

Background: Caveolin-3 facilitates both caveolae formation and a range of cell signaling pathways, including Ca2+ homeostasis.
Results: Caveolin-3 forms a disc-shaped nonamer that binds the Ca2+-release channel, RyR1.
Conclusion: Multiple caveolin-3 nonamers bind to a single RyR1 homotetramer.
Significance: First three-dimensional structural insights into caveolin-3 assembly, interactions with RyR1 suggest a novel role in muscle contraction and/or for channel localization within the membrane.
Keywords: Caveolae, Caveolin, Electron Microscopy (EM), Excitation-Contraction Coupling, Membrane Proteins, Muscle, Ryanodine Receptor, Sarcoplasmic Reticulum (SR), Scaffold Proteins, Single Particle Analysis
Abstract
Caveolin-3 (cav-3), an integral membrane protein, is a building block of caveolae as well as a regulator of a number of physiological processes by facilitating the formation of multiprotein signaling complexes. We report that the expression of cav-3 in insect (Sf9) cells induces caveola formation, comparable in size with those observed in native tissue. We have also purified the recombinant cav-3 determining that it forms an oligomer of ∼220 kDa. We present the first three-dimensional structure for cav-3 (using transmission electron microscopy and single particle analysis methods) and show that nine cav-3 monomers assemble to form a complex that is toroidal in shape, ∼16.5 nm in diameter and ∼ 5.5 nm in height. Labeling experiments and reconstitution of the purified cav-3 into liposomes have allowed a proposal for the orientation of the protein with respect to the membrane. We have identified multiple caveolin-binding motifs within the ryanodine receptor (RyR1) sequence employing a bioinformatic analysis. We have then shown experimentally that there is a direct interaction between recombinant cav-3 nonamers and purified RyR1 homotetramers that would imply that at least one of the predicted cav-3-binding sites is exposed within the fully assembled RyR1 structure. The cav-3 three-dimensional model provides new insights as to how a cav-3 oligomer can bind multiple partners in close proximity to form signaling complexes. Furthermore, a direct interaction with RyR1 suggests a possible role for cav-3 as a modifier of muscle excitation-contraction coupling and/or for localization of the receptor to regions of the sarcoplasmic reticulum.
Introduction
Caveolin (cav)2-1 was first identified 20 years ago (1, 2) with two additional forms, caveolin-2 (cav-2), caveolin-3 (cav-3), later described (3, 4). Two splice variants of cav-1 and three isoforms of cav-2 have also been reported (4–6). Caveolins are the principal structural components of caveola, a type of lipid raft formed by vesicular invaginations of the plasma membrane (7, 8). In addition, caveolins are implicated in a host of cell signaling processes through an association with a myriad of proteins to form macromolecular multiprotein complexes (9). Single amino acid mutations and alterations in expression levels of caveolins have also been linked to the pathogenesis of multiple disease states, including cancers, cardiovascular disorders, diabetes, and a wide range of degenerative muscular dystrophies (10).
cav-1 is the most widely studied protein of the caveolin family, yet there is a dearth of structural data, with no three-dimensional structures currently available for any of the full-length caveolins. It is generally accepted that caveolins self-assemble to form higher polymers, although there are conflicting reports in the literature regarding the number of caveolin monomers that associate to form an oligomer. For example, Fernandez et al. (11) expressed, in Escherichia coli, a recombinant fragment of cav-1 (amino acids 1–101), and by using transmission electron microscopy to examine negatively stained samples, they proposed that the fragment oligomerized to form a heptameric complex ∼11 nm across, although no image analysis was undertaken. In contrast, an earlier study of native cav-1 isolated from adipocyte plasma membranes reported that cav-1 formed complexes that migrated between 200 and 600 kDa when analyzed by gel electrophoresis, with a major band at ∼300–325 kDa and a minor band at 200 kDa (12). Again, no structural analysis of the cav-1 oligomers was conducted.
The primary sequence of cav-3 can be considered to be composed of several structural modules as follows: a cytosolic N-terminal segment (aa 1–73) that contains a caveolin signature motif (aa 41–48, FEDVIAEP) and a scaffolding domain (aa 55–74), with a putative transmembrane region (aa 75–106) that is proposed to form a hairpin structure thereby also placing the C-terminal domain (aa 105–151) in the cytoplasm (13, 14). It is clear from Fig. 1 that large portions of the primary sequence are conserved between the caveolin proteins with 61% sequence similarity between the cav-1 and cav-3 isoforms. However, there are distinct differences in sequence such as the extended N terminus of cav-1 compared with cav-3. There is also substantially less conservation between the sequences of cav-3 and cav-1 with cav-2, 33 and 30%, respectively. A divergence in function between the isoforms has also been established; for example, cav-2, unlike cav-1 and cav-3, appears not to be required for caveolae formation nor does it seem to be involved in vesicular transport (15). Functional differences between caveolin isoforms are likely influenced by the variation in expression location. cav-1 and cav-2 are expressed in most cell types, whereas cav-3 is specific to skeletal, smooth, and cardiac muscles (3, 16–18). Genetic ablation of cav-3 has been reported to effect muscular disorders, including severe cardiac hypertrophy and dilation (19, 20), myocyte degeneration, fibrosis, and impairment of function (21, 22).
FIGURE 1.

Primary sequence alignment of human cav-3 and human cav-1. There is a 61% sequence identity between cav-1 and cav-3 (ClustalW2). The figure shows that there are several regions that are highly conserved between the two caveolin isoforms but also illustrates that there are regions of divergence, for example the N terminus is extended in cav-1 compared with cav-3. Asterisk indicates fully conserved residue; colon indicates conservation of residues with strongly similar properties; period indicates conservation of residues with weakly similar properties. The boxed amino acids indicate the conserved caveolin signature motif, and the underlined residues indicate the putative transmembrane domain. A secondary structure prediction for cav-3 (Jpred) is depicted above the sequence; arrows represent β-sheets; cylinders represent α-helical regions. The absence of either implies the protein in this region is predicted to be largely random coil.
Interestingly, cav-3 null mice have also been shown to develop a markedly disorganized T-tubule system in skeletal muscle (23, 24). T-tubules are invaginations of the sarcolemmal membrane found in excitable cells facilitating the transmission of the action potential from the exterior to the interior of the cell. In brief, depolarization of the plasma membrane leads to the opening of the L-type voltage-gated calcium channels, localized to the T-tubules controlling excitation-contraction coupling in cooperation with the ryanodine receptor (RyR), which resides in the sarcoplasmic reticulum (SR). Although there are a number of reports describing a direct interaction of cav-3 with L-type voltage-gated calcium channels (25), it is not clear whether this association involves only subpopulations of L-type voltage-gated calcium channels associated with caveolae spatially distal from the SR RyR complexes (26). Interestingly, Vassilopoulos et al. (27) have shown that cav-3 is immunoprecipitated from rat microsomes in complex with triadin and the ryanodine receptor skeletal muscle isoform RyR1. In addition, the group also reported that cav-3 could be co-immunoprecipitated in COS-7 cells transfected with a GFP-RyR construct corresponding to the C-terminal domain (4450–5032 aa), implying that the interaction was independent of triadin and that the transmembrane region of the receptor was involved. It was not clear however whether the expressed fragment had oligomerized to form a tetramer in an organization similar to that of the native receptor. Given that this domain would ordinarily be within the SR membrane, it may indicate that for cav-3 to have access to this region cav-3 is located in the SR membrane, although the authors did not comment on this observation. Intriguingly, an earlier study from Li et al. (28) had detected cav-3 in SR membranes.
In this study, we have purified full-length human cav-3 and determined the first three-dimensional structure for this protein. Reconstitution of the isolated cav-3 into lipid vesicles allowed interpretation of the three-dimensional structure in the context of function providing new insights as to how this protein can bind multiple partners in close proximity and thus act as a signaling hub. We have additionally shown that cav-3 is a component of the junctional terminal cisternae (JTC) of SR membranes and that the purified oligomer can directly interact with isolated homotetrameric RyR1 complexes.
EXPERIMENTAL PROCEDURES
cav-3 Expression and Purification
The template DNA for human caveolin-3 (cav-3) was purchased from OriGene. Appropriate primers were designed, and PCR of the full-length cav-3 was carried out. The PCR product was gel-purified (Qiagen) and digested by EcoRI/BamHI enzymes (Fermentas). The pPolh-MAT-2 plasmid was digested with EcoRI/BglII enzymes and then dephosphorylated using calf intestinal alkaline phosphatase enzyme (Roche Applied Science). Vectors and inserts (at a molar ratio of 1:3) were ligated (U. S. Biochemical Corp.) and transformed into competent JM109 cells. Cells were plated onto LBamp plates. Positive colonies were sequence-verified and shown to correspond to full-length cav-3. Small scale pilot studies were undertaken to devise optimal expression conditions. For large scale expression, Sf9 cells were grown to 4 × 106 cells/ml and then passaged to 1 × 106 cells/ml in fresh media containing the baculovirus at a final multiplicity of infection of 5. Virus-infected cells were incubated for 72 h at 28 °C after which time cells expressing cav-3 were harvested by centrifugation at 1000 × g for 5 min. The cell pellets were resuspended in buffer (20 mm Tris, pH 7.7, 150 mm NaCl with protease inhibitors (Roche Applied Science)), including 2% w/v β-dodecyl d-maltoside (DDM), 1.2% v/v Triton X-100, at a ratio of 20 ml of buffer to 400 ml of original cell solution. The resuspended cell pellet solution was sonicated three times at 20% for 30 s, resting for 30 s between cycles. Cells were rotated for 1 h at 4 °C and then centrifuged at 45,000 × g (Sorval SS-34) for 30 min at 4 °C.
