral plasmid (pMX-puro) containing RNAi-resistant Tob mutants, TobF series, or His6-Cdc7 cDNA using TransIT-LT1 (MirusBio). The next day, the U2OS cells were plated. Two days after transfection, the virus-containing medium was collected, filtered (0.45 μm filter, Millipore), and supplemented with Polybrene (Sigma; 5 μg/ml). For infection, the culture medium was replaced by the virus-containing medium and cells were incubated for a further 2 days. The infected cells were selected in medium containing puromycin (Sigma; 2 μg/ml) for 4 days. Double-stranded RNA was transfected into U2OS cells with Lipofectamine RNAiMAX (Invitrogen). The sequences of siRNAs are in supplemental Methods. Cells were irradiated with UV or treated with the indicated chemicals at 2 days post-transfection.
UV Irradiation and Preparation of Cell Extracts
Cells at 80% confluence were exposed to UV-C (30 J/m2 unless otherwise indicated) with a FUNA-UV-linker (FS-800; FUNAKOSHI). Cells were lysed with buffer (50 mm Hepes-NaOH, pH 7.5, 150 mm NaCl, 100 mm NaF, 4 mm EDTA, 1% Triton X-100, 0.1% SDS, 1 mm phenylmethylsulfonyl fluoride) 8 h after UV irradiation unless otherwise indicated. To inhibit proteasome-dependent degradation, cells were pretreated with MG132 (40 μm; Peptide Institute) for 2 h. Total RNA was prepared from cells with ISOGEN (Nippon Gene). Northern blot analysis was performed as described previously (13).
Immunoprecipitation
U2OS or retrovirus-infected U2OS cells were washed three times in phosphate-buffered saline (PBS), treated with 0.1% formaldehyde in PBS for 10 min, and washed again in PBS. The cells were lysed with TNE buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 10 mm sodium fluoride, 10 mm β-glycerophosphate, 2 mm EDTA, 1% Nonidet P-40, 1 mm phenylmethylsulfonyl fluoride). The lysates were immunoprecipitated with anti-FLAG antibody (M2)-conjugated agarose (Sigma) or anti-Tob antibody-conjugated Protein A-Sepharose. The immunoprecipitates were submitted to immunoblotting. For glutathione S-transferase (GST) pulldown experiments, in vitro phosphorylation reaction products were suspended in TNE buffer, and GST-Cdc7 was purified using glutathione-Sepharose 4B resin (GE Healthcare).
In Vitro Ubiquitination Assay
Recombinant Tob (21) (110 nm) was assayed in a 10-μl reaction mixture containing 25 mm Hepes-NaOH (pH 7.8), 100 mm NaCl, 6 mm ATP,10 mm MgCl2, 5 mm DTT, 6 μg of ubiquitin (Biomol), 100 ng of E1 (Wako), 200 ng of E2 (UbcH5c, Biomol), and 300 ng of E3 complex. The Cul4A-DDB1Cdt2 E3 complex was purified as described previously (20). After incubation at 37 °C for 1 h, the reaction was terminated by the addition of SDS-PAGE sample buffer. Ubiquitinated Tob was detected by immunoblotting using an anti-Tob antibody.
Immunoblot Analyses and Immunofluorescence
Immunoblotting and immunofluorescence staining were performed using appropriate antibodies as described previously (12). Apoptotic cells showing the condensed nuclei were visualized by nuclear staining with Hoechst 33342 (Molecular Probe). Differences between groups were examined for statistical significance using Student's t test. Phosphate-affinity gel electrophoresis was performed in gels containing 7.5% acrylamide, 200 μm MnCl2, and 100 μm Phos-tag ligand (NARD Institute).
Kinase Assay
Purified Cdc7-ASK kinase complex (22) was mixed with 2 μg of recombinant protein (Tob, proliferating cell nuclear antigen, or MCM2) in kinase buffer (40 mm Hepes-NaOH, pH 7.4, 10 mm MgCl2, 1 mm β-glycerophosphate, 1 mm sodium fluoride, 2 mm DTT, 200 μm ATP), and reactions were carried out at 30 °C for 30 min after the addition of [γ-32P]ATP (0.2 mCi/ml). The cDNAs corresponding to full-length proliferating cell nuclear antigen and MCM2 (amino acids 1–283) were amplified by polymerase chain reaction and inserted into the pGEX6P-1 vector. The GST-fused proteins were expressed in BL21 Escherichia coli and purified using glutathione-Sepharose beads (GE Healthcare). The GST portion was cleaved by Precision protease (GE Healthcare).
