

Background: NEG2 regulates CFTR gating but the mechanism is unknown.
Results: A putative NEG2-CL3 electrostatic attraction, possibly weakened by Arg-764/Arg-766 of the R domain, prohibited CFTR activation. A charge exchange between NEG2 and CL3 caused misprocessing.
Conclusion: Electrostatic regulation of CFTR activation and processing may be asymmetric at the CL3-R interface.
Significance: The CL3-R interface is optimally designed for multiple regulations of CFTR functions.
Keywords: ABC Transporter, Anion Transport, CFTR, Patch Clamp Electrophysiology, Phosphorylation, Electrostatic Regulation, Gating, Processing, Asymmetry, Interdomain Interaction
Abstract
NEG2, a short C-terminal segment (817–838) of the unique regulatory (R) domain of the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel, has been reported to regulate CFTR gating in response to cAMP-dependent R domain phosphorylation. The underlying mechanism, however, is unclear. Here, Lys-946 of cytoplasmic loop 3 (CL3) is proposed as counter-ion of Asp-835, Asp-836, or Glu-838 of NEG2 to prevent the channel activation by PKA. Arg-764 or Arg-766 of the Ser-768 phosphorylation site of the R domain is proposed to promote the channel activation possibly by weakening the putative CL3-NEG2 electrostatic attraction. First, not only D835A, D836A, and E838A but also K946A reduced the PKA-dependent CFTR activation. Second, both K946D and D835R/D836R/E838R mutants were activated by ATP and curcumin to a different extent. Third, R764A and R766A mutants enhanced the PKA-dependent activation. However, it is very exciting that D835R/D836R/E838R and K946D/H950D and H950R exhibited normal channel processing and activity whereas D835R/D836R/E838R/K946D/H950D was fractionally misprocessed and silent in response to forskolin. Further, D836R and E838R played a critical role in the asymmetric electrostatic regulation of CFTR processing, and Ser-768 phosphorylation may not be involved. Thus, a complex interfacial interaction among CL3, NEG2, and the Ser-768 phosphorylation site may be responsible for the asymmetric electrostatic regulation of CFTR activation and processing.
Introduction
The cystic fibrosis (CF)2 transmembrane conductance regulator (CFTR) chloride channel is a unique member of the ATP-binding cassette family because it has a unique regulatory (R) domain flanked by two intracellular nucleotide-binding domains (NBDs) and two transmembrane domains (TMDs) (Fig. 1A) (1, 2). Both TMDs consist of six membrane-spanning helixes that are extended to the intracellular side to form four long cytoplasmic loops (CLs) (3). These CLs interact not only with NBD1 and NBD2 but also with the R domain (3–10). The pore formed by TMD1 and TMD2 is gated by binding/hydrolysis of ATP at the interface of NBD1 and NBD2 and regulated by R domain phosphorylation/dephosphorylation (11, 12). Although CLs have been found to play a central role in channel gating (6, 7, 9, 10, 13, 14), it is largely unknown how CLs couple ATP actions at NBDs to gating rearrangements of TMDs and how R domain phosphorylation by cAMP-dependent PKA mediates the coupling.
FIGURE 1.

CFTR schematic. A, the R domain is inserted between NBD1 and TMD2. Four cytoplasmic loops (CLs) are extended from TMD1 and TMD2. Residues at the interface of the R domain and CL3 are highlighted with red crosses. B, a putative model defines proximity relationships and the relative orientation of charged residues at the R-CL3 interface. Three helixes are based on three reports (3, 19, 30). NEG2 is a small C-terminal segment (817–838) of the R domain and has a conserved helical region and a net charge of −9 (19).
The R domain is a segment with 193 amino acids (645–838). It has multiple phosphorylation sites, most of which are stimulatory except Ser-768 and Ser-737 (9, 10, 12, 15–18). Recent functional studies revealed that NEG2 (1B), which is a small C-terminal segment (817–838) of the R domain and has a conserved helical region and a net charge of −9, plays a critical role in CFTR gating and trafficking in response to cAMP stimulation (19, 20). Removal of NEG2 suppresses PKA-dependent activation and trafficking of CFTR (19, 20). However, the underlying mechanism is still a mystery.
Recent functional and biochemical studies have shown that correct interdomain interactions of CFTR are required not only for its optimal activation but also for its optimal folding, biosynthesis, and trafficking from endoplasmic reticulum to the plasma membrane (6, 8, 21–23). Our recent investigations uncovered that Ser-768 inhibits the CFTR activity by interacting with CL3 no matter whether it is phosphorylated or not (9, 10). In the nonphosphorylated state, the -OH group of Ser-768 forms a putative H-bond with the imidazole group of His-950 of CL3 (10). Once phosphorylated, Ser-768 may enhance binding of Fe3+ to His-950, His-954, Cys-832, Asp-836, and His-775 at the interface of the R domain and CL3 (9). Because Cys-832 and Asp-836 are residues of NEG2 and close to CL3 (19), we wondered whether the interaction of NEG2 with CL3 modulates CFTR activation and whether the Ser-768 phosphorylation site is involved (Fig. 1B).
