

Background: Laminar flow protects from atherosclerosis in endothelium.
Results: Laminar flow induces Nrf2 activation dependent on ERK5 activation, leading to up-regulation of downstream genes of Nrf2.
Conclusion: ERK5 requires Nrf2 activation to exert cytoprotective effect on HUVEC. ERK5 inhibitor BIX02189 regulates Nrf2 activation in vivo.
Significance: Identifying ERK5 as a molecular target for regulating flow-mediating Nrf2-dependent gene expression may have significant therapeutic potential for treating atherosclerosis.
Keywords: Atherosclerosis, Endothelial Dysfunction, ERK, Kruppel-like Factor (KLF), Nrf2, ERK5, KLF2, Laminar Flow
Abstract
Atherosclerosis is often observed in areas where disturbed flow is formed, whereas atheroprotective region is found in areas where steady laminar flow is developed. It has been reported that some genes activated by blood flow play important roles in vascular function and pathogenesis of atherosclerosis. Extracellular signal-regulated kinase 5 (ERK5) has been reported to regulate endothelial integrity and protect from vascular dysfunction and disease under laminar flow. Krüppel-like factor 2 (KLF2) and NF-E2-related factor 2 (Nrf2) are major transcriptional factors that contribute to anti-atherogenic responses under laminar flow. Implication of ERK5 in laminar flow-mediated regulation of KLF2-dependent gene has been established, whereas the role of ERK5 in laminar flow-mediated activation of Nrf2 pathway has not been addressed yet. In this study, we found that the blockage of ERK5 either by genetic depletion with siRNA or by biochemical inactivation with a specific chemical compound inhibited laminar flow-induced up-regulation of Nrf2-dependent gene expressions, whereas activation of ERK5 increased transcriptional activity and nuclear translocation of Nrf2, which suggests that ERK5 mediates laminar flow-induced up-regulation of Nrf2-dependent gene expression. Further functional studies showed that ERK5 provides protection against oxidative stress-induced cytotoxicity dependent on Nrf2. Molecular interaction between ERK5 and Nrf2 was further induced by laminar flow. Finally, flow-dependent nuclear localization of Nrf2 was inhibited by BIX02189, a specific inhibitor of MEK5, in aorta of mice in vivo. Collectively, these data demonstrate that laminar flow-induced activation of ERK5-Nrf2 signal pathway plays a critical role for anti-inflammatory and anti-apoptotic mechanism in endothelial cells.
Introduction
Steady laminar flow (referred to as flow), a critical regulator of inflammation, apoptosis, and a variety of cellular functions in endothelium, has been studied for protection against atherosclerosis. Previously reported studies suggest that atheroprotective regions contain high, unidirectional, and steady laminar flow, whereas atheroprone regions develop in low and disturbed flow areas (1, 2). A growing body of evidence from both in vitro and in vivo systems has established several lines of transcription factors, which confer the laminar flow-mediated protective role in endothelial cells (ECs).2 NF-E2-related factor 2 (Nrf2) is one of the major transcription factors in laminar flow-mediated cytoprotective responses in ECs (3–5). Flow activation of Nrf2 induces a group of antioxidant genes via the activation of antioxidant response element (ARE) (3, 6). In resting cells, Nrf2 is mainly located in cytoplasm and associated with Kelch-like Ech-associated protein 1 (Keap1), an adaptor protein in Cul3 ubiquitin ligase-mediated ubiquitination and proteasomal degradation of Nrf2 (7, 8). Upon protein modification by electrophiles and kinase-activating signaling pathways, Nrf2 escapes from the Keap1-Cul3 complex and translocates into the nucleus (9, 10). A recent study shows that Nrf2 activation ameliorates inflammatory responses at atherosusceptible sites in wild-type mice, but not in Nrf2−/− mice, suggesting the protective role of Nrf2 in arterial inflammation in vivo (11). However, the underlying molecular mechanism by which laminar flow induces Nrf2 activation has not been fully addressed yet.
Krüppel-like factor 2 (KLF2) is a key mediator in flow-mediated anti-inflammatory responses and a regulator of vascular integrity and also mediates protective gene expression in a flow-dependent manner (12, 13). Parmar et al. (13) reported that KLF2 transcription is induced in endothelium under atheroprotective laminar flow via MAPK/ERK kinase 5 (MEK5)-extracellular signal-regulated protein kinase 5 (ERK5)-MEF2 signaling pathway. Transcriptional activation of KLF2 triggers induction of anti-inflammatory genes including eNOS and reduction of proinflammatory genes including cytokines (13, 14). In addition to KLF2, ERK5 activation induces the expression of KLF4-dependent genes, which are also important in flow-mediated EC-protective responses (15, 16). Taken together, a growing body of evidence suggests ERK5 as an emerging upstream signaling molecule in flow-mediated atheroprotective responses. ERK5 signaling is activated by upstream kinase, MEK5, and plays critical roles in cell proliferation, survival, differentiation, and vascular tone (17). Unlike ERK1/2, ERK5 has C-terminal transactivation domain that regulates transcriptional activation of specific targets for ERK5 (18, 19). It has been reported that ERK5 is involved not only in anti-inflammatory responses but also cytoprotective effect in response to flow in ECs (20). Endothelial apoptosis elicited by serum deprivation is significantly reduced under flow, and flow-mediated anti-apoptotic response is reversed by transducing the dominant negative form of ERK5, suggesting a cytoprotective role of ERK5 in flow signaling pathway (20). Because Nrf2 is a major cytoprotective molecule in flow signaling, we hypothesized that ERK5 activation induced by flow may exert cytoprotective effect via Nrf2-ARE-dependent expression of antioxidant gene independently of KLF2.
In this study, we found that ERK5 activation is required for flow-mediated ARE-dependent gene induction as well as transcriptional activation and nuclear translocation of Nrf2. We also found that ERK5 activation protects ECs against oxidative stress-induced cytotoxicity in a Nrf2 activation- and nuclear translocation-dependent manner. Moreover, we found that flow-mediated ERK5-dependent activation of Nrf2 does not interplay with flow-mediated ERK5-dependent activation of KLF2. Taken together, these data thus demonstrate that laminar flow-mediated activation of ERK5-Nrf2 signal pathway also plays a critical role for anti-atherosclerosis strategies along with the well known ERK5-MEF2-KLF2 pathway.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
BIX02189, a specific inhibitor of MEK5, was purchased from Selleck Chemicals (Houston, TX), and MTT reagents, H2O2, and DMSO were from Sigma. Antibodies were purchased from the following vendors: ERK5 and phospho-ERK5 (Cell Signaling Technology, Danvers, MA); FLAG and tubulin (Sigma); Nrf2, NQO1, and VP16 (Santa Cruz Biotechnology, Santa Cruz, CA); and eNOS (Cayman Chemical, Ann Arbor, MI).
