

Background: IL-18 induces profibrotic changes in TECs independent of TGF-β1 activity.
Results: IL-18 stimulates the TLR4 promoter via AP-1 activation to increase TLR4 expression in TECs and stimulates profibrotic changes in TECs through increased TLR4 expression/signaling.
Conclusion: The profibrotic effect of IL-18 in TECs is mediated through stimulation of TLR4 expression via activation of AP-1.
Significance: This represents a novel fibrotic signaling pathway in TECs independent of TGF-β1.
Keywords: Cytokine, EMT, Fibrosis, Interleukin, Toll-like Receptors (TLR), AP-1, IL-18, TLR4, Tubular Epithelial Cell
Abstract
IL-18 is an important mediator of obstruction-induced renal fibrosis and tubular epithelial cell injury independent of TGF-β1 activity. We sought to determine whether the profibrotic effect of IL-18 is mediated through Toll-like receptor 4 (TLR4). Male C57BL6 wild type and mice transgenic for human IL-18-binding protein were subjected to left unilateral ureteral obstruction versus sham operation. The kidneys were harvested 1 week postoperatively and analyzed for IL-18 production and TLR4 expression. In a separate arm, renal tubular epithelial cells (HK-2) were directly stimulated with IL-18 in the presence or absence of a TLR4 agonist, TLR4 antagonist, or TLR4 siRNA knockdown. Cell lysates were analyzed for TLR4, α-smooth muscle actin, and E-cadherin expression. TLR4 promotor activity, as well as AP-1 activation and the effect of AP-1 knockdown on TLR4 expression, was evaluated in HK-2 cells in response to IL-18 stimulation. The results demonstrate that IL-18 induces TLR4 expression during unilateral ureteral obstruction and induces TLR4 expression in HK-2 cells via AP-1 activation. Inhibition of TLR4 or knockdown of TLR4 gene expression in turn prevents IL-18-induced profibrotic changes in HK-2 cells. These results suggest that IL-18 induces profibrotic changes in tubular epithelial cells via increased TLR4 expression/signaling.
Introduction
Chronic renal obstruction results in progressive tubulointerstitial fibrosis, a process characterized by inflammatory cell infiltration, fibroblast proliferation, and an imbalance in extracellular matrix synthesis, deposition, and degradation (1–4). In the kidney, interstitial fibrosis is characterized by de novo activation of α-smooth muscle actin-positive myofibroblasts, the principal cells responsible for excess extracellular matrix deposition. Evidence suggests that renal tubular epithelial cells (TECs)2 are capable of undergoing a phenotypic transformation into matrix-producing fibroblasts in pathologic states (5–8), and this transformation in the phenotype of the cell (epithelial-mesenchymal transition (EMT)) is thought to contribute greatly to renal fibrosis (9, 10). EMT is characterized by a loss in epithelial cell markers (i.e., E-cadherin) and increased expression of the myofibroblast marker α-smooth muscle actin (α-SMA). Although TGF-β1 can induce renal fibrosis via stimulation of EMT, fibroblast proliferation (11–17), and extracellular matrix synthesis (1, 17–22), IL-18 has recently been identified as an important mediator of obstruction-induced renal fibrosis and EMT independent of downstream TGF-β1 production (23).
IL-18 is a proinflammatory cytokine that is structurally and functionally related to the IL-1 family and a potent stimulator of cytokine gene expression via activation of NFκB (24, 25). Recent studies have demonstrated a significant increase in renal tubular cell IL-18 production in response to obstruction, and an IL-18-mediated increase in collagen deposition, tubulointerstitial fibrosis, EMT, and tubular epithelial cell apoptosis independent of TNF-α or TGF-β1 activity (23, 26). We have recently demonstrated that IL-18 production localizes primarily to renal tubular cells, not infiltrating macrophage, during renal obstruction in mice (27), and studies have shown that circulating IL-18 levels and renal tubular cell IL-18 receptor expression is significantly increased in patients with chronic kidney disease (28). Cumulatively, these studies suggest that IL-18 is an important mediator of renal fibrotic injury and potentially a proximal initiator of obstruction-induced renal fibrosis.
Toll-like receptors (TLRs) are a family of receptors that recognize pathogen-associated molecular patterns and stimulate both immune and nonimmune cells to express inflammatory cytokines and chemokines. TLR4 is present on renal tubular epithelium and provides important host response mechanisms in the control of ascending urinary tract infections (29, 30). Although TLR4 is considered to be the critical component of the LPS receptor complex, TLR4 can also bind a variety of other ligands, including the extracellular matrix breakdown products hyaluronan, heparan sulfate, and fibrinogen that are prevalent during renal obstruction (31–33). We have previously demonstrated that TLR4 expression is increased in response to renal obstruction and that TLR4 is a mediator of obstruction-induced EMT and renal fibrosis. Because IL-18-induced renal fibrosis occurs independent of the well recognized TGF-β1 signaling pathway and both TLR4 and IL-18 appear to be important mediators of obstruction-induced profibrotic signaling, we hypothesized that the profibrotic effect of IL-18 is mediated through alterations in TLR4 expression. To study this, renal cortical IL-18 production and TLR4 expression were examined in male C57Bl6 WT mice and mice transgenic for IL-18-binding protein (IL-18BP Tg; overexpression of soluble IL-18BP reliably binds and inhibits IL-18 activity in these mice) using a well established model of unilateral ureteral obstruction (UUO) (34). In a separate arm, human proximal tubular cells (HK-2) were directly stimulated with IL-18 in the presence or absence of a TLR4 agonist (ultrapure LPS, 1 μg/ml), TLR4 antagonist (Escherichia coli K12 msbB LPS; 1 μg/ml), or siRNA knockdown of the TLR4 gene, and the cell lysates were subsequently examined for TLR4 expression and markers of EMT. TLR4 promoter activity was then evaluated in HK-2 cells in response to IL-18 stimulation, and AP-1 activation was investigated as a potential mechanism of IL-18-induced alterations in TLR4 expression.
EXPERIMENTAL PROCEDURES
The animal protocol was reviewed and accepted by the Indiana University Animal Care and Use Committee. Male C57BL6 mice transgenic for IL-18-binding protein (IL-18BP Tg) were generously donated by Dr. Charles Dinarello (University of Colorado Health Science Center, Denver, CO). These mice overexpress human IL-18-binding protein isoform a and reliably inhibit IL-18 activity but do not have any notable phenotype (34). The genotype of the mice was confirmed with a PCR analysis of extracted DNA samples from tail snips (5′ primer, 5′-ACA CCT GTC TCG CAG ACC AC-3′; 3′ primer, 5′-TCA GCT GCT CCA GCA CCA A-3′) as described by Fantuzzi et al. (34), and overexpression of serum levels of human IL-18BP was confirmed using an ELISA prior to utilization of the animals.