EXPERIMENTAL PROCEDURES
Molecular Biology
Human wild type (WT) CFTR was subcloned into the pCDNA3 mammalian expression vector (Invitrogen). All of the mutants were produced by using the QuikChange site-directed mutagenesis kit from Stratagene and confirmed by automated sequencing and Western blotting. A Cys-free construct without all 18 cysteines was provided by Dr. David Gadsby (Rockefeller University). ΔR-S660A-CFTR was provided by Michael Welsh (University of Iowa, Iowa City, IA).
Cell Culture and Transfection
Human embryonic kidney (HEK)-293T cells were transiently transfected with WT or mutant CFTR cDNA using the Lipofectamine transfection kit (Invitrogen). Cells were cultured in Dulbecco's modified Eagle's medium (Mediatech) supplemented with 10% fetal bovine serum and 1 mm penicillin/streptomycin. For patch clamp recordings, all cells were grown on plastic coverslips and used 1–4 days after seeding.
Patch Clamp Analysis
Some channels expressed in HEK-293T cells were recorded in inside-out configurations (Dagan 3900A; Dagan Corp., Minneapolis, MN) for intracellular application of reagents to the cytoplasmic face. CFTR currents were recorded in symmetrical solutions each containing 140 mm N-methyl-d-glutamine chloride, 3 mm MgCl2, 1 mm EGTA, and 10 mm TES (pH 7.3). The resulting resistance of borosilicate patch pipette was 3–4 megohms in the bath solution. Macroscopic currents were evoked using a ramp protocol from +80 to −80 mV with a 10.75-s time period and filtered at 200 Hz. Single-channel recordings at 60 mV were filtered at 20 Hz. 1.5 mm MgATP and 100 units/ml PKA were used to activate the CFTR channel. For those channels expressed in HEK-293T cells and recorded in whole cell configurations, a ramp protocol from +40 to −40 mV was employed with a 10.75-s time period and filtered at 200 Hz. Moreover, 1.5 mm ATP was introduced in the electrode solution, and 50 μm forskolin and 50 μm curcumin were used to estimate the current density of some CFTR mutants. All experiments were carried out at room temperature (22 ± 1 °C). Data were acquired and analyzed using pCLAMP8.1 software (Axon Instruments). Data are shown as mean ± S.E. Student's t test was used to test statistical significance. Curve fitting was made using Microcal Origin software.
Western Blot Analysis
Transfected HEK-293T cells expressing CFTR WT and mutants were washed in divalent-free PBS (Mediatech, Herndon, VA) and then solubilized in 100 μl of SDS sample buffer for SDS-PAGE analysis. Each sample was running on a 4–15% SDS-polyacrylamide gel. Separated proteins were transferred onto PVDF membranes for 60 min at 24 V (Genie Blotter; Research Products International Corp.). The membranes were blocked overnight with LI-COR blocking buffer (LI-COR, Lincoln, NE) and then Western-blotted with the C-terminal CFTR antibody mAb 24-1 (R&D Systems) at 1:5000 and detected with goat anti-mouse Alexa Fluor 680-conjugated antibody (Molecular Probes, Eugene, OR) at 1:100,000. Blots were extensively washed by a TBS buffer (20 mm Tris base and 135 mm NaCl, pH 7.5) and then scanned to obtain fluorescent images with an Odyssey scanner (LI-COR).
RESULTS
Electrostatic Expulsions between the R Domain and CL3 Reduce the PKA-dependent Channel Activation
To address whether a putative electrostatic attraction between CL3 and NEG2 regulates CFTR gating, we first determined the PKA-dependent activity of several mutants at the CL3-NEG2 interface. Fig. 2A indicates that a low concentration (6 units/ml) of PKA activated WT CFTR slowly and weakly. In contrast, mutation of Lys-946 or Asp-836 to alanine clearly accelerated the channel activation and reduced the PKA dependence (Fig. 2, B and C). Similar observations with D835A and E838A are summarized in Fig. 2D. The K1/2 for PKA activation was reduced from 10 to 5 units/ml once Lys-946 from the CL3, and Asp-835, Asp-836, and Glu-838 from NEG2 were mutated to alanines (Fig. 2D). However, K951A failed to change the PKA-dependent channel activity (Fig. 2D). Thus, a putative electrostatic attraction between Lys-946 and Asp-835, Asp-836, or Glu-838 may weaken the PKA sensitivity of CFTR activation.
FIGURE 2.