RESULTS
Under Mild DNA Damage, Tob Is Protected from Proteasome-dependent Degradation, Suppressing Apoptosis
We have previously shown that low doses of ultraviolet (UV) (20–45 J/m2: UV(l)) irradiation up-regulate the level of Tob expression and induce little apoptosis, whereas the apoptotic response to high doses of UV (>100 J/m2: UV(h)) correlates with the extent of proteasomal degradation of Tob (17). Treatment of cells with actinomycin D completely suppressed the UV(l)-induced increase in Tob, indicating that UV(l) activates transcription of the tob gene (Fig. 1A). Northern blot analysis revealed that activation of tob transcription occurred within a couple of hours after UV(l) irradiation (Fig. 1B). To clarify the underlying mechanisms, we examined the effects of several kinase inhibitors on UV(l)-induced up-regulation of Tob. Treatment of U2OS cells with CGK733, which modulates the ATM and ATR kinase pathways, largely inhibited the UV(l)-induced increase in Tob expression, whereas all other tested inhibitors including SP600125 and U0126, which target Jun N-terminal kinases and extracellular signal-regulated protein kinases, respectively, had no effect (Fig. 1C). Importantly, treatment of UV(l)-irradiated cells with CGK733 promoted apoptotic cell death as monitored by cleavage of PARP and nuclear condensation, supporting our previous conclusion that Tob contributes to the maintenance of cell survival (Fig. 1C). Similar results were obtained from experiments using etoposide, a topoisomerase II inhibitor, as a DNA-damaging reagent. Namely, Tob protein is increased through the transcriptional induction after etoposide treatment to protect cells from apoptosis, but the increase was suppressed concomitant with extensive apoptosis in the presence of CGK733 (supplemental Fig. S1). Next, we examined whether suppression of the UV(l)-induced Tob increase by CGK733 treatment was due to transcriptional repression. Northern blot analysis showed that tob mRNA was increased in both the presence and absence of CGK733 (Fig. 1D), indicating that CGK733 did not affect tob transcription. We then asked whether CGK733 affected a mechanism controlling the stability of Tob. Indeed, when we treated cells with the proteasome inhibitor MG132, the UV(l)-induced Tob increase was largely restored in CGK733-treated cells (Fig. 1E). These data suggest that the CGK733-induced Tob degradation results in apoptosis of DNA-damaged cells. Therefore, we speculated that the Tob protein up-regulated upon UV(l) irradiation should be protected from proteasome-dependent degradation to maintain the viability of the DNA-damaged cells.
FIGURE 1.
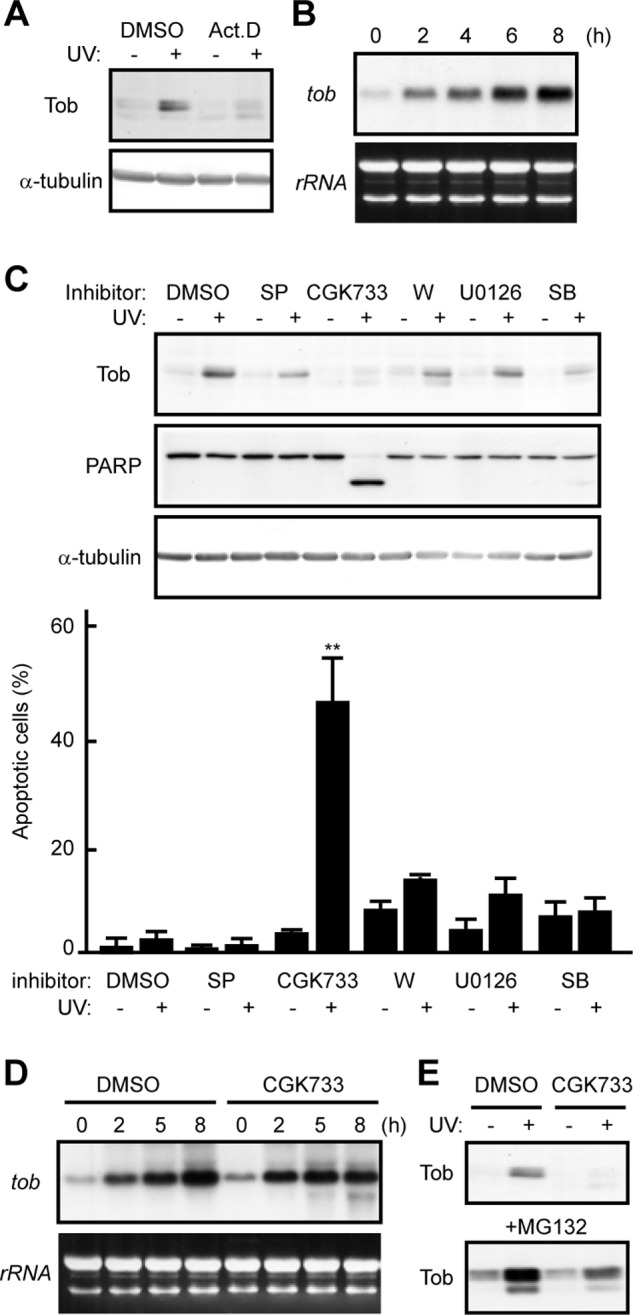
UV-induced Tob is protected from proteasome-dependent degradation. A, U2OS cells were irradiated with UV in the presence of dimethyl sulfoxide (DMSO) or actinomycin D (Act.D). The cell lysates were analyzed using immunoblotting. B, total RNA was prepared from U2OS cells at the indicated times after UV irradiation and subjected to Northern blotting using tob cDNA as a probe. Ethidium bromide-stained agarose gel is shown as a loading control (rRNA). C, U2OS cells pretreated with the indicated chemicals were irradiated with UV. SP, SP600125; W, wortmannin; SB, SB203580. The cell lysates were analyzed using immunoblotting. The graph shows the percentages of apoptotic cells with condensed nuclei in the indicated chemical-treated cells. At least 100 cells are scored for each experiment. Values are shown as the mean ± S.D. from triplicate experiments (**, p < 0.01). D, total RNA was prepared from U2OS cells at the indicated times after UV irradiation in the presence of DMSO or CGK733 and subjected to Northern blotting. E, U2OS cells (upper) or MG132-treated U2OS cells (lower) were irradiated with UV in the presence of DMSO or CGK733. The cell lysates were analyzed using immunoblotting.
