

Background: Bacterial phosphatidylinositol-specific phospholipase C targets PI and glycosylphosphatidylinositol-linked proteins of eukaryotic cells.
Results: Functional relevance of a homodimeric S. aureus PI-PLC crystal structure is supported by enzyme kinetics and mutagenesis. Nonsubstrate phosphatidylcholine increases activity by facilitating enzyme dimerization.
Conclusion: Activating transient dimerization is antagonized by anions binding to a discrete site.
Significance: Interplay of protein oligomerization and anion binding controls enzyme activity.
Keywords: Crystal Structure, Fluorescence Correlation Spectroscopy, Kinetics, Lipolysis, Membrane Enzymes, Phosphatidylinositol, Phospholipase C
Abstract
Staphylococcus aureus phosphatidylinositol-specific phospholipase C (PI-PLC) is a secreted virulence factor for this pathogenic bacterium. A novel crystal structure shows that this PI-PLC can form a dimer via helix B, a structural feature present in all secreted, bacterial PI-PLCs that is important for membrane binding. Despite the small size of this interface, it is critical for optimal enzyme activity. Kinetic evidence, increased enzyme specific activity with increasing enzyme concentration, supports a mechanism where the PI-PLC dimerization is enhanced in membranes containing phosphatidylcholine (PC). Mutagenesis of key residues confirm that the zwitterionic phospholipid acts not by specific binding to the protein, but rather by reducing anionic lipid interactions with a cationic pocket on the surface of the S. aureus enzyme that stabilizes monomeric protein. Despite its structural and sequence similarity to PI-PLCs from other Gram-positive pathogenic bacteria, S. aureus PI-PLC appears to have a unique mechanism where enzyme activity is modulated by competition between binding of soluble anions or anionic lipids to the cationic sensor and transient dimerization on the membrane.
Introduction
Secreted bacterial phosphatidylinositol-specific phospholipase C (PI-PLC)2 enzymes contribute to virulence in pathogenic bacteria. These enzymes catalyze the cleavage of PI or glycan phosphatidylinositol-anchored proteins to diacylglycerol and an inositol 1,2-(cyclic)-phosphate. Several of these enzymes are kinetically activated by the zwitterionic phospholipid phosphatidylcholine (PC) (1–3). Intriguingly, the activation mechanism varies among the PI-PLCs. Bacillus PI-PLC enzymes exhibit high affinity for PC interfaces and specifically bind at least one PC molecule quite tightly (4). The enzyme from Listeria monocytogenes is activated by many amphiphiles in a somewhat nonspecific fashion where the diluting lipid prevents protein-induced vesicle aggregation and the resulting sequestration of the enzyme. This nonspecific activation mechanism also likely reduces penetration of the highly cationic protein into a negatively charged bilayer (3). Consistent with this mode of PC activation, L. monocytogenes PI-PLC has very low affinity for PC small unilamellar vesicles (SUVs) but very high affinity for anionic phospholipid interfaces.
Staphylococcus aureus PI-PLC, a likely virulence factor whose production was shown to be up-regulated in a study of methicillin-resistance S. aureus (5), presents yet another wrinkle in the properties of these exoproteins. Its optimum activity is around pH 6.5, and unlike the other homologues it is strongly inhibited by moderate concentrations of salt (6, 7). A recent structure of the protein (7) showed two features not found in other PI-PLCs: (i) a π-cation latch, which appears important for water-soluble product release at low pH where the protein is tightly bound to vesicle surfaces, and (ii) a surface anion-binding pocket distinct from the active site that could explain the salt sensitivity. S. aureus PI-PLC activity is also enhanced by the presence of PC in assays systems (7).
We have solved a novel crystal structure of S. aureus PI-PLC, formed in the presence of dibutyroyl-PC, with significant inter-monomer contacts at the interface of the hydrophobic side chains of helix B and no anion in the anion binding pocket. This is strikingly different from previous, monomeric crystal structures (7). The relevance of this dimer structure to protein function and regulation has been tested by measuring enzymatic activity and lipid binding of recombinant enzyme and variants with selected mutations. The experimental findings suggest that S. aureus PI-PLC is optimally active as a dimer when, as the crystal structure suggests, the anion binding pocket is occluded. PC activates the enzyme by diluting anionic substrate (which has some affinity for the anion binding pocket as well as the active site), enhancing transient PI-PLC aromatic side chain π/choline cation interactions, and in turn allowing transient dimers to form. This complex mechanism, in which electrostatic interactions, spatially removed from the active site, compete with dimerization facilitated by zwitterionic PC, may be a broadly applicable mechanism allowing differential regulation of peripheral membrane enzymes by specific phospholipids.
EXPERIMENTAL PROCEDURES
Chemicals
l-α-Phosphatidylinositol and other phospholipids, including 1,2-dibutyroyl-sn-glycero-3-phosphocholine (diC4PC), PC, and 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (PG), were purchased from Avanti Polar Lipids, Inc., and used without further purification. All competent cells used in mutagenesis (XL1-Blue) and overexpression (BL21 Codonplus) were obtained from Stratagene. LB media and LB agar were obtained from Fisher Scientific, Inc. The fluorescent dye Alexa Fluor 488 carboxylic acid, succinimidyl ester was purchased from Invitrogen. Nickel-nitrilotriacetic acid-agarose resin was purchased from Qiagen and Q-Sepharose fast-flow anion-exchange resin was purchased from GE Healthcare. Bio-Spin 6 columns were obtained from Bio-Rad Laboratories. All other chemicals were purchased from Sigma.
PI-PLC Enzymes
Recombinant S. aureus PI-PLC was produced and purified as described previously (7). Mutations in the piplc gene were made using site-directed mutagenesis (Stratagene) and complementary primers purchased from Operon to generate the PI-PLC variants V44C, V44W, H86E, H86Y, F249W, Y253K, Y253S, Y253W, Y290A, and Y253S/Y255S. The sequence of each mutated gene was confirmed by Genewiz. Escherichia coli BL21-Codonplus (DE3)-RIL cells were transformed with the plasmid containing the S. aureus piplc gene. Overexpression and purification of the His-tagged recombinant protein followed protocols reported previously (7). The purity of the PI-PLC enzymes was above 95% as monitored by SDS-PAGE; concentrations were measured by the absorption at 280 nm using ϵ280 = 60,280 m−1 cm−1 calculated by ProtParam (8). Secondary structure and thermal stability of S. aureus PI-PLC variants were determined using far-UV CD as described previously for Bacillus thuringiensis PI-PLC (9). Compared with recombinant wild type S. aureus PI-PLC (WT), there are no significant changes in structure and thermal stability for the single mutation enzymes. Although Y253S/Y255S has the same secondary structure content, its thermal denaturation temperature is 5 °C lower than WT. Recombinant B. thuringiensis PI-PLC was generated as described previously (9).
Crystallography
Purified S. aureus PI-PLC was concentrated to 20 mg/ml before being combined with 100 mm myo-inositol and, in the case of WT only, 5 mm diC4PC. Each mixture was diluted to a final concentration of 10 mg/ml using deionized water. The protein was incubated on ice for a minimum of 2 h before setting up crystallization trays. Samples were crystallized at 20 °C by vapor diffusion, using hanging drops of 3 μl. Crystals of the recombinant PI-PLC were obtained in 150 mm ammonium acetate, 100 mm sodium acetate, pH 4.6, with 1 mm Mg(NO3)2, and 20% PEG 4000. Except for the presence of the monomeric diC4PC, these conditions differ from those used previously (7) only in the inclusion of Mg(NO3)2. Y253S crystals were obtained using similar conditions with an increase in the concentration of Mg(NO3)2 to 100 mm, and a slight decrease in PEG 4000 (to 16%). WT S. aureus PI-PLC crystals appeared as large plates and formed in the drop after approximately nine months, whereas the Y253S crystals appeared as plates after 2 weeks. Suitable crystals were mounted in nylon loops and frozen in liquid nitrogen.