The supernatant was diluted to reduce the detergent concentration, filtered and degassed, and then applied to a HisTrapHP column (5-ml column volume, GE Healthcare) using an AKTAprime plus FPLC to purify cav-3 via the C-terminal metal affinity tag (MAT). The column was washed with 5 system volumes of buffer (20 mm Tris, pH 7.7, 150 mm NaCl, 0.02% w/v DDM, and protease inhibitors) to remove any unbound protein at a flow rate of 1 ml/min. The bound cav-3 was eluted with imidazole (flow rate of 0.5 ml/min). All buffers and columns were maintained at 4 °C. The eluted fractions containing partially purified cav-3 (as determined by SDS-PAGE and Western blotting; an anti-MAT, Sigma M6693, and anti-cav-3, Abcam ab2912) were layered on top of a step sucrose gradient as follows: 10% (5 ml), 12.5% (5 ml), 15% (3 ml), 20% (3 ml), 22.5% (3 ml), 30% (6 ml), and 40% (7 ml) in 20 mm Tris, pH 7.7, 150 mm NaCl, 0.02% DDM, and protease inhibitors and ultracentrifuged for 16 h at 132,000 × g (SW28 rotor) at 4 °C. The gradients were fractionated (0.5 ml) and analyzed for cav-3. Pure cav-3 was isolated in the gradient where the sucrose concentration was ∼22.5%. This method typically yielded ∼0.2–0.3 mg of purified cav-3 from a 2.5-liter cell suspension (1 million cells/ml).
Sample Preparation for SDS-PAGE/Western Blotting
Boiling cav-3 samples prior to analysis by SDS-PAGE led to a ladder of cav-3 oligomers as well as smeared aggregated protein at the top of the gel. Omission of the boiling step with incubation of the protein sample in SDS-PAGE buffer overnight at room temperature was necessary for the complete breakdown of the cav-3 oligomers.
Preparation of Thin Sections of Infected Sf9 Cell
Infected cells (multiplicity of infection of 5) were fixed with 2.5% glutaraldehyde in 0.1 m sodium cacodylate buffer, pH 7.2, for 2 h on ice with standard methods for processing, resin embedding, preparation of thin sections, and staining. Sections were examined using an FEI BioTwin Tecnai 12 TEM microscope at an operating voltage of 100 kV. Images were recorded on a Gatan 2K × 2K CCD camera.
Characterization of Molecular Mass of Purified cav-3 Using Multiangle Laser Light Scattering (MALLS)
Purified recombinant cav-3 was applied to a Superose 6 column 10/300GL (GE Healthcare) running at a flow rate of 0.75 ml/min in TBS buffer. Samples eluting from the column passed through an in-line DAWN HELEOS-II laser photometer (laser wavelength 658 nm) and an Optilab rEX refractometer with a QELS dynamic light scattering attachment (Wyatt). Light scattering intensity and eluent refractive index (concentration) were analyzed using ASTRA version 5.3.4.13 software to give a weight-averaged molecular mass.
Electron Microscopy and Three-dimensional Structure Analysis of Purified cav-3
Aliquots of cav-3 taken from the peak fraction from the MALLS were negatively stained with 2% w/v uranyl acetate following standard protocols. Images of cav-3 were recorded on a Polara 300 kV Field Emission Gun transmission electron microscope. Data were collected with the microscope operated at 200 kV and under cryo and low dose conditions, i.e. 10–20 e−/Å2 using a 4K × 4K CCD Gatan camera with a calibrated magnification corresponding to 5.1 Å/pixel at the specimen level. Each micrograph was examined using the CTFIT program within the EMAN package (29) to determine the degree of under focus; only those images between a 0.5- and 1.8-μm defocus were included in the three-dimensional reconstruction. cav-3 particles (5643) were selected using the GUI Boxer software into a 64 × 64 pixel (320 × 320 Å) box. The contrast transfer function was determined for each image using CTFIT with images corrected for phase (30). Classification of the particles, after centering and translational and rotational alignment, produced a set of unbiased reference-free class averages showing different orientations of cav-3. The contour-delineated class averages shown in Fig. 3 were generated using the SPIDER software contouring function (31).
FIGURE 3.

Analysis of purified human cav-3 by electron microscopy and image analysis. A, example area of negatively stained cav-3 taken from the peak fraction shown in Fig. 2C, illustrating that the sample is monodisperse with multiple orientations of cav-3 presented. B, montage of selected raw particles used in the three-dimensional reconstruction. The arrowheads highlight particles with a distinct circular shape, ∼160–170 Å in diameter with a central density. Another distinct orientation is a rod-shaped view of cav-3 that is ∼160 Å in length and ∼30–40 Å wide (arrows). C, contoured projection map of a class average of cav-3 characterized by a ring of protein resolved into nine domains (1–9) with an overall a diameter of ∼165 Å, surrounding a central region of density as indicated by the black arrowhead (n = 87). D, class average of cav-3 that is rod-shaped, ∼165 Å long, and ∼40 Å thick and composed of three protein domains (n = 93). E, this class average shows a partial side view of the complex (n = 111). Box size = 326 × 326 Å. F, power spectrum with a peak indicative of c9 rotational symmetry. Inset, class average presenting a circular view of cav-3 composed of 97 raw images. G, rotational cross-correlation symmetry analysis. A class average presenting a circular view of cav-3 was rotated in increments around the z axis with each of the rotated images compared with the original unrotated image. The plot of the angle of rotation against the cross-correlation coefficient shows nine peaks separated by 40°. Note, the cross-correlation coefficient values remain high because the whole image is self-compared with only discrete changes at the rim of the complex being detected. H, grayscale intensity around the circumference of the class average was measured (Oval Plot) and plotted as a function of angle, finding nine peaks of white density (protein).
Symmetry analysis was conducted employing the software “rotastat” (32), which calculates the rotational power spectrum of individual images forming a class average, presenting a circular view of the complex, which are combined and compared with the intensity contribution from background images. The final power spectrum of the class average (97 raw particles) examined (Fig. 3F) shows a principal peak of c9 rotational symmetry. In another approach, a class average (circular projection) was rotated around the z axis through 360°, with a comparison of the rotated two-dimensional images with the original unrotated class average (SPIDER command CC C) giving clear peaks of correlation at 40° intervals, which would correspond to a complex with c9 symmetry (Fig. 3G). In addition, Oval Plot (ImageJ) was used to measure the grayscale intensities around the circumference of a class average of the circular en face view (the radius of sampling was at 13 pixels). Nine fairly evenly spaced intensity peaks were observed (Fig. 3H), again consistent with a protein complex exhibiting 9-fold rotational symmetry.
As a result of the above analysis, a three-dimensional model was iteratively refined applying c9 symmetry using the EMAN software. Refinement was carried out with an angular step of 5° generating 85 classes containing between 44 and 163 particles per class. Convergence and stabilization of the three-dimensional structure was monitored by examining the Fourier shell correlation of the three-dimensional models generated from each iteration. Following established procedures (33) the resolution was estimated by plotting the Fourier shell correlation coefficient against spatial frequency. An estimation of the resolution limit is taken to be where the Fourier shell correlation value falls below 0.5. All the cav-3 three-dimensional maps and models are displayed using the Chimera software (34).
Labeling the C Terminus of cav-3
Following a previously described protocol (35), an aliquot of purified cav-3 was incubated for 3 h with Ni-NTA-goldTM (Nanoprobes Inc.). Samples were negatively stained and examined in a Tecnai 12 BioTwin TEM at an operating voltage of 100 kV.
Reconstitution of Purified cav-3 into Unilamellar Vesicles
Large unilamellar vesicles were prepared as described by Geertsma et al. (36). In brief, lipids (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine, 1,2-dioleoyl-sn-glycero-3-phospho-rac-(1-glycerol) sodium salt, and 1,2-dioleoyl-sn-glycero-3-phosphocholine, Sigma), were dissolved in diethyl ether, mixed for a 2:1:1 molar ratio, dried under N2, and placed in a vacuum evaporator for 1 h. The lipids were resuspended in 25 mm Tris, pH 7.4, 115 mm NaCl for a final concentration of 20 mg/ml and sonicated (six cycles of 15 s) on ice under a gentle stream of N2 gas to create small unilamellar vesicles. The small unilamellar vesicles were flash-frozen in liquid nitrogen and thawed at room temperature for 20 min. This process was repeated three times, and the vesicles were then passed through an extruder (400-nm polycarbonate filter) 11 times to form large unilamellar vesicles, which were then diluted to 4 mg/ml in 25 mm Tris, pH 7.4, 115 mm NaCl. The glycerol content was then adjusted to 20% (v/v) to stabilize them. The large unilamellar vesicles were titrated with Triton X-100 until a midpoint between membrane saturation and solubilization was reached (as judged by measuring the absorbance at 540 nm). Purified cav-3 was added to the destabilized large unilamellar vesicles over a range of cav-3 to lipid ratios. The samples were then incubated for 15 min at 4 °C. SM-2 polystyrene beads (Bio-Beads, Bio-Rad) were added, incubated for 30 min at 4 °C, and then removed by centrifugation. Samples were prepared for transmission electron microscopy as above.