Cul4-DDB1Cdt2 Induces Ubiquitination and Degradation of Tob
Proteasome-dependent degradation often requires a post-translational modification of target proteins called ubiquitination. The small protein ubiquitin is covalently attached to substrates by the sequential action of three enzymes, E1, E2, and E3 (23). E3 ubiquitin ligases determine target specificity; therefore, we searched for an E3 ligase responsible for Tob degradation in CGK733-treated cells. Because UV irradiation induces several checkpoint responses during the cell cycle, we first examined whether multisubunit E3 ligase complexes related to cell cycle regulation, such as Skp1-Cullin 1-F-box protein (SCF) and anaphase promoting complex/cyclosome (APC/C), were involved. Tob degradation still occurred in CGK733-treated cells after transfecting cells with siRNAs specific to components of the SCF or APC/C E3 ligases, including Skp2, β-TrCP, Cdc4, Cdh1, and Cdc20 (Fig. 2A). We then examined whether the Cul4-DDB1 E3 ligase complexes, which regulate UV-induced responses (24), were involved in Tob degradation. siRNA-mediated suppression of DDB1 resulted in restoration of the UV(l)-induced Tob increase in CGK-treated cells (Fig. 2A). Because DDB1 forms a multiprotein complex with Cul4, Rbx/Roc1, and DDB1-binding WD40 repeat (DWD) proteins, which recognize specific targets (25), we examined the effect of the suppression of several known DWD proteins on Tob degradation. Suppression of Cdt2, but not DDB2, restored the UV(l)-induced Tob increase in CGK-treated cells (Fig. 2B). Again, restoration of the UV(l)-induced Tob increase was clearly correlated with cell viability as monitored by PARP cleavage and nuclear condensation (Fig. 2, A–C). To confirm the involvement of Cdt2 in regulating the level of Tob protein, we examined whether Tob physically interacts with Cdt2. To do so, we established U2OS cells expressing C terminally FLAG-tagged Tob (TobF), which was resistant to proteasome-dependent degradation (17). When TobF was immunoprecipitated from soluble extracts of the cells, endogenous Cdt2 was co-precipitated (Fig. 2D). These data suggest that the Cul4-DDB1Cdt2 E3 complex induced ubiquitination and subsequent degradation of Tob. To test this hypothesis, we purified the Cul4A-DDB1Cdt2 complex from insect cells that were co-infected with Cul4A, DDB1, Cdt2, and Rbx/Roc1-harboring baculoviruses and utilized the purified complex in an in vitro Tob ubiquitination assay. Immunoblot analysis of the reaction products showed a shift in the molecular weight of Tob corresponding to polyubiquitination in the presence of E1, E2 (UbcH5c), E3 (Cul4A-DDB1Cdt2), and ubiquitin (Fig. 2E), which indicated that Cul4A-DDB1Cdt2 specifically ubiquitinated Tob in vitro.
FIGURE 2.

Tob is ubiquitinated by Cul4-DDB1Cdt2. A and B, U2OS cells transfected with the indicated siRNAs were pretreated with CGK733 for 30 min, followed by UV irradiation. The cell lysates were analyzed using immunoblotting. C, apoptotic cells showing the condensed nuclei were visualized by nuclear staining with Hoechst 33342. The graph shows the percentages of apoptotic cells with condensed nuclei in the indicated cells. At least 100 cells are scored for each experiment. Values are shown as the mean ± S.D. from triplicate experiments (**, p < 0.01). D, the interaction between TobF and endogenous Cdt2 was analyzed using immunoprecipitation (IP) and subsequent immunoblotting. Empty virus-infected U2OS (puro/U2OS) or TobF-expressing virus-infected U2OS (TobF/U2OS) cells were used. E, in vitro ubiquitination reactions were performed using the indicated sets of protein components. The reaction mixtures were subjected to immunoblotting using an anti-Tob antibody. The arrow indicates unmodified Tob. DMSO, dimethyl sulfoxide.
