

Background: Halogenated inhaled anesthetics modulate voltage-gated ion channels by unknown mechanisms.
Results: Biophysical analyses revealed novel activation of Kv channels by the inhaled anesthetic sevoflurane.
Conclusion: Kv channel activation by sevoflurane results from the positive allosteric modulation of activation gating.
Significance: The unique activation of Kv channels by sevoflurane demonstrates novel anesthetic specificity and offers new insights into allosteric modulation of channel gating.
Keywords: Anesthesia, Anesthetics, Gating, Ion Channels, Potassium Channels
Abstract
Voltage-gated ion channels are modulated by halogenated inhaled general anesthetics, but the underlying molecular mechanisms are not understood. Alkanols and halogenated inhaled anesthetics such as halothane and isoflurane inhibit the archetypical voltage-gated Kv3 channel homolog K-Shaw2 by stabilizing the resting/closed states. By contrast, sevoflurane, a more heavily fluorinated ether commonly used in general anesthesia, specifically activates K-Shaw2 currents at relevant concentrations (0.05–1 mm) in a rapid and reversible manner. The concentration dependence of this modulation is consistent with the presence of high and low affinity interactions (KD = 0.06 and 4 mm, respectively). Sevoflurane (<1 mm) induces a negative shift in the conductance-voltage relation and increases the maximum conductance. Furthermore, suggesting possible roles in general anesthesia, mammalian Kv1.2 and Kv1.5 channels display similar changes. Quantitative description of the observations by an economical allosteric model indicates that sevoflurane binding favors activation gating and eliminates an unstable inactivated state outside the activation pathway. This study casts light on the mechanism of the novel sevoflurane-dependent activation of Kv channels, which helps explain how closely related inhaled anesthetics achieve specific actions and suggests strategies to develop novel Kv channel activators.
Introduction
Despite the ubiquitous clinical use of general anesthetics, the molecular mechanisms of general anesthesia are not fully understood. Ion channels are among the most relevant targets, as their modulation by general anesthetics could explain both endpoints and side-effects of anesthesia (1–5). Recent studies have strongly supported the presence of diverse general anesthetic sites in ligand-gated, voltage-gated and non-gated ion channels (6–15). However, a role of voltage-gated K+ (Kv)2 channels in general anesthesia is uncertain, and there is still no general structural framework to predict anesthetic specificity and the mechanisms linking drug binding to functional modulation.
The modulatory effects of inhaled general anesthetics on ion channels are also diverse. These anesthetics can both activate and inhibit ion channels through mechanisms that may involve modulation of gating and/or permeation. Most notably, inhaled anesthetics potentiate ionotropic GABAA and glycine receptors and non-gated two-pore domain K+ channels, but inhibit other ionotropic receptors (e.g. nicotinic and glutamate receptors). Serotonin (5-HT3) receptors exhibit both inhibition and activation, which depends on the molecular volume of the anesthetic (6, 7, 16–21). The interactions of voltage-gated ion channels with inhaled anesthetics are less well characterized and, to the best of our knowledge, the available studies of these targets report only inhibition by inhaled anesthetics (10, 22–29). Previously, we characterized the inhibition of the archetypical Drosophila voltage-gated K+ (Kv) channel K-Shaw2 (a Kv3 homolog) by relevant concentrations of n-alcohols and inhaled anesthetics (12, 30–33). Furthermore, these studies indicate that the inhibition results from stabilizing the resting and closed states of the K-Shaw2 channel without affecting ion permeation (12, 31).
To investigate more relevant general anesthetics and eventually dissect the inhaled anesthetic pharmacophore in voltage-gated ion channels, we tested diverse general anesthetics on K-Shaw2 expressed heterologously in Xenopus oocytes and discovered the novel and unique activation of this channel by sevoflurane at relevant concentrations (0.05–1 mm). We hypothesized that sevoflurane induces rearrangements in the activation machinery of K-Shaw2, which changes the stability of the closed and/or open states. To test this hypothesis, we examined the following aspects: 1) time course and concentration dependence of the sevoflurane response; 2) voltage-dependence of the sevoflurane response; and 3) the kinetic basis of the sevoflurane-induced activation at steady-state. Additionally, we asked whether the activation by sevoflurane is conserved in mammalian Kv channels. We report the results of this work and discuss its implications in terms of a quantitative framework that explains the sevoflurane-induced activation and the possible contribution of this response to the mechanisms of general anesthesia.
EXPERIMENTAL PROCEDURES
Molecular Biology
All cDNAs encoding the investigated potassium channels (K-Shaw2, ShakerB (inactivation-removed), rKv1.2, hKv1.3, hKv1.5, rKv2.1, hKv3.4, and rKv4.2) were maintained as previously reported (31). The cDNAs encoding hKv1.3, Shaker-B and rKv1.2 and hKv1.5 were gifts from Drs. C. Deutsch (University of Pennsylvania), T. Hoshi (University of Pennsylvania), D. Minor (University of California, San Francisco) and G. Seebohm (Ruhr University, Bochum, Germany), respectively. The K-Shaw2 F335A mutant was used as the “wild-type” background as it exhibits enhanced expression in Xenopus oocytes with minimal effects on anesthetic sensitivity. In vitro transcription was performed as previously described (12).