Data were collected at 100 K using an in-house Rigaku MicroMax-07 HF high intensity microfocus rotating Cu anode x-ray generator, coupled with Osmic VariMax Optics and a R-Axis IV++ image plate area detector. Data were indexed and reduced using HKL2000 (10). Both the dimer, and the Y253S mutant structures were solved by molecular replacement using PHASER in CCP4 (11, 12) with S. aureus PI-PLC structure 3V16 (7) as a model. The models were refined using Refmac (12) with manual model building in COOT (13). Ligands and ligand restraints were generated using the PRODRG2 server (14). Structural comparisons were made using SSM superposition (15) in COOT and alignment in PyMOL (Schrodinger). ProCheck (16) was used for structure validation and Adit (17) was used for deposition.
Preparation of Phospholipid Aggregates
Enzyme kinetics experiments used PI/PC small unilamellar vesicles (SUVs) prepared by sonication, where XPC is the mole fraction of PC in these SUVs. Aliquots of phospholipids (PI, PG, and PC species) in chloroform were mixed, dried under N2, and then lyophilized overnight. The lipid film was rehydrated with the desired buffer. Small PI/PC vesicles were generated by sonicating lipid mixtures for 10 to 15 min on ice (Branson Sonifier 250). The average radii for PI and PI/PC (XPC = 0.5) vesicles were 130 Å, with 20 and 46% polydispersity, respectively. Similar sizes have also been documented for the PG/PC SUVs used in FCS experiments (18).
31P NMR Assays of PI-PLC Activity
Specific activities of the PI-PLC enzymes were measured by monitoring production of cIP from PI by 31P NMR spectroscopy (Varian VNMRS 600) (1, 2, 9). Most assays were in 50 mm MES, pH 6.5, with 1 mm EDTA and 0.1 mg/ml of bovine serum albumin at 28 °C. However, for assays examining the pH dependence of activity, a mixed buffer system of MES and HEPES (total concentration of 50 mm) with the required pH value was used. Specific activities in the presence of salt were determined using a NaCl (137 mm)/KCl (2.7 mm) mixture. S. aureus PI-PLC concentrations used in assays ranged from 0.2 to 8 μg/ml. The enzyme and SUVs were incubated for fixed times at 28 °C, and the reaction was quenched by addition of acetic acid followed by Triton X-100 to solubilize the remaining lipids in mixed micelles. The relative integrated intensity of the cIP resonance versus the total phospholipid concentration was used to calculate PI-PLC specific activity. For all reactions, the cleavage of PI was kept below 20% (no inositol 1-phosphate was generated under these conditions). The increase in cIP under these conditions is linear with time indicative of an initial rate being measured. Because we are dealing with a vesicle and some fusion of SUVs as diacylglycerol is produced, this is not the same as a steady-state rate with the substrate in solution, but it is operationally useful for comparing rates under different conditions (pH, concentration of enzyme, salt, etc.).
S. aureus PI-PLC Vesicle Binding Measured by Fluorescence Correlation Spectroscopy (FCS)
S. aureus PI-PLC proteins were labeled at pH 7.2 to maximize preferential labeling of the N terminus with Alexa Fluor 488 succinimidyl ester according to the manufacturer's protocol (Invitrogen). After removal of unbound dye using 3 spin columns, the absorption at 280 nm for protein plus dye and at 495 nm for the dye were used to estimate the number of dye molecules incorporated. Labeled S. aureus PI-PLC typically had 93–100% dye incorporation. Electrospray ionization mass spectral data of intact PI-PLC were obtained at the University of Massachusetts Amherst Mass Spectrometry Facility, which is supported in part by the National Science Foundation. Labeling percentages ranged from 80 to 99% with >70% of the protein containing a single Alexa Fluor 488 moiety and the remainder doubly labeled. At 99% labeling efficiency, 25–30% of the labeled protein has 2 labels, whereas this percentage is lower (20–25%) at 80% labeling. The activity of labeled PI-PLC is not significantly different from that of unlabeled PI-PLC.
FCS-based binding assays were performed on a home-built confocal setup described previously (18, 19). The fluorescence was monitored at 22 °C with samples placed in chambered coverglass wells (Lab-Tek, Nunc), containing 10 nm labeled PI-PLC and 1 mg/ml of bovine serum albumin in 300 μl of 50 mm MES buffer, pH 6.5 (the same buffer used in the enzymatic assays). The anionic phospholipid PG was used as the substrate analog. For vesicle binding experiments, Alexa Fluor 488-labeled PI-PLC variants were titrated with unlabeled PG/PC vesicles, where XPC was varied between 0 and 1. For each XPC, FCS titrations were run in duplicate and repeated a second time with different vesicle and protein preparations. Data analysis was based on previous work by Elson, Thompson and others (20–24) and similar to that described for B. thuringiensis PI-PLC (25). The fitted diffusion coefficient for free, Alexa Fluor 488-labeled S. aureus PI-PLC (Dfree) was 50 ± 2 μm2 s−1 and the vesicle diffusion coefficient, Dbound, determined from analysis of vesicles containing fluorescently labeled lipids, was in the range of 12–15 μm2 s−1 (18, 19). To account for the possibility that multiple proteins could bind to the same vesicle, autocorrelations (G(τ)) (obtained in cross-correlation mode using a 50–50 beamsplitter) for samples containing SUVs were fit to Equation 1 (22–24),
where p and v denote free protein and SUVs that are fluorescent due to PI-PLC binding, respectively, and Aj is the amplitude of species j. The correlation function for species j, gj(τ) is shown in Equations 2–4 (20–22),
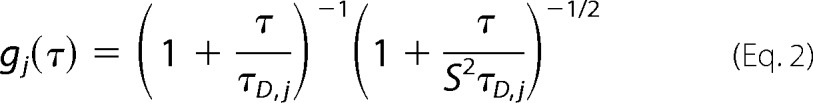 |
 |
 |
where the values of ωo, the radius of the observation volume in the x and y directions, and S, which depends on zo, the extent of the observation volume, were determined from fits to the rhodamine 110 calibration data using D = 280 mm2 s−1 at 22 °C (21)). τD,j is the diffusion time. The fraction of protein bound to the SUVs, f, is determined from Ap and the time-averaged number of proteins in the observation volume, 〈No〉, as described previously (18). The average brightness of the SUVs relative to the protein (22, 24), α = Av/(Apo − Ap), where Apo is the correlation amplitude in the absence of vesicles, had values around 1 for S. aureus PI-PLC, again indicating that for PI-PLC and these small PG/PC vesicles, on average one protein is bound per SUV during the diffusion time of ∼190 ms.
The apparent dissociation constant, Kd, representing PI-PLC partitioning to the vesicle surface, and a cooperativity coefficient, n, was determined using the empirical equation (18, 19),
where f, the fraction of protein bound to the vesicles, is determined for different total lipid concentrations, [PL], at fixed XPC, and fmax is the apparent maximum fraction bound. Although fits to Equation 1 assume that all of the SUVs have the same diffusion coefficient, the SUVs are polydisperse with a rather broad size (18). This assumption does not cause the apparent Kd values to significantly deviate from the actual values, but does result in values for fmax that are less than one, even for computer-generated data where fmax = 1.