Suppression of Cdc7 Induces Cul4-DDB1Cdt2-mediated Tob Degradation and DNA Damage-induced Apoptosis
As CGK733 is inhibitory to the ATM/ATR kinase pathway (supplemental Fig. S2A), we addressed whether, like CGK733 treatment, suppression of their downstream effector Chk1 or Chk2 (26) could antagonize the UV(l)-induced Tob increase and cause apoptotic cell death. However, unlike CGK733 treatment, suppression of Chk1 or Chk2 expression had little effect on the UV(l)-induced Tob increase and apoptosis (supplemental Fig. S3). Then to assess directly the involvement of ATM and ATR, we treated cells with siRNAs specific to ATM, ATR, or both to find that UV(l) irradiation-induced Tob was little affected by the treatment. The degree of induction of apoptosis, monitored by PARP cleavage and nuclear condensation, was little affected in the absence of ATM and ATR (Fig. 3A and supplemental Fig. S3) upon UV(l) irradiation. These data indicate that CGK733 targets not only the ATM/ATR kinases but also another unidentified molecule, which is involved in regulation of the UV(l)-induced Tob increase and apoptosis. To identify the postulated CGK733 targets, we used two-dimensional difference gel electrophoresis in combination with mass spectrometry. Lysates of U2OS cells were prepared after irradiation with UV(l) in the presence or absence of CGK733. Phosphoproteins in the lysates were enriched by column purification (see supplemental Methods for detail). Samples from cells treated with UV(l) and CGK733 together were labeled with Cy5, and those treated with UV(l) alone were labeled with Cy3. Both samples were combined and separated on the same gel. The two-dimensional difference gel electrophoresis image showed several red and green spots, indicating deregulation of several molecules by CGK733 treatment in the UV(l)-induced response (Fig. 3B). Using peptide mass fingerprinting, we identified the Cdc7 and HMGB1 proteins from the spots labeled only with Cy3, indicating that these proteins were specifically suppressed in CGK-treated cells (Fig. 3B). Cdc7 is known to be required for firing of replication origins (3, 4), and it has also been implicated in other chromosome transactions including DNA repair. HMGB1 is involved in the DNA damage response (27, 28). Thus, they may both be potential direct or downstream targets of CGK733. Consistent with this idea, CGK733 treatment suppressed phosphorylation of MCM2, one of the Cdc7 targets (supplemental Fig. S2B). To assess the involvement of Cdc7 and HMGB1 in the UV(l)-induced Tob increase, we transfected U2OS cells with siRNAs targeting Cdc7 or HMGB1, and subsequently examined Tob levels. Immunoblot analysis showed little increase in Tob expression in the cells treated with siRNAs targeting Cdc7 or HMGB1 after UV(l) irradiation (Fig. 3, C and D). As expected, apoptosis, as monitored by both PARP cleavage and nuclear condensation, was induced after UV(l) irradiation in Cdc7- or HMGB1-suppressed cells (Fig. 3, C and D).
FIGURE 3.

Cdc7 regulates Cul4-DDB1Cdt2-mediated Tob degradation and DNA damage-induced apoptosis. A, U2OS cells transfected with the indicated siRNAs were irradiated with UV. The cell lysates were analyzed using immunoblotting. B, the scheme of identification of CGK733 targets (left). The two-dimensional gel image, scanned at different wavelengths to visualize spot patterns corresponding to proteins labeled with Cy3 (red) or Cy5 (green), is shown (right). pI values are indicated above. The indicated spots were identified as Cdc7 and HMGB1 by peptide mass fingerprinting. C and D, U2OS cells transfected with the indicated siRNAs were irradiated with UV. The cell lysates were analyzed using immunoblotting. The graphs show the percentages of apoptotic cells with condensed nuclei. Values are shown as the mean ± S.D. from triplicate experiments. (**, p < 0.01).
Next, we examined whether suppression of the UV(l)-induced Tob increase in the absence of Cdc7 or HMGB1 was due to facilitation of Cul4-DDB1Cdt2 complex-dependent degradation of Tob. Suppression of DDB1 (Fig. 3, C and D) or treatment of cells with MG132 (Fig. 4A) resulted in restoration of the UV(l)-induced Tob increase in Cdc7- or HMGB1-suppressed cells, suggesting that the Cul4-DDB1Cdt2 complex promoted Tob degradation when Cdc7 or HMGB1 was suppressed. The tob mRNA level was increased after UV(l) irradiation of Cdc7- or HMGB1-suppressed cells (Fig. 4B), further suggesting rapid degradation of Tob in those cells. Similar results were obtained using NUGC3, HeLa, and HT1080 cells (supplemental Fig. S4). We conclude that Cdc7 and HMGB1 are responsible for antagonizing UV-induced apoptosis through their ability to block Cul4-DDB1Cdt2 complex-dependent Tob degradation.
FIGURE 4.

HMGB1 regulates Tob levels through Cdc7 stabilization. A, U2OS cells transfected with the indicated siRNAs were irradiated with UV (30 J/m2) in the absence (upper) or presence (lower) of MG132 (50 μm). Cell lysates were analyzed using immunoblotting. B, U2OS cells transfected with the indicated siRNAs were irradiated with UV (30 J/m2). Total RNA was prepared from the cells at the indicated times after UV irradiation and analyzed using Northern blot. C, U2OS cells transfected with the indicated siRNAs were irradiated with UV. Cell lysates were analyzed using immunoblotting. D, U2OS cells transfected with the indicated siRNAs were immunostained with anti-Cdc7 (middle) or anti-HMGB1 (lower) antibody. Upper panels show nuclear staining with Hoechst 33342.
HMGB1 Regulates the Stability of Cdc7
Both Cdc7 and HMGB1 associate with chromatin during the S-phase checkpoint (8, 9, 27, 28). Immunoblot analysis showed that Cdc7 levels decrease upon siRNA-mediated HMGB1 suppression (Fig. 4C), suggesting that stability of Cdc7 is under the control of HMGB1. Single cell analysis by immunofluorescence also showed that Cdc7 was decreased to levels similar to that in Cdc7-suppressed cells in the absence of HMGB1 (Fig. 4D). In contrast, the pattern of HMGB1 was not influenced by Cdc7 suppression (Fig. 4D). These data suggested that HMGB1 is responsible for Tob stability and cell survival functioning as an upstream regulator of Cdc7.