Reagents
Volatile anesthetic stocks were diluted in in ND96 as previously described (12, 33). Final dilutions of all drugs were prepared immediately prior to use. Final sevoflurane dilutions were verified by HPLC to have <15% loss (the data were not corrected for this loss). The anesthetics used were: HPLC grade 1-butanol (Fisher Scientific, Hampton, NH), propofol (SAFC, St Louis, MO), halothane (Halocarbon Laboratories, River Edge, NJ), chloroform (Acros Organics, Somerville, NJ), isoflurane (Minrad, Bethlehem, PA), desflurane (Baxter, Deerfield, IL), and sevoflurane (Abbott Laboratories, North Chicago, IL).
Heterologous Expression
Care and surgery of Xenopus laevis was performed according to a protocol approved by the Thomas Jefferson University Institutional Animal Care and Use Committee. mRNAs encoding the Kv channels of interest were introduced into Xenopus oocytes by microinjection as described previously (31). Recordings were performed 4–72 h post-microinjection.
Electrophysiology
Whole-oocyte currents were recorded at room temperature (20–23 °C) under two-electrode voltage-clamp conditions (OC-725C, Warner Instrument, Hamden, CT) according to established procedures (12, 31, 33). Briefly, oocytes were bathed in ND96 and electrophysiological recordings were performed using borosilicate microelectrodes with tip resistances of 0.2–0.5 MΩ when filled with 3 m KCl. Non-volatile drugs (1-butanol) and bath solution were delivered using a gravity-driven perfusion system. Volatile anesthetics were delivered manually using a Hamilton gastight syringe (Hamilton, Reno, NV) and PTFE tubing. Bath drainage was done by gravity flow. All responses were collected at steady state. Once a stable current baseline was established upon repeating a constant voltage step at 10-s intervals under constant perfusion of ND96, a given concentration of drug was delivered (see above) and the perfusion of drug was maintained until a new stable current level was reached (e.g. Fig. 1D). Then, the drug was washed out and currents were recorded until a new stable baseline was reached. Voltage protocols to generate the peak chord conductance-voltage (G/V) curves were always applied once the responses reached steady state. Because of membrane retention of the lipophilic anesthetics used, only one anesthetic concentration was administered per oocyte and all control data were collected prior to drug administration. Data acquisition, processing, and initial analysis were performed using pClamp 9.x (Molecular Devices, Sunnyvale, CA).
FIGURE 1.

Specific reversible activation of K-Shaw2 channels by sevoflurane. A, semilogarithmic concentration-response relations of various general anesthetics. These anesthetics were tested on K-Shaw2 currents evoked by voltage step to +60 mV. Symbols are means of N>5. Solid lines are the best fits to the Hill equation, which yielded the following best-fit parameters for propofol: K0.5 = 0.06 mm, nH = 1.6; chloroform: K0.5 = 0.70 mm, nH = 1.4; halothane: K0.5 = 0.26 mm, nH = 1.3; isoflurane: K0.5 = 1.69 mm, nH = 2.1; 1-butanol: K0.5 = 10.1 mm, nH = 1.8. Desflurane is also inhibitory at 1 mm (81 ± 0.01% of control, n = 3). B, sevoflurane concentration-activation relation. The response was tested on currents evoked by a voltage step to −20 mV. Symbols are means of N>7. The solid line is the best-fit to a model assuming two independent binding sites (“Experimental Procedures”) with the best-fit parameters indicated in the graph, dashed, and dotted lines show individual binding components for high and low affinity sites, respectively. C, reversible effect of 300 μm sevoflurane on the whole-oocyte K-Shaw2 currents evoked by a voltage step to −20 mV from a holding voltage of −100 mV. The control (C), sevoflurane (S), and washout (W) currents are indicated in the graph. D, time courses of the reversible potentiation of K-Shaw2 by low concentrations of sevoflurane (50 and 300 μm; n ≥ 7). Symbols correspond to averages of four points before/after sevoflurane applications and 4 points before and 7 points after washout. For clarity, other points within the break are not shown.
Data Analysis
Plotting, non-linear curve fitting and statistical evaluations were performed in OriginPro 8.0 (OriginLab, Northampton, MA). A Hill equation assuming two independent low- and high-affinity binding sites (Equation 1) was necessary to describe the concentration-response curve for sevoflurane over a broad concentration range (A = 0.01–10 mm).
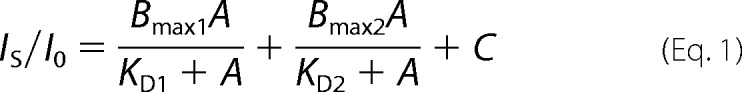 |
Here, IS/I0 is the normalized current (drug/control), Bmax indicates the maximum binding capacity for each site, KD indicates the apparent equilibrium dissociation constants, and C is a constant. Established statistical criteria were used to compare the one-site model against the two-site model (supplemental Fig. S1). This study focuses on the high-affinity interaction. The G-V relations were obtained from current-voltage relations generated by conventional voltage steps. The average reversal potential (VR) was estimated from tail current current-voltage plots to be −67 mV (n = 6). To analyze the G-V curves, the best-fit parameters of the Boltzmann equation (V1/2, z, and Gmax) were obtained as described elsewhere (32). All results are reported as means ± S.E. of the mean.