The PKA-dependent activity of CFTR mutants at the R-CL3 interface. A–C, macroscopic currents across inside-out membrane patches excised from transfected HEK-293T cells expressing WT CFTR (A) and mutants K946A (B) and D836A (C) by using a ramp protocol (±80 mV). In the presence of 1.5 mm ATP, PKA was added gradually until the current was no longer increased. The arrows indicate the final concentrations. 200 μm glibenclamide or 10 μm CFTRinh172 was used to inhibit the channel activity. D, PKA titration curves for CFTR mutants at the R-CL3 interface (n = 3–5). WT CFTR was a control.
To further confirm the putative electrostatic attraction between CL3 and NEG2, we determined the effects of curcumin on the mutants at the CL3-NEG2 interface. In the presence of 1.5 mm ATP, 50 μm curcumin greatly potentiated the K946A mutant activity, which was further increased by PKA (Fig. 3A). If this effect is due to a disruption of the putative electrostatic interaction between CL3 and NEG2, K946D or D835R/D836R/E838R should exhibit a similar effect in the presence of ATP. To support this argument, ATP and curcumin partially increased the K946D activity, and PKA (6 units/ml) completely activated this mutant (Fig. 3B). In contrast, ATP and curcumin dramatically activated the D835R/D836R/E838R mutant. However, addition of PKA decreased the channel activity (Fig. 3C). This decrease in the channel current may be due to the side effect of curcumin (24). It is interesting that ATP and curcumin only activated the K946D mutant by 40% but activated the D835R/D836R/E838R mutant completely (Fig. 3D). Thus, the regulation of CFTR activation by the putative electrostatic attraction between Lys-946 and NEG2 may be limited and asymmetric. The triple arginine mutant of NEG2 may not only disrupt the putative electrostatic interaction with K946 but also weaken the Fe3+ binding at the CL3-R interface because Asp-836 is one of important Fe3+ ligands (9). However, for K946D, His-950 or His-954 may still form the inhibitive Fe3+ bridge with the R domain. To support this argument, K946D/H950D was employed to weaken both the electrostatic attractions and the Fe3+ bridge between CL3 and the R domain. Fig. 3D demonstrates that ATP and curcumin completely activated the K946D/H950D mutant. This high activity may not be due to the H950D mutation because it cannot be activated by ATP and curcumin (10). Thus, the inhibitive Fe3+ bridge at the R-CL3 interface should be weakened when the electrostatic interactions at the same interface are investigated.
FIGURE 3.

Activation of CFTR mutants at the R-CL3 interface by ATP and curcumin. Macroscopic currents across inside-out membrane patches excised from transfected HEK-293T cells expressing CFTR mutants K946A (A), K946D (B) and D835R/D836R/E838R (C) by using a ramp protocol (±80 mV) are shown. A–C, currents were activated with 1.5 mm ATP and 50 μm curcumin followed by 6 units/ml PKA catalytic subunit but inhibited by 200 μm glibenclamide or 10 μm CFTRinh172. The arrows indicate the final concentrations. D, relative current of CFTR mutants induced by ATP and curcumin at the R-CL3 interface is shown. The relative activity of K946D was significantly smaller than that of D835R/D836R/E838R (n = 3–4; *, p < 0.05, unpaired t test); error bars, S.E.
On the other hand, previous studies demonstrated that CFTR can be activated by ATP alone, and PKA does not exert any effect on the channel activation when NEG2 is deleted (19). If this ATP-induced activation results from the disruption of the putative electrostatic attraction between CL3 and the C terminus of NEG2, a similar effect should be observed. However, neither K946D nor D835R/D836R/E838R was fully activated by ATP alone (Fig. 3, B and C).
The Asymmetric Effects of R-CL3 Electrostatic Attractions on the CFTR Activity
To investigate further the ATP-dependent activity of the CFTR mutants at the R-CL3 interface, the whole cell CFTR currents were recorded to maximize the ATP effect on the channel activity when 1.5 mm ATP was introduced in the electrode solution. Even if the activity of the inside-out patch expressing a CFTR construct is very low in the presence of ATP, any ATP-induced effect should be observed in the whole cell configuration. Fig. 4A indicates that when the whole cell current was determined with D835R/D836R/E838R, the application of external forskolin to the bath solution increased the mutant current. It is interesting that external curcumin continued to potentiate the mutant activity. Finally, glibenclamide or CFTRinh172 completely suppressed the mutant current (Fig. 4A). A similar case was observed with H950R (Fig. 4B). Because the D835R/D836R/E838R mutant exhibited a lower current with forskolin than H950R or WT CFTR, a cell capacitance was measured with WT and D835R/D836R/E838R CFTR constructs before and after forskolin was applied to evaluate the contribution of trafficking (20). Table 1 demonstrates that the cell capacitances of both WT and D835R/D836R/E838R were stable upon activation by forskolin. Thus, the low forskolin-stimulated current of the latter may not be due to trafficking.
FIGURE 4.