Cdc7 Negatively Regulates Cul4-DDB1Cdt2-dependent Degradation of Tob
Upon UV(l) irradiation, siRNA-mediated suppression of Cdc7 induced apoptosis of U2OS cells but not of the cells expressing degradation-resistant TobF (17), indicating that cells lacking Cdc7 underwent UV(l)-induced apoptosis due to Tob degradation (Fig. 5A). To understand the relevance of Cdc7 in Tob degradation, we examined whether Tob interacted with Cdc7 in U2OS. Endogenous Cdc7 was detected in anti-FLAG immunoprecipitates of these cells (Fig. 5B). Similarly, co-immunoprecipitation experiments showed that endogenous Tob interacts with both Cdc7 and Cdt2 (Fig. 5C). It should be noted that the amount of Cdt2 present in the anti-Tob precipitates was elevated in Cdc7-suppressed cells. Furthermore, enforced overexpression of Cdc7 suppressed the interaction between Tob and Cdt2 (Fig. 5D). These data suggest that Cdc7 contributes to Tob stabilization by preventing Cdt2 from associating with Tob. Because ubiquitination of Tob by Cul4-DDB1Cdt2 was not affected in the presence of a purified active Cdc7 complex (data not shown), Cdc7 appears to require other molecule(s) to effectively prevent Cul4-DDB1Cdt2 from associating with and ubiquitinating Tob.
FIGURE 5.

Cdc7 prevents Cdt2 from associating with Tob to suppress UV-induced apoptosis. A, puro/U2OS or TobF/U2OS cells transfected with the indicated siRNAs were irradiated with UV (30 J/m2). The cell lysates were analyzed using immunoblotting. The graph (lower) shows the percentages of apoptotic cells with condensed nuclei. Values are shown as the mean ± S.D. from triplicate experiments (**, p < 0.01). B and C, the interaction of TobF (B) or endogenous Tob (C) with endogenous Cdc7 and Cdt2 was analyzed using immunoprecipitation (IP) and subsequent immunoblotting. puro/U2OS, TobF/U2OS, or siRNA-transfected U2OS cells were used. The asterisks indicate heavy chain. The arrows indicate the positions of each protein. D, U2OS cells infected with control, TobF, or both TobF and His-Cdc7 expressing virus were lysed and subjected to IP analysis using anti-FLAG antibody. The lysates and anti-FLAG precipitates were analyzed by immunoblotting.
Tob Stability Is Regulated by Its Cdc7-dependent Phosphorylation
We examined whether Cdc7 kinase activity is involved in regulation of Tob stability. As shown in Fig. 6, A and B, treatment of cells with the Cdc7 inhibitor PHA-767491 (18) or depletion of Cdc7 activators Dbf4 or Drf1 from cells using siRNA treatment resulted in Tob protein degradation. Apoptosis was also induced by inhibition of Cdc7 activity or inhibition of Drf1 or Dbf4 expression (Fig. 6, A and B). Phosphate-affinity gel electrophoresis showed that the Tob protein became multiply phosphorylated in UV(l)-treated cells, and the phosphorylation-dependent mobility shift of endogenous Tob was largely suppressed in the absence of Cdc7 (Fig. 6C), indicating that Tob is phosphorylated in vivo in a manner dependent of Cdc7. Next, we performed an in vitro kinase assay and showed that Cdc7 directly phosphorylated Tob (Fig. 6D). Because in vitro GST pulldown experiments revealed that phosphorylated Tob interacted with Cdc7 (supplemental Fig. S5), the phosphorylation event appears to promote the Cdc7-Tob interaction. To identify Cdc7-mediated phosphorylation sites of Tob, we constructed several Tob mutants. Because Cdc7 tends to target a serine residue followed by a Ser/Thr-Pro motif (29, 30), we speculated that Ser151-Ser152-Pro153 and Thr204-Ser205-Pro206 in the amino acid sequence of Tob would be the candidates. Thus, Ser151 and Thr204 as well as Ser152 and Ser205 were converted to Ala. We performed in vitro kinase assays using Tob mutants (S151A, S152A, S151A,S152A, and T204A,S205A) as substrates and found that all the mutants tested were less phosphorylated than wild-type Tob in the presence of recombinant Cdc7 (Fig. 6E), suggesting that those four residues are phosphorylated by Cdc7 in vitro. To investigate the influence of the phosphorylation event on Tob stability, a series of Tob mutants were expressed in COS7 cells. Immunoblot analysis of the cell lysates showed that the S151A and S151A,S152A mutants were less expressed than the other constructs (Fig. 6F), suggesting that the Cdc7-mediated phosphorylation of Tob at Ser151 contributes to Tob stability. Finally, to show that the role of Cdc7-mediated Tob stabilization in apoptosis is independent from other effects of Cdc7 (such as regulation of DNA replication that might affect apoptosis), we introduced the RNAi-resistant form of wild-type Tob, TobS151A, or TobS151A,S152A into U2OS cells using recombinant retrovirus and compared the extent of apoptosis among the cells after UV(l) irradiation. Endogenous Tob expression was suppressed by the specific siRNA transfection. As shown in Fig. 7A, cells expressing TobS151A or TobS151A,S152A underwent apoptosis after UV(l) irradiation, whereas cells expressing wild-type Tob did not. Upon siRNA-mediated suppression of DDB1 expression, the amount of TobS151A or TobS151A,S152A mutants was significantly increased so that the UV(l)-irradiated cell could escape from apoptosis (Fig. 7B). Furthermore, the C terminally FLAG-tagged, degradation-resistant form of TobS151A and TobS151A,S152A suppressed UV(l)-induced apoptosis as well as wild-type Tob in the absence of Cdc7 (Fig. 7C, see also Fig. 5A). These data suggest that Cdc7 inhibits the Cul4-DDB1Cdt2-dependent degradation of Tob through its ability to phosphorylate Tob. Thus, we propose that Cdc7-mediated Tob stabilization enables cells to survive mild DNA damage.