Kinetic Modeling
Partly based on previous studies by Smith-Maxwell et al. and Ledwell et al. (35, 36), we assumed the following three-state scheme to economically describe K-Shaw2 gating at steady-state (Scheme I).
SCHEME I.
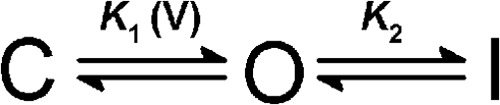
C, O, and I represent resting-closed, open and inactivated states, respectively. The equilibrium constants K1 and K2 are voltage-dependent and voltage-independent, respectively. As explained under “Results,” assuming an unstable inactivated state was sufficient and necessary to account for Gmax changes. Focusing on the relevant high-affinity interaction of sevoflurane (above), Scheme I served as the framework to generate an allosteric model that can explain the sevoflurane-dependent activation of the K-Shaw2 current (Scheme II).
SCHEME II.
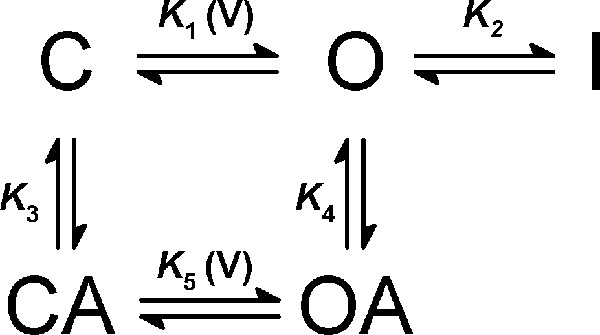
Essentially, Scheme II assumes that high-affinity sevoflurane binding changes the energetics of activation gating. Accordingly, sevoflurane binds to the C and O conformations with equilibrium binding constants K3 and K4, which introduces state-dependent binding cooperativity (K4 > K3) and shifts gating to a mode that promotes opening (K5 > K1) and cannot access I. K5 is voltage-dependent, and the equilibrium open probability (Po) predicted by Scheme II is defined in Equation 2.
 |
A is the sevoflurane concentration and K1K4 = K3K5, as dictated by the principle of detailed balanced.
A three-step process was then used to determine the best-fits of Scheme II to two separate relations: the voltage dependence of the sevoflurane-dependent increase in conductance (G-Sevoflurane/G-Control versus voltage = GS/G0 versus voltage), and the concentration dependence of the sevoflurane-dependent activation (IS/I0 versus sevoflurane concentration). First, to initialize the fit of Scheme II to the GS/G0 relationship, the voltage-dependent equilibrium constant K1 was determined from the best-fit Boltzmann function that describes the G-V relation in the absence of sevoflurane in Equation 3.
Here, K1,0 is the equilibrium constant K1 at 0 mV, z is the equivalent gating charge, V1/2 is the voltage of half-activation, and F, R, and T are Faraday's constant, gas constant and absolute temperature, respectively. K4 was also fixed to the inverse of the apparent high-affinity equilibrium dissociation constant (1/KD1) obtained from the analysis of the concentration-response curve (Fig. 1B). Second, the GS/G0 relationship was evaluated at a concentration of sevoflurane that saturates the high affinity interaction (0.5 mm ∼10KD1). The remaining adjustable parameters of Scheme II, z2, K2, and K3, were allowed to vary to determine the best fit to the GS/G0 versus voltage curve and K5 was calculated from detailed balance (above). The best-fit free parameters exhibited low dependencies (<0.58). Because the GS/G0 versus voltage relationship is only evaluated at a saturating concentration of sevoflurane, modeling of the concentration dependence is poorly constrained. Thus, in the third step of the analysis, the best-fit to the activation-concentration relation (IS/I0 versus A) was optimized by only allowing K1 to vary (all other parameters were fixed to the values determined in the second step), and K4 was estimated from detailed balance (above). This modeling does not allow estimation of the absolute Pomax of the channel. Given that the focus of the analysis is on the high-affinity sevoflurane interaction, the Po estimates are relative to the maximum activation resulting from this interaction.
RESULTS
Specific Concentration-dependent Activation of K-Shaw2 Currents by Sevoflurane
Previous work with K-Shaw2 and halogenated anesthetics (12, 33, 37) prompted us to examine other clinically relevant general anesthetics. Thus, we constructed concentration-response relations to compare the inhibition of K-Shaw2 by 1-butanol and halothane to the effects of chloroform, isoflurane, desflurane, sevoflurane, and propofol (Fig. 1A). With distinct potencies, most anesthetics inhibit K-Shaw2 currents expressed in Xenopus oocytes. Surprisingly, sevoflurane, which is physicochemically related to isoflurane and desflurane, drastically deviates from this behavior. At all concentrations, this heavily fluorinated inhaled anesthetic increases the K-Shaw2 current (Fig. 1A). The onset of this response is rapid (within the exchange time of the oocyte perfusion system) and reversible, even though the washout appeared to be generally slower that the onset (Fig. 1, C and D). To investigate the concentration dependence of the sevoflurane-dependent activation at equilibrium, we plotted the normalized plateau response against the sevoflurane concentration (Fig. 1B). This curve was well described by assuming two independent binding sites with distinct apparent affinities for sevoflurane. Statistical criteria strongly favored the two-site model over the one-site model (supplemental Fig. S1). The apparent high- and low-affinity equilibrium dissociation constants were 60 μm and 4000 μm, respectively (Fig. 1C; at −20 mV, see below). These results indicate that the fast reversible modulation of K-Shaw2 by sevoflurane occurs over a concentration range that is clinically relevant (minimum alveolar concentration is 350 μm) (38). Given that relevant activation occurs in the sub-mm range of sevoflurane concentrations and the apparent equilibrium dissociation constants differ substantially (67-fold), further analysis was focused on the putative high-affinity site.