Effects of electrostatic interactions on current densities of CFTR mutants at the R-CL3 interface. A–C, whole cell currents from transfected HEK-293T cells expressing CFTR mutants D835R/D836R/E838R (A), H950R (B), and K946D/H950D/D835R/D836R/E838R (C) recorded by using a ramp protocol (±40 mV). Cm = 14.25, 16.84, and 24.64 picofarads, respectively. The mutants were activated by 50 μm forskolin and 50 μm curcumin and inhibited by 200 μm glibenclamide or 10 μm CFTRinh172. D, mean whole cell currents normalized to the cell capacitance (n = 3–9). The current mediated by K946D/H950D/D835R/D836R/E838R was statistically smaller than the K946D/H950D and D835R/D836R/E838R and H950R currents (n = 3–5; *, p < 0.05, unpaired t test). The currents mediated by S768D/K946D/H950D/D836R and S768D/K946D/H950D/E838R were statistically smaller than the S768D/K946D/H950D current (n = 3,*, p < 0.05, unpaired t test); error bars, S.E.
TABLE 1.
Whole cell currents Im (picoamperes) and capacitances Cm (picofarads) of HEK-293T cells expressing CFTR constructs in response to forskolin
| Constructs | Stimulated Im | Control Cm | Stimulated Cm | n |
|---|---|---|---|---|
| WT-CFTR | 701.6 ± 9.0 | 17.4 ± 0.9 | 16.4 ± 1.1 | 3 |
| D835R/D836R/E838R | 539.5 ± 5.3 | 15.0 ± 1.0 | 16.2 ± 0.8 | 3 |
Although both D835R/D836R/E838R and H950R were greatly activated by forskolin and curcumin, the introduction of K946D and H950D to D835R/D836R/E838R prevented the channel activation by forskolin and curcumin (Fig. 4, C and D). It is very attractive that the small basic conductance was clearly observed before the inhibition by glibenclamide (Fig. 4C). In contrast, D835R/D836R/E838R and H950R failed to exhibit the basic activity (Fig. 4, A and B). Thus, Asp-835, Asp-836, and Glu-838 may not be responsible for the channel activation by ATP. Other parts of NEG2 may interact with other domains or other parts in the R domain to remove the PKA-dependent channel activation.
Upon normalization of the channel current to the cell capacitance, the insertion of K946D/H950D to D835R/D836R/E838R dramatically reduced the CFTR current density (Fig. 4D). In contrast, D835R/D836R/E838R and K946D/H950D and H950R exhibited comparable current densities (Fig. 4D). Thus, the putative electrostatic attractions between CL3 and the R domain may not be exchangeable to suppress the maximal channel activity. In other words, the NEG2-CL3 electrostatic effects on the maximal channel activity may be asymmetric. The putative electrostatic attraction between K946/H950R and D835/D836/E838 failed to inhibit the maximal channel activity, but the putative electrostatic attraction of D835R/D836R/E838R with K946D/H950D greatly suppressed the maximal channel activity.
The Asymmetric Effects of R-CL3 Electrostatic Attractions on CFTR Processing
To further determine whether the reduction in the current density of K946D/H950D/D835R/D836R/E838R originates from the decrease in the channel expression or the single channel conductance, we carried out Western blot analysis. Fig. 5 shows that both WT and ΔR CFTR constructs had a very strong mature band C but a very weak immature band B, suggesting that the deletion of the R domain had no effect on channel processing. Similar cases were seen with D835R/D836R/E838R and H950R and K946D/H950D (Fig. 5). However, the insertion of K946D/H950D to D835R/D836R/E838R clearly reduced the fractional mature Band C by 50%. Thus, the asymmetric electrostatic regulation of the maximal channel activity at the NEG2-CL3 interface may be due to the asymmetric electrostatic regulation of channel processing. However, the maximal channel activity of D835R/D836R/E838R/K946D/H950D was still very low (Fig. 4D). Therefore, it is also possible that the putative electrostatic CL3-R attraction may also stop the channel opening (10).
FIGURE 5.
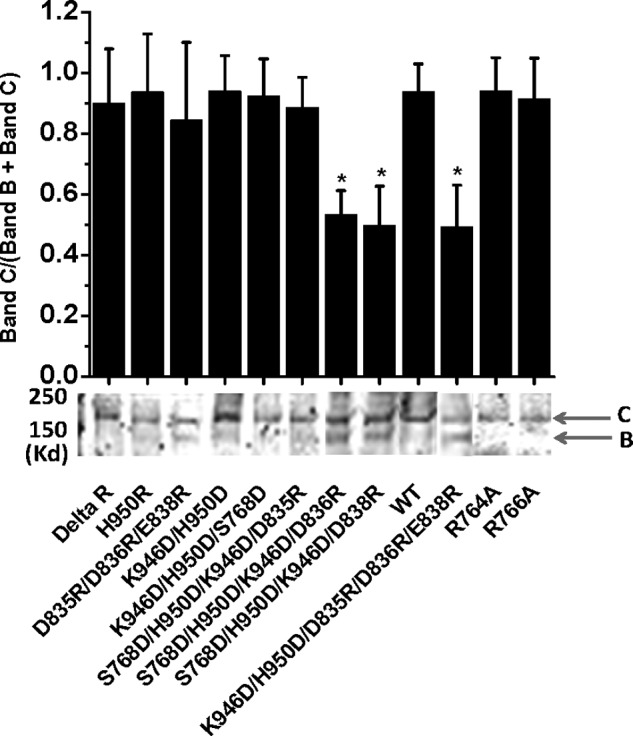
Effects of electrostatic interactions on the processing of CFTR mutants at the R-CL3 interface. Immunoblots indicate their relative intensities of immature Band B and mature Band C (n = 4–7; *, p < 0.05, unpaired t test); error bars, S.E.