FIGURE 6.

Cdc7 activity influences the stability of Tob protein. A, U2OS cells pretreated with dimethyl sulfoxide (DMSO) (−) or Cdc7 inhibitor (+) were irradiated with UV. The cell lysates were analyzed using immunoblotting. B, U2OS cells transfected with the indicated siRNAs were irradiated with UV. The cell lysates were analyzed using immunoblotting. The graphs (shown on the right side in each figure) represent the percentages of apoptotic cells with condensed nuclei. Values are shown as the mean ± S.D. from triplicate experiments (**, p < 0.01). C, U2OS cells transfected with the indicated siRNAs were treated with MG132 for 40 min, followed by UV irradiation. The cell lysates were subjected to Phos-tag SDS-PAGE (top panel) or SDS-PAGE (other panels) followed by immunoblotting. D and E, in vitro kinase assays (KA) of a purified GST-Cdc7 complex were performed with the indicated substrates. The lower panels show the added substrates stained with Coomassie Brilliant Blue (CBB). F, the indicated series of Tob expression vectors were transfected into COS7 cells together with EGFP expression vector (for monitoring transfection efficiency). The cell lysates were analyzed by immunoblotting.
FIGURE 7.

Cdc7-dependent phosphorylation regulates stability of the Tob protein. A–C, U2OS cells infected with retrovirus expressing the indicated constructs were transfected with control siRNA or siRNA against Tob. Tob* represents the RNAi-resistant form of Tob. The cell lysates were prepared after UV irradiation (A–C) or control treatment (A and C) and analyzed by immunoblotting. The graph represents the percentages of apoptotic cells with condensed nuclei. Values are shown as the mean ± S.D. from triplicate experiments (**, p < 0.01). D, proposed model for Cdc7-mediated regulation of Tob levels to keep cell survival upon mild DNA damage.
DISCUSSION
In the present studies we have shown the interplay between the DNA replication-initiating kinase Cdc7 and the proliferation-regulating protein Tob in cellular response to DNA damage. We found that Cdc7 plays a key role in cellular response to DNA damage by interacting with and phosphorylating Tob. The involvement of Cdc7 in controlling the DNA damage checkpoint apart from its well characterized role in DNA replication initiation has been proposed based on observations that Cdc7 activity is maintained even after UV irradiation or etoposide treatment (7–9). It appears that Cdc7 attenuates the S-phase checkpoint response to trigger DNA replication re-initiation during checkpoint recovery (7–9). Our data that Cdc7, in conjunction with Tob, protects cells from apoptosis following mild DNA damage defines an additional role of Cdc7 in the DNA damage checkpoint.
DNA-damaged cells die by apoptosis in the absence of Tob (17). In this report we show that Cdc7 protects Tob from Cul4-DDB1Cdt2-mediated polyubiquitination and degradation, and that cells with mild DNA damage die by apoptosis in the absence of Cdc7 (Fig. 7D). This role of Cdc7 in stabilizing the Tob protein would account for the previously reported susceptibility of cells lacking Cdc7 to DNA damage-induced apoptosis (8). The effect of Cdc7 on Tob stability and the cells' viability was not limited to a specific cell type (supplemental Fig. S4). These findings together lead us to conclude that Cdc7 participates in general pro-survival signaling via the ability of Tob to suppress apoptosis, enabling cells to undergo DNA repair without unnecessary cell death.
HMGB1 has also been implicated in the DNA repair checkpoint. Binding preferentially to damaged DNA, HMGB1 is able to induce DNA bending. This characteristic of HMGB1 and its ability to interact with DNA repair proteins and chromatin remodeling factors render the damaged DNA more accessible to the DNA repair apparatus (27, 28). Cdc7 is also likely to contribute to chromatin remodeling because Cdc7 aids the recruitment of chromatin assembly factor 1 to chromatin by promoting the interaction between chromatin assembly factor 1 and proliferating cell nuclear antigen, the homotrimeric processivity factor for DNA polymerase (31). Intriguingly, we found that HMGB1 functions as an upstream regulator of Cdc7 to suppress DNA damage-induced apoptosis (Fig. 4). The functional interaction among HMGB1, Cdc7, and Tob can be expected to impact cell viability during the repair of damaged DNA as well as regulation of these cells' eventual entry into S-phase. Further investigation into the roles of Cdc7 and Tob in DNA repair and chromatin remodeling would help clarify the whole scenario.
Because almost no cells entered into S-phase after DNA damage irrespective of the presence or absence of Tob (17), Cdc7-Tob axis-dependent cell survival seems to be irrelevant to cell cycle arrest. Nevertheless, our present study led us to propose that cells deal with DNA damage not only by cessation of the cell cycle but also through pro-survival signaling mediated by the Cdc7-Tob axis. The use of a mechanism other than delayed entry into S-phase to suppress DNA damage-induced apoptosis may not be unique. Cells lacking the retinoblastoma protein are deficient in both repair of UV-induced DNA damage and recovery of RNA synthesis, resulting in apoptosis (32, 33).