RESULTS
S. aureus PI-PLC Dimer Structure
Previously reported crystal structures of S. aureus PI-PLC show a monomeric protein with significantly different, pH-dependent conformations of a rim loop near the active site (7) and the structure of the basic form is very similar to that of the Bacillus PI-PLCs (25, 26). However, in the presence of diC4PC and Mg(NO3)2 at pH 4.6, we serendipitously found that the protein crystallized as a complete homodimer in the asymmetric unit (Fig. 1). This protein crystallizes in a variety of unit cells with very different crystal packing (supplemental Fig. S1). However, this particular crystal structure is the only one to date where the dimer is not the result of crystal contacts. This new crystal form was in the P21 space group, with unit cell a = 43.36 Å, b = 133.38 Å, c = 50.19 Å, β = 89.95°. The dataset was twinned and was found to have the twinning operator of H, -K, -L with a fraction of 45.1%. Using twinned refinement in Refmac we have successfully improved the R values from the untwinned refinement (Rcryst = 0.261, Rfree = 0.322) to the final twinned refinement Rcryst = 0.215 and Rfree = 0.250. No distinct density consistent with diC4PC was observed. Crystallization as a dimer in the presence of diC4PC might be indicative of an additional control mechanism at the membrane surface.
FIGURE 1.

S. aureus PI-PLC can form a homodimer. A, S. aureus PI-PLC dimer structure, crystallized at pH 4.6 with the addition of 100 mm myo-inositol and 5 mm diC4PC, with B, a close-up of the dimer interface. In A, residues that coordinate an anion in monomer structures are shown to indicate their relationship to the dimer interface. C, close up view of the anion binding site with key residues labeled.
In the dimer, both monomers have their mobile loops in the compact, acidic-like conformation with Phe-249 forming an intramolecular π-cation interaction with His-258, which pulls the mobile loop in line with the barrel rim. Each monomer contains one molecule of myo-inositol in the active site, and the monomers are oriented in a head to head fashion with respect to each other (Fig. 1A). The dimer interface is formed by the direct interaction of the side chains belonging to helix B of each monomer (Fig. 1B). The area of contact is quite small (primarily consisting of residues 38–44 and adjacent regions, including residues 50, 250–254, 282, and 285, with a total buried surface area of 565.7 Å2, or 4.7% of the total surface area), and held together primarily by hydrophobic interactions, with only 6 hydrogen bonds between the two monomers. Each monomer only gains 3.9 kcal/mol upon complex formation (as evaluated with the PISA service), which would suggest a very transient dimer in solution (27). The main inter-monomer contact is the alignment of Val-41 from one monomer with Val-44 from the opposite monomer to form a hydrophobic box. When viewed as a dimer, an area of hydrophobicity can clearly be seen at the interface (Fig. 1, B and C), as well as along the barrel rim. Besides contributing to dimer formation, the valine box could also directly participate in membrane binding. Several other interactions aid the valine box in holding the monomers together. An ionic bond forms between Lys-38 and Asp-50 from the opposing monomer, and a hydrogen bond occurs between the backbone carbonyl of Thr-36 and the side chain hydroxyl of Tyr-285 from the opposing monomer. This helical valine box, along with several hydrophobic side chains, would orient the active site toward the membrane.
The anion binding pocket adjacent to helix B in the S. aureus PI-PLC monomer structures is made up of the backbone amides of Asp-38 and Lys-39, as well as the cationic side chains of His-86 and Lys-42 (7). This open surface pocket is shaped to snugly fit a tetrahedral anion such as phosphate or sulfate. Anionic phospholipid could also localize in the vicinity of this cationic site. However, in the dimer, helix B of one monomer occludes the pocket of the other (Fig. 2B), limiting water-soluble or membrane-bound anion binding. Thus, in the dimer, this unoccupied positively charged region could contribute to electrostatic steering to anionic phospholipids, but the binding pocket is no longer accessible.
FIGURE 2.

Anion binding destabilizes the dimer interface. A, overlay of monomers A and B from the WT dimer. The backbone structure between the two monomers is very similar. View of the anion binding pocket as seen looking down from the membrane interface in B, the homodimer, where it is unoccupied, and C, the in silico Y253S dimer, generated from the monomer with a sulfate ion bound in the anion site. In both B and C a dashed circle indicates the side chain of residue 253. With an anion bound in the pocket, the distance between the two monomers (indicated by the dashed line, measured from the α carbon of Val-41) widens, destabilizing the dimer interface.
Although the secondary structures of both Bacillus and Listeria proteins are quite similar to that of S. aureus PI-PLC, kinetic evidence suggests that those other proteins do not function as dimers. As will be shown in Fig. 4, the specific activity of B. thuringiensis PI-PLC does not increase with increasing protein concentration (and previous work showed that the enzyme was a monomer in solution (1)). The L. monocytogenes PI-PLC specific activity decreased with increasing protein concentration strongly suggesting it does not function as a dimer (3). Examining crystal structures for each of those would suggest that if they do form transient dimers on a membrane, it is not with the helix B orientation found in the crystal structure for that protein (supplemental Fig. S2). Significant conformational changes in this region would be needed.
FIGURE 4.

S. aureus PI-PLC relative specific activity (R.S.A.), as measured by production of cIP from PI using 31P NMR, increases with enzyme concentration suggesting oligomerization of this protein (but not B. thuringiensis PI-PLC) on interfaces. For S. aureus PI-PLC, specific activities are normalized to the highest specific activity obtained for WT enzyme acting on 4 mm PI SUVs in the absence of added salt (9.8 μmol min−1 mg−1 at pH 6.5). For B. thuringiensis PI-PLC activities are normalized to 164 μmol min−1 mg−1 at pH 7.5, the optimal activity toward 8 mm PI SUVs in the absence of salt. R.S.A. as a function of protein concentration is shown for: A, S. aureus PI-PLC toward PI (4 mm) SUVs in the absence (●) and presence (○) of 140 mm salt; B, B. thuringiensis PI-PLC toward 8 mm PI SUVs in the absence (■) and presence (□) of 140 mm salt; C, S. aureus PI-PLC activity toward PI (8 mm)/PC (8 mm) SUVs in the absence of salt; and D, V44C (Δ) and V44W (▴) compared with the WT S. aureus PI-PLC (●) toward 4 mm PI SUVs.
Although crystallographically elegant, it is unclear whether the S. aureus PI-PLC dimer structure is functionally relevant. If the dimer is the active form of PI-PLC, PI-PLC cleavage of PI in small unilamellar vesicles in the absence and presence of PC should display the following characteristics. (i) Specific activity should increase with increasing enzyme concentration in a saturable manner. (ii) If PC kinetic activation functions to enhance dimer formation, then the dependence of specific activity on enzyme concentration should shift to lower protein concentrations. (iii) If formation of the dimer and occupation of the nearby anion binding pocket are mutually exclusive, the enzyme should show reduced sensitivity to soluble anions under conditions that favor dimerization.
