

Background: l-Glucose, the enantiomer of d-glucose, was believed not to be utilized by any organisms.
Results: An l-glucose-utilizing bacterium was isolated, and its l-glucose catabolic pathway was identified genetically and enzymatically.
Conclusion: l-Glucose was utilized via a novel pathway to pyruvate and d-glyceraldehyde 3-phosphate.
Significance: This might lead to an understanding of homochirality in sugar metabolism.
Keywords: Bacterial Metabolism, Dehydrogenase, Enzyme Kinetics, Enzyme Purification, Gene Structure, l-Glucose, Aldonate Catabolism, α-Proteobacteria, Sugar Catabolism
Abstract
An l-glucose-utilizing bacterium, Paracoccus sp. 43P, was isolated from soil by enrichment cultivation in a minimal medium containing l-glucose as the sole carbon source. In cell-free extracts from this bacterium, NAD+-dependent l-glucose dehydrogenase was detected as having sole activity toward l-glucose. This enzyme, LgdA, was purified, and the lgdA gene was found to be located in a cluster of putative inositol catabolic genes. LgdA showed similar dehydrogenase activity toward scyllo- and myo-inositols. l-Gluconate dehydrogenase activity was also detected in cell-free extracts, which represents the reaction product of LgdA activity toward l-glucose. Enzyme purification and gene cloning revealed that the corresponding gene resides in a nine-gene cluster, the lgn cluster, which may participate in aldonate incorporation and assimilation. Kinetic and reaction product analysis of each gene product in the cluster indicated that they sequentially metabolize l-gluconate to glycolytic intermediates, d-glyceraldehyde-3-phosphate, and pyruvate through reactions of C-5 epimerization by dehydrogenase/reductase, dehydration, phosphorylation, and aldolase reaction, using a pathway similar to l-galactonate catabolism in Escherichia coli. Gene disruption studies indicated that the identified genes are responsible for l-glucose catabolism.
Introduction
All living organisms show homochirality toward organic compounds, such as amino acids, with l- rather than d- amino acids utilized for protein synthesis. The same is true for sugars, where d-sugars are more abundant than l-sugars in natural environments. Accordingly, most d-sugars can be utilized by many organisms, but l-sugar usage is less common, indicating that homochirality also occurs in sugar metabolism. A good example of this is glucose, where d-glucose is the most abundant sugar in nature, and most organisms use d-glucose as an energy source via well known pathways such as the Embden-Meyerhof-Parnas, the pentose phosphate, and the Entner-Doudoroff pathways. Meanwhile, l-glucose is not present in nature, and few instances of l-glucose usage have appeared since Rudney (1) reported that mammalian cells, yeasts, and some bacteria could not utilize this epimer with several reports also confirming that l-glucose was not utilized by other organisms (2, 3). As for enzymes with specificity toward l-glucose, Sasajima and Shinskey (4) reported l-glucose dehydrogenase activity in Pseudomonas caryophilli, but no further reports concerning this finding are currently available.
To determine whether there are organism(s) capable of utilizing l-glucose and to obtain further insights into homochirality in sugar metabolism, isolation of l-glucose-utilizing organisms and analysis of their catabolic pathways are necessary. Here, we describe the isolation of an l-glucose-utilizing bacterium, and its catabolic pathway for l-glucose metabolism is characterized.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, and Media
Escherichia coli DH10B was used as a host for plasmid construction. pGEM-T Easy (Promega) and pUC19 (Takara Bio) were used for gene cloning, and BL21(DE3) and plasmids pET21a(+) and pET28a(+) (Novagen) were for enzyme production. pBBR1MCS2 (5) was used for constructing suicide plasmids. E. coli S17-1 λpir acted as a donor for biparental mating of cells. LB medium supplemented with appropriate antibiotics was used for E. coli cultivation.
For screening of l-glucose-utilizing microorganisms, l-glucose minimal medium (l-Glc MM,2 10 mm NH4Cl, 10 mm potassium phosphate, pH 7.0, 10 mm MgSO4, 10 mm KCl, 0.2% (v/v) Hutner's trace element solution (per liter, 2.2 g of ZnSO4·7H2O, 1.1 g of H3BO3, 0.5 g of MnCl2·4H2O, 0.5 g of FeSO4·7H2O, 0.16 g of CoCl2·6H2O, 0.16 g of CuSO4·5H2O, and 0.11 g of (NH4)6Mo7O24·4H2O) containing 14 mm l-glucose; l-glucose was separately sterilized by filtration and added to the medium) was used, and for l-glucose dehydrogenase (l-GDH) purification, d-galactose minimal medium (20 mm NH4Cl, 20 mm potassium phosphate, pH 7.0, 10 mm MgSO4, 10 mm KCl, 0.2% (v/v) Hutner's trace element solution, 0.1% yeast extract containing 28 mm d-galactose; d-galactose was sterilized separately by filtration) was used.
PCR Amplification and Sequencing
PCR primers used in this study are listed in supplemental Table 1, and amplification was conducted under standard conditions. When appropriate, fragments were cloned into pUC19, pET21a, or pET28a vectors after digestion with restriction enzymes whose sites corresponded to the primers. Nucleotide sequencing was conducted with a CEQ8000XL sequencer (Beckman Coulter).
Enzyme Assay Substrates
l-Glucose and inositol derivatives were purchased from Sigma and Hokko Chemical Industry, respectively. l-Gluconate and d-galactonate were prepared as potassium salts by hypoiodite-in-methanol oxidization (6) of l-glucose and d-galactose, respectively. d-Idonic acid was obtained by base hydrolysis of d-idono-1,4-lactone from Carbosynth. d-2-Keto-3-deoxygalactonate (KDGal) was enzymatically synthesized from d-galactonate by the recombinant d-galactonate dehydratase (DgoD) from E. coli DH10B (7). DgoD was produced and purified as described below. d-2-Keto-3-deoxy-6-phosphogalactonate (KDPGal) was enzymatically synthesized from KDGal using recombinant LgnF cloned in this study.
l-Glucose-utilizing Microorganism Screening
Soil samples were inoculated in 10 ml of l-Glc MM and cultivated at 30 °C for 48 h with shaking at 300 rpm. Enrichment cultivation was conducted by inoculating 1 ml of the cultures to fresh l-Glc MM and cultivating under the same conditions, which was repeated 10 times. The cultures were then serially diluted and plated onto l-Glc MM agar plates to isolate l-glucose-utilizing microorganisms. l-Glucose consumption was monitored using the 3,5-dinitrosalicylic acid (DNS) method to determine the amount of reducing sugars in the culture supernatants (8).