Kinetic Basis of the Allosteric Activation of K-Shaw2 by Sevoflurane
To investigate the mechanism underlying K-Shaw2 activation by sevoflurane, we characterized the voltage dependence of this modulation by comparing the G-V relations in the absence and presence of the anesthetic (0.5 and 1 mm) and described them assuming first-order Boltzmann functions (Fig. 2, A and B). Sevoflurane induces a negative shift in the V1/2, and increases the Gmax (Fig. 2, C and D; supplemental Table S1). Thus, sevoflurane may induce a relative destabilization of closed/resting states. The sevoflurane-induced increase in Gmax additionally suggests a voltage-independent equilibrium outside the activation pathway, which, in the absence of sevoflurane, connects the O state to a non-conducting (“Experimental Procedures”). The latter state can in part be responsible for the low Po of K-Shaw2. Thus, sevoflurane could increase Gmax by eliminating access to an unstable inactivated state.
FIGURE 2.

The effects of sevoflurane on the conductance-voltage relation of K-Shaw2. A, whole-oocyte families of K-Shaw2 currents evoked by the depicted pulse protocol. B, normalized G-V relations in the absence (n = 16) and presence of two concentrations of sevoflurane (0.5 and 1 mm; n = 14 and 9, respectively). In each control-sevoflurane pair, the G-V curve in the presence of sevoflurane is normalized to the control Gmax. Solid lines are the best-fit Boltzmann functions with the following parameters: Gmax (Control) = 1; V1/2 (Control) = 27 mV; z (Control) = 1.1 e0; Gmax (0.5 mm) = 1.3; V1/2 (0.5 mm) = 22 mV; z (0.5 mm) = 1.2 e0; Gmax (1 mm) = 1.4; V1/2 (1 mm) = 8 mV; z (1 mm) = 1.2 e0. C, bar graph summarizing the negative V1/2 shifts induced by two concentrations of sevoflurane. D, bar graph summarizing the increases in Gmax induced by two concentrations of sevoflurane. Descriptive statistics of this analysis is summarized in supplemental Table S1.
To investigate these possibilities more quantitatively and gain biophysical insight into the mechanism of action, we assumed a three-state gating scheme for K-Shaw2 (Scheme I; “Experimental Procedures”). The parsimonious assumption of this simple scheme is based on the following criteria. First, studies of the ShakerB-ILT and Shaker-Shaw S4 mutants by Smith-Maxwell et al. (35) and Ledwell et al. (36), strongly suggest a simple two-state activation pathway for K-Shaw2 channels, which characteristically exhibit weak-voltage dependence and relatively low open probability (39). Second, although macroscopic K-Shaw2 currents exhibit little or no apparent inactivation, the relatively low open probability of the underlying channels may in part be due to the presence of an unstable inactivated state. Third, also in agreement with the conclusions reached by Smith-Maxwell et al. and Ledwell et al. (35, 36), voltage-dependent activation of K-Shaw2 is accurately described by a first-order Boltzmann function associated with a relatively small equivalent gating charge (z∼1 e0) (Fig. 2 and supplemental Table S1); and current activation occurs with no apparent delay. Fourth, current activation and deactivation kinetics exhibit double exponential trajectories consistent with three observable states (data not shown). Fifth, a two-state C-O scheme cannot to account for both the shift in voltage-dependence and the increase in Gmax induced by sevoflurane.
Then, we hypothesized that sevoflurane acts as an allosteric gating modifier that binds only to the C and O states (Fig. 3A). Supporting this hypothesis, nearly identical parameter sets in Scheme II produced excellent quantitative descriptions of both voltage dependence (GS/G0 versus voltage) and concentration dependence (IS/I0 versus concentration) of K-Shaw2 activation by sevoflurane (Fig. 3, B–E). Note that the kinetic modeling was constrained by experimental estimates of the equilibrium constants and, at most, 3/7 adjustable parameters were allowed to vary freely in this analysis (“Experimental Procedures”). Also, note that the non-conducting state I is necessary and sufficient to explain the apparent change in Gmax (Fig. 3B, dashed line versus solid line). Sevoflurane binds tighter to the O state (K4 = 1.7K3), enhances voltage-dependent activation (K5 = 1.7K1 and z2 = 1.14z1) and eliminates access to I. Consequently, sevoflurane binding increases the K-Shaw2 Pomax from 0.74 to 0.98 (“Experimental Procedures”). Overall, this kinetic analysis offers a parsimonious biophysical framework that explains K-Shaw2 channel activation by sevoflurane.