Male mice weighing 25–30 g (5–8 animals/group) were anesthetized with isofluorane, and the left ureter was isolated and completely ligated with 5–0 silk suture. Sham-operated mice underwent an identical surgical procedure without ureteral ligation. This sham model was selected because previous work has demonstrated that unilateral renal injury can induce bilateral renal cytokine production (35). One week postoperatively, the mice were re-anesthetized, the left kidneys were removed and snap frozen in liquid nitrogen, and the animals were subsequently euthanized.
The human proximal tubular cell line HK-2 was cultured in keratinocyte serum-free medium + 5 ng/ml EGF and 40 mg/ml bovine extract + 100 units/ml penicillin and 100 μg/ml of streptomycin. The cells were passaged weekly by trypsinization (0.25% trypsin, 0.02% EDTA) following formation of a confluent monolayer and placed in serum-free medium 24 h prior to stimulation. The cells were exposed to control medium, human recombinant IL-18 (0.1, 1, 10, or 100 ng/ml), a TLR4 agonist (E. coli K, ultrapure LPS-EK, 1 μg/ml; Invivogen, San Diego, CA) ± IL-18 (100 ng/ml), or a TLR4 antagonist (E. coli K12 msbB LPS; 1 μg/ml; Invivogen) ± IL-18 (100 ng/ml) for 3 days. Cell lysates were subsequently collected and analyzed. All of the experiments were run in triplicate with at least three samples/group.
HK-2 cells (2.5 × 105 cells/well) were cultured in a 6-well dish and placed in serum-free medium 18 h prior to transfection. The cells were than reversely transfected with 40 pmol of siRNA (Silencer Select Negative Control #1 siRNA), hTLR4 siRNA (Silencer Select Validated TLR4 siRNA; Ambion, Austin, TX), c-Jun siRNA (Silencer Select Pre-designed JUN; Ambion), or c-Fos siRNA (Silencer Select Pre-designed FOS; Ambion) using 7.5 μl of Lipofectamine 2000 reagent (Invitrogen). After 5 h, the cells were incubated in fresh serum-free medium and treated with either vehicle (Lipofectamine) or 100 ng/ml IL-18 (R & D Systems, Minneapolis, MN) for 3 days. Conditioned medium, total RNA, and protein were then collected for future quantitative PCR and Western blot analysis. All of the experiments were run in triplicate with at least three samples/group.
A portion of each renal cortex was homogenized after the samples had been diluted in 10 volumes of homogenate buffer/g of tissue (10 mm Hepes, pH 7.9, 10 mm KCl, 0.1 mm EGTA, 1 mm DTT, and Complete Protease Inhibitor tabs (Roche Applied Science)) using a VertiShear tissue homogenizer. Renal homogenates were then centrifuged at 3,000 × g for 15 min, and the supernatants were stored at −70 °C until the ELISAs/Western blots could be performed.
Renal homogenate IL-18 levels were determined using an ELISA (mouse IL-18: MBL Int., Woburn, MA). The ELISA was performed by adding 100 μl of each sample to wells in a 96-well plate of a commercially available ELISA kit, and the assay was performed according to the manufacturer's instructions. All of the samples were tested in duplicate. The ELISA results were expressed as pg/mg of protein.
Total RNA was extracted from the renal cortical tissue or cell lysates by homogenization in TRIzol (Invitrogen) and then isolated by precipitation with chloroform and isopropanol as previously described (23). Total RNA (0.5 μg) was subjected to cDNA synthesis using iScript (Bio-Rad). cDNA from each sample was analyzed for TLR4 (human, Hs00152939_m1; mouse, Mm00445274_m1) using a TaqMan gene expression assay (RT-PCR; Applied Biosystems, Foster City, CA). FAM Dye/MGB-labeled probes for either mouse GAPDH (Applied Biosystems) served as endogenous controls.
Protein extracts from homogenized samples (50 μg/lane) or cell lysates (20 μg/lane) were subjected to SDS-PAGE electrophoresis on a Tris-glycine gel and transferred to a nitrocellulose membrane. Immunoblotting was performed for each antibody by incubating each membrane in 5% dry milk overnight at 4 °C, followed by incubation with an anti-α-SMA monoclonal antibody (clone 1A4, 1:500 for 2 h at room temperature; Sigma), an anti-E-cadherin monoclonal antibody (1:200 overnight at 4 °C; BD Transduction Laboratories, San Jose, CA), or anti-TLR4 monoclonal antibody (1:200 for 2 h at room temperature; human (Rockland, Gilbertsville, PA) or mouse (Santa Cruz Biotechnology, Santa Cruz, CA)). After washing three times in TBST, each membrane was incubated for 1 h at room temperature with a peroxidase-conjugated secondary antibody (1:2000 for α-SMA, 1:1,000 for E-cadherin, and 1:1,000 for TLR4). Equivalent protein loading in each lane was confirmed by stripping and reblotting each membrane for GAPDH (1:20,000 for 30 min at room temperature, secondary 1:20,000 for 30 min at room temperature; Biodesign International, Saco, ME). The membranes were developed using enhanced chemiluminescence (Amersham Biosciences), and the density of each band was determined using National Institutes of Health Image analysis software and expressed as percentages of GAPDH density.
Nuclear extracts from renal cortical tissue samples were prepared by homogenizing the samples in 7 volumes of lysis buffer (10 mm Hepes, pH 7.9, 10 mm KCL, 0.1 mm EGTA, 1 mm DTT, and 0.5 mm phenylmethanesulfonyl fluoride) and centrifuged at 3,000 × g for 15 min at 4 °C. The supernatants, which contained the cytosolic fraction, were stored at −70 °C, whereas the crude nuclear pellet was resuspended in 100 μl of ice-cold nuclear extraction buffer (20 mm Hepes, pH 7.9, 0.4 m NaCl, 1 mm EGTA, 1 mm DTT, Complete Protease Inhibitor tabs). The samples were incubated on ice for 30 min with brief, gentle vortexing every 5–10 min. The nuclear extract was centrifuged at 12,000 × g for 10 min at 4 °C, and the supernatant was collected and stored at −70 °C. Protein was quantified in both cytosolic and nuclear extracts with a Coomassie Plus protein assay (Pierce).