Cell Culture and Laminar Flow
Human umbilical vein endothelial cells (HUVECs) were grown on 0.2% gelatin-coated dish with endothelial growth medium supplemented with low serum growth supplement (Cascade Biologics, Portland, OR) and 5% fetal bovine serum (FBS). Confluent HUVECs cultured in 100-mm dishes were exposed to unidirectional laminar flow (12 dynes/cm2) for 16–24 h using the cone flow system with 5% CO2 at 37 °C as described previously (21). Chinese hamster ovary (CHO) cells and HEK293 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). CHO cells were cultured in F12 medium (gibco) supplemented with 10% FBS, 50 units/ml penicillin, and 50 μg/ml streptomycin. HEK293 cells were maintained in DMEM (gibco) supplemented with 10% FBS, 50 units/ml penicillin, and 50 μg/ml streptomycin.
Plasmid Construction and Adenoviral Vectors
ARE reporter gene was used to test the effect of ERK5 activation on Nrf2 transcriptional activity. pGL3-ARE was constructed by PCR cloning as described previously (22). Briefly, three copies of ARE sequence of the NQO-1 promoter, 5′-CAGTCACAGTGACTCAGCAGAATC-3′, were cloned into the SacI-BglII site of pGL3-basic vector (Promega, Madison, WI). The plasmid encoding human Nrf2 (pMyc-Nrf2) was a gift of Y. Xiong (Addgene plasmid 21555) (8). pXpress-ERK5 and pHA-CA-MEK5 were cloned as described previously (23). Various truncated fragments of VP16-fused ERK5 were cloned into the BamHI-NotI site of pACT vector (Promega) by PCR-based amplification as described previously (19). The Gal4-fused Nrf2 was cloned by subcloning pcDNA3-FLAG-Nrf2 digested with BamHI and XbaI into the BamHI-XbaI site of pBIND vector (Promega). All constructs were verified by DNA sequencing. Adenoviral vector encoding constitutive active form of MEK5α (CA-MEK5α) was described previously (24).
Small Interfering RNA (siRNA)
HUVECs were transiently transfected with 100 pm of control or specific siRNAs against ERK5, Nrf2, or KLF2 using Lipofectamine 2000 reagent (Invitrogen) following the manufacturer's instructions. The targeting sequences of siRNA are as follows: ERK5 siRNA, 5′-GGGCCTATATCCAGAGCUU-3′; Nrf2 siRNA, 5′-AAGAGUAUGAGCUGGAAAAAC-3′ (25); and KLF2 siRNA, 5′-AATTTGTACTGTCTGCGGCAT-3′ (26). A nonspecific control siRNA from Invitrogen was used as a negative control. The cells were harvested 48–72 h after siRNA transfection, and the expression levels of protein and mRNA were measured by immunoblotting with specific antibodies and quantitative real-time RT-PCR (qRT-PCR) with the respective primers.
RNA Isolation and qRT-PCR
mRNA expression of Nrf2 or KLF2 target genes was determined by quantitative real-time RT-PCR assay as described previously (27). Briefly, total RNA was isolated by using TRIzol® reagent (Invitrogen), and reverse transcription reaction was conducted by using TaqMan reverse transcription reagents (Applied Biosystems) following the manufacturer's instructions. Quantitative real-time PCR was conducted with 1 μl of template cDNA using Power SYBR Green (Applied Biosystems). Quantification was carried out using the efficiency-corrected ΔΔCq method. The specific oligonucleotide sequences for target genes were as follows: HO-1, forward 5′-ACATCTATGTGGCCCTGGAG-3′ and reverse 5′-GTAGACAGGGGCGAAGAC-3′; NQO1, forward 5′-TTACTATGGGATGGGGTCCA-3′ and reverse 5′-TGCCAAAACTGTTCACCAAA-3′; ferritin heavy chain, forward 5′-TGACAAAAATGACCCCCATT-3′ and reverse 5′-GATGGCTTTCACCTGCTCAT-3′; KLF2, forward 5′-CCTCCCAAACTGTGACTGGT-3′ and reverse 5′-GAGGGAGACCCTCTGTAGCC-3′; eNOS, forward 5′-CCTGGAAAGTTCCCTCATCA-3′ and reverse 5′-CTTCTGGCAGGGAGACAGAC-3′; Nrf2, forward 5′-AAACCACCCTGAAAGCACAG-3′ and reverse 5′-AGTGTTCTGGTGATGCCACA-3′; and GAPDH, forward 5′-GGAGCCAAAAGGGTCATCAT-3′ and reverse 5′-GTGATGGCATGGACTGTGGT-3′.
Western Blot Analysis
HUVECs were lysed, and cell extracts were incubated on ice for 15 min followed by centrifugation (15,000 × g for 15 min at 4 °C). The protein concentrations in samples were determined by the Bradford assay. Proteins separated by SDS-PAGE were immunoblotted with primary antibodies (1:1000) followed by immunoblotting with secondary antibodies (1:5000). Proteins were then visualized by using the enhanced chemiluminescence detection reagents (Amersham Biosciences) according to the manufacturer's instructions.
Assays of Nrf2 Localization
Cellular localization of Nrf2 was determined by Western blotting analysis using cytosolic and nuclear protein fractionations. Cells were lysed with a lysis buffer A (10 mm HEPES, 10 mm KCl, 0.1 mm EDTA, 0.1 mm EGTA, 1 mm DTT, 1 mm PMSF) and incubated on ice for 20 min followed by centrifugation (15,000 × g for 2 min at 4 °C). The cytosolic compartment was collected from the supernatant, and the nuclear compartment was obtained from the precipitated cell pellet by adding lysis buffer B (20 mm HEPES, 0.4 m NaCl, 1 mm EDTA, 1 mm EGTA, 1 mm DTT, 1 mm PMSF) followed by centrifugation for 10 min. Cellular location of Nrf2 was determined by immunoblotting with Nrf2 antibody (Santa Cruz Biotechnology, scH300) from cytosolic and nuclear compartments, and proper fractionation of cytosolic and nuclear compartments was confirmed by specific antibodies against tubulin (Sigma-Aldrich, T5168) and lamin B (Invitrogen), respectively.
In Situ Proximity Ligation Assay (Duolink)
Endogenous protein-protein interaction of ERK5 and Nrf2 was determined by using the Duolink II fluorescence kit (Olink Bioscience, Uppsala, Sweden). HUVECs were washed with cold PBS, fixed with 4% paraformaldehyde for 30 min, and then permeabilized with 0.1% Triton-X 100 for 5 min. 5% goat serum was used for blocking and diluting primary antibodies. Cells were stained with monoclonal anti-mouse ERK5 antibody (Cell Signaling Technology) and rabbit polyclonal Nrf2 antibody (H300; Santa Cruz Biotechnology) at 4 °C overnight. Oligonucleotide probe-conjugated secondary antibodies were added and incubated for 60 min at 37 °C. Then, ligation and amplification reactions were performed with the provided ligase and polymerase for 30 and 100 min at 37 °C, respectively. DAPI cooperative mounting medium was added to detect the nuclear translocation, and protein-protein interaction of ERK5 and Nrf2 was analyzed in a fluorescence microscope (28, 29).