FIGURE 3.

Kinetic modeling of K-Shaw2 gating and its positive modulation by sevoflurane. A, kinetic scheme that economically explains the allosteric activation of K-Shaw2 by sevoflurane, and the corresponding best-fit parameters (“Experimental Procedures” and “Results”). K1 and K5 depend exponentially on membrane potential (Vm) according to this relation: K(Vm) = K(0)exp(zVmF/RT) where z is the equivalent gating charge, and F, R, and T are Faraday's constant, gas constant, and temperature, respectively. The tabulated values of K1 and K5 are at 0 mV and z1 and z2 are the corresponding equivalent gating chargers. B, voltage dependence of the sevoflurane-dependent conductance increase (GS/G0 versus voltage; n = 14). At each voltage, the conductance in the presence of sevoflurane (GS) is divided by the corresponding control conductance (G0). The data (symbols) are calculated from the control and sevoflurane G-V relations in Fig. 2. Solid line is the best-fit to Scheme II and dashed line shows a simulated best-fit G-V curve from a 2-state binding model. C, concentration-activation relation in the low-concentration range of sevoflurane (n = 3–12). Data from Fig. 1B are re-plotted on linear axes. Solid line is the best-fit to Scheme II. To optimize the fit to the IS/I0 versus [Sevoflurane] curve, the values of K1 and K4 were modestly adjusted (K1 = 0.25; K4 = 21.4 mm−1) while maintaining the remaining constants fixed (“Experimental Procedures”). E and F, best-fit residuals from B and C, respectively.
Sevoflurane-dependent Activation of Kv1 Channels
Even though most mammalian Kv channels are resistant to inhibition by n-alcohols and inhaled anesthetics (12, 25, 30, 32), we wondered whether these channels might also be resistant to activation by sevoflurane. Surprisingly, certain Kv1 channels display voltage-dependent activation by sevoflurane (Figs. 4 and 5; supplemental Table S1). Sevoflurane shifts the G-V curves of mammalian Kv1.2, Kv1.5 and Drosophila ShakerB leftwards (−4, −2.2 and −3 mV, respectively) and increases the Gmax (∼13–16% increase) (Fig. 4A–C, supplemental Table S1). By contrast, other representative Shaker-related Kv channels in four distinct subfamilies (Kv1.3, Kv2.1, Kv3.4, and Kv4.2) exhibit little or no modulation by sevoflurane (Fig. 4, D–F, supplemental Table S1).
FIGURE 4.

A–F, effect of 1 mm sevoflurane on the G-V relations of Shaker-related Kv channels. Symbols are means of n = 5, 6, 7, 6, 6, 6 for ShakerB-IR, Kv1.2, Kv1.5, Kv2.1, Kv3.4, and Kv4.2, respectively. The solid lines are the best-fit Boltzmann functions with parameters summarized in supplemental Table S1.
FIGURE 5.

Voltage dependence of the sevoflurane-dependent conductance increase (GS/G0 versus voltage; n = 4–7 for (A) K-Shaw2, (B) Kv1.2, (C) Kv1.5, (D) ShakerB-IR. The GS/G0 was calculated as in Fig. 2 from the average G-V curves shown in Figs. 2B and 4, A–C.
DISCUSSION
We have reported the novel positive allosteric modulation of the Kv channel K-Shaw2 by sevoflurane, an inhaled anesthetic commonly used in general anesthesia. This action is in sharp contrast to the robust inhibition of this channel by the majority of general anesthetics. The specific activation results from high-affinity binding of sevoflurane to the channel, which allosterically favors activation gating and eliminates access to an unstable inactivated state. Moreover, sevoflurane similarly activates the Drosophila ShakerB channel and mammalian Kv1.2 and Kv1.5 channels.
Mechanism of K-Shaw2 Gating and Allosteric Activation by Sevoflurane
In contrast to most Kv channels, which must undergo multiple voltage-dependent closed-state transitions before they can open, K-Shaw2 appears to exhibit a much simpler activation pathway. Based on the analysis of the Shaker-ILT and Shaker-ShawS4 mutants, Smith-Maxwell et al. and Ledwell et al. (35, 36) provided compelling evidence to suggest that K-Shaw2 may behave as a two-state channel and that a relatively small gating charge (∼1 e0) is sufficient to open the pore. K-Shaw2 may thus open by undergoing a weakly voltage-dependent concerted conformational change corresponding to the last opening step of most Kv channels. A two-state activation scheme, however, cannot explain K-Shaw2 gating because sevoflurane increases the Gmax suggesting that the Pomax of the channel is determined by a non-conducting state outside the voltage-dependent activation pathway but connected to the O state. This non-conducting state may behave as the “flicker” state in the ShakerB Kv channel (40). Thus, we propose that a three-state scheme assuming unstable voltage-independent inactivation from the open state to economically explain gating of K-Shaw2 (Scheme I).This simple framework is furthermore sufficient to explain the activation of K-Shaw2 by sevoflurane through binding to both the C and O states (Scheme II). With nearly identical parameter sets, Scheme II quantitatively accounts for both voltage- and concentration-dependent activation (Fig. 3). In the allosteric loop, sevoflurane binds with higher affinity (1/K4 ≈ 60 μm) to the open state, which favors voltage-dependent activation. Simultaneously, sevoflurane binding increases Pomax because the favored sevoflurane-bound open channels do not enter I. Alternatively or additionally, the increase in Gmax might have resulted from an increase in the number of active channels and/or an increase in unitary conductance. The first possibility is unlikely because the reversible activation by sevoflurane is too fast (Fig. 1) to account for the insertion and removal of channels in the membrane. Data do not rule out the second possibility; however, the physical-chemical properties of sevoflurane do not suggest an obvious mechanism through which the non-polar anesthetic might directly increase the throughput of K+ in the pore. Also, to the best of our knowledge, there is no precedent for changes in unitary conductance induced by inhaled anesthetics and related drugs in other ion channels.