For the EMSA, synthetic double-stranded oligonucleotides of the sequence AP-1 (5′-CGCTTGATGACTCAGCCGGAA-3′) were fill-in labeled with [α-32P]dATP using Sequenase DNA polymerase. The DNA binding reaction was conducted at room temperature for 30 min in a volume of 20 μl. The reaction mixture contained 2 μg of each sample, 10 mm Tris-HCl, pH 7.5, 50 mm NaCl, 0.5 mm EDTA, 0.5 mm DTT, 1 mm MgCl2, 4% glycerol, 0.5 μl of poly(dI-dC)·poly(dI-dC), and 1 μl of 32P-labeled double-stranded oligonucleotides. After incubation, the samples were run on an 8% polyacrylamide gel. Each gel was then dried and subject to autoradiography. The density of each band was determined using National Institutes of Health Image analysis software.
A 920-base pair TLR4 promoter fragment (−745/+178) was amplified from human genomic DNA (Clontech) using the Advantage 2 PCR kit (Clontech) and the primers CTC GGT ACC AGT AAA GCT TAG CGG TTT ACA TG (sense) and CTC CCA TGG CCT CAC TGC TTC TGT GAG CAG CA (antisense), which were designed according to the sequence of the TLR4 gene published in GenBankTM (accession number NG_011475). This PCR product was then cloned into the PCR cloning vector pCR2.1-TOPO (Promega). To generate the luciferase reporter construct, the TLR4 promoter fragment was released from pCR2.1-TOPO by digesting with KpnI/NcoI and subcloned into the pGL3-basic vector (Promega) between KpnI/NcoI sites. This 560 bp (−385/+178) plasmid construct was named pGL-hTLR4-luc. The plasmid was isolated and purified using a Qiafilter plasmid midi kit (Qiagen) and confirmed by DNA sequencing analysis.
HK-2 cells were transfected in triplicate with pGL3-basic vector (Promega, Madison, WI) or the TLR4 promoter reporter plasmid using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Briefly, 2 × 105 cells were seeded into each well of 12-well plates a day before transfection. 0.5 μg of pGL3-basic vector or TLR4 reporter plasmid together with 20 ng of Renilla plasmid were added to each well. 100 ng/ml of IL-18 or solvent were added after 5 h of incubation. Cell lysates were collected after 3 days and assayed for firefly and Renilla luciferase activity using a dual luciferase reporter assay system (Promega) on a Femtomaster FB12 (Zylux corporation). Firefly luciferase activity was normalized to Renilla luciferase activity for each sample. The results are representative of at least three independent experiments.
The data are presented as the mean values ± S.E. Differences at the 95% confidence level were considered significant. The experiment groups were compared using one-way analysis of variance with post-hoc Bonferroni-Dunn (JMP 5.0.1). For cell culture experiments using varying concentrations of IL-18, a multiple comparisons analysis was performed in all pairs using Tukey-Kramer HSD.
RESULTS
Renal cortical tissue obtained from sham-operated animals revealed low levels of IL-18; however, IL-18 levels increased significantly in response to 1 week of obstruction (Fig. 1). Renal cortical TLR4 protein and mRNA expression were low in sham treated animals but increased significantly in response to 1 week of obstruction (Figs. 2). A significant reduction in TLR4 protein and mRNA expression, however, was detected in IL-18BP Tg mice exposed to the same degree of obstruction.
FIGURE 1.
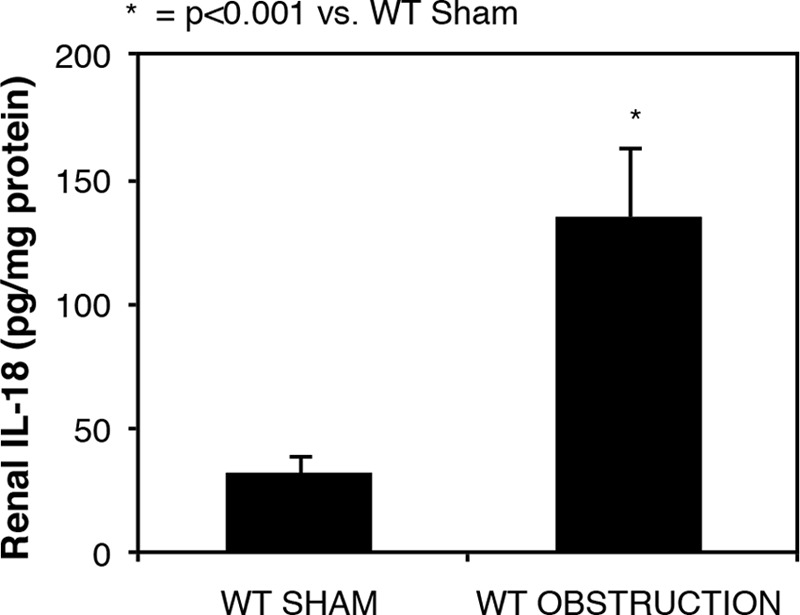
Renal cortical IL-18 protein levels following UUO. Renal cortical IL-18 protein levels in WT sham and 1-week obstructed kidneys are shown.
FIGURE 2.
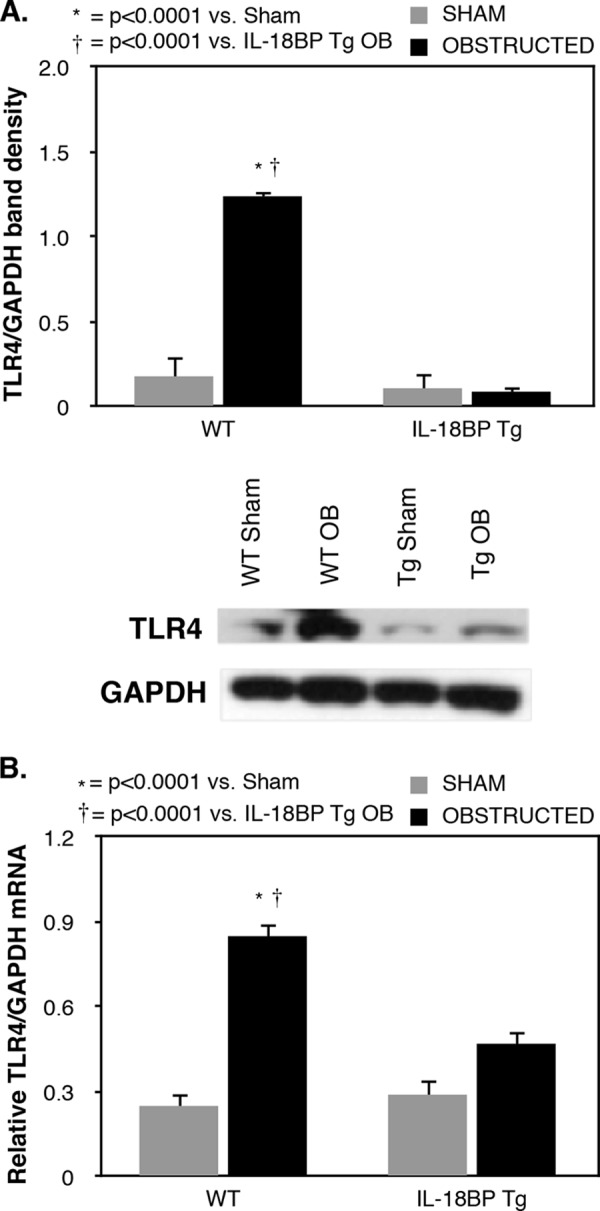
Renal cortical TLR4 expression following UUO. A, gel photograph and densitometric analysis of TLR4 expression represented as percentages of GAPDH in WT and IL-18BP transgenic animals (IL-18BP Tg) exposed to sham operation or 1 week of UUO. B, quantitative TLR4 mRNA expression represented as percentages of GAPDH in WT and IL-18BP Tg animals exposed to sham operation or 1 week of UUO.