Mice and en Face Experiments
C57BL/6-specific pathogen-free mice were purchased from Samtaco (Seoul, Korea) and housed in accordance with the procedures outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals under an animal study proposal approved by the Institutional Animal Care and Use Committee at Yeungnam University College of Medicine. To determine the role of ERK5 on laminar flow-dependent Nrf2 nuclear translocation in vivo, 6-week-old male C57BL/6 mice were intraperitoneally treated with BIX02189 (10 mg/kg of body weight in 25% DMSO) or vehicle control. Following euthanization, vascular perfusion was performed with saline for 5 min followed by fixation with 4% paraformaldehyde for 5 min. Isolated aorta was incubated with 0.1% PBS with Tween, and then fat was removed. 5% goat serum was used for blocking and antibody diluents. Aortic endothelial cells were stained with anti-vascular endothelial-cadherin antibody and Topro3 for endothelial cell junction and nuclear, respectively. Cellular localization of Nrf2 was determined by immunofluorescence staining with anti-Nrf2 antibody under the Confocal microscope.
In Vitro Kinase Assay
In vitro kinase assay was performed to examine the ability of ERK5 to phosphorylate Nrf2. Cells were transfected with pFlag-ERK5 in the presence or absence of pHA-CA-MEK5α. Cell lysates were incubated with M2 antibody (Sigma-Aldrich, F3165) overnight at 4 °C followed by immunoprecipitation with A/G-agarose beads. Then, MOSLB (50 mm NaCl, 50 mm NaF, 100 μm Na3VO4, 5 mm EDTA, 5 mm EGTA) and LiCl (500 mm LiCl, 100 mm Tris-HCl, 0.1% Triton X-100, 1 mm DTT) buffers were used for washing, and unbound proteins were removed by centrifugation. The complex was used for in vitro kinase assay with GST-fused Nrf2 recombinant protein in the presence of ATP for 30 min at 30 °C. The phosphorylation of Nrf2 by ERK5 was determined by immunoblotting with anti-MPM2 antibody (Millipore, 05-368), which recognizes the phosphorylation of consensus sequence of MAPK.
GST Pulldown assay
HUVECs were transfected with various fragment mutants of ERK5 cloned into pACT vector for 4 h, washed with PBS, changed with fresh medium, and then incubated for 24 h. The cell lysates were incubated with GST-fused recombinant protein of Nrf2 (GST-Nrf2), and ERK5-Nrf2 complex was captured by centrifugation and eluted with SDS-PAGE sample buffer. ERK5-Nrf2 complex was detected by immunoblotting with respective antibodies. Electrophoresed SDS-polyacrylamide gel was incubated with Coomassie Blue staining solution for 30 min at room temperature.
Immunoprecipitation Analysis
Cell lysates were incubated with anti-ERK5 antibody (Cell Signaling Technology) or anti-rabbit IgG (Cell Signaling Technology) overnight at 4 °C as described previously (14). Protein A/G-agarose beads were added and incubated for 1 h at 4 °C, and unbound proteins were removed by centrifugation. Protein A/G bead-bound ERK5-Nrf2 complex was detected by immunoblotting with anti-Nrf2 antibody.
MTT Assay
Cytotoxicity was measured by MTT assay. HUVECs were treated 100 μm H2O2 for 8 h and then incubated with MTT reagent for an additional 2 h in the 37 °C incubator. Precipitants were collected by centrifugation, washed with PBS, and eluted by adding 500 μl of DMSO. Cell viability was measured using a microplate reader (Bio-Rad) at 570 nm.
TUNEL Assay
HUVECs transduced by Ad-LacZ or Ad-CA-MEK5α at 50 multiplicity of infection were transfected with 60 pm of control or siRNA against Nrf2. Two days after transfection, cells were exposed to 100 μm H2O2 for 8 h and then washed with PBS briefly and fixed with 4% paraformaldehyde for 30 min. To detect apoptotic cells, TUNEL assay kit (Roche Applied Science) was used, and nuclei were stained with DAPI. TUNEL-positive cells were observed under immunofluorescence microscope.
Statistical Analysis
Data in the bar graph and table were expressed as mean ± S.D. Statistical significance of difference was measured by Student's t test. A probability value of <0.05 was considered significant. p values less than 0.05 are indicated by *, and p values less than 0.01 are indicated by **.
RESULTS
Nrf2 and KLF2 Target Genes Are Induced by Laminar Flow but Have a Specific Regulatory Pathway
We first examined the role of Nrf2 and KLF2 in laminar flow-mediated gene expression by using Nrf2- and KLF2-specific siRNAs. HUVECs were transfected with either Nrf2 or KLF2 siRNA for 3 days and treated with 12 dynes/cm2 laminar flow for 16–24 h. As shown in Fig. 1A, flow increased protein expression of HO-1 and NQO1, well known downstream target genes of Nrf2, and also eNOS, a well known downstream target of KLF2. Flow-induced protein expression of Nrf2 and KLF2 target genes was markedly inhibited by knockdown with Nrf2 and KLF2 siRNA, respectively. However, the expression of Nrf2 target genes, HO-1 and NQO1, was not affected by knockdown with KLF2 siRNA and vice versa, which suggests that both Nrf2 and KLF2 show flow dependence for regulating downstream target genes but have independent regulatory mechanisms for downstream pathways (Fig. 1A). Similar results were also observed at the mRNA level by performing qRT-PCR (Fig. 1, B–F). These results led us to look for an emerging upstream signaling molecule in laminar flow-induced Nrf2- and KLF2-specific gene expressions.
FIGURE 1.

Depletion of KLF2 fails to inhibit laminar flow-induced Nrf2-dependent gene expression. A, HUVECs transfected with control, Nrf2, or KLF2 siRNA (100 pm) for 3 days were exposed to atheroprotective flow (12 dynes/cm2) for 16–24 h. Protein expression of HO-1, NQO1, Nrf2, eNOS, and tubulin was determined by immunoblotting with specific antibodies. B–F, mRNA expression of KLF2, eNOS, Nrf2, HO-1, and ferritin heavy chain (Ferritin HC) was determined by qRT-PCR analysis as follows. Total RNA was isolated by using TRIzol® reagent (Invitrogen), and reverse transcription reaction was conducted by using TaqMan reverse transcription reagents (Applied Biosystems) following the manufacturer's instructions. Quantitative real-time PCR was conducted with 1 μl of template cDNA using Power SYBR Green (Applied Biosystems) as described under “Experimental Procedures.” Data are expressed as mean ± S.D. from three independent experiments (n = 2–3). **, p < 0.01; NS, not significant.