Detection of Enzyme Activities toward l-Glucose in Strain 43P Cell-free Extracts
Strain 43P was cultured in l-Glc MM at 30 °C for 20 h, and the cells were collected by centrifugation at 17,400 × g for 10 min. The cells were then resuspended in 100 mm Tris-HCl, pH 7.0, containing 10% (v/v) glycerol and 1 mm DTT (Buffer A) and disrupted by sonication with a Sonifier 250 (Branson). After centrifugation at 17,400 × g for 10 min, the cell-free extracts were used to detect enzyme activities toward l-glucose. For isomerase activity, extracts were incubated with 10 mm l-glucose in 100 mm Tris-HCl, pH 7.5, for 2 h at 30 °C, and the reaction was terminated by addition of trichloroacetic acid. Formation of ketose was analyzed by TLC according to the method described by Lobanok et al. (9). For kinase activity, the reaction was performed as described above with the buffer containing 1 mm MgCl2 and 10 mm ATP, and the reaction mixture was analyzed by TLC with silica gel plates (Merck) as described by Hemker et al. (10). Dehydrogenase activity was assayed as described below.
Purification of Native l-Glucose Dehydrogenase (l-GDH) and l-Gluconate Dehydrogenase (l-GnDH)
Strain 43P was pre-cultured in 200 ml of LB medium at 30 °C for 20 h. After collecting the cells by centrifugation at 5800 × g for 15 min, they were transferred to 1 liter of d-galactose minimal medium, and cultivation was continued for 24 h. The cells were then harvested by centrifugation, washed, and resuspended with Buffer A. Cell-free extracts were prepared by sonication and centrifugation at 27,000 × g for 30 min at 4 °C and applied to a DEAE-cellulose column (DE52, Whatman International; 2.5-cm inner diameter × 15-cm height) equilibrated with Buffer A. After washing the column with Buffer A, proteins were eluted with a linear gradient of 0–500 mm NaCl in Buffer A. The active fractions were eluted at 80–120 mm NaCl. To the fractions containing l-GDH activity, (NH4)2SO4 was added to the solution to 20% saturation. This solution was then applied to a Butyl-Toyopearl 650 m column (Tosoh Corp; 2.5-cm inner diameter × 15-cm height) equilibrated with Buffer A containing 1.5 m (NH4)2SO4 and washed with the same buffer. Proteins were eluted with a linear gradient of 1.5–0 m (NH4)2SO4 in Buffer A, and the active fractions eluted at 0.6–0.5 m (NH4)2SO4 were dialyzed against Buffer A. The solution was loaded onto a Mono Q 5/50 GL column (GE Healthcare) equilibrated with Buffer A and washed with the same buffer. Proteins were eluted with a linear gradient of 0–250 mm NaCl in Buffer A. The active fractions eluted at 150 mm NaCl were concentrated by osmotic ultrafiltration (11) against dialysis membrane, size 20 (Wako Chemical), with PEG 20,000 at 4 °C for 3 h, applied to a Superdex 200 10/300 GL column (GE Healthcare) pre-equilibrated with Buffer A containing 150 mm NaCl, and eluted at a flow rate of 0.5 ml min−1. Fractions containing l-GDH activity were dialyzed against 100 mm Tris-HCl, pH 8.0, containing 50% (v/v) glycerol and 1 mm DTT.
For purification of l-GnDH, strain 43P was cultivated similar to that described above, except the second cultivation was conducted with 200 ml of l-Glc MM containing 0.1% yeast extract for 6 h. Cell-free extracts were prepared with Buffer B (100 mm Tris-HCl, pH 8.5, containing 20% (v/v) glycerol and 2.5 mm DTT), as described above and applied to a DEAE-cellulose column (DE52; 1-cm inner diameter × 15-cm height) pre-equilibrated with Buffer B. After washing the column with Buffer B containing 50 mm NaCl, proteins were eluted with a linear gradient of 50–500 mm NaCl in Buffer B. The fractions containing l-GnDH activity eluted at 170–200 mm NaCl were dialyzed against Buffer B and applied again to the same column equilibrated with Buffer B. After washing the column with Buffer B containing 120 mm NaCl, proteins were eluted with a linear gradient of 120–250 mm NaCl in Buffer B. The active fractions were directly loaded onto a Mono Q 5/50 GL column equilibrated with Buffer B containing 200 mm NaCl and washed with the same buffer. Proteins were eluted with a linear gradient of 200–350 mm NaCl in Buffer B. The active fractions were eluted at 280 mm NaCl, which were concentrated by Amicon Ultra-0.5 ml 10 K (Millipore) at 12,000 × g for 7 min at 4 °C. The concentrated proteins were applied to a Superdex 200 10/300 GL column pre-equilibrated with Buffer B containing 150 mm NaCl and eluted at a flow rate of 0.5 ml min−1. Fractions containing l-GnDH activity were dialyzed against 100 mm Tris-HCl, pH 8.5, containing 50% (v/v) glycerol and 1 mm DTT. Purification steps were monitored by SDS-PAGE as described by Laemmli (12).
Determination of N-terminal Amino Acid Sequences
Purified l-GDH and l-GnDH were resolved by 10% SDS-PAGE and electroblotted onto a polyvinylidene difluoride membrane. The bands corresponding to the enzymes were excised and used for Edman degradation and sequencing that was carried out by Hokkaido System Science.
Sequencing Analyses of Genes Encoding l-GDH and l-GnDH and Their Regions
Based on the N-terminal amino acid sequence of l-GDH and an internal conserved amino acid sequence derived from Pden_1680, two degenerate primers, l-GDH_Nter and l-GDH_midR, were synthesized and used for PCR amplification from strain 43P genomic DNA. The amplified 500-bp fragment was cloned into the pGEM-T Easy vector and sequenced. Based on the obtained sequence, two primers, 43P-LGDH-inverseF and 43P-l-GDH-inverseR, were designed and used for inverse PCR (13) with a mixture of EcoRI-digested and self-ligated strain 43P genomic DNA. The amplified 3.5-kb fragment was cloned into the pGEM-T Easy vector and sequenced.
Cloning and sequencing of the l-GnDH gene was conducted as described above except for the primers used as follows: l-GnDH_Nter and l-GnDH_midR for the first PCR, and 43P-LGnDH-inverseF and 43P-LGnDH-inverseR for the inverse PCR. For inverse PCR, PstI-digested, and self-ligated genomic DNA was used as a template.
To obtain sequence information for the regions around the l-GDH and l-GnDH genes, inverse-PCR and primer walking were conducted using the primers listed in supplemental Table 1. The nucleotide sequences of the resulting fragments were then determined.
Recombinant Enzyme Production and Purification
The ORFs of lgdA, lgnE, lgnF, lgnG, lgnH, and lgnI of strain 43P and E. coli dgoD were PCR-amplified with the appropriate primers from genomic DNA of strain 43P and E. coli DH10B, respectively (supplemental Table 1). After digestion with restriction enzymes whose sites were located in the primers, lgdA was cloned into pET21a(+), and the remaining genes were cloned in pET28a(+). The His6 tags in the vectors were attached at the LgdA C terminus and at the N termini for all other genes.