The ability of IL-18 to stimulate tubular epithelial cell expression of TLR4 was subsequently evaluated by exposing human HK-2 cells to increasing concentrations of IL-18 in vitro. A significant increase in TLR4 expression was detected in cells directly stimulated with IL-18 in a dose-dependent fashion (Fig. 3). This observation provides valuable interspecies concordance with our in vivo results.
FIGURE 3.
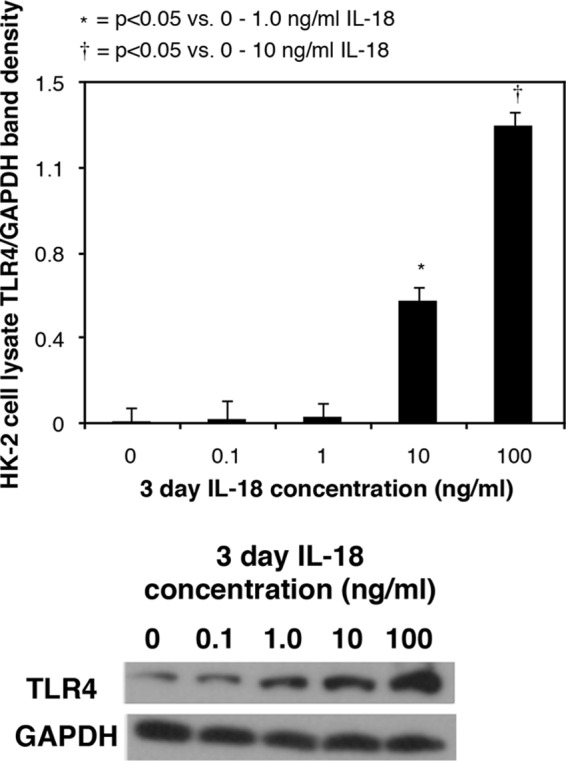
TLR4 expression in HK-2 cells in response to IL-18 stimulation in vitro. Shown are a gel photograph and densitometric analysis of cell lysate TLR4 expression represented as percentages of GAPDH following 3 days of cell stimulation with varying concentrations of recombinant human IL-18 (0–100 ng/ml).
To elucidate the effect of TLR4 modulation on IL-18-induced EMT, HK-2 cells were exposed to a TLR4 agonist, a TLR4 antagonist, or control medium prior to stimulation with IL-18. Cell lysate α-SMA and E-cadherin expression were analyzed as markers of EMT, and cell lysate TLR4 expression was analyzed as well. TLR4 expression increased significantly in HK-2 cells in the presence of either IL-18, a TLR4 agonist, or the combination of IL-18 and a TLR4 agonist when compared with control medium alone (Fig. 4A). α-SMA expression increased significantly in HK-2 cells in the presence of either IL-18 or a TLR4 agonist as compared with control media, whereas the combination of IL-18 and a TLR4 agonist resulted in significantly higher levels of α-SMA expression as compared with IL-18 stimulation alone (Fig. 4B). Conversely, E-cadherin expression decreased significantly in response to either IL-18, a TLR4 agonist, or IL-18 + TLR4 agonist, as compared with control cells (Fig. 4C).
FIGURE 4.

TLR4, E-cadherin, and α-SMA expression in HK-2 cells exposed to TLR4 agonist and IL-18 stimulation in vitro. A–C, densitometric analysis of cell lysate TLR4 (A), α-SMA (B), and E-cadherin (C) expression represented as percentages of GAPDH following 3 days of cell exposure to recombinant human IL-18 (100 ng/ml) in the presence or absence of a TLR4 agonist (E. coli K, ultrapure LPS-EK, 1 μg/ml). D, gel photograph of cell lysate TLR4, E-cadherin, α-SMA, and GAPDH expression following 3 days of cell exposure to recombinant human IL-18 (100 ng/ml) in the presence or absence of a TLR4 agonist (E. coli K, ultrapure LPS-EK, 1 μg/ml) or antagonist (E. coli K12 msbB LPS; 1 μg/ml).
HK-2 cells were subsequently exposed to a TLR4 antagonist versus control medium 24 h prior to stimulation with IL-18. TLR4 expression increased significantly in HK-2 cells in the presence of IL-18 (Fig. 5A). A significant decrease in IL-18-induced TLR4 and α-SMA expression occurred in response to the TLR4 antagonist (Fig. 5, A and B), and a significant increase in E-cadherin expression occurred following IL-18 exposure in the presence of the TLR4 antagonist (Fig. 5C). These results demonstrate that direct TLR4 stimulation induces cellular changes consistent with EMT, similar to that observed with IL-18 stimulation, and provides evidence that the downstream profibrotic effect of IL-18 is mediated in part via increased TLR4 expression/activation.
FIGURE 5.

TLR4, E-cadherin, and α-SMA expression in HK-2 cells exposed to TLR4 antagonist and IL-18 stimulation in vitro. A–C, densitometric analysis of cell lysate TLR4 (A), α-SMA (B), and E-cadherin (C) expression represented as percentages of GAPDH following 3 days of cell exposure to recombinant human IL-18 (100 ng/ml) in the presence or absence of a TLR4 antagonist (E. coli K12 msbB LPS; 1 μg/ml).
To evaluate the effect of TLR4 gene knockdown on TLR4 expression and EMT, HK-2 cells were exposed to TLR4 siRNA versus control medium in the presence or absence of IL-18. TLR4 mRNA expression increased significantly in HK-2 cells in the presence of IL-18 but remained at control levels in the presence of TLR4 gene knockdown despite IL-18 stimulation (Fig. 6A). IL-18-induced HK-2 cell EMT was also dramatically inhibited in the presence of TLR4 gene knockdown, as evidenced by suppression of α-SMA expression and preservation of E-cadherin expression during IL-18 stimulation (Fig. 6B). This finding provides further evidence that the injurious effect of IL-18 on TEC is mediated through alterations in TLR4 expression.