Cul4-DDB1Cdt2 is involved in UV-induced degradation of the licensing factor Cdt1 and the CDK inhibitor p21 (20, 24). Although Tob is ubiquitinated by the Cul4-DDB1Cdt2 complex, degradation of Tob is not induced by UV(l) irradiation alone. Our results indicate that Cdc7 prevents the Cul4-DDB1Cdt2 complex from associating with Tob (Fig. 5, C and D) and this ability of Cdc7 depends on its kinase activity (Figs. 6 and 7). However, the purified active Cdc7 complex did not inhibit Cul4-DDB1Cdt2-mediated Tob ubiquitination in vitro (data not shown), suggesting that Cdc7 requires other molecule(s) to prevent Cdt2 from interacting with Tob. Because phosphorylation of Tob by Cdc7 promotes Cdc7-Tob interaction at least in vitro (supplemental Fig. S5), it is possible that Cdc7-dependent phosphorylation promotes the formation of a multiprotein complex including Tob and Cdc7 to ensure protection of Tob from the Tob-Cdt2 interaction and subsequent ubiquitination of Tob in vivo.
CGK733-treated U2OS cells underwent apoptosis even after mild DNA damage, which otherwise induces little apoptosis (Fig. 1 and supplemental Fig. S1). We show that the effect of CGK733 on cell viability is due to Tob destabilization but not due to suppression of ATM/ATR kinase-mediated signaling. These data suggest that the pro-survival effects of Tob may provide a tumor escape mechanism against chemo- and radiation therapies, and lower doses of anticancer agents in combination with Tob suppression might effectively induce apoptosis in human cancer cells. A Tob inhibitor might therefore be a potentially effective adjuvant to such treatments. It is important to confirm the effect of Tob suppression on the sensitivity to anticancer agents in several types of human cancer cells.
Supplementary Material
Acknowledgments
We thank T. Kitamura for providing retrovirus packaging cells. We thank Y. Nakajima for mass spectrometry. We also thank R. Whittier for useful discussion and comments on the manuscript.
This work was supported by a grant for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and the NFAT project of the New Energy and Industrial Technology Development Organization.

This article contains supplemental Methods and Figs. S1–S5.
- Z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- siRNA
- small interfering RNA
- PARP
- poly(ADP-ribose) polymerase
- ATM
- ataxia telangiectasia mutated
- ATR
- ATM-Rad3-related
- HMGB1
- high mobility group B1
- EGFP
- enhanced green fluorescent protein
- SCF
- Skp1-Cullin 1-F-box.
REFERENCES
- 1. Zhou B. B., Elledge S. J. (2000) The DNA damage response. Putting checkpoints in perspective. Nature 408, 433–439 [DOI] [PubMed] [Google Scholar]
- 2. Clarke P. R., Allan L. A. (2009) Cell-cycle control in the face of damage. A matter of life or death. Trends Cell Biol. 19, 89–98 [DOI] [PubMed] [Google Scholar]
- 3. Labib K. (2010) How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev. 24, 1208–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Masai H., Matsumoto S., You Z., Yoshizawa-Sugata N., Oda M. (2010) Eukaryotic chromosome DNA replication. Where, when and how? Annu. Rev. Biochem. 79, 89–130 [DOI] [PubMed] [Google Scholar]
- 5. Costanzo V., Shechter D., Lupardus P. J., Cimprich K. A., Gottesman M., Gautier J. (2003) An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol. Cell 11, 203–213 [DOI] [PubMed] [Google Scholar]
- 6. Dierov J., Dierova R., Carroll M. (2004) BCR/ABL translocates to the nucleus and disrupts an ATR-dependent intra-S phase checkpoint. Cancer Cell 5, 275–285 [DOI] [PubMed] [Google Scholar]
- 7. Heffernan T. P., Unsal-Kaçmaz K., Heinloth A. N., Simpson D. A., Paules R. S., Sancar A., Cordeiro-Stone M., Kaufmann W. K. (2007) Cdc7-Dbf4 and the human S checkpoint response to UVC. J. Biol. Chem. 282, 9458–9468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tenca P., Brotherton D., Montagnoli A., Rainoldi S., Albanese C., Santocanale C. (2007) Cdc7 is an active kinase in human cancer cells undergoing replication stress. J. Biol. Chem. 282, 208–215 [DOI] [PubMed] [Google Scholar]
- 9. Tsuji T., Lau E., Chiang G. G., Jiang W. (2008) The role of Dbf4/Drf1-dependent kinase Cdc7 in DNA-damage checkpoint control. Mol. Cell 32, 862–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Winkler G. S. (2010) The mammalian anti-proliferative BTG/Tob protein family. J. Cell Physiol. 222, 66–72 [DOI] [PubMed] [Google Scholar]