E. coli BL21(DE3) harboring these plasmids was cultured in LB medium containing appropriate antibiotics at 37 °C until the optical densities of the cultures reached 0.5. Isopropyl β-d-thiogalactopyranoside (final concentration 0.1 mm) was then added, and culturing was continued for 3 h. For lgnE, culturing was carried out at 28 °C, and the cultivation time after isopropyl β-d-thiogalactopyranoside addition was prolonged to 12 h. Cells were harvested by centrifugation and disrupted by sonication in 50 mm Tris-HCl, pH 8.0, 10% (v/v) glycerol, 0.5 m NaCl, and 10 mm imidazole. The resulting cell-free extracts were loaded onto a HisTrap HP column (GE Healthcare), and His6-tagged proteins were eluted with 0.5 m imidazole. The purified proteins were dialyzed against 100 mm Tris-HCl, pH 8.0, containing 50% (v/v) glycerol and 1 mm DTT.
Enzyme Assays
l-GDH, l-GnDH, and l-5-keto-gluconate reductase activities were assayed using a DU 800 spectrophotometer (Beckman Coulter) to measure reduction/oxidation of NAD+ and NADPH at 340 nm and 25 °C. The reactions were conducted in 28 mm l-glucose and 1 mm NAD+ in 100 mm Tris-HCl, pH 8.0, 2 mm potassium l-gluconate and 1 mm NAD+ in 100 mm Tris-HCl, pH 9.0, and 0.5 mm l-5-keto-gluconate and 0.2 mm NADPH in 100 mm HEPES-KOH, pH 7.0, respectively.
d-Idonate dehydratase activity was assayed by measuring the reducing activity of the product using DNS assay as described by Kuorelahtib et al. (14). The enzyme was mixed with 10 mm d-idonate, 0.5 mm FeCl2 and 20 mm DTT, in 100 mm Tris-HCl, pH 7.5, in a total volume of 100 μl, and the mixture was incubated at 30 °C for 15 min. The reaction was terminated by addition of 200 μl of DNS reagent and heated for 5 min in boiling water. After dilution with 900 μl of water, absorbance at 535 nm was measured. The molar absorption coefficient (ϵ = 91 m−1 cm−1) was obtained from a standard curve with KDGal that was produced by recombinant DgoD.
KDGal kinase activity was assayed at 25 °C by measuring the amount of ADP produced using a coupled assay with pyruvate kinase (PK) and lactate dehydrogenase (LDH). The enzyme was mixed with 2 mm KDGal, 1 mm ATP, 10 mm MgCl2, 0.2 mm NADH, 1 mm phosphoenolpyruvate containing 1.5 units of LDH and 2 units of PK (Sigma) in 100 mm HEPES-KOH, pH 7.5. The ADP produced by the kinase was used for production of pyruvate by PK, which was converted to lactate by LDH with concomitant oxidation of NADH.
KDPGal aldolase activity was assayed by measuring the reaction products pyruvate and d-glyceraldehyde 3-phosphate (GAP) by LDH and GAPDH reactions, respectively, based on the method described by Wong and Yao (15). The enzyme was mixed with 1 mm KDPGal in 100 mm potassium phosphate, pH 7.5, containing 0.2 mm NADH and 1.5 units of LDH or 2 mm NAD+ and 1.5 units of GAPDH (Sigma). The mixture was incubated at 25 °C, and the pyruvate or GAP produced was quantified by concomitant oxidation/reduction of NADH or NAD+, respectively. One unit of each enzyme activity was defined as the amount of the enzyme that reduced/oxidized 1 μmol of NAD(P)+/NADPH for LgdA, LgnH, LgnI, LgnF and LgnG and that which produced 1 μmol of KDGal for LgnE per min, under the assay condition.
Kinetic parameters Km and kcat were calculated using the fitting tool of Origin version 6.0 (OriginLab) with the basic Michaelis-Menten equation. Protein concentrations were determined by the method of Lowry et al. (16) with bovine serum albumin as a standard.
TLC and HPLC Analyses of Enzyme Reaction Products
Enzyme reaction products were routinely analyzed by TLC with silica gel plates (Merck) using ethyl acetate/methanol/acetate/water (6:1.5:1.5:1) as the mobile phase. Spots were detected by diphenylamine/aniline/phosphoric acid reagent (17). HPLC analysis was performed with a LC-2000 plus system (Jasco). Samples were applied to an IC Sep COREGEL 87H column (7.8 × 300 mm, Tokyo Chemical Industry) and eluted with 4 mm H2SO4 at a flow rate of 0.6 ml/min and 30 °C. Detection was conducted using a refractive index detector (RI-2031 plus, Jasco) and a chiral detector (OR-2090 plus, Jasco).
Purification of Reaction Products
The LgdA reaction product was produced in a 10-ml reaction mixture containing 2 units of LgdA, 100 mm Tris-HCl, pH 8.0, 50 mm l-glucose, 2 mm NAD+, with 100 mm pyruvate and 2 units of d-lactate dehydrogenase (Oriental Yeast) to regenerate NAD+. The reaction mixture was incubated at 30 °C for 8 h and treated with activated charcoal to remove NAD+ and NADH. The supernatant was then applied to a Dowex 1-X8 column (formate form, 200–400 mesh, Wako Pure Chemical). After washing with deionized water, the product was eluted with 100 mm formate. The eluted sample was concentrated by lyophilization and further purified by preparative TLC with a preparative silica gel plate (Merck) as above.
The LgnH reaction product was produced in a 10-ml reaction mixture containing 1 unit of LgnH, 100 mm Tris-HCl, pH 8.0, 20 mm potassium l-gluconate, 2 mm NAD+, and 40 mm scyllo-inosose (Hokko Chemical Industry) and 2 units of myo-inositol dehydrogenase (Sigma) for regeneration of NAD+. After 8 h of incubation at 30 °C, the reaction mixture was treated with activated charcoal and applied to a Dowex 1-X8 column (Cl− form). The column was washed with water, and the reaction product was eluted with 200 mm HCl. Then a 10% (w/v) slurry of calcium carbonate was added to the eluted fraction until the solution pH was neutral. After excess amounts of calcium carbonate were removed, the solution was lyophilized, and the resulting pellet was washed twice with 70% ethanol to remove CaCl2. The precipitate was resuspended in water, and an appropriate amount of Dowex 50W (H+ form, 200–400 mesh) was added to the solution and incubated for 30 min with shaking at room temperature so that it was dissolved in water as a free acid. Finally, the resin was removed, and the sample was lyophilized.
For production of LgnI reaction products, a reaction mixture (20 ml) containing 1 unit of LgnI, 100 mm potassium acetate, pH 5.0, 20 mm LgnH reaction product prepared as above, and 30 mm NADPH was incubated at 30 °C for 2 h with periodic pH adjustment to <6.0 by acetic acid addition. The reaction mixture was then treated with activated charcoal and loaded onto a Dowex 50W column. The column was washed with deionized water, and the flow-through fraction was concentrated with a rotary evaporator to remove the acetic acid. The dried sample was dissolved in water and loaded onto a Dowex 1-X8 column (formate form, 200–400 mesh). After the column was washed with deionized water, the product was eluted with 100 mm formate and lyophilized.