FIGURE 6.
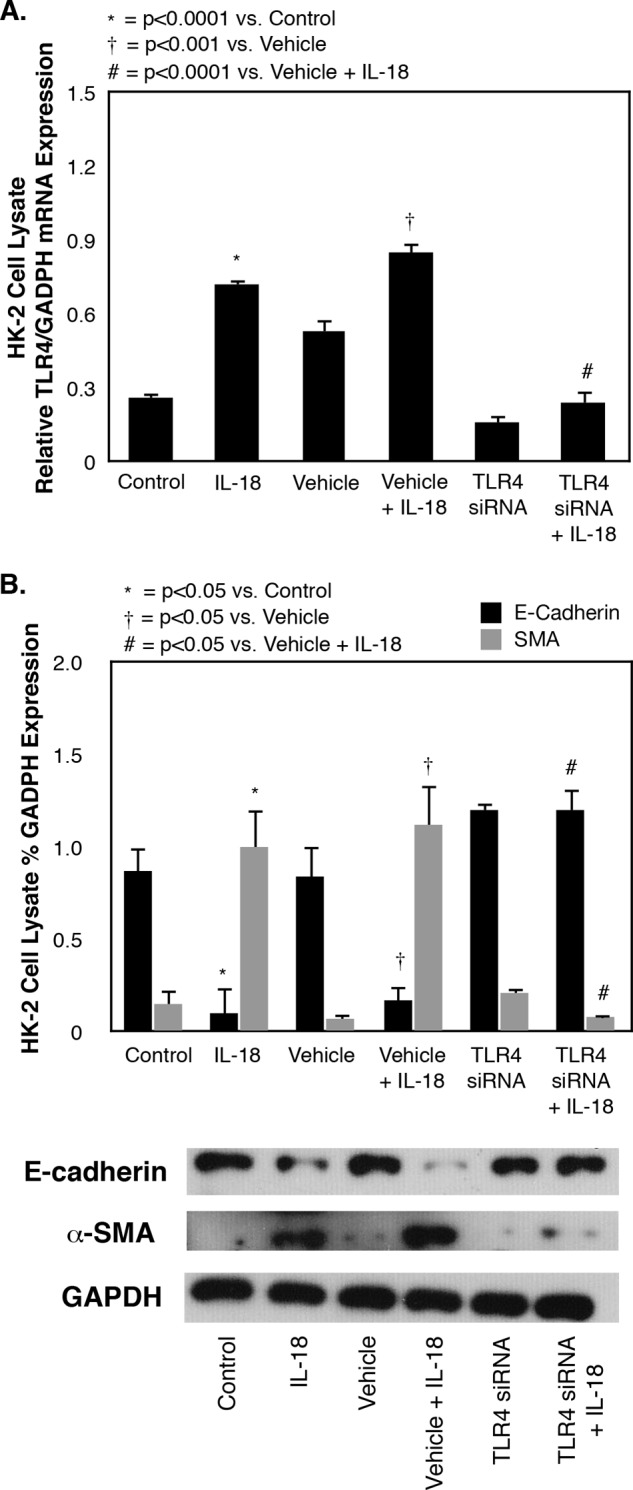
TLR4, E-cadherin, and α-SMA expression in HK-2 cells exposed to TLR4 siRNA knockdown and IL-18 stimulation in vitro. A, quantitative cell lysate TLR4 mRNA expression represented as percentages of GAPDH following 3 days of cell exposure to recombinant human IL-18 (100 ng/ml) in the presence or absence of a TLR4 siRNA (40 pmol). B, gel photograph and densitometric analysis of cell lysate E-cadherin and α-SMA expression represented as percentages of GAPDH following 3 days of cell exposure to recombinant human IL-18 (100 ng/ml) in the presence or absence of a TLR4 siRNA (40 pmol).
To further evaluate the effect of IL-18 on TLR4 gene expression in HK-2 cells in vitro, HK-2 cells were transfected either with a reporter plasmid containing the TLR4 promoter (pTLR4) or with empty vector and stimulated with IL-18. The results demonstrate a 2-fold increase in TLR4 promoter activity in response to direct IL-18 stimulation (Fig. 7) as compared with control cells.
FIGURE 7.
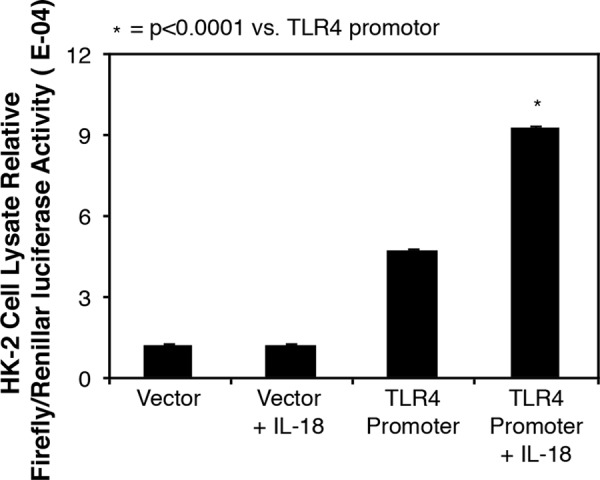
TLR4 promoter reporter activity in HK-2 cells in response to IL-18 stimulation. Cell lysate firefly luciferase activity is represented as percentages of Renilla luciferase activity following cell transfection with a TLR4 promoter reporter plasmid versus empty vector and 3 days of cell exposure to recombinant human IL-18 (100 ng/ml).
AP-1 activation was subsequently examined in HK-2 cells in response to cell stimulation with IL-18. Minimal AP-1 DNA binding was detected in response to low concentrations of IL-18; however, a dose-dependent increase in AP-1 DNA binding/activation was observed in response to increasing concentrations of IL-18 (Fig. 8A). The role of AP-1 as a mediator of IL-18-induced TLR4 expression was subsequently evaluated using c-Jun and c-Fos siRNA knockdown. IL-18-induced TLR4 expression in HK-2 cells was significantly reduced in the presence of both c-Jun and c-Fos gene knockdown as compared with vehicle + IL-18 or IL-18 alone (Fig. 8B), suggesting that IL-18-induced activation of AP-1 is the mechanism by which IL-18 stimulates TLR4 expression in HK-2 cells.
FIGURE 8.