Effects of ERK5 Inhibition on Laminar Flow-mediated Induction of Nrf2 and KLF2 Downstream Target Genes
ERK5 is a well known upstream signaling molecule for mediated flow-induced expression of KLF2 target genes. We thus examined whether ERK5 acts as an emerging upstream modulator on flow-induced activation of Nrf2 as it does on flow-induced activation of KLF2. ERK5 was either knocked down with ERK5-specific siRNA or inhibited by using BIX02159, a MEK5-specific chemical inhibitor, and protein expression of HO-1 and eNOS was measured by performing Western blotting analysis. Depletion of ERK5 with ERK5 siRNA markedly inhibited laminar flow-induced expression of eNOS and HO-1 in HUVECs (Fig. 2A), and flow-induced expression of HO-1, NQO1, and eNOS was also inhibited by pretreatment of BIX02189 (Fig. 2B). Similar results were also observed at the expression level of mRNA (data not shown). Taken together, these data revealed ERK5 as an important signaling molecule for flow-mediated Nrf2-dependent gene expression in HUVECs.
FIGURE 2.
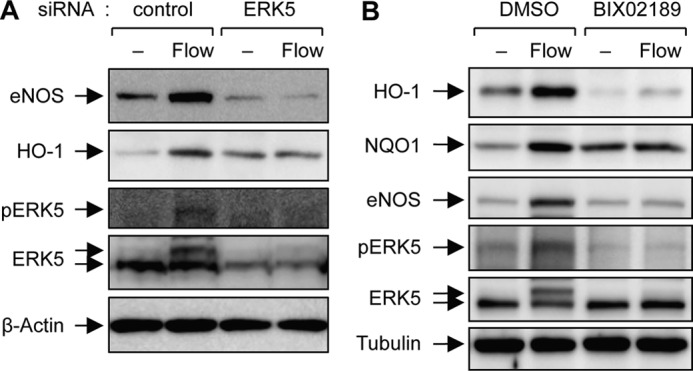
Inhibition of ERK5 suppresses laminar flow-mediated Nrf2-dependent gene expression. A, HUVECs transfected with either control or ERK5 siRNA for 3 days were exposed to atheroprotective flow (12 dynes/cm2). Protein expression of HO-1, eNOS, pERK5, ERK5, and β-actin was determined by immunoblotting with specific antibodies. B, HUVECs were exposed to atheroprotective flow for 16–24 h in the presence of either DMSO or BIX02189 (10 μm). Protein expression of HO-1, NQO1, eNOS, pERK5, ERK5, and tubulin was determined by immunoblotting with specific antibodies. Data are representative from three independent experiments.
ERK5 Induces ARE-dependent Gene Expression
Because ERK5 was found to be involved in flow-mediated Nrf2-dependent gene expressions, we next assessed whether ERK5 regulates Nrf2 transcriptional activity by using ARE-responsive luciferase vector. Cells were transfected with control or ERK5 siRNA, and flow-induced Nrf2-ARE activation was measured. As shown in Fig. 3A, flow-induced Nrf2-ARE activation was significantly inhibited by ERK5 knockdown. To further assess the potential role of ERK5 in Nrf2 transcriptional activation, cells were co-transfected with Gal4-fused Nrf2 construct and pXpress-ERK5 in the presence or absence of pHA-CA-MEK5α encoding the constitutive active form of MEK5α. The combination of pGal4-Nrf2 and pXpress-ERK5 did not increase Nrf2 transcriptional activity, whereas luciferase activity was significantly increased in the presence of pCA-MEK5α (Fig. 3B). These results suggest that ERK5 activation is required for Nrf2-dependent transcriptional activation. Next, we conducted qRT-PCR analysis with adenovirus of CA-MEK5α (Ad-CA-MEK5α) or Ad-LacZ to observe mRNA changes by ERK5 activation. Transducing Ad-CA-MEK5 significantly elevated not only KLF2-dependent genes but also Nrf2-depedent genes (Table 1). These data suggest that ERK5 activation is responsible for flow-induced Nrf2 transcriptional activation and that ERK5 activation is sufficient to induce Nrf2 transcriptional activation without flow stimulation. Thus, ERK5-activating signaling plays an important role in flow-dependent cytoprotective responses via Nrf2-ARE pathway.
FIGURE 3.
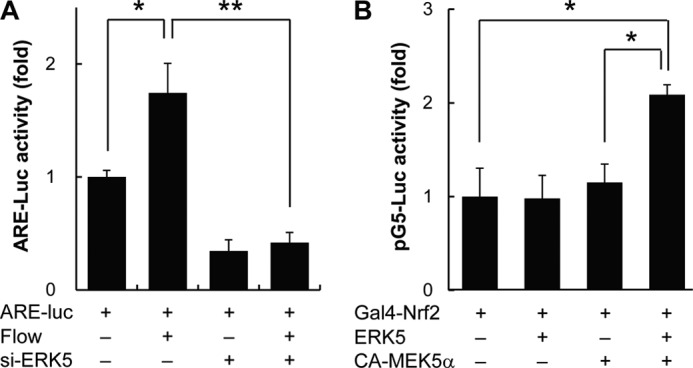
ERK5 activation increases transcriptional activity of Nrf2. A, HUVECs were co-transfected with either control or ERK5 siRNA (si-ERK5) (100 pm) and pGL3-ARE-luciferase (ARE-luc) for 2 days. Cells were then exposed to atheroprotective flow for 16–24 h, and luciferase activity was measured by using the Dual-Luciferase reporter assay kit (Promega) and GloMax 20/20 luminometer (Promega). Transfection efficiency was normalized by Renilla luciferase activity derived from pRL-TK construct. B, cells were co-transfected with pBIND-Nrf2, pG5-luciferase, and pERK5 in the presence or absence of pCA-MEK5α as indicated in the figure. Luciferase activity was measured by using Dual-Luciferase reporter assay kit (Promega) and GloMax 20/20 luminometer (Promega). Transfection efficiency was normalized by Renilla luciferase activity derived from pRL-TK construct. Data are expressed as mean ± S.D. from three independent experiments. *, p < 0.05; **, p < 0.01.
TABLE 1.
ERK5 activation induced mRNA levels of Nrf2-dependent genes
HUVECs were transduced with 50 multiplicity of infection of Ad-CA-MEK5α or control (Ad-LacZ) for 40 h. Relative expression levels of respective genes were determined by qRT-PCR. Relative expression levels from samples were normalized by GAPDH. Data are expressed as means ± S.D.; n = 3 per group. Data are representative of three independent experiments with similar results
| Ad-LacZ | Ad-CAMEK5α | |
|---|---|---|
| NQO1 | 1.00 ± 0.97 | 2.41 ± 2.57 |
| HO-1 | 1.00 ± 0.51 | 5.64 ± 1.97 |
| Ferritin HC | 1.00 ± 0.69 | 3.91 ± 1.43 |
| eNOS | 1.00 ± 0.60 | 13.04 ± 3.04 |
| KLF2 | 1.00 ± 0.42 | 2.92 ± 0.52 |
| Nrf2 | 1.00 ± 0.53 | 2.24 ± 0.239 |
ERK5 Is Involved in Flow-induced Nrf2 Nuclear Translocation
In resting state, Nrf2 is suppressed by Keap1, which sequestrates and induces proteasomal degradation of Nrf2 (7). Nrf2 is activated by PI3K or stimuli such reactive oxygen species, which results in dissociation of the Nrf2-Keap1 complex and subsequent translocation of Nrf2 to the nucleus (16, 17). We thus tested whether ERK5 activation affects flow-induced Nrf2 nuclear translocation by using BIX02189 compound. Localization of Nrf2 into nuclei was increased by flow, and pretreatment of BIX02189 strongly suppressed Nrf2 nuclear translocation (Fig. 4A). In addition, we found that ERK5 also translocated into nuclei under flow condition.