Likewise, the LgnE reaction product was produced in a 10-ml reaction mixture containing 2 units of LgnE, 100 mm Tris acetate, pH 7.5, 50 mm d-idonate, 20 mm DTT, and 0.5 mm FeCl2. After 5 h of incubation at 30 °C, the reaction mixture was filtered through a 0.22-μm filter to remove iron precipitates and passed over a AG 501-X8 column (Bio-Rad, 20–50 mesh) to remove proteins and negatively charged iron complexes. The flow-through fraction was loaded onto a Dowex 1-X8 column (acetate form, 200–400 mesh), and the column was washed with 80 mm acetate. The product was eluted with 200 mm potassium acetate, pH 7.0, and concentrated to ∼300 μl by lyophilization. One ml of ethanol was added to the solution, and the precipitates were washed twice with ethanol to remove potassium acetate (18).
NMR Analyses and Assignment of Reaction Products
NMR analyses were carried out at 25 °C with an Avance-500 apparatus (500 MHz, Bruker) at the University of Tsukuba Research Facility Center of Science and Technology with the purified reaction products dissolved in deuterium oxide. For samples of free acids and aldonolactones, the pH was adjusted to 8.0–9.0 by sodium deuteroxide. All chemical shifts were referenced to a 4,4-dimethyl-4-silapentane-1-sulfonic acid internal standard (0 ppm). LgdA and LgnE reaction product spectra were assigned by comparison with those available on the SDBS web of the National Institute of Advanced Industrial Science and Technology, Japan, and that reported by Kuorelahti et al. (14), respectively. Spectra for other compounds were assigned by HH-COSY and hetero-nuclear multiple quantum coherence analyses.
Construction of Gene Disruption Mutants of Strain 43P
Gene disruption was carried out by inserting a kanamycin-resistant gene (Kmr) into the target genes by homologous recombination. For construction of a mobile suicide vector, the mob region of pBBR1MCS2 was amplified with a primer set of Mob_F and Mob_R and cloned into the pGEM-T Easy vector to generate pGEM-mob, which was subsequently digested with SphI and SalI. The resultant fragment containing mob was cloned into pUC19 to generate pUC19-mob. Homologous regions for target genes were amplified by PCR with corresponding primer sets as follows: lgdA, LgdA1700_EcoF and LgdA1700_BamR; lgnE, LgnE2000_HinF and LgnE2000_XbaR; lgnH, LgnH1500_EcoF and LgnH1500_BamR; and lgnI, LgnI1500_XbaF and LgnI1500_SacIR. Each amplified fragment was digested with appropriate restriction enzymes and cloned into pUC19 to generate pUC19-lgdA, -lgnE, -lgnH and -lgnI, respectively. The Kmr cassette amplified by PCR from pBBR1MCS2 with primers PkanF and KanR was introduced into the EcoRV and the SmaI sites of pUC19-lgnE and -lgnI, respectively. Alternatively, primers Pkan_SacIIF and Kan_SacIIR were used for amplification of the Kmr cassette, which was digested with SacII and cloned into the SacII sites of pUC19-lgdA and pUC19-lgnH. Finally, the mob cassette from pUC19-mob was introduced into the EcoRI site of pUC19-lgdA::kan, -lgnE::kan, -lgnH::kan and the HindIII-XbaI site of pUC19-lgnI::kan. Introduction of the suicide plasmids into strain 43P cells was achieved by biparental mating with E. coli S17–1 λpir as a donor (19). Mating was carried out at a ratio of 1:10 (donor/recipient) for 8 h at 30 °C on a cellulose membrane filter placed on an LB agar plate without antibiotics. Transconjugants were selected by LB medium containing 100 μg/ml kanamycin and 15 μg/ml nalidixic acid, and proper integration was confirmed by PCR amplification with primers listed in supplemental Table S1.
Phylogenetic Analyses
The amino acid sequences of the l-glucose catabolic genes and related sequences from the KEGG genome database, together with those of bacterial genes in the same protein families having known functions (Gfo/Idh/MocA, zinc-containing ADH, short chain dehydrogenase, UxaA, and Aldolase_KDPG_KHG_2 for LgdA, LgnH, LgnI, LgnE and LgnG, respectively) that were selected from the “reviewed” entries from the UniProt KB database, were aligned with ClustalX2 software (20), and unrooted phylogenetic trees were visualized using TreeView software (21).
RESULTS
Isolation of l-Glucose-utilizing Bacteria
Several l-glucose-utilizing bacteria were isolated from soil samples by enrichment cultivation in l-Glc MM, on which strain 43P showed a maximum growth rate with a concomitant decrease of reducing sugars in the medium (Fig. 1). Phylogenetic analysis based on 16S rRNA gene sequences showed that strain 43P is included in the genus Paracoccus cluster, with the closest related species being Paracoccus denitrificans (96.8%, supplemental Fig. 1), although it should be noted that the P. denitrificans strain NBRC 102528T did not grow on l-Glc MM (Fig. 1). These results indicate that strain 43P assimilates l-glucose, and this strain should be classified in the genus Paracoccus. Therefore, we sought to further analyze l-glucose catabolism in strain 43P.
FIGURE 1.

Growth (open symbols) and l-glucose consumption (closed symbols) of strains 43P (circles) and NBRC 102528T (triangles) in l-Glc MM. Growth was monitored by measuring the absorbance at 600 nm. The DNS method was used to determine the l-glucose concentration by measuring the reducing sugars in the culture medium of strains 43P and NBRC 102528T. Cultivation was conducted in three independent cultures, and average values ± S.D. are shown.
l-GDH, the First Enzyme in the l-Glucose Catabolic Pathway
To determine the l-glucose catabolic pathway in strain 43P, we used cell-free extracts prepared from cells grown in l-Glc MM as an enzyme source and l-glucose as a substrate to analyze enzyme activities known to occur in the first reactions in sugar catabolism, such as kinase, isomerase, and dehydrogenase activity. No activities beyond NAD+-dependent l-GDH activity were detected, suggesting that the first reaction in this pathway is l-glucose oxidation. Because this activity was also detected when the strain was cultivated with d-galactose as the sole carbon source, we purified l-GDH from cells grown in a minimal medium containing d-galactose (supplemental Table 2). The purified enzyme showed a single band of ∼40 kDa on SDS-PAGE (supplemental Fig. 2A), and its N-terminal amino acid sequence MSNAEKALGVALIGTGFMGK exhibited 80% identity with that of P. denitrificans PD1222 Pden_1680, a putative oxidoreductase that is a member of the Gfo/Idh/MocA family. Therefore, we assumed that the gene encoding l-GDH in strain 43P is an ortholog of Pden_1680, and we then identified this gene by PCR and DNA sequencing as described under “Experimental Procedures.”
As expected, this gene, termed lgdA, showed 84% amino acid sequence identity with Pden_1680, and it was located in a cluster of putative inositol catabolic genes (Fig. 2A), the organization of which was quite similar to that of strain PD1222 with 76–89% identities, except that Pden_1673 encodes a hypothetical protein of 61 amino acids that was not present in strain 43P.