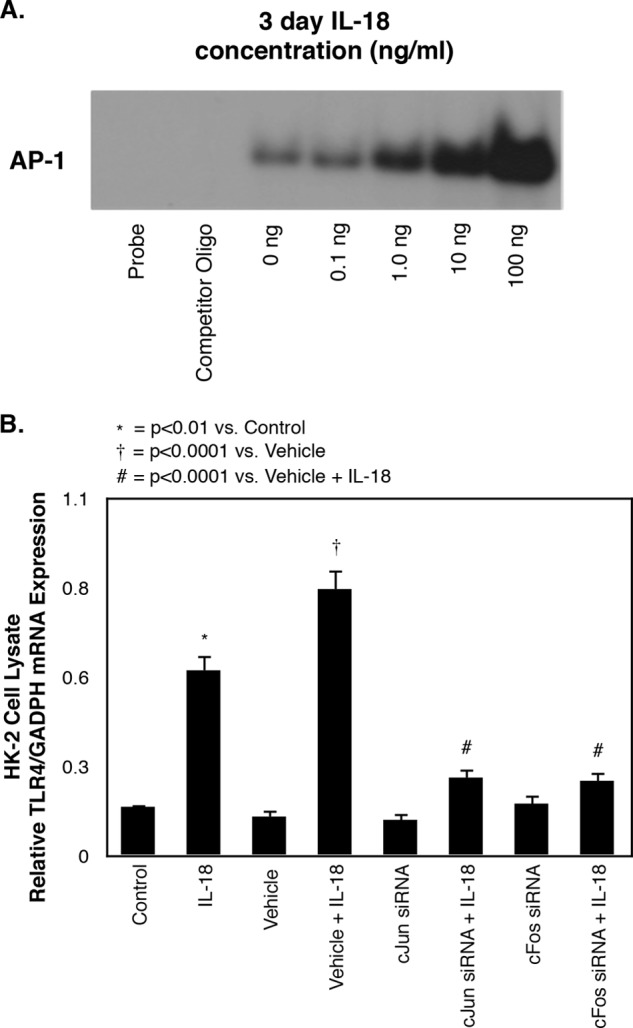
AP-1 activity and TLR4 expression in HK-2 cells exposed to c-Jun and c-Fos siRNA knockdown and IL-18 stimulation in vitro. A, cell lysate AP-1 DNA binding activity following 3 days of cell exposure to varying concentrations of recombinant human IL-18 (0–100 ng/ml). B, quantitative cell lysate TLR4 mRNA expression represented as percentages of GAPDH following 3 days of cell exposure to recombinant human IL-18 (100 ng/ml) in the presence or absence of a c-Jun siRNA (40 pmol) or c-Fos siRNA (40 pmol).
DISCUSSION
IL-18 is a proinflammatory cytokine that has recently been implicated in the pathophysiology of obstructive renal injury (23). Interestingly, whereas increased TGF-β1 production has long been regarded as the final common pathway for renal fibrotic injury, the profibrotic effect of IL-18 both in vivo and in tubular epithelial cells in vitro occurs independent of any alterations to TGF-β1 activity. Given the prominent expression of TLR4 in tubular epithelial cells and the role of TLR4 in obstructive renal injury (36, 37), alterations in TLR4 expression/activation were investigated as a possible mechanism of IL-18-induced profibrotic TEC injury. This is the first study to demonstrate that IL-18 induces TLR4 expression during renal obstruction and in renal tubular epithelial cells in vitro and further demonstrates that the downstream profibrotic effects of IL-18 in TECs are mediated through alterations in TLR4 expression/signaling via activation of AP-1.
Toll-like receptors are a key component of the innate immune system and function by recognizing pathogen-associated molecular patterns. Engagement of a TLR with its specific pathogen-associated molecular pattern initiates an intracellular signaling cascade culminating in the release of cytokines and the expression of cell surface co-stimulatory molecules capable of activating T cells and initiating an adaptive immune response (38). Although pathogen-associated molecular patterns were initially studied in the context of identifying bacterial, viral, parasitic, and fungal products, certain endogenous molecules are now understood to function as TLR4 agonists, including necrotic cells, heat shock proteins, and elements of the extracellular matrix (33, 39, 40). TLR4 is expressed in renal tubular epithelial cells and can bind a variety of extracellular breakdown products, such as fibrinogen and hyaluronan, that are more abundant during fibrotic renal injury (32, 33). Through this probable mechanism, TLR4 has previously been demonstrated to contribute to obstruction-induced fibrotic renal injury (37). Our data reveal that renal cortical TLR4 levels are significantly increased in response to obstruction and, further, that obstruction-induced renal TLR4 expression remains at sham treatment levels in the presence of IL-18 neutralization. This suggests that IL-18 is an important mediator of TLR4 expression/activity during obstructive renal injury.
To evaluate the effects of IL-18 stimulation on TLR4 expression in TECs, HK-2 cells were directly stimulated with IL-18 in vitro at a variety of concentrations. The cells exhibited a significant increase in TLR4 expression in response to direct IL-18 stimulation in a dose-dependent fashion. A previous study has demonstrated that IL-18 induces up-regulation of TLR4 on monocytes obtained from patients with rheumatoid arthritis; however, the response was found to be indirect and dependent on the simultaneous presence of IFN-γ (41). This study therefore provides new evidence that IL-18 can directly up-regulate TLR4 expression in renal epithelial cells and thereby elucidates a potential mechanism for IL-18-induced tubular cell injury.
The effect of TLR4 modulation on IL-18-induced TEC injury was evaluated by exposing the cells to a TLR4 agonist, a TLR4 antagonist, or TLR4 siRNA prior to stimulation with IL-18. The cells were subsequently analyzed for markers of EMT as an indicator of profibrotic injury. α-SMA expression increased significantly, and E-cadherin expression decreased significantly, in HK-2 cells exposed to either IL-18 or a TLR4 agonist when compared with control media, while the combination of IL-18 and a TLR4 agonist resulted in significantly higher levels of α-SMA expression, and persistently low levels of E-cadherin expression, as compared with IL-18 or TLR4 agonist stimulation alone. Conversely, a significant decrease in IL-18-induced α-SMA expression and a significant increase in E-cadherin expression occurred following cell exposure to either the TLR4 antagonist or TLR4 gene knockdown. These results demonstrate that IL-18-induced profibrotic injury in tubular epithelial cells is mediated, in part, through increased TLR4 expression and signaling.