- 11. Guardavaccaro D., Corrente G., Covone F., Micheli L., D'Agnano I., Starace G., Caruso M., Tirone F. (2000) Arrest of G1-S progression by the p53-inducible gene PC3 is Rb dependent and relies on the inhibition of cyclin D1 transcription. Mol. Cell Biol. 20, 1797–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Suzuki T., Tsuzuku J., Ajima R., Nakamura T., Yoshida Y., Yamamoto T. (2002) Phosphorylation of three regulatory serines of Tob by Erk1 and Erk2 is required for Ras-mediated cell proliferation and transformation. Genes Dev. 16, 1357–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoshida Y., Nakamura T., Komoda M., Satoh H., Suzuki T., Tsuzuku J. K., Miyasaka T., Yoshida E. H., Umemori H., Kunisaki R. K., Tani K., Ishii S., Mori S., Suganuma M., Noda T., Yamamoto T. (2003) Mice lacking a transcriptional corepressor Tob are predisposed to cancer. Genes Dev. 17, 1201–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boiko A. D., Porteous S., Razorenova O. V., Krivokrysenko V. I., Williams B. R., Gudkov A. V. (2006) A systematic search for downstream mediators of tumor suppressor function of p53 reveals a major role of BTG2 in suppression of Ras-induced transformation. Genes Dev. 20, 236–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ou Y. H., Chung P. H., Hsu F. F., Sun T. P., Chang W. Y., Shieh S. Y. (2007) The candidate tumor suppressor Btg3 is a transcriptional target of p53 that inhibits E2F1. EMBO J. 26, 3968–3980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rouault J. P., Falette N., Guéhenneux F., Guillot C., Rimokh R., Wang Q., Berthet C., Moyret-Lalle C., Savatier P., Pain B., Shaw P., Berger R., Samarut J., Magaud J. P., Ozturk M., Samarut C., Puisieux A. (1996) Identification of BTG2, an antiproliferative p53-dependent component of the DNA damage cellular response pathway. Nat. Genet. 14, 482–486 [DOI] [PubMed] [Google Scholar]
- 17. Suzuki T., Tsuzuku J., Kawakami K., Miyasaka T., Yamamoto T. (2009) Proteasome-mediated degradation of Tob is pivotal for triggering UV-induced apoptosis. Oncogene 28, 401–411 [DOI] [PubMed] [Google Scholar]
- 18. Montagnoli A., Valsasina B., Croci V., Menichincheri M., Rainoldi S., Marchesi V., Tibolla M., Tenca P., Brotherton D., Albanese C., Patton V., Alzani R., Ciavolella A., Sola F., Molinari A., Volpi D., Avanzi N., Fiorentini F., Cattoni M., Healy S., Ballinari D., Pesenti E., Isacchi A., Moll J., Bensimon A., Vanotti E., Santocanale C. (2008) A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat. Chem. Biol. 4, 357–365 [DOI] [PubMed] [Google Scholar]
- 19. Tanaka T., Takeno T., Watanabe Y., Uchiyama Y., Murakami T., Yamashita H., Suzuki A., Aoi R., Iwanari H., Jiang S. Y., Naito M., Tachibana K., Doi T., Shulman A. I., Mangelsdorf D. J., Reiter R., Auwerx J., Hamakubo T., Kodama T. (2002) The generation of monoclonal antibodies against human peroxisome proliferator-activated receptors (PPARs). J. Atheroscler. Thromb. 9, 233–242 [DOI] [PubMed] [Google Scholar]
- 20. Nishitani H., Shiomi Y., Iida H., Michishita M., Takami T., Tsurimoto T. (2008) CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J. Biol. Chem. 283, 29045–29052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Suzuki T., Kim M., Kozuka-Hata H., Watanabe M., Oyama M., Tsumoto K., Yamamoto T. (2011) Monoubiquitination of Tob/BTG family proteins competes with degradation-targeting polyubiquitination. Biochem. Biophys. Res. Commun. 409, 70–74 [DOI] [PubMed] [Google Scholar]
- 22. Masai H., Matsui E., You Z., Ishimi Y., Tamai K., Arai K. (2000) Human Cdc7-related kinase complex. J. Biol. Chem. 275, 29042–29052 [DOI] [PubMed] [Google Scholar]
- 23. Hershko A., Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 24. O'Connell B. C., Harper J. W. (2007) Ubiquitin proteasome system (UPS). What can chromatin do for you? Curr. Opin. Cell Biol. 19, 206–214 [DOI] [PubMed] [Google Scholar]
- 25. Jackson S., Xiong Y. (2009) CRL4s. The CUL4-RING E3 ubiquitin ligases. Trends Biochem. Sci. 34, 562–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abraham R. T. (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 15, 2177–2196 [DOI] [PubMed] [Google Scholar]
- 27. Lange S. S., Mitchell D. L., Vasquez K. M. (2008) High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc. Natl. Acad. Sci. U.S.A. 105, 10320–10325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lange S. S., Vasquez K. M. (2009) HMGB1. The jack-of-all-trades protein is a master DNA repair mechanic. Mol. Carcinog. 48, 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sasanuma H., Hirota K., Fukuda T., Kakusho N., Kugou K., Kawasaki Y., Shibata T., Masai H., Ohta K. (2008) Cdc7-dependent phosphorylation of Mer2 facilitates initiation of yeast meiotic recombination. Genes Dev. 22, 398–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Randell J. C., Fan A., Chan C., Francis L. I., Heller R. C., Galani K., Bell S. P. (2010) Mec1 is one of multiple kinases that prime the Mcm2–7 helicase for phosphorylation by Cdc7. Mol. Cell 40, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gérard A., Koundrioukoff S., Ramillon V., Sergère J. C., Mailand N., Quivy J. P., Almouzni G. (2006) The replication kinase Cdc7-Dbf4 promotes the interaction of the p150 subunit of chromatin assembly factor 1 with proliferative cell nuclear antigen. EMBO Rep. 7, 817–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Carlson C. A., Ethier S. P. (2000) Lack of RB protein correlates with increased sensitivity to UV-radiation-induced apoptosis in human breast cancer cells. Radiat. Res. 154, 590–599 [DOI] [PubMed] [Google Scholar]
- 33. Billecke C. A., Ljungman M. E., McKay B. C., Rehemtulla A., Taneja N., Ethier S. P. (2002) Lack of functional pRb results in attenuated recovery of mRNA synthesis and increased apoptosis following UV radiation in human breast cancer cells. Oncogene 21, 4481–4489 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.