FIGURE 2.

Gene organization of the clusters containing lgdA (A) and lgn (B). Genes encoding the enzymes for l-glucose catabolism are shaded, and sequence identities to the corresponding P. denitrificans PD1222 genes are shown below.
Recombinant LgdA was produced with an E. coli pET system and purified (supplemental Fig. 2C). Purified LgdA showed NAD+-dependent l-GDH activity (Table 1), which is comparable with the native enzyme (kcat = 710 ± 24 min−1 and Km = 44.4 ± 6.3 mm) and also had dehydrogenase activity toward myo-inositol, scyllo-inositol, and d-glucose (Table 1), with the highest activity observed for scyllo-inositol. The reaction products of LgdA from an l-glucose source were analyzed by NMR and HPLC, which showed 1H and 13C chemical shifts that were identical to potassium d-gluconate (supplemental Fig. 3A) and optical rotation that was identical to potassium l-gluconate (Fig. 3A). These data indicate that LgdA catalyzes dehydrogenation of the l-glucose C1-hydroxyl group to form l-glucono-1,5-lactone, which would in turn be spontaneously hydrolyzed to l-gluconate.
TABLE 1.
Kinetic parameters of the l-glucose catabolic enzymes
The assays were conducted in triplicate, and average values ± S.D. are shown.
| Enzyme | Substrate | Km | kcat | kcat/Km |
|---|---|---|---|---|
| mm | min−1 | min−1 mm−1 | ||
| LgdA | l-Glucosea | 59.7 ± 5.7 | 1040 ± 28 | 17.4 |
| d-Glucosea | 88.7 ± 8.3 | 215 ± 36 | 2.42 | |
| scyllo-Inositola | 3.70 ± 0.4 | 705 ± 12 | 190 | |
| myo-Inositola | 53.3 ± 8.6 | 572 ± 30 | 10.7 | |
| NAD+b | 2.24 ± 0.4 | 742 ± 20 | 309 | |
| NADP+b | NDc | |||
| LgnH | l-Gluconated | 0.659 ± 0.023 | 573 ± 5 | 869 |
| l-Galactonated | 0.224 ± 0.011 | 178 ± 2 | 828 | |
| NAD+e | 0.155 ± 0.006 | 543 ± 4 | 3500 | |
| NADP+e | NDc | |||
| LgnI | l-5-Keto-gluconatef | 0.180 ± 0.020 | 1200 ± 33 | 6640 |
| NADPHg | 0.085 ± 0.004 | 1910 ± 90 | 22,500 | |
| NADHg | NDh | |||
| LgnE | d-Idonate | 0.454 ± 0.072 | 376 ± 12 | 828 |
| LgnF | KDGal | 0.629 ± 0.054 | 1710 ± 34 | 2720 |
| LgnG | KDPGali | 0.587 ± 0.023 | 4290 ± 270 | 7310 |
| KDPGalj | 0.466 ± 0.012 | 3900 ± 31 | 8370 |
a The activity was measured with a fixed concentration of 10 mm NAD+.
b The activity was measured with a fixed concentration of 20 mm scyllo-inositol.
c Activity was not detected with 10 mm NADP+.
d The activity was measured with a fixed concentration of 2 mm NAD+.
e The activity was measured with a fixed concentration of 10 mm l-gluconate.
f The activity was measured with a fixed concentration of 0.2 mm NADPH.
g The activity was measured with a fixed concentration of 2 mm l-5-keto-gluconate.
h Activity was not detected with 0.2 mm NADH.
i The activity was measured by pyruvate production.
j The activity was measured by GAP production.
FIGURE 3.
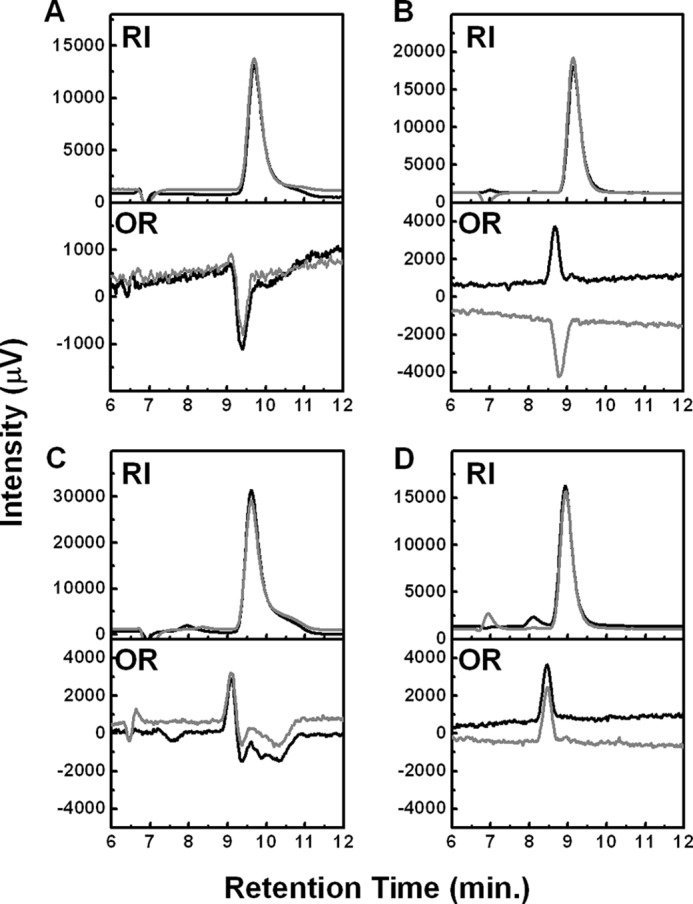
HPLC analysis of LgdA (A), LgnH (B), LgnI (C), and LgnE (D) reaction products detected by a refractive index (RI) and chiral detector (OR). Black and gray lines indicate chromatograms of the reaction products and authentic compounds l-gluconate (A), d-5-keto-gluconate (B), d-idonate (C), and KDGal (D), respectively.
Pathway Downstream of l-Gluconate
Next, we evaluated enzyme activity toward l-gluconate with cell-free extracts of strain 43P cultivated in l-Glc MM, and again an NAD+-dependent l-GnDH activity was detected. The enzyme catalyzing the reaction was partially purified by about 100-fold (supplemental Table 3) and showed a 35-kDa band on SDS-PAGE that corresponded to the activity (supplemental Fig. 2B). This protein had an N-terminal amino acid sequence of MKALIIEKEGHTVIGEISEP, which has 100% identity with strain PD1222 Pden_4931, a putative oxidoreductase that is a member of the zinc-containing alcohol dehydrogenase family. As such, the same PCR-based strategy used to identify lgdA was adopted to obtain sequence information for the gene for l-GnDH and its surrounding region.