The Effects of Ser-768 Phosphorylation on the Asymmetric NEG2-CL3 Electrostatic Regulation of the CFTR Activity and Processing
Although previous studies demonstrated that Ser-768 phosphorylation had no electrostatic contribution to the channel gating, nonphosphorylated Ser-768 may form a strong putative H-bond with H950D to affect the CL3-NEG2 electrostatic interaction (10). Therefore, we prepared a control mutant S768D/K946D/H950D to exclude this putative H-bond. Fig. 4D indicates a high current density of S768D/K946D/H950D based on a whole cell patch recording. The introduction of D835R to this mutant failed to change the current density. However, the insertion of D836R or E838R to S768D/K946D/H950D dramatically reduced the CFTR current density (Fig. 4D). Fig. 5 further demonstrates that the fractions of the mature Band C of S768D/K946D/H950D/D836R and S768D/K946D/H950D/E838R were also significantly decreased by 45%. Thus, the reduction of their current densities may result from the decrease in the channel processing and opening (10). Taken together, the asymmetric electrostatic regulation of CFTR activation and processing was not due to the putative H-bond. However, because S768D mimics Ser-768 phosphorylation and H950R/S768D also exhibited high channel activity (10), Ser-768 phosphorylation failed to change the asymmetric electrostatic regulation of CFTR activation and processing.
The Effects of R764A and R766A on the PKA-dependent Channel Activation
Because both Ser-768 and Asp-836 are close to His-950 of CL3, it is fitting to ask whether Arg-764, Arg-765, and Arg-766 form a salt bridge with the C terminus of NEG2 to cause the asymmetry of the electrostatic regulation. Because R764X and R766M were reported with CF patients, we investigated whether missense alanine mutation of Arg-764 and Arg-766 alters the channel processing and activity. Fig. 4D demonstrates that both R764A and R766A exhibited a normal channel density. Fig. 5 further shows that they were also processed normally. However, Fig. 6 demonstrates that the PKA-dependent activity of these two mutants was greatly enhanced. The K1/2 for PKA activation of R764A and R766A increased from 10 units/ml to 25 and 44 units/ml, respectively. Thus, a putative electrostatic attraction between Arg-764 or Arg-766 and Asp-835, Asp-836, or Glu-838 may weaken the putative electrostatic attraction between Lys-946 and Asp-835, Asp-836, or Glu-838. To support this hypothesis, we determined whether S832C is closed enough to S768C to form a detectable disulfide bond cross-linking based on the Cys-free CFTR construct. Fig. 7 demonstrates that 10 μm diamide greatly suppressed the channel activity of S768C/S832C/V510A but failed to affect the channel activities of both S768C/V510A and S832C/V510A. Thus, the Ser-768 phosphorylation site may be close to the C terminus of NEG2. Taken together, these observations clearly suggest that a putative salt bridge between Arg-766 or Arg-764 and Asp-836 or Glu-838 may weaken the putative inhibitory electrostatic attraction between Lys-946 and Asp-835 or Asp-836 or Glu-838.
FIGURE 6.

The PKA-dependent activity of CFTR mutants R764A and R766A. A, unit channel currents across inside-out membrane patches excised from transfected HEK-293T cells expressing R766A by using a holding potential (+60 mV). In the presence of 1.5 mm ATP, PKA was added gradually until the current was no longer increased. The arrows indicate the final concentrations. N-Ethylmaleimide (NEM) was used to increase the channel activity (9). 200 μm glibenclamide or 10 μm CFTRinh172 was used to inhibit the channel activity. B, PKA titration curves for CFTR mutants R764A and R766A (n = 3–5). WT CFTR was a control.
FIGURE 7.

Effects of diamide on CFTR mutants at the R-CL3 interface. A–C, macroscopic currents across inside-out membrane patches excised from transfected HEK-293T cells expressing CFTR mutants S768C/V510A (A), S832C/V510A (B), and S768C/S832C/V510A (C) by using a ramp protocol (±80 mV). NPPB-AM is a CFTR-specific opener (25). D, fractional inhibition of the current by diamide for three CFTR mutants (n = 3–5; *, p < 0.05, unpaired t test); error bars, S.E.