Isolation of Skeletal Muscle Sarcoplasmic Reticulum JTC
The protocol employed was adapted from Refs. 37, 38. In brief, 200 g of sheep skeletal muscle were homogenized (Moulinex® Optiblend 2000) in 1 liter of buffer (300 mm sucrose, 10 mm Hepes, pH 7.0, 0.5 mm EDTA). The homogenate was centrifuged for 15 min at 11,000 × g; the pellet was discarded, and the supernatant was centrifuged at 110,000 × g for 60 min to sediment the mixed membrane population. The mixed membrane population was resuspended in 200 ml of buffer (650 mm KCl, 300 mm sucrose, 10 mm Hepes, pH 7.0, 0.5 mm EDTA) and stirred at 4 °C for 60 min before being repelleted by centrifugation at 200,000 × g for 60 min. The pellet was collected and resuspended (using a hand homogenizer) in 30 ml of buffer (650 mm KCl, 300 mm sucrose, 10 mm Hepes, pH 7.0, 0.5 mm EDTA), layered onto a sucrose gradient, 45, 38, 32, and 27% (w/v) sucrose steps (in the same buffer as the membranes), and centrifuged at 100,000 × g in an SW28 rotor for 16 h. This process yielded sarcoplasmic reticulum JTC at the 38–45% interface. The membranes were collected, diluted 2-fold with buffer (300 mm sucrose, 10 mm Hepes, pH 7.0, 0.5 mm EDTA), and recentrifuged at 110,000 × g for 60 min. The pellets were resuspended in buffer (300 mm sucrose, 10 mm Hepes, pH 7.0), then snap-frozen in liquid nitrogen, and stored at −80 °C.
Purification of RyR1 from JTCs
The method described in Ref. 39 was followed with minor adaptations. JTC membranes (∼20 mg) were solubilized at 2.0 mg/ml in buffer (50 mm MOPS, pH 7.4, 185 mm NaCl, 100 mm EGTA, 2.0 mm dithiothreitol (DTT), 2% (w/v) CHAPS, 1% (w/v) soybean lecithin) for 60 min on ice. To remove the insoluble material, the mixture was then centrifuged at 100,000 × g for 30 min at 4 °C. The supernatant was layered onto a stepwise sucrose gradient (5–25% (w/v)) on top of a 35% (w/v) sucrose cushion. Sucrose gradient solutions were prepared in buffer (50 mm MOPS, pH 7.4, 185 mm NaCl, 100 mm EGTA, 2.0 mm DTT, 0.5% (w/v) CHAPS) and centrifuged at 125,000 × g in an SW28 rotor for 16 h, after which each of the gradients was carefully fractionation into ∼0.5-ml portions. Aliquots (100 μl) from each fraction were analyzed by protein assay and SDS-PAGE/Western blotting. Fractions were quick frozen in liquid nitrogen and stored at −80 °C until required. All buffers contained the following protease inhibitors: 2 mm PMSF, 1 μm pepstatin A, 1 μm E64, 1 mm benzamidine or complete mini EDTA-free protease inhibitor mixture tablets.
Bioinformatic Analysis
Hydrophobicity plots and transmembrane helices were calculated using the SPLIT tool (40). Alignments were completed using ClustalW (41). Primary sequences were scanned for caveolin-binding motifs (FWY)X(FWY)XXXX(FWY) and (FWY)XXXX(FWY)XX(FWY) (42) using ScanProsite (43). The RyR1 (AAA60294) and the RyR2 (NP_001026.2) sequence files were taken from the NCBI database.
Interaction of Recombinant cav-3 with Purified RyR1
Two approaches were taken to investigate the association of cav-3 nonamers with RyR1 homotetramers. For method A, a cobalt-chelate resin spin column (Thermo Scientific) was washed four times in buffer (25 mm Tris, pH 7.5, 115 mm NaCl, 0.075% (w/v) DDM). cav-3 (bait) was loaded onto the column and incubated with rotation for 30 min at 4 °C; unbound protein was removed by centrifugation (1250 × g for 1 min). The cav-3 bound to the cobalt resin was washed five times with buffer (composition as above) after which the “prey” protein (RyR1) was applied and incubated with gentle agitation for 1 h at 4 °C. The column was then centrifuged for 30 s at 1250 × g to remove any unbound prey. The prey-bait-bound cobalt resin was washed five times with buffer (eight times the column volume per wash) (25 mm Tris, pH 7.2, 150 mm NaCl), after which the bound protein was eluted in imidazole buffer (25 mm Tris, pH 7.2, 150 mm NaCl, 300 mm imidazole) followed by centrifugation at 1250 × g to collect the eluted fraction. The eluted protein was analyzed by SDS-PAGE Western blotting. Both 12 and 5% gels were run to identify the presence of cav-3 and RyR1, respectively. Control blots illustrated that the antibodies did not hybridize nonspecifically with either partner protein. Additional control experiments also showed that RyR1 did not associate with the cobalt-resin in the absence of cav-3. For method B, an aliquot of recombinant cav-3 was mixed with purified RyR1 in roughly equimolar concentrations. After 30 min, 20 μl was taken and examined by TEM after negative staining as described earlier. For comparative purposes, purified RyR1 was also analyzed by negative staining TEM.
[3H]Ryanodine Binding Assay
Following the method in Ref. 44, solubilized cav-3 was added to skeletal SR membranes (0.03 mg/ml) in buffer (300 mm KCl, 50 mm Mops, 100 μm CaCl2, 5 mm MgCl2, 1 mm AMP-PCP, 0.1% w/v CHAPS, and protease inhibitors, pH 7.4) in molar excess. After 10 min of incubation, 20 nm [3H]ryanodine was added, and the sample was incubated for 100 min. 100-μl aliquots were filtered through a Millipore vacuum filtration apparatus fitted with Whatman GF/F glass fiber filters (pre-soaked in 2% polyethyleneimine). Filters were washed five times with ice-cold buffer (300 mm KCl, 100 μm CaCl2, and 10 mm MOPS, pH 7.4). Filters were added to scintillation tubes containing 4 ml of scintillation fluid and shaken for 60 min, and radioactivity was measured by liquid scintillation counting.
RESULTS
Previous studies of canine cav-1 have shown that heterologous expression in Sf21 insect cells produces caveola-type organelles comparable in size to those observed in mammalian cells (45). We therefore examined thin sections of cav-3-infected Sf9 cells by TEM, and we found that caveola-type vesicles ∼50–140 nm in diameter were also formed (Fig. 2A). Although the caveolae were clearly apparent, the cav-3 expression did not induce “hundreds” of these vesicles per cell as described for cav-1. Because Sf9 cells are a subclone of Sf21 and are considered virtually indistinguishable, the prevalence of caveolae may reflect the level of cav-3 protein expression and/or the caveolin isoform. Indeed, Une and co-workers (6) have reported that different isoforms of cav-1 induce caveolae with a morphology characterized by either deep or shallowly structured invaginations. The presence of caveolae after infection with cav-3 would suggest that the recombinant protein used in this study is functional at the cellular level.
FIGURE 2.

Characterization of caveolin-3 expressed in baculovirus Sf9 cells. A, electron micrograph of a thin section of Sf9 cells infected with cav-3 illustrating formation of caveolae; scale bar, 200 nm. Inset, Western blot of Sf9 cells (anti-MAT) demonstrates expression of cav-3. B, purified cav-3 analyzed by SDS-PAGE. Lane 1, Coomassie-stained 10% SDS-PAGE of purified cav-3 showing a single band at ∼20 kDa; lane 2, silver-stained gel (12%) demonstrates the purity of the sample at a higher sensitivity compared with Coomassie staining, illustrating that there are no contaminating bands. Lane 3, Western blot confirming the identity of the single polypeptide band as cav-3. C, multiangle laser light scattering and elution profiles of purified cav-3. The molecular mass of the eluted peak is ∼200–220 kDa. Inset, presence of cav-3 in the peak fractions confirmed by SDS-PAGE (silver-stained) in row 1 and Western blotting (anti-cav-3, Abcam) in row 2.
Purification of Human cav-3
cav-3 was purified from Sf9 cells by a combination of Ni-NTA affinity chromatography and sucrose gradient centrifugation. It can be seen from Fig. 2B, lane 1, that purified cav-3 migrates as a single band at ∼20–22 kDa. Mass spectrometric analysis (trypsin digest and peptide fingerprinting) confirmed the identity of the protein band as cav-3 (data not shown). Samples analyzed by SDS-PAGE were also silver-stained (with a sensitivity level typically in the range of 5–0.5 ng of protein), which revealed that the cav-3 samples were pure with no minor protein components present in the preparations with Western blotting confirming the presence of cav-3 (Fig. 2B, lanes 2 and 3, respectively). We employed native gel electrophoresis to probe whether the cav-3 purified here had polymerized. A single band at ∼190 kDa was observed (data not shown) thereby indicating oligomerization of cav-3. For a more accurate determination of the cav-3 complex molecular mass, we employed MALLS. The purified cav-3 eluted in a single protein peak with a corresponding molecular mass between 200 and 220 kDa as shown in Fig. 2C, which closely agreed with the findings from the native gel electrophoresis experiments. SDS-PAGE/Western blotting of peak fractions confirmed the identity of the eluting protein as cav-3 (inset to Fig. 2C). An aliquot of the peak fraction was then negatively stained and examined by TEM with an example area showing a field of cav-3 particles presented in Fig. 3A. It can be seen that purified cav-3 forms a monodisperse, homogeneous, and well separated population of particles. A montage of some of the selected cav-3 complexes is shown in Fig. 3B. Examination of the raw particles finds that there are distinct views of cav-3 as follows: particles that are almost circular (∼160–170 Å in diameter) characterized by a central density, and complexes that are rod-like in appearance with the long axis measuring ∼160–170 Å and a thickness of ∼ 40–50 Å.