The gene for l-GnDH was found in a nine-gene cluster corresponding to Pden_4924 to Pden_4932 in strain PD1222 with amino acid sequence identities of 71–91% (Fig. 2B), and it was designated lgnA-lgnI. At the cluster's upstream end, we also found a gene corresponding to Pden_4923, termed lgnR, which may function as a transcriptional regulator of the cluster. Based on the Pden gene annotations, the first four genes of the cluster, lgnA–lgnD, presumably encode subunits of an ABC transporter for polar amino acids, whereas lgnE–lgnI encode, respectively, a galactarate dehydratase, a KDGal kinase, a KDPGal aldolase, an alcohol dehydrogenase (l-GnDH), and a short chain dehydrogenase/reductase. This information led us to consider the possibility that l-gluconate can be assimilated by enzymes encoded in this cluster, and this was assessed by analyzing each enzyme activity using purified recombinant enzymes (supplemental Fig. 2C).
l-Gluconate Dehydrogenation by LgnH
As expected, LgnH showed l-GnDH activity (Table 1). The activity was solely dependent on NAD+, not NADP+. The enzyme also showed dehydrogenase activity toward l-galactonate, the C-4 epimer of l-gluconate, whereas no activity was observed toward other epimers, such as d-idonate (C-5) and l-mannonate (C-2) and uronic acids such as d-glucuronate and d-galacturonate. NMR analysis of the reaction product from l-gluconate showed chemical shifts identical to that of commercial d-5-keto-gluconate (supplemental Fig. 3B), whereas HPLC analysis showed opposite optical rotation to d-5-keto-gluconate (Fig. 3B). These data clearly indicate that LgnH catalyzes dehydrogenation of the l-gluconate C-5 hydroxyl group to form l-5-keto-gluconate.
LgnI-mediated l-5-Keto-gluconate Reduction
LgnI is a member of the short chain dehydrogenase/reductase family and is therefore predicted to catalyze NAD(P)+/NAD(P)H-dependent oxidoreductase activity. Consequently, reduction of the purified reaction product of LgnH, l-5-keto-gluconate, was observed with LgnI in an NADPH-dependent manner (Table 1). The reaction product showed the same chemical shifts and peak in NMR and HPLC analyses, respectively, as an authentic d-idonate, the C-5 epimer of l-gluconate (supplemental Fig. 3C and Fig. 3C). Therefore, we concluded that LgnI is responsible for the reduction of l-5-keto-gluconate to form d-idonate.
LgnE-mediated Dehydration of d-Idonate
When LgnE was incubated with d-idonate in the presence of FeCl2 and DTT, the formation of reducing sugars was detected (Table 1). We did not detect any activity toward meso-galactarate, although LgnE showed sequence similarity to galactarate dehydratase (GarD) from E. coli K-12 (38%). NMR and HPLC analyses of the reaction products showed, respectively, the same chemical shifts and peaks as KDGal, which was prepared from d-galactonate using recombinant E. coli d-galactonate dehydratase (DgoD) (supplemental Fig. 3D and Fig. 3D). These data indicate that LgnE catalyzes dehydration of d-idonate to produce KDGal.
Phosphorylation and Aldol Cleavage of KDGal by LgnF and LgnG
Because LgnF and LgnG showed amino acid sequence similarity to E. coli DgoK (33%) and DgoA (43%), which catalyze ATP-dependent phosphorylation of KDGal to form KDPGal and aldol-cleavage of KDPGal to form pyruvate and GAP, respectively, we tested their corresponding activities.
As expected, LgnF produced ADP from ATP only in the presence of KDGal, indicating that it phosphorylates KDGal (Table 1). Also, pyruvate and GAP formation was enzymatically detected when LgnG was incubated with KDPGal. These data clearly show that LgnF catalyzes phosphorylation of KDGal to form KDPGal, which is then converted to pyruvate and GAP by LgnG.
Involvement of the Identified Genes in the l-Glucose Catabolic Pathway
To determine how the identified genes participate in the l-glucose catabolic pathway, we constructed gene disruption mutants of lgdA, lgnE, lgnH, and lgnI, based on strain 43P, by inserting a Kmr cassette into the respective genes (supplemental Fig. 4). Growth of cells harboring these disrupted genes in minimal medium containing l-glucose or related compounds was observed. As expected, the ΔlgdA, ΔlgnH, and ΔlgnI mutants did not grow in the presence of “upstream” compounds of the pathway, such as l-glucose for ΔlgdA and l-glucose and l-gluconate for ΔlgnH and ΔlgnI mutants, whereas they could grow using “downstream” compounds (Fig. 4). These results strongly indicate that these genes are responsible for l-glucose catabolism.
FIGURE 4.

Growth of strain 43P and gene disruption mutants in minimal media containing l-glucose (A), l-gluconate (B), d-idonate (C), l-galactonate (D), scyllo-inositol (E), and myo-inositol (F). Open circles, strain 43P; closed circles, ΔlgdA strain; open triangles, ΔlgnE strain; closed triangles, ΔlgnH strain; open squares, ΔlgnI strain. B, the growth of strain NBRC 102528T is also shown (closed squares). Cultivation was conducted in three independent cultures, and average values ± S.D. are shown.
In contrast, the ΔlgnE mutant showed rather weak but distinct growth in the presence of each compound tested. It may be possible that strain 43P possesses a paralog to lgnE, as the related strain, PD1222, possesses such a paralog, Pden_4671, which exhibits 53% identity.
Although LgnH utilized l-galactonate as a substrate as well as l-gluconate, none of the gene-disrupted mutants, including the ΔlgnH mutant, showed growth defects with l-galactonate (Fig. 4D), suggesting that these genes are not involved in l-galactonate catabolism (see below). Also, none of the mutants showed growth defects with scyllo- and myo-inositols, except the ΔlgdA mutation affected the growth with scyllo-inositol severely (Fig. 4, E and F). These results indicate that lgdA is required for both l-glucose and scyllo-inositol utilization, whereas the genes in the lgn cluster are not involved in inositol catabolism.
DISCUSSION
This study sought to describe the l-glucose catabolic pathway in Paracoccus sp. 43P, a model for which is shown in Fig. 5. Based on the gene organization, this pathway could be divided into two parts, oxidation of l-glucose by LgdA and l-gluconate catabolism by enzymes encoded by the lgn cluster.
FIGURE 5.

Model of the l-glucose catabolic pathway in strain 43P based on the results of this study. Altered moieties in each enzyme reaction are shown in gray.
LgdA showed sequence similarity to the Gfo/IDH/MocA family proteins, and its gene was located in a putative inositol utilization gene cluster of strain 43P. Kinetic analysis of purified LgdA showed efficient oxidation of scyllo-inositol rather than l-glucose, and the ΔlgdA mutation affected the growth with scyllo-inositol. Together, these results strongly indicate that the physiological role of LgdA involves scyllo-inositol catabolism. Among inositol dehydrogenases (IDHs), myo-IDH of Bacillus subtilis (IolG) is the best characterized enzyme, and it was reported to catalyze oxidation of the axial hydroxyl group of myo-inositol and d-chiro-inositol (22). IolG also catalyzes oxidation of α-d-glucose and α-d-xylose (23), which possess the same stereo-configuration of myo-inositol hydroxyl groups. A β-anomer of l-glucose possesses the same stereo-configuration as scyllo-inositol, i.e. hydroxyl groups occupy all equatorial positions, so it is possible that LgdA utilizes both l-glucose and scyllo-inositol as substrates. There are two scyllo-IDHs that have been characterized at an enzyme level, IolW and IolX from B. subtilis (24), and these enzymes showed low sequence identity with LgdA (22.1 and 26.3%, respectively). IolX showed no activity toward l-glucose, whereas IolW had only low activity (kcat/Km = 0.37 min−1 mm−1)3 that was about 1/50th that of LgdA. These findings may indicate that LgdA has a unique substrate recognition mechanism.