FIGURE 4.
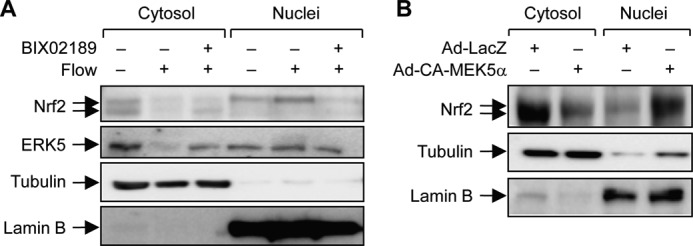
ERK5 is required for laminar flow-induced Nrf2 nuclear translocation. A, HUVECs were pretreated with BIX02189 (10 μm) for 16–24 h and then exposed to laminar flow (12 dynes/cm2) or static condition. Both cytosolic and nuclear compartments were obtained by cellular fractionation as described under “Experimental Procedures.” Cellular localization of Nrf2 and ERK5 was determined by immunoblotting with anti-Nrf2 and anti-ERK5 antibodies, respectively, and proper fractionation of cytosolic and nuclear compartments was confirmed by specific antibodies against tubulin and lamin B, respectively. B, HEK293 cells were infected with adenovirus LacZ or CA-MEK5α for 40 h, and cellular localization of Nrf2 was determined by immunoblotting with anti-Nrf2 antibody. Data are representative of three independent experiments with similar results.
Next, we determined the molecular interaction between ERK5 and Nrf2 in nuclear translocation of Nrf2 by using Ad-CA-MEK5α, upstream activator for ERK5. Cells were transduced with Ad-CA-MEK5α or Ad-LacZ for 2 days, and cellular fractionation was performed to determine the cellular localization of Nrf2. Nrf2 in nuclei was significantly increased in the presence of Ad-CA-MEK5α, whereas Nrf2 still mainly remained in cytosol in cells transduced with Ad-LacZ (Fig. 4B). These results demonstrate that upon activation, ERK5 induces Nrf2 nuclear translocation and subsequent activation of Nrf2-dependent genes. Thus, ERK5 is a strong candidate for the upstream kinase to regulate transcriptional activity of Nrf2 in laminar flow-mediated EC signaling pathway.
Identification of ERK5 Binding Site to Nrf2
Given the potential role of ERK5 in Nrf2 translocation and transcriptional activation, we investigated the molecular interaction mechanism between Nrf2 and ERK5 by performing an immunocoprecipitation assay. Cells were overexpressed with Xpres-ERK5 and Myc-Nrf2 in the presence or absence of CA-MEK5α, pulled down with anti-Myc antibody, and immunoblotted against Nrf2 and ERK5. As shown in Fig. 5A, the protein-protein interaction between ERK5-Nrf2 was detected in the in vitro overexpression system, and this interaction was enhanced in the presence of CA-MEK5α. Next, we determined whether protein-protein interaction of ERK5 and Nrf2 occurs in endogenous condition and is induced by flow stimulation. Immunoprecipitation analysis shows that ERK5 binds to Nrf2 and that protein-protein interaction of ERK5 and Nrf2 is further enhanced by flow in endogenous condition (Fig. 5B). Using the immunostaining-based Duolink system, we confirmed the endogenous protein-protein interaction in vivo in HUVECs. ERK5-Nrf2 interaction was visualized under the fluorescence microscope with specific antibodies or control IgG. The protein-protein interaction of ERK5 and Nrf2 was increased as early as 1 h under flow condition and further enhanced in a time-dependent manner (Fig. 5C). Taken together, these data indicate that ERK5 interacts with Nrf2 to induce its nuclear translocation and transcriptional activity. Because we confirmed interaction between ERK5 and Nrf2, we next examined which domain of ERK5 mediates ERK5-Nrf2 interaction by performing a pulldown assay. Cell lysates were collected from the cells transfected with various pACT(VP16)-ERK5 fragments and incubated with GST-fused Nrf2 recombinant protein for 1 h followed by precipitation with Sepharose 4B beads (Fig. 5D). As shown in Fig. 5D, ERK5 fragment A, an N-terminal domain of ERK5, showed the strongest binding affinity to Nrf2. Because ERK5 fragment A contains an entire kinase domain of ERK5, it has been assumed that ERK5-Nrf2 interaction might be dependent on the kinase activity or active state of ERK5. To assess the role of kinase activity in ERK5-Nrf2 interaction, we utilized the kinase activity-deficient mutant of ERK5 (ERK5-AEF), which mutates the TEY motif in the kinase domain. As shown in Fig. 5E, binding affinity of ERK5 to Nrf2 was diminished in ERK5-AEF mutant when compared with that in ERK5 wild type, indicating that ERK5 interacts with Nrf2 dependent on kinase activity of ERK5. In addition, an in vitro kinase assay showed that ERK5 directly phosphorylates Nrf2, suggesting that Nrf2 is a novel substrate of ERK5 (data not shown). However the role of phosphorylation of Nrf2 in ERK5-mediated Nrf2 activation remains to be determined.
FIGURE 5.

ERK5 binds to Nrf2 in a kinase activity-dependent manner. A, cells were co-transfected with pMyc-Nrf2 and pXpress-ERK5 in the presence or absence of pHA-CA-MEK5α. Cell lysates were used for immunoprecipitation (IP) analysis with α-Myc antibody 24 h after transfection and then blotted with anti-Xpress and anti-Nrf2 antibodies as indicated in the figure. The total amount of ERK5 and CA-MEK5 from whole cell lysates was determined by immunoblotting (IB). B, HUVECs were exposed to laminar flow for the indicated times or static condition. Cell lysates were then immunoprecipitated with anti-ERK5 antibody followed by immunoblotting with anti-Nrf2 antibody. The expression of ERK5 and Nrf2 was determined by immunoblotting with specific antibodies. Asterisk indicates nonspecific band. ON, overnight. C, HUVECs were exposed to laminar flow for the indicated times or static condition, and in vivo protein-protein interaction of ERK5 and Nrf2 was determined using the in vivo fluorescence protein-protein interaction detection kit (Duolink, Olink) following the manufacturer's instructions. Flow-induced ERK5-Nrf2 interaction was detected as red dots. Cells were stained with anti-ERK5 antibody and anti-Nrf2 antibody, and nucleus was stained with DAPI (×200). D, cells were transfected with various pACT-ERK5 fragments as indicated in the figure, and cell lysates were then incubated with recombinant GST-Nrf2 followed by immunoblotting with anti-VP16 antibody (left panel). The expression of various ERK5 fragments from whole cell lysates was determined by immunoblotting with anti-VP16 antibody. Data are representative of three independent experiments. Right panel, schematic model showing the length and legion of each fragments of ERK5 (frag-A to frag-D). P, phosphorylation. E, cells were transfected with pERK5-WT or pERK5-AEF in the presence or absence of pCA-MEK5α, and cell lysates were then incubated with recombinant GST-Nrf2. The binding affinity of ERK5 to GST-Nrf2 was determined by immunoblotting with anti-ERK5 antibody (top panel). The equal amount of GST-Nrf2 was verified in Coomassie Blue staining (middle panel). The expression of ERK5 from whole cell lysates was determined by immunoblotting with anti-ERK5 antibody (bottom panel). Data are representative of three independent experiments.