DISCUSSION
Since Ma's group reported the regulation of PKA-dependent CFTR gating by NEG2 (19), it is obscure which part interacts with NEG2 mediating gating. Based on the findings that both Ser-768 and Asp-836 are close to CL3 (9, 10), the current study further uncovered the regulation role of the complex electrostatic interaction between the R domain and CL3 in channel activation and processing. Our results strongly suggest that the putative electrostatic attractions of CL3 with the C terminus of NEG2 in the whole CFTR prohibited the channel activation by PKA. More importantly, Arg-764 and Arg-766 of the Ser-768 phosphorylation site may regulate the CL3-NEG2 interaction by a putative salt bridge with NEG2. Finally, the putative electrostatic attractions of CL3 with both NEG2 and the Ser-768 phosphorylation site may lead to CFTR misprocessing.
Asymmetric Electrostatic Regulation of CFTR Activation and Processing
Unlike CFTR, other ATP-binding cassette transporters do not have a regulatory domain but express and function well on the cell membrane. However, the deletion of the R domain does not affect CFTR expression (10, 24–27). Moreover, the location of the R domain was found not to be essential for the function (28). Finally, the R domain is unstructured and disordered (29, 30). Thus, the R domain seems unnecessary for the CFTR activation until Ma's group found that a reduction in the negative charge of NEG2 of the R domain promotes the channel activation (19). Our study further suggests that the putative electrostatic interactions between NEG2 and CL3 regulate the CFTR activation. First, the PKA sensitivity of channel activation was significantly enhanced for K946A, D835A, D836A, and E838A mutants (Fig. 2). Second, the curcumin sensitivity was also increased for K946A, K946D, and D835R/D836R/E838R (Fig. 3). Finally, it is very exciting that the putative electrostatic attraction between K946D/H950D and D835R/D836R/E838R dramatically suppressed the maximal channel activity by stopping the channel processing or opening (10). However, the putative electrostatic attraction between K946/H950R and D835/D836/E838 failed to exert this inhibitory effect (Figs. 4 and 5). Moreover, the putative Fe3+ bridge, H-bond, or Ser-768 phosphorylation at the R-CL3 interface failed to affect this asymmetric electrostatic regulation. Therefore, the third charged motif may be involved.
The Role of the Ser-768 Phosphorylation Site
Because both Ser-768 and Asp-836 are close to His-950 (9, 10), and Ser-768 may be closed to Cys-832 (Fig. 7), we examined whether Arg-764 or Arg-766 interacts with NEG2 and thus destroys the symmetry of the putative electrostatic attraction between CL3 and NEG2. Our finding shows that R764A and R766A suppressed not the channel processing and the current density but the channel activation by PKA (Figs. 4–6). Thus, we propose two models of the electrostatic regulation. First, Asp-835, Asp-836, or Glu-838 may interact electrostatically with both Lys-946 and Arg-764 or Arg-766 (Fig. 8A). The former may prohibit the channel activation by PKA whereas the latter may promote the channel activation by PKA possibly by weakening the former interaction. However, the putative electrostatic attraction between CL3 and NEG2 may not be exchangeable. Otherwise, a putative electrostatic attraction of CL3 (K946D/H950D) with both NEG2 (D836R or E838R) and the Ser-768 phosphorylation site (Arg-764 or Arg-766) may result in misprocessing and low channel activity (Fig. 8B). Therefore, a complex electrostatic balance among CL3, NEG2, and the Ser-768 phosphorylation site may ensure the normal channel processing and activity.
FIGURE 8.

Tentative electrostatic regulation mechanisms of CFTR activation and processing. A, a putative electrostatic attraction between Lys-946 and Asp-835 or Asp-836 or Glu-838 may prevent the channel opening by PKA. However, a putative electrostatic attraction between Arg-764 or Arg-766 and Asp-835 or Asp-836 or Glu-838 may promote the channel opening by weakening the putative NEG2-CL3 electrostatic attraction. B, a putative electrostatic attraction of CL3 with both NEG2 and the Ser-768 phosphorylation site may cause CFTR misprocessing.
However, S945L and H949Y, found in patients with CF, are close to Lys-946 and His-950 of CL3 and stop maturation of the protein (31). What is more, CL3 also involves ubiquitination-dependent CFTR trafficking (32). Finally, D979A in CF patients causes misprocessing (33). Thus, the interaction of Lys-946 with the COMMD1 protein (32) or Asp-979 (33) may also result in the asymmetric electrostatic regulation by orchestrating the integral folding to enhance or stabilize CFTR expression on the membrane, allowing more proteins to exit from endoplasmic reticulum, or accelerating the fuse of stabilized CFTR-containing vesicles with the cell membrane.