However, results for the ΔlgdA mutant showed a clear involvement of this gene in l-glucose catabolism. Although l-glucose is an unnatural sugar, these results indicate that, at least for strain 43P, bacterial inositol dehydrogenases may be involved in both inositol and sugar catabolism.
The reactions downstream of the LgdA reaction product, l-gluconate, are analogous to those of the E. coli l-galactonate catabolic pathway, which is the C-4 epimer of l-gluconate (25, 26). Both of the l-aldonates are epimerized at the C-5 position by dehydrogenase/reductase reactions (d/l conversion) to produce d-aldonates, converted to d-2-keto-3-deoxyaldonates by dehydratases, phosphorylated at C-6 position by kinases, and then converted by aldolases to the glycolysis substrates pyruvate and GAP. Comparison of the amino acid sequences of the lgn enzymes, LgnH, LgnI, LgnE, LgnF, and LgnG, with those of corresponding enzymes in the E. coli l-galactonate pathway, YjjN, UxaB, UxaA, KdgK and Eda, respectively, showed limited sequence identities of about 30%, with LgnI and LgnF in particular showing no similarities to their corresponding proteins. Moreover, the mutant deficient in LgnH or LgnI could not grow on l-gluconate, and it did not show any growth defects on l-galactonate (Fig. 4D). Taken together, we conclude that the lgn cluster enzymes are specific to l-gluconate rather than l-galactonate.
The d/l conversion by the dehydrogenase/reductase reactions are also seen in the catabolism of other six-carbon l-aldonates, such as l-gulonate and l-idonate, in E. coli (27, 28). Like the pathway described in this study, the final products of these catabolic pathways are pyruvate and GAP, where GAP is used as a key intermediate for substrate level phosphorylation in the downstream portion of the Embden-Meyerhof-Parnas pathway. Because l-glyceraldehyde 3-phosphate is not utilized as a substrate by GAPDH, but rather shows a bactericidal effect (29), the d/l conversion may be important for obtaining the GAP d-form following cleavage reactions that are catalyzed by aldolases. In contrast, the fungal l-galactonate catabolic pathway does not involve this process, with l-2-keto-3-deoxygalactonate, formed by l-galactonate dehydratase, being directly cleaved to form pyruvate and l-glyceraldehyde. However, the latter product is not utilized directly but is reduced to an achiral compound, glycerol (30, 31). Therefore, a homochirality issue is likely present in the catabolism of glyceraldehyde, the minimum chiral compound, not in six-carbon sugars. This notion agrees with the recent findings that d-glyceraldehyde might have been selectively synthesized in the prebiotic era, via a formose reaction in the presence of l-amino acids (32).
Phylogenetic analyses based on the amino acid sequences of the enzymes in this pathway and their related sequences showed that they are located in deeply branched lineages that are distinct from those of enzymes in the corresponding protein families whose functions are known and from KEGG genome database sequences, except for those from P. denitrificans (supplemental Fig. 5). The lineages containing LgdA, LgnF, and LgnG were included in the clusters of Alphaproteobacteria, whereas those of LgnH, LgnI, and LgnE diverged at branch points with clusters including sequences from other bacterial affiliations, i.e. clusters containing sequences mainly from Bacteroidetes for LgnH and LgnI and Betaproteobacteria for LgnE. This divergence may indicate a unique evolutionary origin of this catabolic pathway.
There are several microorganisms in the class Alpha-, Beta-, and Gammaproteobacteria that possess sets of all six orthologs, or five orthologs corresponding to the enzymes for l-gluconate assimilation, which can be assumed as “potential” l-glucose or l-gluconate utilizing organisms from a metabolic pathway standpoint. These orthologs were widely distributed within the clusters of those from the genome sequences, and there are no organisms other than P. denitrificans that possess all such orthologs found in strain 43P in the same or nearby clusters. Most of these organisms likely cannot assimilate l-glucose or l-gluconate, because there are no previous reports on the assimilation of such compounds. Rather, it may be that either no other organisms can utilize such compounds or different catabolic pathways for l-glucose or l-gluconate exist if any of these organisms can indeed utilize them.
P. denitrificans utilized l-gluconate (Fig. 4B) but not l-glucose (Fig. 1). In our preliminary experiments, recombinant Pden_1680 from strain NBRC 102528T showed l-GDH activity similar to LgdA (kcat = 836 ± 12 min−1, Km = 78.0 ± 8.2 mm), indicating that this strain possesses the ability to utilize l-glucose. The reason why P. denitrificans cannot assimilate l-glucose is not known, but it could be because the gene cluster, including Pden_1680, is not transcribed or l-glucose cannot be incorporated into the cells, at least under our culture conditions.
l-Glucose is not found in natural environment, and therefore the physiological role of this catabolic pathway is unknown. However, our data clearly show that the lgn gene cluster is specifically responsible for l-gluconate utilization. This finding may indicate the presence of l-gluconate in nature, which is believed to not exist like l-glucose. We are now attempting to analyze the l-glucose catabolic pathway of the other isolates we obtained, which will lead to a better understanding of l-glucose catabolism.
Supplementary Material
This work was supported by NISR Research Grant from the Noda Institute for Scientific Research, Japan, and grants-in-aid for scientific research (B) from Japan Society for Promotion of Science.

This article contains supplemental Figs. 1–5 and Tables 1–3.
The nucleotide sequence(s) reported in this paper has been submitted to the DDBJ/GenBankTM/EBI Data Bank with accession number(s) AB727354, AB727355, and AB727356.
Dong-min Kang and Ken-ichi Yoshida, Kobe University, personal communication.
- l-Glc MM
- l-glucose minimal medium
- l-GDH
- l-glucose dehydrogenase
- DNS
- 3,5-dinitrosalicylic acid
- l-GnDH
- l-gluconate dehydrogenase
- KDGal
- d-2-keto-3-deoxygalactonate
- KDPGal
- d-2-keto-3-deoxy-6-phosphogalactonate
- GAP
- d-glyceraldehyde-3-phosphate
- LDH
- l-lactate dehydrogenase
- PK
- pyruvate kinase
- OR
- optical rotation
- IDH
- inositol dehydrogenase.