ERK5-Nrf2 Cascade Contributes to Protective Responses from Oxidative Stress-induced Cytotoxicity
To assess the biological relevance of flow-induced ERK5 activation, we examined its protective role against oxidative stress. An MTT assay was conducted to examine the viability of HUVECs in the presence or absence of 100 μm H2O2. As shown in Fig. 6A, flow protected HUVECs from oxidative stress-induced cytotoxicity, and its protective effect was abolished by ERK5 knockdown with ERK5 siRNA. Next, we examined the role of Nrf2 in ERK5-induced cytoprotective effect. HUVECs were transduced with Ad-CA-MEK5α in the presence or absence of Nrf2 siRNA, and cell viability was measured by the MTT assay. ERK5 activation protecting against H2O2-induced cell death was dramatically induced, and Ad-CA-MEK5α protects against H2O2-induced cell death; these effects are abrogated by depletion of Nrf2 (Fig. 6B). These data show that ERK5 activation confers a cytoprotective effect on oxidative stress-induced cytotoxicity through Nrf2-dependent pathway.
FIGURE 6.

ERK5 activation protects against oxidative stress-induced cytotoxicity dependent on Nrf2. A, HUVECs transfected with either control or ERK5 siRNA (siERK5, 100 pm) for 3 days were exposed to laminar flow overnight and then exposed to 100 μm H2O2 for 8 h to induce oxidative stress. Cell viability was determined by MTT analysis as described under “Experimental Procedures.” B–D, HUVECs transfected with either control or Nrf2 siRNA (100 pm) were infected with either Ad-CA-MEK5α or Ad-LacZ for 2 days as indicated and then exposed to 100 μm H2O2 for 8 h. Cell viability and apoptosis were determined by MTT analysis (B), TUNEL staining (C), and Western blotting assay (D). TUNEL-positive cells were observed under the fluorescence microscope. The numbers of TUNEL-positive cells are presented by bar graph. Cell lysates were used for immunoblotting with anti-PARP1, anti-caspase 3 (Casp3) (against for cleaved form of caspase 3), and anti-tubulin antibodies. Data are expressed as mean ± S.D. from three independent experiments. **, p < 0.01; NS, not significant.
To give more direct information of the anti-apoptotic effect of ERK5-Nrf2 signal pathway, we performed a TUNEL assay. Consistent with MTT assay data in Fig. 6B, transducing CA-MKE5α decreased the TUNEL-positive apoptotic cell number, and knockdown Nrf2 with Nrf2 siRNA abrogated the CA-MEK5α-mediated anti-apoptotic effect (Fig. 6C). In addition to TUNEL assay, protein expression of apoptotic markers was measured and is presented in Fig. 6D. The cleaved forms of caspase 3 and PARP1 cleavage were inhibited by ERK5 activation, which was abrogated by depletion of Nrf2. Taken together, these data clearly indicate that the cytoprotective effect of ERK5 against oxidative stress-induced endothelial cell death requires Nrf2.
The Role of ERK5 on Nrf2 Nuclear Translocation in Vivo
Because we found that ERK5 activation is able to enhance flow-induced Nrf2 nuclear translocation in vitro in a cell system (Fig. 4B), we further determined whether ERK5 activation is required for flow-induced Nrf2 nuclear translocation in vivo in an animal model. Intracellular localization of Nrf2 was observed in the atheroprotective region, which has steady laminar flow in vasculature. As shown in Fig. 7, endothelial Nrf2 of mouse aorta mainly locates in both the cytoplasm and the nucleus with greater intensity of Nrf2 in the nucleus than in the cytosol as reported previously (30). However, the nuclear localization of Nrf2 was inhibited in aortic endothelial cells from mice treated with BIX02189. These data suggest that ERK5 plays a critical role in Nrf2 translocation not only in cell systems but also in vivo in an animal model.
FIGURE 7.

Inhibition of ERK5 suppresses Nrf2 nuclear translocation in mouse aorta in vivo. Nrf2 nuclear translocation in mouse aortic endothelium was analyzed by en face immunofluorescence staining assay using anti-Nrf2 antibody (red). Mice were treated with either 10 mg/kg of BIX02189 (in 25% DMSO) or vehicle control (same volume of 25% DMSO) by intraperitoneal injection. Endothelium of thoracic aorta was stained with anti-vascular endothelial-cadherin (VE-cadherin) antibody for endothelial cell-cell junction staining (green), Topro3 for nuclear staining (blue), and anti-Nrf2 antibody (red) and photographed under a confocal microscope.
DISCUSSION
Recent studies have revealed that expression of cytoprotective genes is altered in response to laminar flow, but underlying molecular mechanisms that contribute to atheroprotective effects are only partially understood. In this study, we demonstrated that flow-induced ERK5 activation exerts cytoprotective responses via Nrf2-dependent activation of atheroprotective signaling pathway. In the present study, we provide evidence that in HUVECs, ERK5 activation increases Nrf2-up-regulated genes involved in cytoprotective responses, and Nrf2 transcriptional activity was lost in cases where ERK5 expression was depleted or inhibited by either siRNA or a biochemical inhibitor compound of ERK5. It has been previously reported that Keap1 binding to Nrf2 results in the inhibition of nuclear translocation of Nrf2 and subsequent inhibition of Nrf2 activation (7, 8). However, stimuli activating MAPKs, PI3K, or PKC dissociate Nrf2 from the Nrf2-Keap1 complex and lead to its translocation into nucleus (31–33). Interestingly, a biochemical experiment showed that flow-induced Nrf2 nuclear translocation was diminished by inhibiting ERK5 with BIX02189 (Fig. 4A), indicating that the kinase activity of ERK5 may be involved in ERK5-induced Nrf2 transcriptional activation and nuclear translocation. Furthermore, overexpression of CA-MEK5α using an adenoviral system induced Nrf2 transcriptional activation and nuclear translocation without flow stimulation (Fig. 4B), suggesting that ERK5 activates Nrf2 in a kinase activity-dependent manner.