S. aureus PI-PLC Enzymatic Activity: Testing for a Functional Oligomer
The pH optimum for S. aureus PI-PLC enzymatic activity depends on the SUV composition (7). Toward pure PI SUVs, the pH optimum is pH 5.5–6, whereas for vesicles with 0.5 XPC, the optimum is shifted to pH 6.5–7 (Fig. 3A). Thus, most assays were carried out at pH 6.5. Specific activity increases dramatically when PC is present in the bilayer (Fig. 3B). Similar PC enhanced activity and a shift in the pH profile were observed with PI dispersed in micelles of diheptanoyl-PC versus Triton X-100 (Fig. 3C). Unlike B. thuringiensis PI-PLC, where specific activity decreases when XPC > 0.5 (18), no surface dilution inhibition is observed for S. aureus PI-PLC with increasing PC content up to 0.8 XPC, either for fixed total phospholipid (as shown in Fig. 3B) or fixed PI with increasing amounts of PC. Because the dimer structure with its occluded anion binding site was obtained in the presence of PC, S. aureus PI-PLC salt sensitivity might be ameliorated by PC. Indeed, the presence of PC rescues the loss of activity observed at moderate salt concentrations (Fig. 3B), and higher XPC results in lower salt sensitivity. This is consistent with a transient dimer that has lost the ability to bind soluble anions in the anion binding pocket. That this is an interfacial phenomenon is show by cIP kinetics (Fig. 3D), which exhibit both only a small activation by diheptanoyl-PC micelles and only about a 2-fold drop in specific activity with added salt.
FIGURE 3.

PC modulates the pH and salt dependence of S. aureus PI-PLC activity as measured by production of cIP from PI using 31P NMR. A, the pH dependence for S. aureus PI-PLC cleavage of PI in pure PI SUVs (4 mm) (○) and PI/PC (4 mm/4 mm) (●) SUVs. B, specific activity at pH 6.5 increases with increasing XPC and is not as sensitive to the addition of 140 mm salt. Filled bars, no salt; hatched bars, plus 140 mm salt. The total phospholipid concentration was held constant at 4 mm for the indicated XPC. The enzyme concentration was 4 μg/ml for pure PI SUVs and 0.3 μg/ml for PI/PC SUVs. C, specific activity for cleavage of 4 mm PI in 8 mm Triton X-100 (○) or 16 mm diheptanoyl-PC (●) micelles. D, specific activity toward 5 mm cIP alone or with 5 mm diheptanoyl-PC and in the absence (grey column) or presence of 140 mm salt (▨). Error bars in each plot represent S.D. from multiple assays.
A more direct indication of a functional dimer is the observation that, in the absence of salt, S. aureus PI-PLC specific activity is dependent on enzyme concentration, particularly when the substrate is pure PI SUVs (Fig. 4A). For comparison, the specific activity of B. thuringiensis PI-PLC, which is unlikely to form the same dimer through helix B, does not depend on enzyme concentration and is only weakly salt dependent (Fig. 4B). If PC is present in the bilayer, there is still an increase in S. aureus PI-PLC-specific activity with increasing enzyme concentration, but this reaches an optimal value at a much lower enzyme concentration (Fig. 4C). This dependence of PI-PLC-specific activity on protein concentration strongly suggests that the optimally active form of the enzyme is a dimer. The presence of PC in the membrane must shift the equilibrium between protein monomer and oligomer to lower protein concentrations.
As a further test of this hypothesis we mutated Val-44, one of the key components of the dimer interface in the crystal structure. This valine was replaced with: (i) a cysteine, a small amino acid capable of hydrogen bonding that is likely to destabilize the dimer interface (a large excess of DTT was present to prevent any disulfide formation) or (ii) a tryptophan, a large side chain with a propensity to partition into membranes that might be expected to strengthen the transient dimer interaction. For PI SUVs, the specific activity of V44C was lower than that of the WT enzyme and increased at most 2-fold (compared with 5-fold for WT) over the protein concentration range examined (Fig. 4D). In contrast, the specific activity of V44W was higher than wild type and appeared to reach a maximum value at lower enzyme concentrations. These data clearly indicate that S. aureus PI-PLC functions optimally as a transient dimer on the SUVs. They strongly suggest that enhanced protein dimerization on the surface is part of the PC activation mechanism for S. aureus PI-PLC.
The Effect of PC on S. aureus PI-PLC Vesicle Binding
For B. thuringiensis PI-PLC, increasing XPC up to 0.8 also increases the affinity of the enzyme for SUVs, and tight binding (Kd < 10 μm) sequesters the enzyme on the vesicles inhibiting enzyme activity (19). S. aureus PI-PLC behaves quite differently. As shown in Fig. 5A, S. aureus PI-PLC binds well, with mm Kd values, to either PG or PG/PC (1:1) SUVs at acidic pH, but binding at pH 7.5 is much weaker, indicating that the primary interaction is electrostatic. At pH 6.5, where optimal activity is observed, the enzyme has high affinity for anionic phospholipid-rich bilayers (XPC ≤ 0.5), with fairly low affinity for PC-rich bilayers (Fig. 5B). Indeed, binding to pure PC SUVs could not be measured. The apparent Kd for pure PG vesicles is 0.38 ± 0.14 mm at pH 6.5, and this value corresponds well with the apparent Km value (0.23 ± 0.03 mm) for cleavage of PI in pure PI SUVs (Fig. 5C). For XPC = 0.5, again the apparent Kd determined by FCS (2.5 ± 0.5 mm) is approximately the same as the kinetic apparent Km for PI/PC SUVs (1.9 ± 0.5 mm). S. aureus PI-PLC apparent Kd values increase with PC content and the enzyme binds much less tightly at high PC. For these compositions, and in the absence of salt, the Km reflects the Kd for vesicle binding as measured by FCS.
FIGURE 5.