Implication for CFTR Activation by PKA Phosphorylation
The important finding with NEG2 is that the deletion of NEG2 activates the CFTR channel in the presence of ATP but PKA does not continue to increase the channel activity (19). However, the current study indicates that mutation of Asp-835, Asp-836, and Arg-838 to alanine or arginine failed to greatly promote the channel activation by ATP although the PKA dependence was reduced. Thus, other parts of NEG2 may interact with other stimulatory RRXS motifs in the R domain. Phosphorylation of these sites or removal of those parts of NEG2 may weaken their electrostatic attractions and thus activate the channel.
Acknowledgments
We thank Drs. Joseph Hume, Nathanael Heyman, and Lih Chyuan Ng for technical support and assistance and Drs. David Gadsby and Xuehong Liu for helpful discussion.
This work was supported, in whole or in part, by National Institutes of Health Grant R21 HL 106256 (to D. D. D). This work was also supported by American Heart Association (AHA) Grant 10SDG4120011 (to G. W.) and AHA Western State Affiliate Grant-in-aid 11GRNT7610161 (to D. D. D.).
- CF
- cystic fibrosis
- CFTR
- CF transmembrane conductance regulator
- CL
- cytoplasmic loop
- NBD
- nucleotide binding domain
- R
- regulatory
- TES
- N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid
- TMD
- transmembrane domain.
REFERENCES
- 1. Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J. L., Drumm M. L., Iannuzzi M. C., Collins F. C., Tsui L. C. (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245, 1066–1073 [DOI] [PubMed] [Google Scholar]
- 2. Bear C. E., Li C. H., Kartner N., Bridges R. J., Jensen T. J., Ramjeesingh M., Riordan J. R. (1992) Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell 68, 809–918 [DOI] [PubMed] [Google Scholar]
- 3. Dawson R. J., Locher K. P. (2006) Structure of a bacterial multidrug ABC transporter. Nature 443, 180–185 [DOI] [PubMed] [Google Scholar]
- 4. Zhang L., Aleksandrov L. A., Zhao Z., Birtley J. R., Riordan J. R., Ford R. C. (2009) Architecture of the cystic fibrosis transmembrane conductance regulator protein and structural changes associated with phosphorylation and nucleotide binding. J. Struct. Biol. 167, 242–251 [DOI] [PubMed] [Google Scholar]
- 5. Zhang L., Aleksandrov L. A., Riordan J. R., Ford R. C. (2011) Domain location within the cystic fibrosis transmembrane conductance regulator protein investigated by electron microscopy and gold labelling. Biochim. Biophys. Acta 1808, 399–404 [DOI] [PubMed] [Google Scholar]
- 6. Serohijos A. W., Hegedus T., Aleksandrov A. A., He L., Cui L., Dokholyan N. V., Riordan J. R. (2008) Phenylalanine 508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc. Natl. Acad. Sci. U.S.A. 105, 3256–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. He L., Aleksandrov A. A., Serohijos A. W., Hegedus T., Aleksandrov L. A., Cui L., Dokholyan N. V., Riordan J. R. (2008) Multiple membrane-cytoplasmic domain contacts in the cystic fibrosis transmembrane conductance regulator (CFTR) mediate regulation of channel gating. J. Biol. Chem. 283, 26383–26390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loo T. W., Bartlett M. C., Clarke D. M. (2008) Processing mutations disrupt interactions between the nucleotide binding and transmembrane domains of P-glycoprotein and the cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem. 283, 28190–28197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang G. (2010) State-dependent regulation of cystic fibrosis transmembrane conductance regulator (CFTR) gating by a high affinity Fe3+ bridge between the regulatory domain and cytoplasmic loop 3. J. Biol. Chem. 285, 40438–40447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang G. (2011) The inhibition mechanism of non-phosphorylated Ser-768 in the regulatory domain of cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 286, 2171–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gadsby D. C., Vergani P., Csanády L. (2006) The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 440, 477–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cheng S. H., Rich D. P., Marshall J., Gregory R. J., Welsh M. J., Smith A. E. (1991) Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell 66, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 13. Wang W, Wu J., Bernard K., Li G., Wang G., Bevensee M. O., Kirk K. L. (2010) ATP-independent CFTR channel gating and allosteric modulation by phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 107, 3888–3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. El Hiani Y, Linsdell P. (2012) Role of the juxtamembrane region of cytoplasmic loop 3 in the gating and conductance of the cystic fibrosis transmembrane conductance regulator chloride channel. Biochemistry 51, 3971–3981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wilkinson D. J., Strong T. V., Mansoura M. K., Wood D. L., Smith S. S., Collins F. S., Dawson D. C. (1997) CFTR activation: additive effects of stimulatory and inhibitory phosphorylation sites in the R domain. Am. J. Physiol. 273, L127–33 [DOI] [PubMed] [Google Scholar]