Direct protein-protein interaction between ERK5 and Nrf2 was determined using in vitro GST pulldown analysis and also an in vivo endogenous fluorescence interaction assay (Fig. 5). ERK5 interacts with Nrf2 under flow condition dependent on the active state of ERK5, and endogenous interaction of ERK5 and Nrf2 was further enhanced by flow in a time-dependent manner. In the analysis of Duolink, protein complex of ERK5 and Nrf2 appears in cytosol in static condition but is greatly enhanced under flow condition in a time-dependent manner (Fig. 5C). Thus, we believe that activated ERK5 binds to and phosphorylates Nrf2, which may result in dissociation of Nrf2 from Keap1-Cul3 complex. Furthermore, ERK5 binds to Nrf2 via its N-terminal kinase domain, and the binding affinity is diminished in the kinase activity-deficient mutant of ERK5, indicating that the kinase activity of ERK5 has an important role for liberating Nrf2 from the Nrf2-Keap1 complex. Several studies suggested the positive role of MAPK-mediated Nrf2 phosphorylation in a Nrf2-dependent signaling pathway (33, 34). Actually, we observed several potential sites for phosphorylation modification on the Nrf2 amino acid sequence. Some of them are closely located to the nuclear localization signal sequence of Nrf2, implying the potential role of ERK5-mediated Nrf2 phosphorylation in Nrf2 nuclear translocation. However, the exact molecular mechanism underlying ERK5-induced Nrf2 activation is unclear in the present study and should be addressed by further investigations.
Flow-induced ERK5 activation plays an important role on vascular protection via KLF2-dependent anti-inflammatory responses (13). ERK5 was also found as a critical factor for endothelial survival as ERK5-null mice showed vascular leakage (35, 36). In addition, ERK5 is also involved in flow-mediated EC protection from cell death induced by serum deprivation (20). Indeed, our data showed that H2O2-induced oxidative stress was greatly diminished by ERK5 activation, whereas ERK5-mediated protection against H2O2-induced oxidative stress was abrogated by depletion of Nrf2, suggesting that a novel cascade ERK5-Nrf2 is functionally effective in flow-induced endothelial protection. Thus, flow-induced ERK5 activation may contribute to the prevention of vascular dysfunction atherosclerosis via KLF2- as well as Nrf2-dependent pathway.
In conclusion, we have identified ERK5 as a novel positive regulator of Nrf2 signaling in ECs in response to laminar flow. ERK5 activation exerts Nrf2 transcriptional activation and nuclear translocation via potential protein-protein interaction in a kinase activity-dependent manner. Additionally, the ERK5-Nrf2 cascade links to flow-mediated EC protection from oxidative stress-induced cytotoxicity. Our results suggest that the ERK5-Nrf2 signal module could be a potential target for pharmacological intervention to prevent vascular diseases including atherosclerosis.
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Korea government (Grants 2011-0006184 and 2011-0026075) and a grant from the Korea Health Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (Grant A111345).
- EC
- endothelial cell
- HUVEC
- human umbilical vein endothelial cell
- ARE
- antioxidant response element
- KLF2
- Krüppel-like factor 2
- Nrf2
- NF-E2-related factor 2
- Keap1
- Kelch-like ECH-associated protein 1
- eNOS
- endothelial nitric-oxide synthase
- qRT-PCR
- quantitative real-time RT-PCR
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- DMSO
- dimethyl sulfoxide
- Ad
- adenovirus
- CA
- constitutively active
- pERK5
- phospho-ERK5.
REFERENCES
- 1. Malek A. M., Alper S. L., Izumo S. (1999) Hemodynamic shear stress and its role in atherosclerosis. JAMA 282, 2035–2042 [DOI] [PubMed] [Google Scholar]
- 2. Dai G., Kaazempur-Mofrad M. R., Natarajan S., Zhang Y., Vaughn S., Blackman B. R., Kamm R. D., García-Cardeña G., Gimbrone M. A., Jr. (2004) Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proc. Natl. Acad. Sci. U.S.A. 101, 14871–14876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Warabi E., Wada Y., Kajiwara H., Kobayashi M., Koshiba N., Hisada T., Shibata M., Ando J., Tsuchiya M., Kodama T., Noguchi N. (2004) Effect on endothelial cell gene expression of shear stress, oxygen concentration, and low-density lipoprotein as studied by a novel flow cell culture system. Free Radic. Biol. Med. 37, 682–694 [DOI] [PubMed] [Google Scholar]
- 4. Takabe W., Jen N., Ai L., Hamilton R., Wang S., Holmes K., Dharbandi F., Khalsa B., Bressler S., Barr M. L., Li R., Hsiai T. K. (2011) Oscillatory shear stress induces mitochondrial superoxide production: implication of NADPH oxidase and c-Jun NH2-terminal kinase signaling. Antioxid. Redox. Signal. 15, 1379–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Warabi E., Takabe W., Minami T., Inoue K., Itoh K., Yamamoto M., Ishii T., Kodama T., Noguchi N. (2007) Shear stress stabilizes NF-E2-related factor 2 and induces antioxidant genes in endothelial cells: role of reactive oxygen/nitrogen species. Free Radic. Biol. Med. 42, 260–269 [DOI] [PubMed] [Google Scholar]
- 6. Hosoya T., Maruyama A., Kang M. I., Kawatani Y., Shibata T., Uchida K., Warabi E., Noguchi N., Itoh K., Yamamoto M. (2005) Differential responses of the Nrf2-Keap1 system to laminar and oscillatory shear stresses in endothelial cells. J. Biol. Chem. 280, 27244–27250 [DOI] [PubMed] [Google Scholar]
- 7. Kobayashi A., Kang M. I., Okawa H., Ohtsuji M., Zenke Y., Chiba T., Igarashi K., Yamamoto M. (2004) Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 24, 7130–7139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Furukawa M., Xiong Y. (2005) BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol. Cell. Biol. 25, 162–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maher J., Yamamoto M. (2010) The rise of antioxidant signaling–the evolution and hormetic actions of Nrf2. Toxicol. Appl. Pharmacol. 244, 4–15 [DOI] [PubMed] [Google Scholar]
- 10. Osburn W. O., Kensler T. W. (2008) Nrf2 signaling: an adaptive response pathway for protection against environmental toxic insults. Mutat. Res. 659, 31–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zakkar M., Van der Heiden K., Luong le A., Chaudhury H., Cuhlmann S., Hamdulay S. S., Krams R., Edirisinghe I., Rahman I., Carlsen H., Haskard D. O., Mason J. C., Evans P. C. (2009) Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Arterioscler. Thromb. Vasc. Biol. 29, 1851–1857 [DOI] [PubMed] [Google Scholar]
- 12. Dekker R. J., Boon R. A., Rondaij M. G., Kragt A., Volger O. L., Elderkamp Y. W., Meijers J. C., Voorberg J., Pannekoek H., Horrevoets A. J. (2006) KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood 107, 4354–4363 [DOI] [PubMed] [Google Scholar]