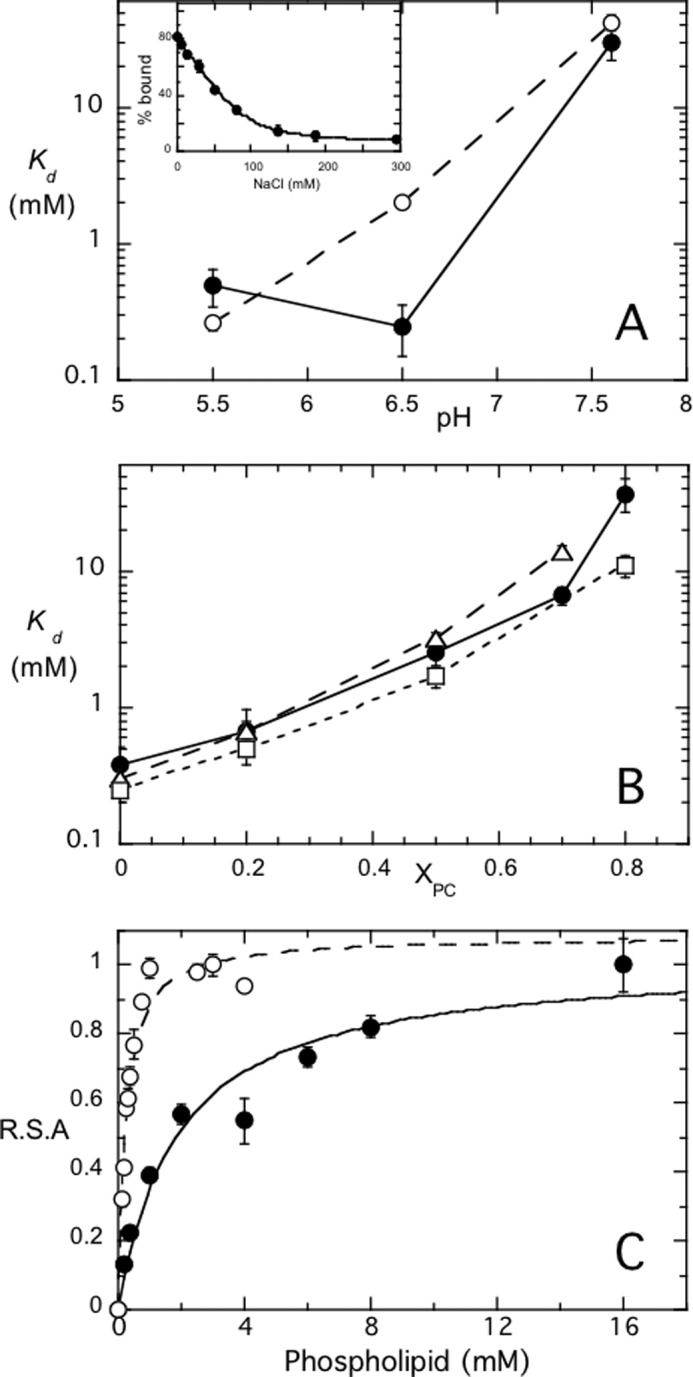
S. aureus PI-PLC binding affinity, measured by FCS, increases with pH and PC content and is similar to the apparent Km for the enzyme. Variation of the PI-PLC apparent dissociation constant, Kd, for SUVs as a function of: A, pH for PG (●) and PG/PC (XPC = 0.5) (○), and B, mole fraction PC, XPC, at pH 6.5, for WT (●), F249W (□), and Y253S/Y255S (Δ). The inset in A shows the fraction of WT protein bound to pure PG (18 mm) SUVs at pH 6.5 as the concentration of salt is increased. C, relative specific activity of S. aureus PI-PLC as a function of total phospholipid concentration ([PI] + [PC]) at XPC = 0 (○) and 0.5 (●) at pH 6.5. The amount of enzyme used was 0.1 μg/ml for pure PI SUVs and 0.2 μg/ml for experiments with PI/PC SUVs. For each system the observed activity was normalized to the maximum observed activity, 11.2 μmol min−1 mg−1 for pure PI SUVs and 282 μmol min−1 mg−1 for the PI/PC SUVs. The error bars are the S.D. from independent experiments.
Added salt reduces the affinity for PG-rich SUVs to such an extent that the apparent Kd for pure PG SUVs could not be measured. To explore this further we examined the fraction of protein bound to a large excess of vesicles (18 mm PG) in the absence and presence of NaCl (Fig. 5A, inset). Without salt, all of the protein partitioned onto the vesicles, whereas increasing the ionic strength dramatically reduced protein binding. With ∼130 mm NaCl, only ∼20% of the protein was bound. Given the high bulk phospholipid concentration (18 mm), in the presence of salt the apparent Kd for PG vesicles must be very high (>50 mm). This result suggests that S. aureus PI-PLC binding to pure PG vesicles is almost entirely mediated by electrostatic interactions. However, with PC in the vesicle, the much higher residual activity indicates that binding of the enzyme to the bilayer now has a more substantial hydrophobic component.
Perturbing Hydrophobic Interactions with the Membrane
In the Bacillus enzymes, a number of aromatic residues are critical for tight binding to PC-rich membranes, including Trp-242, which partitions into the bilayer (9, 25), and a strip of four surface Tyr residues in helix G (26). In S. aureus PI-PLC, Trp-242 is replaced by Phe-249 and this enzyme has only two of the four tyrsoine residues (Tyr-253 and Tyr-255) in helix G. It is also unclear if Phe-249 interacts with the membrane because it is tucked into the protein and away from the surface in the monomeric acidic S. aureus PI-PLC crystal structure (7) and in the dimer structure described here. To test whether these residues interact with the membrane in S. aureus PI-PLC, we constructed the following variants: F249W and F249I, Y253S, and Y253S/Y255S. Tyr-290, a single Tyr residue that is unlikely to interact with the membrane, was also mutated to serine as a control. As shown in Fig. 6, the activity of the control, Y290S, is similar to WT. However, both Y253S and the double Tyr mutant enzyme are much less active under all conditions. Increasing PC content has a very modest effect on PI cleavage by Y253S, whereas Y253S/Y255S is virtually insensitive to the presence of PC. In contrast, mutating Phe-249 to a tryptophan significantly enhanced enzyme activity for XPC ≤ 0.5, whereas the activity of F249I is equivalent to that of the WT. The pH dependence was varied for several of these variants, and whereas F249I and Y290S still behave essentially like WT, Y253S exhibits much lower activity regardless of pH (data not shown). The loss of activity and PC activation for Y253S and Y253S/Y255S suggest that these residues play a role in the conformational change(s) that activate the protein in the presence of PC.
FIGURE 6.
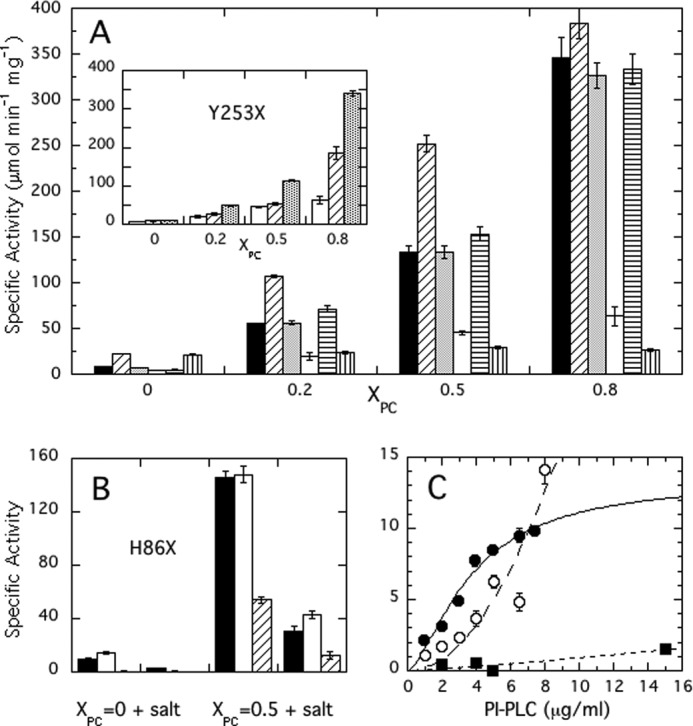
Specific activities of S. aureus PI-PLC variants as a function of XPC at pH 6.5.
A, WT (■), F249W (▨), F249I (grey column), Y253S (□), Y290S (▤), and Y253S/Y255S ( ). The inset shows the activity of Y253 variants: Y253S (□), Y253K (▨), and Y253W (▥). B, specific activities for H86 variants toward pure PI SUVs and PI/PC (1:1) SUVs in the absence and presence of added salt: H86Y (□) and H86E (▨) are compared with WT (■). The kinetic experiments in A and B kept the PI concentration at 4 mm and increased the PC to yield the indicated XPC. C, dependence of PI-PLC specific activity on protein concentration: WT (●), H86Y (○), and H86E (■). Error bars are the S.D. of activities measured in triplicate.
