

Background: Uric acid is an independent risk factor in fructose-induced fatty liver, but whether it is a marker or a cause remains unknown.
Results: Hepatocytes exposed to uric acid developed mitochondrial dysfunction and increased de novo lipogenesis, and its blockade prevented fructose-induced lipogenesis.
Conclusion: Rather than a consequence, uric acid induces fatty liver
Significance: Hyperuricemic people are more prone to develop fructose-induced fatty liver.
Keywords: Fructose, Lipogenesis, Mitochondrial Metabolism, Nox, Uric Acid, Fatty Liver, Metabolic Syndrome, NOX4
Abstract
Metabolic syndrome represents a collection of abnormalities that includes fatty liver, and it currently affects one-third of the United States population and has become a major health concern worldwide. Fructose intake, primarily from added sugars in soft drinks, can induce fatty liver in animals and is epidemiologically associated with nonalcoholic fatty liver disease in humans. Fructose is considered lipogenic due to its ability to generate triglycerides as a direct consequence of the metabolism of the fructose molecule. Here, we show that fructose also stimulates triglyceride synthesis via a purine-degrading pathway that is triggered from the rapid phosphorylation of fructose by fructokinase. Generated AMP enters into the purine degradation pathway through the activation of AMP deaminase resulting in uric acid production and the generation of mitochondrial oxidants. Mitochondrial oxidative stress results in the inhibition of aconitase in the Krebs cycle, resulting in the accumulation of citrate and the stimulation of ATP citrate lyase and fatty-acid synthase leading to de novo lipogeneis. These studies provide new insights into the pathogenesis of hepatic fat accumulation under normal and diseased states.
Introduction
Nonalcoholic fatty liver disease (NAFLD)2 is a common condition, present in 20–30% of adults in the United States, and is associated with increased mortality (1). A recently recognized risk factor of NAFLD is fructose, especially that present in soft drinks (2–5). Fructose is known to be lipogenic (6), and the feeding of fructose to rats causes the rapid development of fatty liver (7). This is mediated by the initial rapid phosphorylation of fructose to fructose 1-phosphate by fructokinase (ketohexokinase, KHK) with paralleled ATP depletion and uric acid generation. Therefore, one unique aspect of fructose compared with glucose and other sugars is that it generates uric acid inside the hepatocyte during its metabolism (8). Serum uric acid rises acutely after the ingestion of fructose (9). Chronic administration of diets enriched in fructose also raises both fasting and 24-h uric acid levels (10, 11), and studies of the general population have linked soft drink intake with increased serum uric acid levels (12). Given this information, it is not surprising that an elevated serum uric acid has been repeatedly shown to both predict as well as to be common in subjects with NAFLD (13–22).
The possibility that uric acid may have a causal role in NAFLD has to our knowledge not been studied in detail. There is a report that lowering of uric acid can prevent hepatic steatosis in the Mongolian gerbil (23), but the mechanism was not elucidated. However, recent studies have shown that uric acid can induce intracellular oxidative stress and proinflammatory effects in various cell types (24–28). This led us to hypothesize that uric acid may have a role in fructose-induced fatty liver disease and possibly in other models of NAFLD. In this study, we identify a mechanism by which uric acid may mediate fatty liver, involving a pathway in which uric acid alters mitochondrial function with the alteration in fat synthesis.
EXPERIMENTAL PROCEDURES
Materials
Cell culture medium (RPMI 1640 medium), fetal calf serum, and antibiotics were from Invitrogen. Rabbit polyclonal antibody to KHK (HPA00740) was purchased from Sigma; the antibody to NOX4 (SC-30141) was from Santa Cruz Biotechnology Inc. (Santa Cruz, CA); antibodies to β-actin (3700) was from Cell Signaling (Danvers, MA), and antibody to aldolase B (H00000229-B01) was from Novus Biologicals (Littleton, CO). Secondary antibodies conjugated with horseradish peroxidase were from Cell Signaling, and those conjugated with Alexa dyes were purchased from Molecular Probes (Eugene, OR). Fructose, glucose, mannitol, uric acid, and allopurinol were purchased from Sigma, and probenecid was obtained from Molecular Probes.