Three-dimensional Structure of cav-3
More than 5000 cav-3 oligomers were selected, contrast transfer function corrected, centered, and aligned by employing established programs within the EMAN image processing software suite (29). Reference free class averaging of the dataset revealed that in addition to the circular and rod-shaped views of the complex, a range of orientations of cav-3 had been imaged, and thus three-dimensional reconstruction using the angular reconstitution method was feasible. An example of a class average presenting a “top view” of the cav-3 complex shows an oligomer that is almost circular in shape (Fig. 3C), and thus it reflects the features of some of the raw images highlighted by an arrowhead in Fig. 3B. The protein density, which is white under negative staining conditions, can be seen to form a ring around the perimeter of the complex with an overall diameter of ∼165 Å and is resolved into nine protein domains as highlighted on Fig. 3C. These nine densities are connected to a central protein domain indicated by an arrowhead (Fig. 3C). Fig. 3D shows a rod-shaped cav-3 complex that is ∼ 160 Å long and ∼ 40 Å wide, which we have termed a side-view, and is composed of three protein domains; this projection average is consistent with the raw images in Fig. 3B highlighted by the white arrows. The projection map in Fig. 3E presents an intermediate orientation, i.e. between the top and side views. To investigate cav-3 oligomerization and symmetry, the method described by Kocsis et al. (32) was employed generating a power spectrum indicative of a complex with a 9-fold rotational symmetry as shown in Fig. 3F. Cross-correlation methods and density analysis were also employed to analyze the symmetry as shown in Fig. 3, G and H, with the results supporting the c9 assignment, i.e. cav-3 has assembled to form a nonamer, roughly equating to a cav-3 monomer with an approximate molecular mass of 22–24 kDa. The calculated mass of human cav-3 from the primary sequence was 17,259 Da; however, with post-translational modifications and the inclusion of the C-terminal MAT tag, the protein is likely to be larger, and indeed SDS-PAGE analysis of the purified protein typically showed a migration at ∼20–22 kDa. In addition, because the recombinant cav-3 has been purified in the presence of β-dodecyl d-maltoside, there will be detergent associated with the protein. Previous studies of membrane proteins have found that depending upon the detergent employed, the mass of the protein can potentially be increased by between 30 and 150% (47).
Presented in Fig. 4 is the three-dimensional volume of cav-3 refined with c9 symmetry with the surface-rendered volume corresponding to an oligomer of mass of ∼220 kDa. The cav-3 complex (Fig. 4A) is toroidal in shape with a diameter of 165 Å. There is an outer rim of protein ∼45 Å wide and connected through a series of narrow “spoke-like” densities to a central domain that is ∼70 Å across. The view of the opposite face of the complex shows that the central density extends out from the base of the structure and forms a “cone-shaped” domain (Fig. 4B). Fig. 4C shows the side view of the complex revealing that the outer rim of the volume extends downward from the relatively flat base by about 55 Å. Removal of the front portion of the volume when viewed from the side provides a cross-section of the “cone” domain therefore finding that it has a height of ∼45 Å (Fig. 4D). The resolution of the cav-3 volume approaches ∼17 Å (Fig. 4E). A comparison of the class averages and back projections of the final three-dimensional volume shows good correlation in terms of structural features and details (Fig. 4F). Further comparison with the reference-free class averages (Fig. 3, C--E) and raw images (Fig. 3B) additionally demonstrates that the dimensions and features are all in agreement.
FIGURE 4.

Three-dimensional structure of human cav-3 at ∼17 Å resolution. The three-dimensional structure of the cav-3 nonamer is disc-shaped with a diameter of ∼165 Å. A, “base” of the cav-3 oligomer, showing that it is formed by an outer ring of protein ∼45 Å wide. The outer protein rim is connected to a central density by narrow spoke-like domains. B, rotation of this view about the x axis presents the opposite face of the complex showing that the central protein density extends away from the base of the complex and is cone-shaped in appearance. C, side-view (orthogonal to the orientation in A) reveals that the overall height of the cav-3 nonamer is ∼55 Å and that the upper surface (base) of the complex is relatively flat. D, removal of the front portion of the complex when viewed from the side shows that the central cone density has a base of ∼70 Å diameter and height of ∼45 Å. E, Fourier shell correlation plot illustrating that at a cutoff of 0.5 the resolution of the cav-3 nonamer is ∼17 Å, which is perhaps optimistic, although as can be seen in Fig. 7 the three-dimensional volume can be segmented into individual monomers using the software Segger. F, column 1 presents back-projections of the final three-dimensional volume with the corresponding class averages in column 2. A comparison of the images in each row finds a good correlation between the two (box size 326 × 326 Å).
C-terminal Domains of cav-3 Form the Central Cone-shaped Density
Following established methods (35), the purified cav-3 was incubated with Ni-NTA-gold (1.8 nm) exploiting the presence of the C-terminal MAT tag, which if sterically exposed within the oligomeric structure would bind the Ni-NTA-gold conjugate. The sample mix was negatively stained and examined by TEM with the electron-dense gold visualized as black and protein imaged as white density. The location of the bound gold gives an indication as to the position of the C-terminal domain within the three-dimensional volume. A montage of cav-3 gold-labeled complexes is shown in Fig. 5, from which it can be seen that only a single gold density decorates each cav-3 nonamer. If the C-terminal domain formed part of the outer rim of protein, then it might be expected to observe cav-3 oligomers with multiple gold particles attached around the perimeter of the complex (depending upon the orientation of the oligomer and binding efficiency). Instead, we find that only a single electron-dense (gold) particle is associated with each cav-3 complex, which would indicate that the C-terminal domain of each monomer unit is closely associated, and we suggest they form the central cone region.
FIGURE 5.
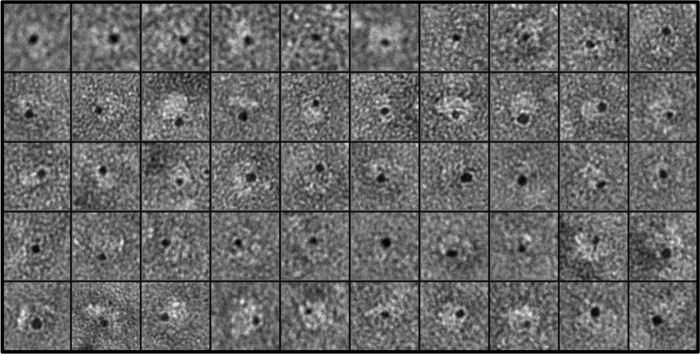
Labeling of the C-terminal domain of cav-3. A montage of negatively stained cav-3 oligomers after incubation with Ni-NTA-gold; the MAT tag at the C terminus of cav-3 binds the Ni-NTA-gold conjugate. The gold is visualized under these conditions as electron-dense (black) particles. In each example, a cav-3 nonamer (white) is decorated with only one Ni-NTA-gold conjugate (black). Box size = 326 × 326 Å.
Reconstitution of cav-3 into Lipid Vesicles
Following the method described by Geertsma et al. (36), we successfully reconstituted the purified cav-3 into large unilamellar vesicles as shown in Fig. 6. Upon reconstitution, cav-3 can be seen to present a circular view of the complex that is ∼150–160 Å in diameter with a defined central density surrounded by a ring of protein. There will be surface curvature variations in the angle of view of the reconstituted protein due to the flattening and collapse of the three-dimensional vesicles upon adhering to the carbon support film; however, visual inspection finds that the features and dimensions of the reconstituted protein clearly match those of the three-dimensional structure of the purified cav-3 in solution (Fig. 4). cav-3 complexes that were well separated from the neighboring protein were selected and subjected to reference-free class averaging with two representative classes shown in Fig. 6B. The circular shape of the cav-3 oligomer is clearly apparent in each average, as is the central cone domain. In addition, the protein densities forming the outer ring were resolved into multiple domains, which again is consistent with the calculated three-dimensional volume. Given that these features are well contrasted by the negative stain, it would suggest that a portion of the outer rim of protein and central cone extend beyond the bilayer. Importantly these experiments illustrate that the purified protein can be reconstituted back into lipid vesicles, and thus it may represent a physiologically functional complex and further, permit a proposed orientation of the cav-3 EM volume with respect to the lipid bilayer as shown in Fig. 7.
FIGURE 6.

Reconstitution of purified cav-3 into large unilamellar vesicles. A, cav-3 is inserted into the lipid vesicle so that the circular view of the complex is presented, with the central cone-shaped density facing out of the plane of the membrane. B, two representative reference-free class averages (composed of 47 and 35 particles) of reconstituted cav-3 confirm the visual analysis. The projection images show a complex ∼160 Å in diameter with a central cone domain (indicated by the solid arrowhead) surrounded by an outer ring of protein that can be resolved into multiple domains as indicated by the arrows. The high contrast staining would infer that these features of the complex extend beyond the bilayer. Box size = 275 × 275 Å.