). The inset shows the activity of Y253 variants: Y253S (□), Y253K (▨), and Y253W (▥). B, specific activities for H86 variants toward pure PI SUVs and PI/PC (1:1) SUVs in the absence and presence of added salt: H86Y (□) and H86E (▨) are compared with WT (■). The kinetic experiments in A and B kept the PI concentration at 4 mm and increased the PC to yield the indicated XPC. C, dependence of PI-PLC specific activity on protein concentration: WT (●), H86Y (○), and H86E (■). Error bars are the S.D. of activities measured in triplicate.
Because both F249W and Y253S/Y255S have activities that are significantly different from WT, protein binding to PG/PC SUVs was measured by FCS (Fig. 5B). The Kd values for F249W are comparable with WT at XPC = 0 and 0.2 but significantly smaller at 0.8 XPC, indicative of enhanced interactions with the membrane as might be expected when the larger tryptophan side chain replaces a phenyalanine that inserts into the membrane. However, this higher affinity for PC-rich SUVs does not translate into significantly higher activity. For comparison, Y253S/Y255S has similar affinities to WT up to 0.7 XPC, but much lower activity. For higher PC content, the fraction of mutant protein bound was small and linear up to 50 mm phospholipid, and the apparent Kd could not be estimated. These surface tyrosine residues are important for PC enhancement of activity; however, except at 0.8 XPC, the loss of activity is not a result of reduced binding. Thus, unlike Bacillus PI-PLC where Kd values for SUVs vary by orders of magnitude (from micromolar to >100 mm), as a function of XPC and/or enzyme mutations, the range of Kd for S. aureus PI-PLC is much more limited and less easily perturbed.
Occupation of the Anion Binding Pocket versus Dimerization
Compared with WT, Y253S has little activation by PC but similar membrane affinity. To help explain this behavior, a 2.3-Å structure of Y253S was obtained at pH 4.6. This variant crystallized as a monomer in the P212121 space group, with a unit cell of a = 86.1 Å, b = 55.96Å, and c = 61.68 Å. The Y253S structure was refined down to a final Rcryst value of 0.187 (Rfree of 0.241). Statistics for the structure are given in Table 1. In the homodimer structure, the α-carbon of residue 253 is 14.7 Å away from the active site, and, whereas not a direct part of the anion binding pocket, Tyr-253 aids in occluding the anion-binding site. When compared with the S. aureus monomeric PI-PLC structures, all catalytic residues in Y253S appear unchanged. The structure is essentially identical to the WT monomeric structure at the same pH except around the mutation site. This mutation also has no effect on the acid form of the mobile rim loop. The monomer structure for Y253S, therefore, offers little explanation for the loss of PC activation in Y253S. However, when the native homodimer structure is used as a model to create an in silico Y253S homodimer, there is a significant perturbation in the region of the anion binding pocket, which is empty in the homodimer. Tyr-253 aids in formation of a lid for the pocket in the opposing monomer of the homodimer structure (Fig. 2B). Replacement of the Tyr side chain with a serine leaves an opening in the adjacent monomer, thus increasing anion access to the anion binding pocket in any Y253S homodimer (Fig. 2C). This increased accessibility could facilitate soluble anion or interfacial anionic phospholipid binding to the site. Binding of ligand to the anion binding pocket in turn would destabilize the dimer interface by shifting helix B slightly away from the surface and lengthening distances between side chains that make up the tenuous dimer interface. This is visualized in Fig. 7 by comparing two monomer structures with bound chloride overlaid on the dimer. There are slight changes in the opposing helix B moieties. Helix B and the section of the adjacent loop move slightly to bind the ion. This slight shift would pull Val-41 back from the dimer interface. This separation of the valines would destabilize the already small dimer interface.
TABLE 1.
Crystallographic summary for the S. aureus PI-PLC dimer and for monomeric Y253S
| Protein | WT dimer | Y253S |
|---|---|---|
| PDB ID | 4F2B | 4F2T |
| Diffraction data | ||
| Resolution range (Å) | 66.7–2.16 | 29.89–2.30 |
| Reflections | 27,485 | 13,541 |
| Reflections in free set | 1,534 | 678 |
| Space group | P21 | P212121 |
| Unit cell | ||
| a (Å) | 43.36 | 86.1 |
| b (Å) | 133.38 | 55.96 |
| c (Å) | 50.19 | 61.68 |
| β (°) | 89.95 | 90 |
| Monomers in the A.U. | 2 | 1 |
| Completeness | 95.4 | 98.4 |
| Rmerge | 0.063 | 0.063 |
| Refinement | ||
| Rcrysta | 0.215 | 0.187 |
| Rfreeb | 0.250 | 0.241 |
| Residues | 606 | 302 |
| Non-H protein atoms | 4844 | 2420 |
| Non-H inositol atoms | 24 | 0 |
| H2O molecules | 123 | 132 |
| Ions | 0 | 3 |
| Root mean square deviation bonds (Å) | 0.024 | 0.008 |
| Root mean square deviation angles (Å) | 2.1 | 1.2 |
| Ramachandran plot (%) | ||
| Most favored | 85.8 | 90 |
| Additionally allowed | 14 | 9.6 |
| Generously allowed | 0.2 | 0.4 |
| Disallowed | 0 | 0 |
| Average B-factor (Å2) | 42.46 | 44.64 |
a Rcryst = {Σ (‖Fo| − |Fc‖)/|Fo|}, where |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively.
b Ref. 30.
FIGURE 7.

View of the helix B dimer interface as seen looking down from the membrane interface (A), and from the side of the protein viewing the anion binding pocket (B) (the membrane surface is at the top of the two helix B moieties). In both structures, the homodimer is seen in red and blue, whereas two green acidic monomer structures are overlaid on the dimer subunits. The homodimer has no salt bound in the anionic binding pocket, whereas the acidic monomers contain a chloride ion (green sphere).
To further explore the role of the anion binding pocket and activity, two other mutant enzymes were generated: Y253W, where the “lid” for the pocket is now considerably larger, likely preventing anion binding and favoring dimerization, and Y253K, where the positive charge might increase and enhance anion binding electrostatically, although this could be balanced by the methylene chain occluding the pocket. As seen in the inset in Fig. 6A, Y253W has specific activity much higher than Y253S and comparable with WT. Y253K has lower activity than Y253W at all XPC examined. However, it is not as compromised as Y253S, suggesting that the bulk of the lysine alkyl chain may be blocking access to the anion site.
As a further test of how modulating the anion binding pocket can affect activity we also prepared H86Y and H86E. The His-86 imidazole forms part of the anion binding pocket. H86Y will remove the side chain positive charge but will keep the lid in place in the dimer. H86E should antagonize anion binding because we are replacing a positive charge with a negative one. As shown in Fig. 6B, H86Y exhibits higher activity toward pure PI SUVs (14.1 ± 1.0 versus 9.8 ± 0.3 μmol min−1 mg−1 for WT) and activity comparable with WT with PI/PC SUVs. In contrast, H86E has very low activity toward pure PI SUVs (electrostatic repulsion would impede strong binding to SUVs), but is dramatically enhanced with PC present (a 135-fold increase compared with the more typical 10–15-fold for H86Y and WT). Consistent with the low activity toward PI SUVs, H86E showed only a small increase in activity with enzyme concentration compared with the other proteins, reflecting its difficulty in binding to the target membrane. With XPC = 0.5 SUVs, the relative loss of activity for H86E in the presence of salt (29% residual activity) was also less than WT (21% residual activity). This behavior also suggests that the anion binding pocket is intertwined with dimer formation.