To investigate the relationship between IL-18 and TLR4 expression in vitro, HK-2 cells were transfected with a reporter plasmid containing the human TLR4 promoter prior to stimulation with IL-18. TLR4 promoter activity was found to be 2-fold higher in cells stimulated with IL-18 as compared with control cells, suggesting that IL-18 increases TLR4 expression by stimulating TLR4 promoter activity. IL-18 binding to its receptor activates a signaling cascade leading to the activation of AP-1 in T-cells (42), and the TLR4 promoter has previously been determined to have AP-1-binding sites (43). We therefore investigated AP-1 as potential mechanism of IL-18-induced TLR4 expression in HK-2 cells. Following siRNA knockdown of both the c-Jun and c-Fos components of AP-1, HK-2 cells were stimulated with IL-18, and the effect on TLR4 expression was examined. Both c-Jun and c-Fos knockdown reduced IL-18-induced TLR4 expression to near control levels. These data cumulatively suggest that IL-18 stimulation of HK-2 cells activates AP-1, which in turn stimulates TLR4 promoter activity and TLR4 gene expression.
Upper urinary tract obstruction remains an important cause of renal dysfunction in both adults and children, leading to progressive tubulointerstitial fibrosis and apoptotic cell death. IL-18 has recently been implicated in the pathogenesis of chronic kidney disease and has been found to be a critical mediator of obstruction-induced renal fibrosis and EMT independent of TGF-β1. This is the first study to demonstrate that IL-18 stimulates TLR4 expression in response to renal obstruction and to reveal that the downstream profibrotic effects of IL-18 in TECs are mediated through alterations in TLR4 expression/signaling via activation of AP-1. The observation that renal cortical TLR4 levels are significantly increased in response to obstruction increases the importance of this signaling system as a potential therapeutic target.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 DK 081695.
- TEC
- tubular epithelial cell
- UUO
- unilateral ureteral obstruction
- α-SMA
- α-smooth muscle actin
- EMT
- epithelial-mesenchymal transition
- TLR
- Toll-like receptor.
REFERENCES
- 1. Eddy A. A. (1996) Molecular insights into renal interstitial fibrosis. J. Am. Soc. Nephrol. 7, 2495–2508 [DOI] [PubMed] [Google Scholar]
- 2. Klahr S. (2001) Progression of chronic renal disease. Heart Dis. 3, 205–209 [DOI] [PubMed] [Google Scholar]
- 3. Remuzzi G., Bertani T. (1998) Pathophysiology of progressive nephropathies. N. Engl. J. Med. 339, 1448–1456 [DOI] [PubMed] [Google Scholar]
- 4. Faust J., Menke J., Kriegsmann J., Kelley V. R., Mayet W. J., Galle P. R., Schwarting A. (2002) Correlation of renal tubular epithelial cell-derived interleukin-18 up-regulation with disease activity in MRL-Faslpr mice with autoimmune lupus nephritis. Arthritis Rheum. 46, 3083–3095 [DOI] [PubMed] [Google Scholar]
- 5. Zeisberg M., Bonner G., Maeshima Y., Colorado P., Muller G. A., Strutz F., Kalluri R. (2001) Renal fibrosis. Collagen composition and assembly regulates epithelial-mesenchymal transdifferentiation. Am. J. Pathol. 159, 1313–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Strutz F., Okada H., Lo C. W., Danoff T., Carone R. L., Tomaszewski J. E., Neilson E. G. (1995) Identification and characterization of a fibroblast marker. FSP1. J. Cell Biol. 130, 393–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Strutz F., Müller G. A., Neilson E. G. (1996) Transdifferentiation. A new angle on renal fibrosis. Exp. Nephrol. 4, 267–270 [PubMed] [Google Scholar]
- 8. Healy E., Brady H. R. (1998) Role of tubule epithelial cells in the pathogenesis of tubulointerstitial fibrosis induced by glomerular disease. Curr. Opin. Nephrol. Hypertens. 7, 525–530 [DOI] [PubMed] [Google Scholar]
- 9. Iwano M., Plieth D., Danoff T. M., Xue C., Okada H., Neilson E. G. (2002) Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest. 110, 341–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iwano M., Neilson E. G. (2004) Mechanisms of tubulointerstitial fibrosis. Curr Opin. Nephrol. Hypertens. 13, 279–284 [DOI] [PubMed] [Google Scholar]
- 11. Rastaldi M. P. (2006) Epithelial-mesenchymal transition and its implications for the development of renal tubulointerstitial fibrosis. J. Nephrol. 19, 407–412 [PubMed] [Google Scholar]
- 12. Zeisberg M., Hanai J., Sugimoto H., Mammoto T., Charytan D., Strutz F., Kalluri R. (2003) BMP-7 counteracts TGF-β1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 9, 964–968 [DOI] [PubMed] [Google Scholar]
- 13. Li Y., Yang J., Dai C., Wu C., Liu Y. (2003) Role for integrin-linked kinase in mediating tubular epithelial to mesenchymal transition and renal interstitial fibrogenesis. J. Clin. Invest. 112, 503–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fan J. M., Ng Y. Y., Hill P. A., Nikolic-Paterson D. J., Mu W., Atkins R. C., Lan H. Y. (1999) Transforming growth factor-β regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int. 56, 1455–1467 [DOI] [PubMed] [Google Scholar]
- 15. Kuncio G. S., Neilson E. G., Haverty T. (1991) Mechanisms of tubulointerstitial fibrosis. Kidney Int. 39, 550–556 [DOI] [PubMed] [Google Scholar]
- 16. Postlethwaite A. E., Keski-Oja J., Moses H. L., Kang A. H. (1987) Stimulation of the chemotactic migration of human fibroblasts by transforming growth factor beta. J. Exp. Med. 165, 251–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roberts A. B., McCune B. K., Sporn M. B. (1992) TGF-β. Regulation of extracellular matrix. Kidney Int. 41, 557–559 [DOI] [PubMed] [Google Scholar]
- 18. Alvarez R. J., Sun M. J., Haverty T. P., Iozzo R. V., Myers J. C., Neilson E. G. (1992) Biosynthetic and proliferative characteristics of tubulointerstitial fibroblasts probed with paracrine cytokines. Kidney Int. 41, 14–23 [DOI] [PubMed] [Google Scholar]
- 19. Border W. A., Noble N. A. (1994) Transforming growth factor β in tissue fibrosis. N. Engl. J. Med. 331, 1286–1292 [DOI] [PubMed] [Google Scholar]