FIGURE 7.
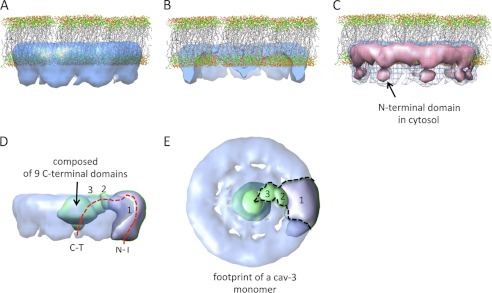
Orientation of the cav-3 nonamer with respect to the lipid bilayer. A, cav-3 spans a monolayer placing the N- and C-terminal regions within the cytoplasm. The lipid Protein Data Bank coordinates were downloaded from the website of Dr. Scott Feller, Wabash College. B, removal of the front portion of the cav-3 complex shows that the base of the central cone domain is submerged within the lipid layer, whereas the apex extends into the cytoplasm. C, cav-3 oligomer is displayed at two thresholds. The blue mesh encompasses a protein mass corresponding to ∼220 kDa (as shown in Fig. 4). Increasing the threshold (equating to a volume of ∼170 kDa) leads to the separation of the outer ring into an upper and lower region shown by the pink surface rendering. We suggest that the lower globular densities correspond to the soluble N-terminal domains from each cav-3 monomer. D, using the program Segger (Chimera UCSF software), the three-dimensional volume was segmented. The side view of the cav-3 nonamer (transparency setting 0.6) is shown after segmentation of the outer ring of the cav-3 complex into nine parts; for clarity, only one segment is displayed. Further segmentation of the spoke and central cone domain identified two further elements that would form a cav-3 monomer in the 9-fold symmetric organization. The outer ring segment is labeled 1; the spoke domain is labeled 2, and a portion of the central cone is labeled 3. The very center of the “central cone” could not be computationally segmented further. The dashed red line shows the putative positions of the N-terminal (N-T) and C-terminal (C-T) terminal domains of a cav-3 monomer. E, rotation of the view in D by 90° shows the domain organization of a cav-3 monomer with the dashed outline giving an impression of the volume occupied by a cav-3 monomer and roughly divides up the core of the central cone.
The three-dimensional cav-3 EM volume is modeled in Fig. 7A to reside within a monolayer in agreement with previous studies (48–50). In this orientation, the putative N terminus of each monomer extends into the cytosol by ∼28 Å. It can be seen from Fig. 7B that a portion of the central cone density lies within the lipid environment, thus agreeing with predictions that the C-terminal domain is palmitoylated and is likely to be hydrophobic associating/inserting within the lipid bilayer (51). However, the apex of the cone protrudes out of the membrane by ∼18 Å, and thus we propose that the hydrophilic segments of the N- and C-terminal domains are located within the cytosol and positioned ∼70 Å apart. When displayed at a higher threshold, the outer ring of cav-3 can be resolved into two domains when viewed perpendicular to the plane of the membrane (Fig. 7C). We propose that the lower globular densities correspond to the hydrophilic N-terminal domains of each contributing cav-3 monomer. Segmentation of the three-dimensional volume using the software Segger (an extension to the UCSF Chimera software) reveals the approximate footprint of a cav-3 monomer as shown in Fig. 7, D and E.
Interactions of cav-3 and RyR1
As discussed earlier, one study has reported a putative interaction between cav-3 and RyR1, although this has not yet been demonstrated between the two purified proteins. We examined the primary sequence of both RyR1 and RyR2 for caveolin-binding motifs (CBMs) as described under “Experimental Procedures.” As shown in Table 1, there are six predicted CBMs within RyR1 with five of these located in the putative transmembrane region. Interestingly, it can also be seen that six motifs are well conserved in the cardiac isoform, RyR2, with three additional CBMs (7, 8, and 9) unique to the cardiac receptor sequence.
TABLE 1.
Aligned CBMs identified in the primary sequences of RyR1 and RyR2 labeled 1–6
There are also three distinct CBMs, 7–9, that are unique to RyR2. Motifs 1–6 were aligned to show the degree of similarity. Asterisk indicates fully conserved residue; colon indicates conservation of residue with strongly similar properties; period indicates conservation of residue with weakly similar properties. Capital letters indicate key amino acid residues within the motif. CBM 1 is found in the putative cytosolic assembly, and motifs 2–6 are housed in the transmembrane domain. It can be seen that several of the CBMs in the transmembrane region overlap, e.g. 3–5 in both isoforms.
| CBM | Sequence alignment | Region CBM located to within RyR | |
|---|---|---|---|
| 1 | (RyR1)1431–1438, | YyYsvrvF | Cytosolic |
| (RyR2)1426–1433, | YyYsvriF | ||
| ******:* | |||
| 2 | (RyR1)4712–4720, | FpsnyWdkF | Transmembrane |
| (RyR2)4640–4648, | FpnnyWdkF | ||
| **.****** | |||
| 3 | (RyR1)4850–4857, | YlYtvvaF | Transmembrane |
| (RyR2)4779–4786, | YlYtvvaF | ||
| ******** | |||
| 4 | (RyR1)4852–4860, | YtvvaFnfF | Transmembrane |
| (RyR2)4781–4789, | YtvvaFnfF | ||
| ********* | |||
| 5 | (RyR1)4857–4864, | FnFfrkfY | Transmembrane |
| (RyR2)4786–4793, | FnFfrkfY | ||
| ******** | |||
| 6 | (RyR1)5015–5023, | YqercWdfF | Transmembrane |
| (RyR2)4944–4952, | YqercWefF | ||
| ******:** | |||
| 7 | (RyR2)1445–1453, | WitsdFhqY | Cytosolic |
| 8 | (RyR2)4283–4291, | FfssyWsiF | Transmembrane |
| 9 | (RyR2)4297–4305, | FvasvFrgF | Transmembrane |
Cryo-EM studies have shown that the RyR1 transmembrane “foot” is ∼7 nm tall, which makes it thicker than the SR membrane (52), which would suggest that portions of the region termed the transmembrane (TM) domain are not necessarily completely within the membrane. Based on the EM volume, the TM region has been predicted to accommodate between 24 and 32 transmembrane α-helices (or 6–8 per subunit) (53). To correlate the identified CBMs to the regions within the RyR1 TM region, we calculated the hydrophobicity of the sequence and likelihood of α-helices to form within this region. As can be seen in Fig. 8A, two of the CBMs identified from the primary sequence in the TM segment ( Table 1, CBMs 2 and 6) are located in regions that are predicted not to be within the membrane and do not adopt a helical structure unlike CBMs 3–5, which overlap at a site that is both predicted to be an α-helix and within the membrane. The two additional CBMs that were located within the transmembrane domain of RyR2, CBMs 8 and 9, but not RyR1, were located in a region that was predicted to be both helical and within the membrane (data not shown). The nature of the hydrophobicity and secondary structure prediction algorithms do not facilitate identification of which side of the membrane the CBMs are located; moreover, and importantly, we do not know whether these sites are exposed or occluded because RyRs assemble to form homotetramers.
FIGURE 8.

Nonameric cav-3 binds purified RyR1 homotetramers. A, regions predicted to form transmembrane α-helices and the location of CBMs are displayed (data analyzed using SPLIT tool). The regions that are predicted to form TM α-helices are indicated by the position of the solid gray blocks. Superimposed are the locations of the CBMs, hatched box. The plot shows that two of the CBMs (Table 1, CBMs 2 and 6) are located at regions that are not predicted to be hydrophobic or form α-helical structures. B, Western blotting of JTC SR membrane proteins separated by SDS-PAGE identifying cav-3 as a constituent. C, Coomassie-stained SDS-PAGE (5%) illustrating the purity of RyR1 with Western blotting of each fraction (lower panel) confirming the identity of the isolated protein as RyR1. The 12% SDS-PAGE shows no accessory proteins have co-purified with RyR1. D, TEM image of a field of purified RyR1 complexes shows that it has been purified as a homotetramer. E, Western blot of cav-3 and RyR1 showing an interaction. Lane 1, loaded protein cav-3, top; RyR1, bottom. Lane 2, unbound protein. It can be seen that the majority of cav-3 bound to the resin and that a portion of RyR1 did not bind to the immobilized cav-3. Lane 3 shows that after multiple washing steps there was no nonspecifically bound protein present. Lane 4, both cav-3 and RyR1 were detected in the eluent indicative of RyR1 having bound to cav-3.