These results suggests a very intriguing proposal: S. aureus PI-PLC is optimally active as a dimer, but an appropriate phospholipid interface and/or high protein concentration is needed to at least transiently stabilize this structure. Soluble anions compete for the anion binding pocket, and prevent dimer formation as well as weakening interfacial binding. An anionic phospholipid might interact with this site, but in doing so, it would also destabilize the dimer, leading to lower activity while aiding in binding the protein to the bilayer. The observation that the double mutant Y253S/Y255S behaves similarly to Y253S supports the importance of this single aromatic group in indirectly promoting interfacial dimerization.
DISCUSSION
S. aureus PI-PLC Activity, Dimer Formation, and the Membrane PC Content
The kinetic and structural data strongly suggest that S. aureus PI-PLC is optimally active as a dimer, and that this dimer is transiently stabilized by the membrane surface. The small dimer interface in the crystal structure suggests that dimers are likely to be transient, making them difficult to observe. S. aureus PI-PLC might aggregate to a small extent in solution, but obtaining accurate sizes for bacterial PI-PLCs in solution is difficult because they have an affinity for carbohydrates. At the protein concentrations required for gel filtration or analytical ultracentrifugation, these PI-PLCs interact with the resin or sucrose leading to aberrant sizes (28). Furthermore, increased ionic strength, which suppresses interaction of the B. thuringiensis enzyme with resins (1), would favor monomeric S. aureus PI-PLC. However, the simple membrane-induced dimer model does not explain why S. aureus PI-PLC activity increases with increasing PC content despite its lower affinity for PC-rich membranes. The presence of PC in the vesicle also rescues activity lost by the enzyme in moderate concentrations of salt (Fig. 4), consistent with a switch from electrostatic to hydrophobic interactions.
A better understanding of S. aureus PI-PLC might be gained by comparing it to the well studied PI-PLC from B. thuringiensis, which has a specific binding site for PC near helices F and G (4). For B. thuringiensis PI-PLC, apparent Kd values range from ∼4 mm for anionic SUVs to ∼2 μm at XPC = 0.75 with apparent Kd values of 200–30 μm from 0.2 to 0.5 XPC where enzyme activity is optimal (18). S. aureus PI-PLC binds more weakly to PC containing SUVs with apparent Kd values of 0.3 to ∼50 mm for 0 to 0.8 XPC, likely due to its lack of a PC specific binding site near the F and G helices (4, 7).
Why then does S. aureus PI-PLC activity increase with PC content when the affinity for vesicles is decreasing? Incorporation of PC may dilute the surface concentration of the anionic phospholipids in vesicles, and that in turn could enhance S. aureus PI-PLC dimer formation. This mechanism is consistent with the observation that much lower enzyme concentrations are required to increase PI-PLC specific activity in the presence of PC (Fig. 4C) even though the apparent Km increases as PC is increased. Alternatively, there are a number of aromatic residues in loops and N-terminal portions of helices around the barrel rim that could form transient π-cation interactions with the choline headgroup (7). This could alter the conformation of loops or side chains and facilitate transient protein dimerization. It should also be noted that the π-cation latch is still observed in the dimer structure (obtained at pH 4.6), and its presence is likely to aid in soluble cIP release, at least at acidic pH. However, the altered pH profile in the presence of PC might suggest that the zwitterionic phospholipid leads to alterations of pKa values for the many histidine residues in and around the active site. This could be the result of a conformational change or change in the surface charge of the membrane. Additionally, interactions with PC and transient dimerization could better orient S. aureus PI-PLC relative to the membrane surface, facilitating enzyme activity by increasing substrate access.
The Anion Binding Pocket, a Key to Inhibiting Dimer Formation
One of the unusual features of S. aureus PI-PLC is the very electropositive anion binding pocket just below the rim of the barrel, adjacent to helix B. Neither the Bacillus enzymes nor L. monocytogenes PI-PLC have such a pocket. In S. aureus PI-PLC the anion pocket is composed of the backbone amides of Lys-38 and Asp-39, as well as the cationic side chains of Lys-42 and His-86. In all solved S. aureus PI-PLC structures (7),3 except the dimer, there is electron density for an anion in this cationic pocket. Anion binding leads to a shift in the pocket residues, as well as nearby helix B. Lys-38 and Asp-39 move ∼1 Å closer to the anion, as do His-86 and Lys-42 (Fig. 7). These shifts pull the entirety of helix B (Val-41 to Ala-46) down, closer to the anion binding pocket, and thus away from the dimer interface by ∼1.3 Å. The shift in helix B residues upon salt binding to the anion binding pocket would, in turn, pull Val-41 and Val-44 farther away from the valines on the opposing monomer increasing the distance of the valine pairs from 4.5 Å in the dimer to 6.7 Å. This would weaken the hydrophobic interactions between the valine residues, destabilizing the dimer interface. The dimer interface itself is quite small consisting of only 14 residues. With an anion bound in the pocket, 10 of these residues show shifts of up to 1.5 Å, small movements, but enough to weaken the dimer interactions. Thus anion binding, probable at high salt concentrations, and dimer formation appear mutually exclusive.
Conclusions
We hypothesize that competition between anion binding and dimer formation is the key regulator of S. aureus PI-PLC activity as summarized in Fig. 8. Soluble anion binding weakens protein affinity for PI-rich vesicles, inhibiting S. aureus PI-PLC even at moderate salt concentrations. An anionic phospholipid might also bind to the pocket electrostatically, and thus aid in anchoring the protein to the surface. However, the presence of an anionic phospholipid such as PI near this site would favor the monomer and lead to low specific activity. High protein concentrations allow dimerization to compete with anionic lipid binding to the pocket leading to some dimers on the membrane, but activities are still low. PC then activates S. aureus PI-PLC, not by specific interactions, but by lowering the interfacial anionic lipid concentration and emptying the anion binding pocket, facilitating dimer formation. Occlusion of the pocket by the dimer interface makes the membrane-bound dimer much less sensitive to physiological salt concentrations. However, there is a trade-off: dimers have higher activity, likely because of a conformational change that enhances catalysis (7) and/or because they optimize the protein orientation on the membrane for catalysis, but they bind more weakly than monomers. On PC-rich bilayers they have lost the electrostatic interactions of the regulatory site.
FIGURE 8.

A model for S. aureus PI-PLC-membrane interactions. Monomeric PI-PLC partitions from solution to the membrane via electrostatic interactions, and a molecule of PI diffuses to the active site (step 1). The enzyme then either binds a soluble anion, and falls off the membrane, or may bind an auxiliary anionic lipid in/near the anion binding pocket (step 2). PI-PLC dimerization is promoted by PC, which dilutes the interfacial concentration of the auxiliary anionic lipid, and releases it from the enzyme (step 3). Finally, in step 4, the dimer dissociates into monomers or dissociates from the membrane. PI-PLC is represented in a semitransparent space filling view, with the active site highlighted in red, and the anion binding pocket highlighted in cyan. PI lipids are modeled as blue spheres, the membrane is shown as a blue gradient, and a water-soluble anion is shown as a purple sphere.