Cell Culture and Silencing
The established human hepatocyte cell line HepG2 was maintained as described elsewhere (29). Expression of KHK, aldolase B, and NOX4 in HepG2 cells was stably silenced. Briefly, lentiviral particles codifying for a silencing sequence were obtained from Open Biosystems (KHK, Huntsville, AL) and Santa Cruz Biotechnology (aldolase B and NOX4). In both cases, HepG2 cells previously treated with Polybrene (10 μg/ml) were exposed to the lentiviral particles for 24 h for transduction. After exposure, medium was removed, and cells were incubated in normal media in the presence of puromycin (2 μg/ml) to select transduced cells. Clones with greater than 90% silencing as assessed by Western blot analysis were selected from colonies growing in plates from a 10-fold dilution series in media prepared with 2 μg/ml puromycin antibiotic. In experiments involving allopurinol and probenecid, cells were preincubated with these compounds for 8 h prior exposure to fructose or uric acid.
Determination of Intracellular, Intrahepatic, and Serum Uric Acid and Triglycerides
Cell lysates obtained with MAPK lysis buffer as well as serum collected from 8-h fasting rats were analyzed using a VetAce autoanalyzer (Alfa Wassermann, West Caldwell, NJ), as described previously (3). For triglyceride determination in liver and HepG2 cells, fat was solubilized by homogenization in 1 ml of solution containing 5% Nonidet P-40 in water; the samples were slowly exposed to an 80-100 °C water bath for 2-5 min until the Nonidet P-40 became cloudy and then cooled down to room temperature. Samples were then centrifuged for 2 min to remove any insoluble material. Triglyceride determination with the VetAce autoanalyzer was used in the initial breakdown into fatty acids and glycerol. Glycerol was then oxidized to generate a product that reacts with the probe to generate color at 570 nm. Similarly, uric acid determination was based on the conversion of uric acid to allantoin, hydrogen peroxide (H2O2), and carbon dioxide by uricase. The H2O2 was then determined by its reaction with the probe to generate color at ∼571 nm. Values obtained were normalized per mg of soluble protein in the lysates. Because rat livers contained endogenous uricase, intrahepatic uric acid in liver samples were analyzed employing the quantichrom uric acid determination kit (Bioassay Systems, Hayward, CA), which is a non-uricase-based system.
Protein Extraction and Western Blotting
Protein lysates were prepared from confluent cell cultures and rat tissue employing MAPK lysis buffer as described previously (10). Sample protein content was determined by the BCA protein assay (Pierce). 50 μg of total protein was loaded per lane for SDS-PAGE (10% w/v) analysis and then transferred to PVDF membranes. Membranes were incubated with primary antibodies and visualized using a horseradish peroxidase secondary antibody and the HRP Immun StAR® detection kit (Bio-Rad). Chemiluminescence was recorded with an Image Station 440CF, and results were analyzed with the 1D Image Software (Kodak Digital Science, Rochester, NY).
Determination of AMPD2 Activity
AMP deaminase activity was determined by estimating the production of ammonia by a modification of the method described by Chaney and Marbach (30) from cells collected in a buffer containing 150 mm KCl, 20 mm Tris-HCl, 1 mm EDTA, and 1 mm dithiothreitol. Briefly, the reaction mixture consisted of 25 mm sodium citrate, pH 6.0, 50 mm potassium chloride, and different concentration of AMP. The enzyme reaction was initiated by the addition of the enzyme solution and incubated at 37 °C for 15 min. For determination of AMPD activation by fructose and fructose 1-phosphate, these compounds (1–5 mm) were added to the lysates 30 min before the assay for preincubation. The reaction was stopped with the addition of the phenol/hypochlorite reagents as follows: reagent A (100 mm phenol and 0.050 g/liter sodium nitroprusside in H2O) was added, followed by reagent B (125 mm sodium hydroxide, 200 mm dibasic sodium phosphate, and 0.1% sodium hypochlorite in H2O), and incubated for 30 min at 25 °C. The absorbance of the samples was measured at 625 nm with a spectrophotometer. To determine the absolute specific activity of ammonia production (micromoles of ammonia/min), a calibration curve was determined in the range of 5 μm to 1 mm ammonia.