To experimentally examine whether homotetrameric RyR1 can bind full-length cav-3 nonamers, we also purified the receptor from sheep skeletal muscle. The first stage of this preparation involved isolating SR membranes. Interestingly, Western blotting of the SR proteins separated by SDS-PAGE identified cav-3 (Fig. 8B) as a constituent that would agree with the findings from Li et al. (28). Following isolation of the SR membranes, RyR1 was purified as described under “Experimental Procedures.” The purity of the isolated RyR1 was assessed by SDS-PAGE as shown in Fig. 8C. To determine whether the purified receptor had been isolated as a homotetramer (native organization), an aliquot was examined by TEM (Fig. 8D) showing that the RyR1 prepared here exhibited the characteristic four-lobed arrangement. cav-3 was immobilized via the C-terminal MAT tag onto a metal affinity column with RyR1 subsequently applied. The Western blot in Fig. 8E shows the protein profiles of the binding, washing, and elution stages revealing that both cav-3 and RyR1 were eluted from the column with imidazole, thereby suggesting a complex had formed between the two full-length folded proteins (lane 4). In a second approach to examine the interaction of the two full-length proteins, aliquots of purified RyR1 and recombinant cav-3 were combined, incubated, and then examined by TEM. Examples of control RyR1 images presenting the cytoplasmic assembly in projection measuring ∼30 × 30 nm are shown for comparative purposes in Fig. 9A. A side view of RyR1 is also shown in Fig. 9A, far-right panel. Two example areas showing a field of RyR1/cav-3 complexes are shown in Fig. 9B, although it was found that the addition of recombinant cav-3 led to much of the sample being aggregated (possibly due to the presence of two different detergents). However, some RyR1·cav-3 complexes showing square-shaped RyR1 tetramers with additional protein densities bound were identified as shown in Fig. 9C. It can be seen from the montage of particles that the protein densities attached to the square-shaped RyR1 complexes have dimensions and features that match those of cav-3 nonamers. In addition, the number of cav-3 oligomers associating with an individual RyR1 tetramer varies considerably as does the orientation when viewed in projection, and as such, single particle averaging and classification were not feasible. Possible future attempts to separate out the various RyR1·cav-3 complexes by size-exclusion methods, for example, may aid the EM analysis of the complex formation, although low yields of purified cav-3 are currently prohibitive. However, what we can infer from the raw images is that there is clearly an association between the RyR1 homotetramers and full-length cav-3 nonamers, although caution should be taken when interpreting noisy single images. Thus, we were not able to draw confident conclusions as to which regions of RyR1 bind cav-3 but can simply infer an interaction has occurred.
FIGURE 9.

Interaction of solubilized recombinant cav-3 with RyR1. A, control RyR1 complexes presenting the characteristic four-lobed appearance corresponding to a homotetramer with dimensions ∼300 × 300 Å. An EM volume for RyR1 (emd_1275.map) has been modeled to illustrate the RyR1 structure. Although negative staining and adsorption of RyR1 onto the carbon support film is known to lead to a preferred orientation and flattening of the structure and the occasional side view of the protein was observed, an example is shown in the far-right column. B, field of particles with arrows indicating cav-3 bound to RyR1 homotetramers. Scale bar, 50 nm. C, montage of individual RyR1 complexes with cav-3 bound as indicated by the arrows. There is a range of orientations of the RyR1 homotetramers each with at least one cav-3 nonamer attached. In some examples, several cav-3 complexes are associated with a single RyR1. For example, image 2 has three cav-3 nonamers bound, stacked side by side along one edge of a RyR1 tetramer. Image 4 shows one RyR1 complex with a cav-3 nonamer bound to one corner with possibly another cav-3 oligomer attached and linked to several other cav-3 particles. Image 19 shows cav-3, with the circular view of the complex and central cone domain visible, bound to one side of RyR1. The far-right complex (image 36) shows a partial side view of RyR1 with a disc-shaped cav-3 attached to the upper surface. The EM volumes of cav-3 and RyR1 have been modeled in the panel below image 36 to illustrate the association. D, recombinant solubilized full-length cav-3 does not alter [3H]ryanodine binding to skeletal muscle SR membranes. cav-3 was added in molar excess (25:1), with all experiments conducted in triplicate.
Is cav-3 a Modulator of RyR1 Channel Activity?
To investigate whether cav-3 binds to RyR1 via the cytoplasmic domain (CBM 1), we employed a [3H]ryanodine binding assay (44). Ryanodine preferentially interacts with the open state of RyR1 with a reduction or increase in binding linked to an inactivation or activation, respectively (54). We found that the addition of solubilized cav-3 nonamers did not significantly alter [3H] ryanodine binding to SR membranes (Fig. 9D). We interpreted these data to have two possible meanings as follows: (i) that CBM 1 is occluded in the RyR1 homotetramer cytoplasmic assembly, or (ii) there is an interaction, but it does not result in any change to the open state of the channel. This method only tests the availability of CBM 1 for binding, because the other motifs would be occluded as they are in, or near, the TM region.
DISCUSSION
Our data show for the first time a direct interaction of cav-3 with homotetrameric RyR1 and thus indicate that at least one or more of the predicted CBMs are exposed within the quaternary organization of the receptor. Interestingly, it has been reported that in cases of central core disease aa 4860 (RyR1) is missing (55). This residue forms part of CBM 4, and thus the deletion of the aromatic phenylalanine, a key residue forming the CBM, would likely perturb this region. In addition, we note that there are reports linking a mutation of aa 4950 (RyR2, glutamic acid to lysine; forming part of CBM 6) to catecholaminergic polymorphic ventricular tachycardia type 1, an electrical disorder of the heart that often results in sudden cardiac death. Further analysis of links between the putative interaction sites in the context of genetic determinants of disease also found that two of the CBMs unique to RyR2 (CBMs 8 and 9) are located within the segment termed divergent region 1, DR1 (aa 4210–4562) (56), which likely explains the loss of conservation with RyR1. However, recent medium/high resolution cryo-EM three-dimensional structures of RyR1 (57) have allowed mapping of the secondary structure features within the TM region identifying six putative helical domains. This assignment places the CBM motifs designated 3, 4, and 5 (spanning aa 4850–4864) very close to the selectivity filter (aa 4891–4900) and hence within the core of the RyR1 TM domain. Therefore, based upon the EM model, these motifs would not be accessible for binding to cav-3. However, a similar analysis would place CBM 2 at the perimeter of the RyR1 volume, with the bioinformatics analysis (Fig. 8A) also predicting that it is not within a TM helix and consequently may represent an exposed site for binding cav-3. In addition, the CBM motif 6 is situated within the short C-terminal tail that would extend just beyond the bilayer into the cytosol, and so we suggest it also may represent a viable binding site for cav-3.
As shown in Table 1, CBM 1 is located to the cytoplasmic domain of RyR1 and RyR2, but despite the fact that we found that the addition of cav-3 did not modulate [3H]ryanodine binding to SR membranes, we cannot rule out that a direct interaction between the two proteins has occurred especially because in the heart a distinct population of cav-3 has been shown to co-localize in the sarcolemma in apposition to clusters of extra-dyadic RyR2s (58). It is generally accepted that RyRs almost bridge the cytosolic space between 12 and 14 nm but fall short of contacting the plasma membrane. We can see from the three-dimensional structure of cav-3 presented here (Fig. 7) that the N-terminal regions of the cav-3 nonamer extend beyond the bilayer into the cytosol by nearly 3 nm and thus would have the potential to form contacts with the large cytoplasmic receptor assembly; therefore, an association between cav-3 and RyR via CBM 1 may be important for localization of the channel within different regions of the SR. Future higher resolution EM studies may enable clarification as to the RyR domains involved.
A further intriguing result from this study was the identification of a population of cav-3 in isolated SR membranes. Li et al. (28) have also identified cav-3 in the SR, although they describe SR preparations that likely equate to the longitudinal SR fractions inferred by the high concentration of sarco/endoplasmic reticulum Ca2+-ATPase, cholesterol, and GM1, all markers of the longitudinal SR. Importantly, the group also showed that there were no protein markers to indicate plasma membrane contamination. One possibility for the presence of cav-3 in both the junctional (prepared here) and longitudinal regions of the SR is that neither of the purification protocols resulted in 100% fractionation or that cav-3 populations exist throughout the SR. A detailed analysis of the localization of cav-3 in skeletal and cardiac myocytes is clearly required to unravel the physiological relevance of an SR population of cav-3. An SR population of cav-3 would also have the potential to interact with RyR1 CBMs 2 and 6. Future approaches to investigate this association may consider, for example, co-reconstitution of cav-3 and RyR1 into bilayers to examine effects upon channel opening using electrophysiology techniques.
The interpretation of the three-dimensional structure of nonameric cav-3 suggests that the C-terminal domain from each contributing monomer cluster together to form the central cone region that is consistent with reports showing that deletion of the C-terminal tail of cav-1 impairs oligomerization (60). Furthermore, the exposure of the putative N-terminal domain, within the cytosol, as shown in Fig. 7C agrees well with studies identifying this region as important for mediating interactions with a wide range of proteins (61). Overall, the three-dimensional cav-3 structure permits visualization as to how the N- and C-terminal domains of cav-3 could independently associate with a number of proteins or interact with the same partner but bind to different regions, and thereby promote assembly of macromolecular signaling complexes. Negative staining methods can lead to deformation of the tertiary and quaternary structure of a protein; however, based upon the modeling presented in Fig. 7, we suggest that there has not been significant flattening of the structure.