- 20. Humes H. D., Cieslinski D. A. (1992) Interaction between growth factors and retinoic acid in the induction of kidney tubulogenesis in tissue culture. Exp. Cell Res. 201, 8–15 [DOI] [PubMed] [Google Scholar]
- 21. Miyajima A., Asano T., Yoshimura I., Seta K., Hayakawa M. (2001) Tranilast ameliorates renal tubular damage in unilateral ureteral obstruction. J. Urol. 165, 1714–1718 [PubMed] [Google Scholar]
- 22. Miyajima A., Chen J., Lawrence C., Ledbetter S., Soslow R. A., Stern J., Jha S., Pigato J., Lemer M. L., Poppas D. P., Vaughan E. D., Felsen D. (2000) Antibody to transforming growth factor-β ameliorates tubular apoptosis in unilateral ureteral obstruction. Kidney Int. 58, 2301–2313 [DOI] [PubMed] [Google Scholar]
- 23. Bani-Hani A. H., Leslie J. A., Asanuma H., Dinarello C. A., Campbell M. T., Meldrum D. R., Zhang H., Hile K., Meldrum K. K. (2009) IL-18 neutralization ameliorates obstruction-induced epithelial-mesenchymal transition and renal fibrosis. Kidney Int. 76, 500–511 [DOI] [PubMed] [Google Scholar]
- 24. Dinarello C. A. (1999) IL-18. A TH1-inducing, proinflammatory cytokine and new member of the IL-1 family. J. Allergy Clin. Immunol. 103, 11–24 [DOI] [PubMed] [Google Scholar]
- 25. Dinarello C. A. (2007) Interleukin-18 and the pathogenesis of inflammatory diseases. Semin. Nephrol. 27, 98–114 [DOI] [PubMed] [Google Scholar]
- 26. Zhang H., Hile K. L., Asanuma H., Vanderbrink B., Franke E. I., Campbell M. T., Meldrum K. K. (2011) IL-18 mediates proapoptotic signaling in renal tubular cells through a Fas ligand-dependent mechanism. Am. J. Physiol. Renal Physiol. 301, F171–F178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. VanderBrink B. A., Asanuma H., Hile K., Zhang H., Rink R. C., Meldrum K. K. (2011) Interleukin-18 stimulates a positive feedback loop during renal obstruction via interleukin-18 receptor. J. Urol. 186, 1502–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liang D., Liu H. F., Yao C. W., Liu H. Y., Huang-Fu C. M., Chen X. W., Du S. H., Chen X. W. (2007) Effects of interleukin 18 on injury and activation of human proximal tubular epithelial cells. Nephrology (Carlton) 12, 53–61 [DOI] [PubMed] [Google Scholar]
- 29. Anders H. J., Banas B., Schlondorff D. (2004) Signaling danger. Toll-like receptors and their potential roles in kidney disease. J. Am. Soc. Nephrol. 15, 854–867 [DOI] [PubMed] [Google Scholar]
- 30. Chowdhury P., Sacks S. H., Sheerin N. S. (2006) Toll-like receptors TLR2 and TLR4 initiate the innate immune response of the renal tubular epithelium to bacterial products. Clin. Exp. Immunol. 145, 346–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson G. B., Brunn G. J., Kodaira Y., Platt J. L. (2002) Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J. Immunol. 168, 5233–5239 [DOI] [PubMed] [Google Scholar]
- 32. Okamura Y., Watari M., Jerud E. S., Young D. W., Ishizaka S. T., Rose J., Chow J. C., Strauss J. F., 3rd (2001) The extra domain A of fibronectin activates Toll-like receptor 4. J. Biol. Chem. 276, 10229–10233 [DOI] [PubMed] [Google Scholar]
- 33. Termeer C., Benedix F., Sleeman J., Fieber C., Voith U., Ahrens T., Miyake K., Freudenberg M., Galanos C., Simon J. C. (2002) Oligosaccharides of hyaluronan activate dendritic cells via toll-like receptor 4. J. Exp. Med. 195, 99–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fantuzzi G., Banda N. K., Guthridge C., Vondracek A., Kim S. H., Siegmund B., Azam T., Sennello J. A., Dinarello C. A., Arend W. P. (2003) Generation and characterization of mice transgenic for human IL-18-binding protein isoform a. J. Leukocyte Biol. 74, 889–896 [DOI] [PubMed] [Google Scholar]
- 35. Meldrum K. K., Meldrum D. R., Meng X., Ao L., Harken A. H. (2002) TNF-alpha-dependent bilateral renal injury is induced by unilateral renal ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 282, H540–H546 [DOI] [PubMed] [Google Scholar]
- 36. Samuelsson P., Hang L., Wullt B., Irjala H., Svanborg C. (2004) Toll-like receptor 4 expression and cytokine responses in the human urinary tract mucosa. Infect. Immun. 72, 3179–3186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Campbell M. T., Hile K. L., Zhang H., Asanuma H., Vanderbrink B. A., Rink R. R., Meldrum K. K. (2011) Toll-like receptor 4. A novel signaling pathway during renal fibrogenesis. J. Surg. Res. 168, e61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gay N. J., Gangloff M. (2007) Structure and function of Toll receptors and their ligands. Annu. Rev. Biochem. 76, 141–165 [DOI] [PubMed] [Google Scholar]
- 39. Li M., Carpio D. F., Zheng Y., Bruzzo P., Singh V., Ouaaz F., Medzhitov R. M., Beg A. A. (2001) An essential role of the NF-κB/Toll-like receptor pathway in induction of inflammatory and tissue-repair gene expression by necrotic cells. J. Immunol. 166, 7128–7135 [DOI] [PubMed] [Google Scholar]
- 40. Ohashi K., Burkart V., Flohé S., Kolb H. (2000) Cutting edge. Heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J. Immunol. 164, 558–561 [DOI] [PubMed] [Google Scholar]
- 41. Radstake T. R., Roelofs M. F., Jenniskens Y. M., Oppers-Walgreen B., van Riel P. L., Barrera P., Joosten L. A., van den Berg W. B. (2004) Expression of toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-γ. Arthritis Rheum. 50, 3856–3865 [DOI] [PubMed] [Google Scholar]
- 42. Nakahira M., Ahn H. J., Park W. R., Gao P., Tomura M., Park C. S., Hamaoka T., Ohta T., Kurimoto M., Fujiwara H. (2002) Synergy of IL-12 and IL-18 for IFN-γ gene expression. IL-12-induced STAT4 contributes to IFN-γ promoter activation by up-regulating the binding activity of IL-18-induced activator protein 1. J. Immunol. 168, 1146–1153 [DOI] [PubMed] [Google Scholar]
- 43. Roger T., Miconnet I., Schiesser A. L., Kai H., Miyake K., Calandra T. (2005) Critical role for Ets, AP-1 and GATA-like transcription factors in regulating mouse Toll-like receptor 4 (Tlr4) gene expression. Biochem. J. 387, 355–365 [DOI] [PMC free article] [PubMed] [Google Scholar]