- 16. Vais H., Zhang R., Reenstra W. W. (2004) Dibasic phosphorylation sites in the R domain of CFTR have stimulatory and inhibitory effects on channel activation. Am. J. Physiol. Cell Physiol. 287, C737–745 [DOI] [PubMed] [Google Scholar]
- 17. Csanády L., Seto-Young D., Chan K. W., Cenciarelli C., Angel B. B., Qin J., McLachlin D. T., Krutchinsky A. N., Chait B. T., Nairn A. C., Gadsby D. C. (2005) Preferential phosphorylation of R-domain serine 768 dampens activation of CFTR channels by PKA. J. Gen. Physiol. 125, 171–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kongsuphol P., Cassidy D., Hieke B., Treharne K. J., Schreiber R., Mehta A., Kunzelmann K. (2009) Mechanistic insight into control of CFTR by AMPK. J. Biol. Chem. 284, 5645–5653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xie J., Adams L. M., Zhao J., Gerken T. A., Davis P. B., Ma J. (2002) A short segment of the R domain of cystic fibrosis transmembrane conductance regulator contains channel stimulatory and inhibitory activities that are separable by sequence modification. J. Biol. Chem. 277, 23019–23027 [DOI] [PubMed] [Google Scholar]
- 20. Lewarchik C. M., Peters K. W., Qi J., Frizzell R. A. (2008) Regulation of CFTR trafficking by its R domain. J. Biol. Chem. 283, 28401–28412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cui L., Aleksandrov L., Chang X.-B., Hou Y.-X., He L., Hegedus T., Gentzsch M., Aleksandrov A., Balch W. E., Riordan J. R. (2007) Domain interdependence in the biosynthetic assembly of CFTR. J. Mol. Biol. 365, 981–994 [DOI] [PubMed] [Google Scholar]
- 22. Du K., Lukacs G. L. (2009) Cooperative assembly and misfolding of CFTR domains in vivo. Mol. Biol. Cell 20, 1903–1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lukacs G. L., Verkman A. S. (2012) CFTR: folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol. Med. 18, 81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang W., Bernard K., Li G., Kirk K. L. (2007) Curcumin opens cystic fibrosis transmembrane conductance regulator channels by a novel mechanism that requires neither ATP binding nor dimerization of the nucleotide-binding domains. J. Biol. Chem. 282, 4533–4544 [DOI] [PubMed] [Google Scholar]
- 25. Wang W., Li G., Clancy J. P., Kirk K. L. (2005) Activating cystic fibrosis transmembrane conductance regulator channels with pore blocker analogs. J. Biol. Chem. 280, 23622–23630 [DOI] [PubMed] [Google Scholar]
- 26. Rich D. P., Gregory R. J., Anderson M. P., Manavalan P., Smith A. E., Welsh M. J. (1991) Effect of deleting the R domain on CFTR-generated chloride channels. Science 253, 205–207 [DOI] [PubMed] [Google Scholar]
- 27. Winter M. C., Welsh M. J. (1997) Stimulation of CFTR activity by its phosphorylated R domain. Nature 389, 294–296 [DOI] [PubMed] [Google Scholar]
- 28. Ostedgaard L. S., Baldursson O., Welsh M. J. (2001) Regulation of the cystic fibrosis transmembrane conductance regulator Cl− channel by its R domain. J. Biol. Chem. 276, 7689–7692 [DOI] [PubMed] [Google Scholar]
- 29. Ostedgaard L. S., Baldursson O., Vermeer D. W., Welsh M. J., Robertson A. D. (2000) A functional R domain from cystic fibrosis transmembrane conductance regulator is predominantly unstructured in solution. Proc. Natl. Acad. Sci. U.S.A. 97, 5657–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baker J. M., Hudson R. P., Kanelis V., Choy W. Y., Thibodeau P. H., Thomas P. J., Forman-Kay J. D. (2007) CFTR regulatory region interacts with NBD1 predominantly via multiple transient helices. Nat. Struct. Mol. Biol. 14, 738–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Seibert F. S., Linsdell P., Loo T. W., Hanrahan J. W., Riordan J. R., Clarke D. M. (1996) Cytoplasmic loop three of cystic fibrosis transmembrane conductance regulator contributes to regulation of chloride channel activity. J. Biol. Chem. 271, 27493–27499 [DOI] [PubMed] [Google Scholar]
- 32. Drévillon L., Tanguy G., Hinzpeter A., Arous N., de Becdelièvre A., Aissat A., Tarze A., Goossens M., Fanen P. (2011) COMMD1-mediated ubiquitination regulates CFTR trafficking. PLoS One 6, e18334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Clain J., Fritsch J., Lehmann-Che J., Bali M., Arous N., Goossens M., Edelman A., Fanen P. (2001) Two mild cystic fibrosis-associated mutations result in severe cystic fibrosis when combined in cis and reveal a residue important for cystic fibrosis transmembrane conductance regulator processing and function. J. Biol. Chem. 276, 9045–9049 [DOI] [PubMed] [Google Scholar]