The association of an auxiliary anion binding site with S. aureus PI-PLC but not with homologues from other Gram-positive bacterial species would argue that it is related to the biology of S. aureus. Because abscesses are acidic (29), the slightly more acidic optimum of S. aureus PI-PLC enzymatic activity would make it more effective. In a study of methicillin-resistant S. aureus, expression of the PI-PLC, as well as other exoproteins and cytoplasmic proteins, was significantly increased (5). Although this protein has a moderate affinity for anionic surfaces in the absence of salt, once the enzyme is secreted into the moderate ionic strength of the extracellular milieu, it will not interact with other S. aureus organisms. It is then poised to interact with the PC or sphingomyelin-containing membranes of eukaryotic target cells via the valine box of the dimer. The still weak interaction with pure PC surfaces may allow it to hone in on negatively charged regions (the cationic character of the anion binding pocket is still available), which include GPI-anchors, the targets of this exoprotein. Clearly, this small soluble phospholipase has evolved in a complex, apparently unique way to control its access to the phosphatidylinositol moieties that are its substrates. (i) A π-cation latch allows it to work on membranes under acidic conditions and more easily release soluble product without dissociating from the surface (7). (ii) An anion binding pocket near the region that binds to the membrane likely modulates the location of the protein, whether it is bound to a negatively charged surface, one with significant zwitterionic lipid content, or in solution. (iii) High activity requires dimerization of the enzyme and dimerization is enhanced by PC content similar to that found in the external leaflet of mammalian plasma membranes. This interplay between protein dimerization and an anion sensing site could be a way of regulating other peripheral (or even integral) membrane enzymes. Juxtamembrane cationic regions, whereas favoring anionic membrane regions, could prohibit protein/protein interactions. However, an appropriately modified bilayer composition might change this balance.
Supplementary Material
Acknowledgment
We thank Dr. Rinat Abzalimov of the University of Massachusetts Amherst Mass Spectrometry Facility for analysis of the mass spectra.
This work was supported, by National Institutes of Health Grant R01 GM60418 (to M. F. R.).

This article contains supplemental Figs. S1 and S2.
R. Goldstein, unpublished data.
- PI-PLC
- phosphatidylinositol-specific phospholipase C
- cIP
- inositol 1,2-(cyclic)phosphate
- dIC4PC
- 1,2-dibutyroyl-sn-glycero-3-phosphocholine
- FCS
- fluorescence correlation spectroscopy
- PC
- 1-palmitoyl-2-oleoylphosphatidylcholine
- PG
- 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- PI
- l-α-phosphatidylinositol
- SUV
- small unilamellar vesicle
- WT
- wild type recombinant S. aureus PI-PLC
- XPC
- mole fraction PC.
REFERENCES
- 1. Zhou C., Qian X., Roberts M. F. (1997) Allosteric activation of phosphatidylinositol specific phospholipase C. Specific phospholipid binding anchors the enzyme to the interface. Biochemistry 36, 10089–10097 [DOI] [PubMed] [Google Scholar]
- 2. Qian X., Zhou C., Roberts M. F. (1998) Phosphatidylcholine activation of bacterial phosphatidylinositiol phospholipase C toward PI vesicles. Biochemistry 37, 6513–6522 [DOI] [PubMed] [Google Scholar]
- 3. Chen W., Goldfine H., Ananthanarayanan B., Cho W., Roberts M. F. (2009) Listeria monocytogenes phosphatidylinositol-specific phospholipase C. Kinetic activation and homing in on different interfaces. Biochemistry 48, 3578–3592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pu M., Orr A., Redfield A. G., Roberts M. F. (2010) Defining specific lipid binding sites for a peripheral membrane protein in situ using subtesla field-cycling NMR. J. Biol. Chem. 285, 26916–26922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burlak C., Hammer C. H., Robinson M. A., Whitney A. R., McGavin M. J., Kreiswirth B. N., Deleo F. R. (2007) Global analysis of community-associated methicillin-resistant Staphylococcus aureus exoproteins reveals molecules produced in vitro and during infection. Cell Microbiol. 9, 1172–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Daugherty S., Low M. G. (1993) Cloning, expression and mutagenesis of phosphatidylinositol-specific phospholipase C from Staphylococcus aureus. A potential staphylococcal virulence factor. Infect. Immun. 61, 5078–5089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goldstein R., Cheng J., Stec B., Roberts M. F. (2012) Structure of the S. aureus PI-specific phospholipase C reveals modulation of active site access by a titratable π-cation latched loop. Biochemistry 51, 2579–2587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., Bairoch A. (2005) in The Proteomics Protocols Handbook (Walker J. M., ed) pp. 571–607, Humana Press, New York [Google Scholar]
- 9. Feng J., Wehbi H., Roberts M. F. (2002) Role of tryptophan residues in interfacial binding of phosphatidylinositol-specific phospholipase C. J. Biol. Chem. 277, 19867–19875 [DOI] [PubMed] [Google Scholar]
- 10. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 11. Collaborative Computational Project, Number 4 (1994) The CCP4 Suite. Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 12. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 14. Schüttelkopf A. W., van Aalten D. M. (2004) PRODRG. A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
- 15. Krissinel E., Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 16. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK. A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 17. Yang H., Guranovic V., Dutta S., Feng Z., Berman H. M., Westbrook J. D. (2004) Automated and accurate deposition of structures solved by x-ray diffraction to the Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 60, 1833–1839 [DOI] [PubMed] [Google Scholar]
- 18. Pu M., Roberts M. F., Gershenson A. (2009) Fluorescence correlation spectroscopy of phosphatidylinositol-specific phospholipase C monitors the interplay of substrate and activator binding sites. Biochemistry 48, 6835–6845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pu M., Fang X., Redfield A. G., Gershenson A., Roberts M. F. (2009) Correlation of vesicle binding and phospholipid dynamics with phospholipase C activity. Insights into phosphatidylcholine activation and surface dilution inhibition. J. Biol. Chem. 284, 16099–16107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elson E. L., Magde D. (1974) Fluorescence correlation spectroscopy. I. Conceptual basis and theory. Biopolymers 13, 1–27 [DOI] [PubMed] [Google Scholar]
- 21. Magde D., Elson E. L., Webb W. W. (1974) Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers 13, 29–61 [DOI] [PubMed] [Google Scholar]
- 22. Thompson N. L. (1991) in Topics in Fluorescence Microscopy (Lakowicz J., ed) pp. 337–378, Plenum Press, New York [Google Scholar]
- 23. Rusu L., Gambhir A., McLaughlin S., Rädler J. (2004) Fluorescence correlation spectroscopy studies of peptide and protein binding to phospholipid vesicles. Biophys. J. 87, 1044–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Middleton E. R., Rhoades E. (2010) Effects of curvature and composition on α-synuclein binding to lipid vesicles. Biophys. J. 99, 2279–2288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heinz D. W., Ryan M., Bullock T. L., Griffith O. H. (1995) Crystal structure of the phosphatidylinositol-specific phospholipase C from Bacillus cereus in complex with myo-inositol. EMBO J. 14, 3855–3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shi X., Shao C., Zhang X., Zambonelli C., Redfield A. G., Head J. F., Seaton B. A., Roberts M. F. (2009) Modulation of Bacillus thuringiensis phosphatidylinositol-specific phospholipase C activity by mutations in the putative dimerization interface. J. Biol. Chem. 284, 15607–15618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 28. Berg O. G., Yu B. Z., Apitz-Castro R. J., Jain M. K. (2004) Phosphatidylinositol-specific phospholipase C forms different complexes with monodisperse and micellar phosphatidylcholine. Biochemistry 43, 2080–2090 [DOI] [PubMed] [Google Scholar]
- 29. Dye E. S., Kapral F. A. (1980) Characterization of a bactericidal system in staphlococcal abscesses. Infect. Immun. 30, 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brunger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography and NMR System. A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905-921 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.