Limitations of Kinetic Modeling
Despite its simplicity and success in explaining K-Shaw2 voltage-dependent gating and its activation by sevoflurane, Scheme II as a detailed general mechanism needs corroboration by additional measurements and analyses. The kinetic analysis so far is only based on measurements of macroscopic whole-cell currents and the derived chord conductances, which cannot directly rule out changes in unitary conductance and number of active channels. Furthermore, the connectivity of the proposed states needs more investigation. Detailed single channel recordings designed to examine the voltage and concentration dependences of K-Shaw2 activation by sevoflurane will be necessary to directly address these limitations and alternative hypotheses.
Are Mammalian Kv Channels Physiological Targets of Sevoflurane?
We propose that the positive allosteric mechanism reported here for K-Shaw2 applies generally to the modulation of mammalian Kv1 channels by sevoflurane, which may act on the opening equilibrium at the end of the activation pathway. There are, however, relevant quantitative differences. Compared with the effects of 1 mm sevoflurane on K-Shaw2, Kv1.2, and Kv1.5 exhibit smaller changes in activation V1/2 and Gmax (Figs. 3 and 4). However, Kv1.2, Kv1.5, and ShakerB exhibit a comparable increase in relative conductance (GS/G0) over the voltage range where these channels undergo steep voltage-dependent gating (Fig. 5). This is a direct consequence of their greater sensitivity to voltage (associated with a large equivalent gating charge), such that a relatively small leftward shift of the G-V curve can have a major enhancing effect on the conductance within the most voltage sensitive region of the curve. Over a physiological range of membrane potentials (−60 to −40 mV), the actual sevoflurane-induced increase in K+ conductance in the mammalian brain might thus have a substantial negative impact on neuronal excitability. Keeping in mind that Kv channels only exhibit activation by sevoflurane (other inhaled anesthetics at relevant concentrations have no effect on Kv channels), this specific action might be associated with hypoexcitability through modulation of the resting membrane potential and/or action potential firing. The specificity of sevoflurane action is nevertheless intriguing because anesthesia induced by different inhaled anesthetics is generally similar. In any event, it critically shows that specific proteins can differentially react to inhaled anesthetics despite the high similarity in their chemical structures and physicochemical properties. The dissection of an inhaled anesthetic pharmcophore and the design of improved inhaled anesthetics will have to take this specificity into consideration.
Relationship to Other Ion Channels
Resembling the pharmacological profile of K-Shaw2, the ionotropic 5-HT3 receptor also exhibits differential dual modulation by inhaled anesthetics (16, 20, 42–43). However, these anesthetics appear to interact with this receptor in a non-competitive manner and the anesthetic effect correlates with the molecular size of the drug used. These authors conclude that potentiating and inhibitory drugs may share a binding site, with their functional effect determined by molecular volume (43). K-Shaw2 channels do not seem to conform to this correlation. Although K-Shaw2 exhibits an apparent n-alcohol potency cut-off at C7, all n-alcohols (C2-C12) examined in a previous work induced inhibition (44). Thus, sevoflurane is unique in its specific ability to activate K-Shaw2 currents. Determining whether its larger volume and heavier fluorination are responsible for this special property will require additional biochemical and structural work to systematically map the sevoflurane binding and allosteric sites in K-Shaw2 and the interactions involved.
Concluding Remarks and Implications
Toward understanding the molecular mechanisms of general anesthesia, recent reports emphasized the need to investigate how halogenated inhaled anesthetics with very similar chemical properties can achieve functional specificity (34, 41). This study offers insight into this problem by suggesting that allosteric interactions affecting activation gating of Kv channels can explain the novel positive allosteric modulation of an archetypical Kv channel by the inhaled anesthetic sevoflurane. This conclusion strongly supports the general notion of targeting the Kv channel activation gate for therapeutic applications with special emphasis on discovering novel allosteric activators, which may serve as specific anesthetics, antiarrhythmics, and anticonvulsants.
Supplementary Material
Acknowledgments
We thank Brian Urbani for technical assistance, and Drs. Roderic Eckenhoff and Richard Horn, and Mr. David M. Ritter for their critical reading of this manuscript.
This work was supported, in whole or in part, by NIAAA, National Institutes of Health Grants AA010615, AA007463 (to M. C.) and NINDS, National Institutes of Health Grant F31 NS077689 (to A. F. B.).

This article contains supplemental Table S1 and Fig. S1.