REFERENCES
- 1. Rudney H. (1940) The utilization of l-glucose by mammalian tissue and bacteria. Science 92, 112–113 [DOI] [PubMed] [Google Scholar]
- 2. Livesey G., Brown J. C. (1995) Whole body metabolism is not restricted to d-sugars because energy metabolism of l-sugars fits a computational model in rats. J. Nutr. 125, 3020–3029 [DOI] [PubMed] [Google Scholar]
- 3. Sun H. J., Saccomanno V., Hedlund B., Mckay C. P. (2009) Stereo-specific glucose consumption may be used to distinguish between chemical and biological reactivity on Mars: a preliminary test on Earth. Astrobiol. 9, 443–446 [DOI] [PubMed] [Google Scholar]
- 4. Sasajima K. I., Sinskey A. J. (1979) Oxidation of lglucose by a Pseudomonad. Biochim. Biophys. Acta 571, 120–126 [DOI] [PubMed] [Google Scholar]
- 5. Kovach M. E., Elzer P. H., Hill D. S., Robertson G. T., Farris M. A., Roop R. M., 2nd, Peterson K. M. (1995) Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic resistance cassettes. Gene 166, 175–176 [DOI] [PubMed] [Google Scholar]
- 6. Moore S., Link K. P. (1940) Carbohydrate characterization. J. Biol. Chem. 133, 293–311 [Google Scholar]
- 7. Babbitt P. C., Mrachko G. T., Hasson M. S., Huisman G. W., Kolter R., Ringe D., Petsko G. A., Kenyon G. L., Gerlt J. A. (1995) A functionally diverse enzyme superfamily that abstracts the α protons of carboxylic acid. Science 267, 1159–1161 [DOI] [PubMed] [Google Scholar]
- 8. Miller G. L. (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 31, 426–428 [Google Scholar]
- 9. Lobanok A. G., Sapunova L. I., Dikhtievski Y. O., Kazakevich I. O. (1998) Screening of glucose isomerase-producing microorganisms. World J. Microbiol. Biotechnol. 14, 259–262 [Google Scholar]
- 10. Hemker M., Stratmann A., Goeke K., Schröder W., Lenz J., Piepersberg W., Pape H. (2001) Identification, cloning, expression, and characterization of the extracellular acarbose-modifying glycosyltransferase, AcbD, from Actinoplanes sp. strain SE50. J. Bacteriol. 183, 4484–4492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Howe A. F., Groom T, Carter R. G. (1964) Use of polyethylene glycol in the concentration of protein solution. Anal. Biochem. 9, 443–453 [DOI] [PubMed] [Google Scholar]
- 12. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 13. Ochman H., Gerber A. S., Hartl D. L. (1988) Genetic application of an inverse polymerase chain reaction. Genetics 120, 621–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuorelahti S., Jouhten P., Maaheimo H., Penttilä M., Richard P. (2006) l-Galactonate dehydratase is a part of the fungal path for d-galacturonic acid catabolism. Mol. Microbiol. 61, 1060–1068 [DOI] [PubMed] [Google Scholar]
- 15. Wong T. Y., Yao X. T. (1994) The Dely-Doudoroff pathway of galactose metabolism in Azotobacter vinelandii. Appl. Environ. Microbiol. 60, 2065–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 17. Anderson K., Li S. C., Li Y. T. (2000) Diphenylamine-aniline-phosphoric acid reagent, a versatile spray reagent for revealing glycoconjugates on thin layer chromatography plates. Anal. Biochem. 287, 337–339 [DOI] [PubMed] [Google Scholar]
- 18. De Groeve M. R., Depreitere V., Desmet T., Soetaert W. (2009) Enzymatic production of α-d-galactose 1-phosphate by lactose phosphorolysis. Biotechnol. Lett. 31, 1873–1877 [DOI] [PubMed] [Google Scholar]
- 19. Simon R., Priefer U., Puhler A. (1983) A broad host range mobilization system for in vivo genetic engineering. Transposon mutagenesis in Gram-negative bacteria. Nat. Biotechnol. 1, 784–791 [Google Scholar]
- 20. Larkinn M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) CLUSTAL W and CLUSTAL_X version 2.0. Bioinfomatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 21. Page R. D. (1996) TreeView. An application to display phylogenetic tree on personal computers. Comput. Appl. Biosci. 12, 357–358 [DOI] [PubMed] [Google Scholar]
- 22. Yoshida K., Yamaguchi M., Morinaga T., Kinehara M., Ikeuchi M., Ashida H., Fujita Y. (2008) myo-Inositol catabolism in Bacillus subtilis. J. Biol. Chem. 283, 10415–10424 [DOI] [PubMed] [Google Scholar]
- 23. Ramaley R., Fujita Y., Freese E. (1979) Purification and properties of Bacillus subtilis inositol dehydrogenase. J. Biol. Chem. 254, 7684–7690 [PubMed] [Google Scholar]
- 24. Morinaga T., Ashida H., Yoshida K. (2010) Identification of two scyllo-inositol dehydrogenase from Bacillus subtilis. Microbiology 156, 1538–1546 [DOI] [PubMed] [Google Scholar]
- 25. Cooper R. A. (1979) The pathway for l-galactonate catabolism in Escherichia coli K-12. FEBS Lett. 103, 216–220 [DOI] [PubMed] [Google Scholar]
- 26. Reed J. L., Patel T. R., Chen K. H., Joyce A. R., Applebee M. K., Herring C. D., Bui O. T., Knight E. M., Fong S. S., Palsson B. O. (2006) System approach to refining genome annotation. Proc. Natl. Acad. Sci. U.S.A. 103, 17480–17484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cooper R. A. (1980) The pathway for l-gulonate catabolism in Escherichia coli K-12 and Salmonella typhimurium LT-2. FEBS Lett. 115, 63–67 [DOI] [PubMed] [Google Scholar]
- 28. Bausch C., Peekhaus N., Utz U., Blais T., Murray E., Lowary T., Conway T. (1998) Sequence analysis of the GntII (subsidiary) system for gluconate metabolism reveals novel pathway for l-idonic acid catabolism in Escherichia coli. J. Bacteriol. 180, 3704–3710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kalyananda M. K., Engel R., Tropp B. E. (1987) Metabolism of l-glyceraldehyde 3-phosphate in Escherichia coli. J. Bacteriol. 169, 2488–2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang L., Thiewes H., van Kan J. A. (2011) d-Galacturonic acid catabolic pathway in Botrytis cinerea. Fungal Genet. Biol. 48, 990–997 [DOI] [PubMed] [Google Scholar]
- 31. Hilditch S., Berghäll S., Kalkkinen N., Penttilä M., Richard P. (2007) The missing link in the fungal d-galacturonate pathway: identification of the l-threo-3-deoxy-hexulosonate aldolase. J. Biol. Chem. 282, 26195–26201 [DOI] [PubMed] [Google Scholar]
- 32. Breslow R., Cheng Z. L. (2010) l-Amino acid catalyze the formation of an excess of d-glyceraldehyde, and thus of other d-sugars, under credible prebiotic conditions. Proc. Natl. Acad. Sci. U.S.A. 107, 5723–5725 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.