- 13. Parmar K. M., Larman H. B., Dai G., Zhang Y., Wang E. T., Moorthy S. N., Kratz J. R., Lin Z., Jain M. K., Gimbrone M. A., Jr., García-Cardeña G. (2006) Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J. Clin. Invest. 116, 49–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Woo C. H., Shishido T., McClain C., Lim J. H., Li J. D., Yang J., Yan C., Abe J. (2008) Extracellular signal-regulated kinase 5 SUMOylation antagonizes shear stress-induced antiinflammatory response and endothelial nitric oxide synthase expression in endothelial cells. Circ. Res. 102, 538–545 [DOI] [PubMed] [Google Scholar]
- 15. Ohnesorge N., Viemann D., Schmidt N., Czymai T., Spiering D., Schmolke M., Ludwig S., Roth J., Goebeler M., Schmidt M. (2010) Erk5 activation elicits a vasoprotective endothelial phenotype via induction of Kruppel-like factor 4 (KLF4). J. Biol. Chem. 285, 26199–26210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clark P. R., Jensen T. J., Kluger M. S., Morelock M., Hanidu A., Qi Z., Tatake R. J., Pober J. S. (2011) MEK5 is activated by shear stress, activates ERK5, and induces KLF4 to modulate TNF responses in human dermal microvascular endothelial cells. Microcirculation 18, 102–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X., Tournier C. (2006) Regulation of cellular functions by the ERK5 signalling pathway. Cell. Signal. 18, 753–760 [DOI] [PubMed] [Google Scholar]
- 18. Buschbeck M., Ullrich A. (2005) The unique C-terminal tail of the mitogen-activated protein kinase ERK5 regulates its activation and nuclear shuttling. J. Biol. Chem. 280, 2659–2667 [DOI] [PubMed] [Google Scholar]
- 19. Akaike M., Che W., Marmarosh N. L., Ohta S., Osawa M., Ding B., Berk B. C., Yan C., Abe J. (2004) The hinge-helix 1 region of peroxisome proliferator-activated receptor γ1 (PPARγ1) mediates interaction with extracellular signal-regulated kinase 5 and PPARγ1 transcriptional activation: involvement in flow-induced PPARγ activation in endothelial cells. Mol. Cell. Biol. 24, 8691–8704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pi X., Yan C., Berk B. C. (2004) Big mitogen-activated protein kinase (BMK1)/ERK5 protects endothelial cells from apoptosis. Circ. Res. 94, 362–369 [DOI] [PubMed] [Google Scholar]
- 21. Fleming I., Bauersachs J., Fisslthaler B., Busse R. (1998) Ca2+-independent activation of the endothelial nitric oxide synthase in response to tyrosine phosphatase inhibitors and fluid shear stress. Circ. Res. 82, 686–695 [DOI] [PubMed] [Google Scholar]
- 22. Chen X. L., Varner S. E., Rao A. S., Grey J. Y., Thomas S., Cook C. K., Wasserman M. A., Medford R. M., Jaiswal A. K., Kunsch C. (2003) Laminar flow induction of antioxidant response element-mediated genes in endothelial cells: a novel anti-inflammatory mechanism. J. Biol. Chem. 278, 703–711 [DOI] [PubMed] [Google Scholar]
- 23. Yan C., Luo H., Lee J. D., Abe J., Berk B. C. (2001) Molecular cloning of mouse ERK5/BMK1 splice variants and characterization of ERK5 functional domains. J. Biol. Chem. 276, 10870–10878 [DOI] [PubMed] [Google Scholar]
- 24. Woo C. H., Massett M. P., Shishido T., Itoh S., Ding B., McClain C., Che W., Vulapalli S. R., Yan C., Abe J. (2006) ERK5 activation inhibits inflammatory responses via peroxisome proliferator-activated receptor δ (PPARδ) stimulation. J. Biol. Chem. 281, 32164–32174 [DOI] [PubMed] [Google Scholar]
- 25. Lee H. H., Park S. A., Almazari I., Kim E. H., Na H. K., Surh Y. J. (2010) Piceatannol induces heme oxygenase-1 expression in human mammary epithelial cells through activation of ARE-driven Nrf2 signaling. Arch. Biochem. Biophys. 501, 142–150 [DOI] [PubMed] [Google Scholar]
- 26. Young A., Wu W., Sun W., Benjamin Larman H., Wang N., Li Y. S., Shyy J. Y., Chien S., García-Cardeña G. (2009) Flow activation of AMP-activated protein kinase in vascular endothelium leads to Krüppel-like factor 2 expression. Arterioscler. Thromb. Vasc. Biol. 29, 1902–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nigro P., Abe J., Woo C. H., Satoh K., McClain C., O'Dell M. R., Lee H., Lim J. H., Li J. D., Heo K. S., Fujiwara K., Berk B. C. (2010) PKCζ decreases eNOS protein stability via inhibitory phosphorylation of ERK5. Blood 116, 1971–1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. E G., Cao Y., Bhattacharya S., Dutta S., Wang E., Mukhopadhyay D. (2012) Endogenous vascular endothelial growth factor-A (VEGF-A) maintains endothelial cell homeostasis by regulating VEGF receptor-2 transcription. J. Biol. Chem. 287, 3029–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lim J. H., Jono H., Komatsu K., Woo C. H., Lee J., Miyata M., Matsuno T., Xu X., Huang Y., Zhang W., Park S. H., Kim Y. I., Choi Y. D., Shen H., Heo K. S., Xu H., Bourne P., Koga T., Xu H., Yan C., Wang B., Chen L. F., Feng X. H., Li J. D. (2012) CYLD negatively regulates transforming growth factor-β-signalling via deubiquitinating Akt. Nat. Commun. 3, 771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ali F., Zakkar M., Karu K., Lidington E. A., Hamdulay S. S., Boyle J. J., Zloh M., Bauer A., Haskard D. O., Evans P. C., Mason J. C. (2009) Induction of the cytoprotective enzyme heme oxygenase-1 by statins is enhanced in vascular endothelium exposed to laminar shear stress and impaired by disturbed flow. J. Biol. Chem. 284, 18882–18892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dai G., Vaughn S., Zhang Y., Wang E. T., Garcia-Cardena G., Gimbrone M. A., Jr. (2007) Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/Akt-dependent activation of Nrf2. Circ. Res. 101, 723–733 [DOI] [PubMed] [Google Scholar]
- 32. Huang H. C., Nguyen T., Pickett C. B. (2002) Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 277, 42769–42774 [DOI] [PubMed] [Google Scholar]
- 33. Sun Z., Huang Z., Zhang D. D. (2009) Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2-dependent antioxidant response. PLoS One 4, e6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shen G., Hebbar V., Nair S., Xu C., Li W., Lin W., Keum Y. S., Han J., Gallo M. A., Kong A. N. (2004) Regulation of Nrf2 transactivation domain activity: the differential effects of mitogen-activated protein kinase cascades and synergistic stimulatory effect of Raf and CREB-binding protein. J. Biol. Chem. 279, 23052–23060 [DOI] [PubMed] [Google Scholar]
- 35. Hayashi M., Kim S. W., Imanaka-Yoshida K., Yoshida T., Abel E. D., Eliceiri B., Yang Y., Ulevitch R. J., Lee J. D. (2004) Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J. Clin. Invest. 113, 1138–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yan L., Carr J., Ashby P. R., Murry-Tait V., Thompson C., Arthur J. S. (2003) Knockout of ERK5 causes multiple defects in placental and embryonic development. BMC Dev. Biol. 3, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]