- Kv
- voltage-gated K+ channel
- 5-HT3
- serotonin
- G/V
- conductance-voltage.
REFERENCES
- 1. Franks N. P. (2006) Molecular targets underlying general anaesthesia. Br. J. Pharmacol. 147, S72–S81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Franks N. P. (2008) General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat. Rev. Neurosci. 9, 370–386 [DOI] [PubMed] [Google Scholar]
- 3. Hemmings H. C., Jr., Akabas M. H., Goldstein P. A., Trudell J. R., Orser B. A., Harrison N. L. (2005) Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol. Sci. 26, 503–510 [DOI] [PubMed] [Google Scholar]
- 4. Urban B. W. (2008) The site of anesthetic action. Modern Anesthetics, 3–29 [DOI] [PubMed] [Google Scholar]
- 5. Vedula L. S., Brannigan G., Economou N. J., Xi J., Hall M. A., Liu R., Rossi M. J., Dailey W. P., Grasty K. C., Klein M. L., Eckenhoff R. G., Loll P. J. (2009) A unitary anesthetic binding site at high resolution. J. Biol. Chem. 284, 24176–24184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krasowski M. D., Harrison N. L. (1999) General anaesthetic actions on ligand-gated ion channels. Cell Mol. Life Sci. 55, 1278–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Patel A. J., Honoré E., Lesage F., Fink M., Romey G., Lazdunski M. (1999) Inhalational anesthetics activate two-pore-domain background K+ channels. Nat. Neurosci. 2, 422–426 [DOI] [PubMed] [Google Scholar]
- 8. Putzke C., Hanley P. J., Schlichthörl G., Preisig-Müller R., Rinné S., Anetseder M., Eckenhoff R., Berkowitz C., Vassiliou T., Wulf H., Eberhart L. (2007) Differential effects of volatile and intravenous anesthetics on the activity of human TASK-1. Am. J. Physiol. Cell Physiol. 293, C1319–C1326 [DOI] [PubMed] [Google Scholar]
- 9. Cacheaux L. P., Topf N., Tibbs G. R., Schaefer U. R., Levi R., Harrison N. L., Abbott G. W., Goldstein P. A. (2005) Impairment of hyperpolarization-activated, cyclic nucleotide-gated channel function by the intravenous general anesthetic propofol. J. Pharmacol. Exp. Ther. 315, 517–525 [DOI] [PubMed] [Google Scholar]
- 10. Horishita T., Eger E. I., 2nd, Harris R. A. (2008) The effects of volatile aromatic anesthetics on voltage-gated Na+ channels expressed in Xenopus oocytes. Anesth. Analg. 107, 1579–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brannigan G., LeBard D. N., Hénin J., Eckenhoff R. G., Klein M. L. (2010) Multiple binding sites for the general anesthetic isoflurane identified in the nicotinic acetylcholine receptor transmembrane domain. Proc. Natl. Acad. Sci. U.S.A. 107, 14122–14127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barber A. F., Liang Q., Amaral C., Treptow W., Covarrubias M. (2011) Molecular mapping of general anesthetic sites in a voltage-gated ion channel. Biophys. J. 101, 1613–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nury H., Van Renterghem C., Weng Y., Tran A., Baaden M., Dufresne V., Changeux J. P., Sonner J. M., Delarue M., Corringer P. J. (2011) X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature 469, 428–431 [DOI] [PubMed] [Google Scholar]
- 14. Forman S. A. (2011) Clinical and molecular pharmacology of etomidate. Anesthesiology 114, 695–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Willenbring D., Liu L. T., Mowrey D., Xu Y., Tang P. (2011) Isoflurane alters the structure and dynamics of GLIC. Biophys. J. 101, 1905–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jenkins A., Franks N. P., Lieb W. R. (1996) Actions of general anaesthetics on 5-HT3 receptors in N1E-115 neuroblastoma cells. Br. J. Pharmacol. 117, 1507–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Downie D. L., Hall A. C., Lieb W. R., Franks N. P. (1996) Effects of inhalational general anaesthetics on native glycine receptors in rat medullary neurones and recombinant glycine receptors in Xenopus oocytes. Br. J. Pharmacol. 118, 493–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Scheller M., Bufler J., Schneck H., Kochs E., Franke C. (1997) Isoflurane and sevoflurane interact with the nicotinic acetylcholine receptor channels in micromolar concentrations. Anesthesiology 86, 118–127 [DOI] [PubMed] [Google Scholar]
- 19. Hapfelmeier G., Schneck H., Kochs E. (2001) Sevoflurane potentiates and blocks GABA-induced currents through recombinant α1β2γ2 GABAA receptors: implications for an enhanced GABAergic transmission. Eur. J. Anaesthesiol. 18, 377–383 [DOI] [PubMed] [Google Scholar]