Finally, the finding that overexpression of human cav-3 appeared to induce caveolae formation in Sf9 cells was intriguing because insect cell membranes typically have a very low cholesterol content compared with mammalian cells and a different phospholipid profile (62). The formation of caveolae would therefore perhaps be unexpected as both the structure and function are intrinsically linked to cholesterol (63), with caveolins also shown to directly bind cholesterol (64). There is also growing evidence that polymerase I and transcript release factor (PTRF)-Cavin is also required for the formation, organization, and function of caveolae (22, 46, 59), but as yet a homolog has not been identified in insects. With respect to cholesterol, there is perhaps a minimum concentration that is required for caveolae biogenesis. Another possibility is that cholesterol content may influence the propensity of caveolae to form, which may provide an alternate explanation as to why we do not observe “hundreds” forming in Sf9 cells. Similarly, because caveolins have been shown to directly bind cholesterol, concentration may also influence the size of the caveolin oligomers formed and thus may explain the variation between reports in the literature due to differences in the cell type or expression systems studied. Another interesting observation was that cav-3 was reconstituted into liposomes devoid of cholesterol. However, because we did not observe the formation of caveolae upon insertion of the protein, this would support the requirement of minimal cholesterol content for induction of caveolae. In conclusion, these data provide a new understanding as to how cav-3 can act as a signaling hub binding multiple protein partners per oligomer and facilitate protein-protein interactions. Our data showing a direct interaction between RyR1 and cav-3 is novel, and future studies are now needed to investigate whether this association has a role in regulating muscle excitation-contraction coupling and/or influences the organization of RyR populations within the SR or has a role in stabilization of the T-tubule/SR configuration.
Acknowledgments
We acknowledge Marge Howard and Emma Keevill in the Biomolecular Analysis Facility, Dr. Edward Mckenzie in the Protein Expression Unit, and Dr. Aleksandr Mironov and Samantha Forbes in the EM Facility at the University of Manchester. We also extend thanks to Prof. Andrew Trafford for providing access to the sheep skeletal muscle tissue and Dr. Ahmed Handhle for advice for isolating the SR membranes and the tritium ryanodine binding assay. We thank Dr. Jaqui Ohanian for critique of this manuscript.
This work was supported by British Heart Foundation Grant FS/08/038/25262 (to G. W. and A. K.).
- cav
- caveolin
- aa
- amino acid
- RyR2
- cardiac ryanodine receptor
- CBM
- caveolin-binding motif
- JTC
- junctional terminal cisternae
- SR
- sarcoplasmic reticulum
- RyR1
- skeletal muscle ryanodine receptor
- TM
- transmembrane
- TEM
- transmission electron microscopy
- DDM
- β-dodecyl d-maltoside
- MALLS
- multiangle laser light scattering
- Ni-NTA
- nickel-nitrilotriacetic acid
- AMP-PCP
- adenosine 5′-(β,γ-methylenetriphosphate)
- MAT
- metal affinity tag.
REFERENCES
- 1. Rothberg K. G., Heuser J. E., Donzell W. C., Ying Y. S., Glenney J. R., Anderson R. G. (1992) Caveolin, a protein component of caveolae membrane coats. Cell 68, 673–682 [DOI] [PubMed] [Google Scholar]
- 2. Glenney J. R. (1992) The sequence of human caveolin reveals identity with VIP21, a component of transport vesicles. FEBS Lett. 314, 45–48 [DOI] [PubMed] [Google Scholar]
- 3. Tang Z., Scherer P. E., Okamoto T., Song K., Chu C., Kohtz D. S., Nishimoto I., Lodish H. F., Lisanti M. P. (1996) Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J. Biol. Chem. 271, 2255–2261 [DOI] [PubMed] [Google Scholar]
- 4. Scherer P. E., Tang Z., Chun M., Sargiacomo M., Lodish H. F., Lisanti M. P. (1995) Caveolin isoforms differ in their N-terminal protein sequence and subcellular distribution. Identification and epitope mapping of an isoform-specific monoclonal antibody probe. J. Biol. Chem. 270, 16395–16401 [DOI] [PubMed] [Google Scholar]
- 5. Kogo H., Ishiguro K., Kuwaki S., Fujimoto T. (2002) Identification of a splice variant of mouse caveolin-2 mRNA encoding an isoform lacking the C-terminal domain. Arch Biochem. Biophys. 401, 108–114 [DOI] [PubMed] [Google Scholar]
- 6. Fujimoto T., Kogo H., Nomura R., Une T. (2000) Isoforms of caveolin-1 and caveolar structure. J. Cell Sci. 113, 3509–3517 [DOI] [PubMed] [Google Scholar]
- 7. Lisanti M. P., Scherer P. E., Tang Z., Sargiacomo M. (1994) Caveolae, caveolin, and caveolin-rich membrane domains: a signaling hypothesis. Trends Cell Biol. 4, 231–235 [DOI] [PubMed] [Google Scholar]
- 8. Razani B., Woodman S. E., Lisanti M. P. (2002) Caveolae. From cell biology to animal physiology. Pharmacol. Rev. 54, 431–467 [DOI] [PubMed] [Google Scholar]
- 9. Boscher C., Nabi I. R. (2012) Caveolin-1. Role in cell signaling. Adv. Exp. Med. Biol. 729, 29–50 [DOI] [PubMed] [Google Scholar]
- 10. Cohen A. W., Hnasko R., Schubert W., Lisanti M. P. (2004) Role of caveolae and caveolins in health and disease. Physiol. Rev. 84, 1341–1379 [DOI] [PubMed] [Google Scholar]
- 11. Fernandez I., Ying Y., Albanesi J., Anderson R. G. (2002) Mechanism of caveolin filament assembly. Proc. Natl. Acad. Sci. U.S.A. 99, 11193–11198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sargiacomo M., Scherer P. E., Tang Z., Kübler E., Song K. S., Sanders M. C., Lisanti M. P. (1995) Oligomeric structure of caveolin. Implications for caveolae membrane organization. Proc. Natl. Acad. Sci. U.S.A. 92, 9407–9411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schlegel A., Lisanti M. P. (2000) A molecular dissection of caveolin-1 membrane attachment and oligomerization. Two separate regions of the caveolin-1 C-terminal domain mediate membrane binding and oligomer/oligomer interactions in vivo. J. Biol. Chem. 275, 21605–21617 [DOI] [PubMed] [Google Scholar]
- 14. Schlegel A., Schwab R. B., Scherer P. E., Lisanti M. P. (1999) A role for the caveolin scaffolding domain in mediating the membrane attachment of caveolin-1. The caveolin scaffolding domain is both necessary and sufficient for membrane binding in vitro. J. Biol. Chem. 274, 22660–22667 [DOI] [PubMed] [Google Scholar]
- 15. Razani B., Wang X. B., Engelman J. A., Battista M., Lagaud G., Zhang X. L., Kneitz B., Hou H., Jr., Christ G. J., Edelmann W., Lisanti M. P. (2002) Caveolin-2-deficient mice show evidence of severe pulmonary dysfunction without disruption of caveolae. Mol. Cell. Biol. 22, 2329–2344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Song K. S., Li Shengwen, Okamoto T., Quilliam L. A., Sargiacomo M., Lisanti M. P. (1996) Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent-free purification of caveolae microdomains. J. Biol. Chem. 271, 9690–9697 [DOI] [PubMed] [Google Scholar]
- 17. Way M., Parton R. G. (1996) M-caveolin, a muscle-specific caveolin-related protein. FEBS Lett. 378, 108–112 [DOI] [PubMed] [Google Scholar]
- 18. Song K. S., Scherer P. E., Tang Z., Okamoto T., Li S., Chafel M., Chu C., Kohtz D. S., Lisanti M. P. (1996) Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J. Biol. Chem. 271, 15160–15165 [DOI] [PubMed] [Google Scholar]
- 19. Park D. S., Woodman S. E., Schubert W., Cohen A. W., Frank P. G., Chandra M., Shirani J., Razani B., Tang B., Jelicks L. A., Factor S. M., Weiss L. M., Tanowitz H. B., Lisanti M. P. (2002) Caveolin-1/3 double-knockout mice are viable, but lack both muscle and non-muscle caveolae, and develop a severe cardiomyopathic phenotype. Am. J. Path. 160, 2207–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Woodman S. E., Park D. S., Cohen A. W., Cheung M. W., Chandra M., Shirani J., Tang B., Jelicks L. A., Kitsis R. N., Christ G. J., Factor S. M., Tanowitz H. B., Lisanti M. P. (2002) Caveolin-3 knock-out mice develop a progressive cardiomyopathy and show hyperactivation of the p42/44 MAPK cascade. J. Biol. Chem. 277, 38988–38997 [DOI] [PubMed] [Google Scholar]
- 21. De Souza A. P., Cohen A. W., Park D. S., Woodman S. E., Tang B., Gutstein D. E., Factor S. M., Tanowitz H. B., Lisanti M. P., Jelicks L. A. (2005) MR imaging of caveolin gene-specific alterations in right ventricular wall thickness. Magn. Reson. Imaging 23, 61–68 [DOI] [PubMed] [Google Scholar]
- 22. Liu L., Brown D., McKee M., Lebrasseur N. K., Yang D., Albrecht K. H., Ravid K., Pilch P. F. (2008) Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab. 8, 310–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Galbiati F., Engelman J. A., Volonte D., Zhang X. L., Minetti C., Li M., Hou H., Jr., Kneitz B., Edelmann W., Lisanti M. P. (2001) Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J. Biol. Chem. 276, 21425–21433 [DOI] [PubMed] [Google Scholar]