- 20. Stevens R., Rüsch D., Solt K., Raines D. E., Davies P. A. (2005) Modulation of human 5-hydroxytryptamine type 3AB receptors by volatile anesthetics and n-alcohols. J. Pharmacol. Exp. Ther. 314, 338–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Franks N. P. (2006) Molecular targets underlying general anaesthesia. Br. J. Pharmacol. 147, S72–S81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takenoshita M., Steinbach J. H. (1991) Halothane blocks low-voltage-activated calcium current in rat sensory neurons. J. Neurosci. 11, 1404–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Herrington J., Stern R. C., Evers A. S., Lingle C. J. (1991) Halothane inhibits two components of calcium current in clonal (GH3) pituitary cells. J. Neurosci. 11, 2226–2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Todorovic S. M., Perez-Reyes E., Lingle C. J. (2000) Anticonvulsants but not general anesthetics have differential blocking effects on different T-type current variants. Mol. Pharmacol. 58, 98–108 [DOI] [PubMed] [Google Scholar]
- 25. Friederich P., Benzenberg D., Trellakis S., Urban B. W. (2001) Interaction of volatile anesthetics with human Kv channels in relation to clinical concentrations. Anesthesiology 95, 954–958 [DOI] [PubMed] [Google Scholar]
- 26. Shiraishi M., Harris R. A. (2004) Effects of alcohols and anesthetics on recombinant voltage-gated Na+ channels. J. Pharmacol. Exp. Ther. 309, 987–994 [DOI] [PubMed] [Google Scholar]
- 27. OuYang W., Hemmings H. C. (2007) Isoform-selective effects of isoflurane on voltage-gated Na+ channels. Anesthesiology 107, 91–98 [DOI] [PubMed] [Google Scholar]
- 28. Ouyang W., Herold K. F., Hemmings H. C. (2009) Comparative effects of halogenated inhaled anesthetics on voltage-gated Na+ channel function. Anesthesiology 110, 582–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Herold K. F., Nau C., Ouyang W., Hemmings H. C. (2009) Isoflurane inhibits the tetrodotoxin-resistant voltage-gated sodium channel Nav1.8. Anesthesiology 111, 591–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Covarrubias M., Rubin E. (1993) Ethanol selectively blocks a noninactivating K+ current expressed in Xenopus oocytes. Proc. Natl. Acad. Sci. U.S.A. 90, 6957–6960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Covarrubias M., Vyas T. B., Escobar L., Wei A. (1995) Alcohols inhibit a cloned potassium channel at a discrete saturable site. Insights into the molecular basis of general anesthesia. J. Biol. Chem. 270, 19408–19416 [DOI] [PubMed] [Google Scholar]
- 32. Bhattacharji A., Kaplan B., Harris T., Qu X., Germann M. W., Covarrubias M. (2006) The concerted contribution of the S4-S5 linker and the S6 segment to the modulation of a Kv channel by 1-alkanols. Mol. Pharmacol. 70, 1542–1554 [DOI] [PubMed] [Google Scholar]
- 33. Bhattacharji A., Klett N., Go R. C., Covarrubias M. (2010) Inhalational anaesthetics and n-alcohols share a site of action in the neuronal Shaw2 Kv channel. Br. J. Pharmacol. 159, 1475–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Singaram V. K., Somerlot B. H., Falk S. A., Falk M. J., Sedensky M. M., Morgan P. G. (2011) Optical reversal of halothane-induced immobility in C. elegans. Curr. Biol. 21, 2070–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Smith-Maxwell C. J., Ledwell J. L., Aldrich R. W. (1998) Role of the S4 in cooperativity of voltage-dependent potassium channel activation. J. Gen. Physiol. 111, 399–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ledwell J. L., Aldrich R. W. (1999) Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J. Gen. Physiol. 113, 389–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eckenhoff R. G., Xi J., Shimaoka M., Bhattacharji A., Covarrubias M., Dailey W. P. (2010) Azi-isoflurane, a Photolabel Analog of the Commonly Used Inhaled General Anesthetic Isoflurane. ACS Chem. Neurosci. 1, 139–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Franks N. P., Lieb W. R. (1996) Temperature dependence of the potency of volatile general anesthetics: implications for in vitro experiments. Anesthesiology 84, 716–720 [DOI] [PubMed] [Google Scholar]
- 39. Tsunoda S., Salkoff L. (1995) Genetic analysis of Drosophila neurons: Shal, Shaw, and Shab encode most embryonic potassium currents. J. Neurosci. 15, 1741–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zagotta W. N., Hoshi T., Aldrich R. W. (1994) Shaker potassium channel gating. III: Evaluation of kinetic models for activation. J. Gen. Physiol. 103, 321–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eckenhoff R. G. (2011) Anesthetic mechanisms: worms light the way. Curr. Biol. 21, R985–R986 [DOI] [PubMed] [Google Scholar]
- 42. Suzuki T., Koyama H., Sugimoto M., Uchida I., Mashimo T. (2002) The diverse actions of volatile and gaseous anesthetics on human-cloned 5-hydroxytryptamine3 receptors expressed in Xenopus oocytes. Anesthesiology 96, 699–704 [DOI] [PubMed] [Google Scholar]
- 43. Stevens R. J., Rüsch D., Davies P. A., Raines D. E. (2005) Molecular properties important for inhaled anesthetic action on human 5-HT3A receptors. Anesth. Analg. 100, 1696–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Harris T., Shahidullah M., Ellingson J. S., Covarrubias M. (2000) General anesthetic action at an internal protein site involving the S4-S5 cytoplasmic loop of a neuronal K+ channel. J. Biol. Chem. 275, 4928–4936 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.