- 24. Minetti C., Bado M., Broda P., Sotgia F., Bruno C., Galbiati F., Volonte D., Lucania G., Pavan A., Bonilla E., Lisanti M. P., Cordone G. (2002) Impairment of caveolae formation and T-system disorganization in human muscular dystrophy with caveolin-3 deficiency. Am. J. Pathol. 160, 265–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Couchoux H., Allard B., Legrand C., Jacquemond V., Berthier C. (2007) Loss of caveolin-3 induced by the dystrophy-associated P104L mutation impairs L-type calcium channel function in mouse skeletal muscle cells. J. Physiol. 580, 745–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Balijepalli R. C., Foell J. D., Hall D. D., Hell J. W., Kamp T. J. (2006) Localization of cardiac L-type Ca2+ channels to a caveolar macromolecular signaling complex is required for β2-adrenergic regulation. Proc. Natl. Acad. Sci. U.S.A. 103, 7500–7505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vassilopoulos S., Oddoux S., Groh S., Cacheux M., Fauré J., Brocard J., Campbell K. P., Marty I. (2010) Caveolin 3 is associated with the calcium release complex and is modified via in vivo triadin modification. Biochemistry 49, 6130–6135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li C., Duan W., Yang F., Zhang X. (2006) Caveolin-3-anchored microdomains at the rabbit sarcoplasmic reticulum membranes. Biochem. Biophys. Res. Commun. 344, 1135–1140 [DOI] [PubMed] [Google Scholar]
- 29. Ludtke S. J., Baldwin P. R., Chiu W. (1999) EMAN. Semiautomated software for high resolution single-particle reconstructions. J. Struct. Biol. 128, 82–97 [DOI] [PubMed] [Google Scholar]
- 30. Adair B., Nunn R., Lewis S., Dukes I., Philipson L., Yeager M. (2008) Single particle image reconstruction of the human recombinant Kv2.1 channel. Biophys. J. 94, 2106–2114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frank J., Radermacher M., Penczek P., Zhu J., Li Y., Ladjadj M., Leith A. (1996) SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 116, 190–199 [DOI] [PubMed] [Google Scholar]
- 32. Kocsis E., Cerritelli M. E., Trus B. L., Cheng N., Steven A. C. (1995) Improved methods for determination of rotational symmetries in macromolecules. Ultramicroscopy 60, 219–228 [DOI] [PubMed] [Google Scholar]
- 33. Böttcher B., Wynne S. A., Crowther R. A. (1997) Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature 386, 88–91 [DOI] [PubMed] [Google Scholar]
- 34. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 35. Walsh C. P., Davies A., Butcher A. J., Dolphin A. C., Kitmitto A. (2009) Three-dimensional structure of CaV3.1. Comparison with the cardiac L-type voltage-gated calcium channel monomer architecture. J. Biol. Chem. 284, 22310–22321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Geertsma E. R., Nik Mahmood N. A., Schuurman-Wolters G. K., Poolman B. (2008) Membrane reconstitution of ABC transporters and assays of translocator function. Nat. Protoc. 3, 256–266 [DOI] [PubMed] [Google Scholar]
- 37. Chu A., Dixon M. C., Saito A., Seiler S., Fleischer S. (1988) Isolation of sarcoplasmic reticulum fractions referable to longitudinal tubules and junctional terminal cisternae from rabbit skeletal muscle. Methods Enzymol. 157, 36–46 [DOI] [PubMed] [Google Scholar]
- 38. Meng X., Wang G., Viero C., Wang Q., Mi W., Su X. D., Wagenknecht T., Williams A. J., Liu Z., Yin C. C. (2009) CLIC2-RyR1 interaction and structural characterization by cryo-electron microscopy. J. Mol. Biol. 387, 320–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Serysheva I. I., Schatz M., van Heel M., Chiu W., Hamilton S. L. (1999) Structure of the skeletal muscle calcium release channel activated with Ca2+ and AMP-PCP. Biophys. J. 77, 1936–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Juretić D., Zoranić L., Zucić D. (2002) Basic charge clusters and predictions of membrane protein topology. J. Chem. Inf. Comput. Sci. 42, 620–632 [DOI] [PubMed] [Google Scholar]
- 41. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 42. Couet J., Li S., Okamoto T., Ikezu T., Lisanti M. P. (1997) Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J. Biol. Chem. 272, 6525–6533 [DOI] [PubMed] [Google Scholar]
- 43. de Castro E., Sigrist C. J., Gattiker A., Bulliard V., Langendijk-Genevaux P. S., Gasteiger E., Bairoch A., Hulo N. (2006) ScanProsite. Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, W362-W365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Needleman D. H., Hamilton S. L. (1997) Factors influencing [3H]ryanodine binding to the skeletal muscle Ca2+ release channel. Anal. Biochem. 248, 173–179 [DOI] [PubMed] [Google Scholar]
- 45. Li S., Song K. S., Koh S. S., Kikuchi A., Lisanti M. P. (1996) Baculovirus-based expression of mammalian caveolin in Sf21 insect cells. A model system for the biochemical and morphological study of caveolae biogenesis. J. Biol. Chem. 271, 28647–28654 [DOI] [PubMed] [Google Scholar]
- 46. Hill M. M., Bastiani M., Luetterforst R., Kirkham M., Kirkham A., Nixon S. J., Walser P., Abankwa D., Oorschot V. M., Martin S., Hancock J. F., Parton R. G. (2008) PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 132, 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gennis R. B. (1989) in Biomembranes: Molecular Structure and Function (Cantor C. R., ed) pp. 91–105, Springer-Verlag, New York [Google Scholar]
- 48. Aoki S., Thomas A., Decaffmeyer M., Brasseur R., Epand R. M. (2010) The role of proline in the membrane re-entrant helix of caveolin-1. J. Biol. Chem. 285, 33371–33380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ostermeyer A. G., Ramcharan L. T., Zeng Y., Lublin D. M., Brown D. A. (2004) Role of the hydrophobic domain in targeting caveolin-1 to lipid droplets. J. Cell Biol. 164, 69–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Parton R. G., Simons K. (2007) The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 8, 185–194 [DOI] [PubMed] [Google Scholar]
- 51. Dietzen D. J., Hastings W. R., Lublin D. M. (1995) Caveolin is palmitoylated on multiple cysteine residues. Palmitoylation is not necessary for localization of caveolin to caveolae. J. Biol. Chem. 270, 6838–6842 [DOI] [PubMed] [Google Scholar]
- 52. Serysheva I. I., Orlova E. V., Chiu W., Sherman M. B., Hamilton S. L., van Heel M. (1995) Electron cryomicroscopy and angular reconstitution used to visualize the skeletal muscle calcium release channel. Nat. Struct. Mol. Biol. 2, 18. [DOI] [PubMed] [Google Scholar]
- 53. Samsó M., Wagenknecht T., Allen P. D. (2005) Internal structure and visualization of transmembrane domains of the RyR1 calcium release channel by cryo-EM. Nat. Struct. Mol. Biol. 12, 539–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chu A., Díaz-Muñoz M., Hawkes M. J., Brush K., Hamilton S. L. (1990) Ryanodine as a probe for the functional state of the skeletal muscle sarcoplasmic reticulum calcium release channel. Mol. Pharmacol. 37, 735–741 [PubMed] [Google Scholar]
- 55. Monnier N., Romero N. B., Lerale J., Landrieu P., Nivoche Y., Fardeau M., Lunardi J. (2001) Familial and sporadic forms of central core disease are associated with mutations in the C-terminal domain of the skeletal muscle ryanodine receptor. Hum. Mol. Genet. 10, 2581–2592 [DOI] [PubMed] [Google Scholar]
- 56. Sorrentino V., Volpe P. (1993) Ryanodine receptors: how many, where, and why? Trends Pharmacol. Sci. 14, 98–103 [DOI] [PubMed] [Google Scholar]
- 57. Samsó M., Feng W., Pessah I. N., Allen P. D. (2009) Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLoS Biol. 7, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Scriven D. R., Klimek A., Asghari P., Bellve K., Moore E. D. (2005) Caveolin-3 is adjacent to a group of extradyadic ryanodine receptors. Biophys. J. 89, 1893–1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu L., Pilch P. F. (2008) A critical role of cavin (polymerase I and transcript release factor) in caveolae formation and organization. J. Biol. Chem. 283, 4314–4322 [DOI] [PubMed] [Google Scholar]
- 60. Song K. S., Tang Z., Li S., Lisanti M. P. (1997) Mutational analysis of the properties of caveolin-1. A novel role for the C-terminal domain in mediating homo-typic caveolin-caveolin interactions. J. Biol. Chem. 272, 4398–4403 [DOI] [PubMed] [Google Scholar]
- 61. Markandeya Y. S., Fahey J. M., Pluteanu F., Cribbs L. L., Balijepalli R. C. (2011) Caveolin-3 regulates protein kinase A modulation of the Ca(V)3.2 (α1H) T-type Ca2+ channels. J. Biol. Chem. 286, 2433–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gerbal M., Fournier P., Barry P., Mariller M., Odier F., Devauchelle G., Duonor-Cerutti M. (2000) Adaptation of an insect cell line of Spodoptera frugiperda to grow at 37°C: characterization of an endodiploid clone. In Vitro Cell. Dev. Biol. Anim. 36, 117–124 [DOI] [PubMed] [Google Scholar]
- 63. Ikonen E., Parton R. G. (2000) Caveolins and cellular cholesterol balance. Traffic 1, 212–217 [DOI] [PubMed] [Google Scholar]
- 64. Murata M., Peränen J., Schreiner R., Wieland F., Kurzchalia T. V., Simons K. (1995) VIP21/caveolin is a cholesterol-binding protein. Proc. Natl. Acad. Sci. U.S.A. 92, 10339–10343 [DOI] [PMC free article] [PubMed] [Google Scholar]