Determination of Intracellular Oxidative Stress
Reactive oxygen species in live HepG2 cells were determined employing the Image-iTTM LIVE green reactive oxygen species detection kit I36007 (Molecular Probes) as per the manufacturer's instructions. Briefly, HepG2 control cells and cells exposed to fructose, uric acid, or fructose and allopurinol for 24 h were washed with warm PBS, incubated with 5 μm of the cell-permeant fluorescent probe carboxyl H2-dichlorodihydrofluorescein for 30 min at 37 °C, and protected from light. After incubation, cells were washed with warm buffer, and total fluorescence was determined employing a multimode microplate reader with the following settings: excitation, 458 ± 20 nm; emission, 528 ± 20 nm. DCF fluorescence signal was expressed as total fluorescence intensity normalized to milligram of soluble protein.
Immunofluorescence of NOX4 and ACAA2 and Confocal Microscopy
HepG2 cells were grown on coverslips to 60% confluency, fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100 in PBS before overnight incubation with NOX4 and ACAA2 antibodies (1:50 dilution in PBS/Triton X-100 with 3% milk). The next day, cell were washed three times with PBS/Triton X-100 and incubated for 1 h with appropriate secondary antibodies conjugated with the following fluorochromes: Alexa488 anti-rabbit antibody for detection of NOX4 and Alexa568 anti-mouse antibody for detection of ACAA2. After incubation, cells were washed, mounted, and imaged with a confocal microscope for colocalization studies. Imaging and analysis were performed with a laser-scanning confocal microscope (LSM510, Carl Zeiss, Thornwood, NY) with a ×40 water immersion objective and the corresponding post-acquisition software. Immunofluorescence analysis was performed from sections of three animals and by evaluation of greater than 10 random fields each. Imaging experiments were performed at the University of Colorado Anschutz Medical Campus Advance Light Microscopy Core supported in part by National Institutes of Health/NCRR Colorado CTSI Grant UL1 RR025780.
Isolation of Pure Mitochondrial Fractions from HepG2 and Liver Tissues
Pure mitochondrial fractions were isolated from liver and cultured cells using the mitochondria isolation kit for tissue (89801) and mitochondria isolation kit for cultured cells (89874), respectively, as per manufacturer's instructions (Thermo Scientific, Rockford, IL). Purity of mitochondrial and cytoplasmic fractions was analyzed by Western blot with specific markers (COX4-mitochondria, and tubulin-cytosol).
Determination of Mitochondrial Activity in HepG2 Cells
Mitochondrial activity was determined by JC-1 staining (Invitrogen). HepG2 cells were incubated with 10 μg/ml JC-1 in complete RPMI 1640 medium for 10 min at 37 °C, and the cells were then washed and examined using a plate reader employing the following settings: for green signal excitation, 485 ± 15 nm, and emission, 530 ± 15 nm; for red signal excitation, 535 ± 17.5 nm, and emission 590 ± 17.5 nm. A green fluorescence emission was detected in the presence of low mitochondrial membrane potential, and an orange/red fluorescence was emitted in mitochondria with high membrane potential.
Determination of NOX4 Activity in Purified Mitochondrial Fractions
NADPH oxidase activity in pure intact mitochondrial fractions was performed similarly as described previously (31). Briefly, mitochondria were incubated with a 50 mm phosphate buffer, pH 7.0, containing 1 mm EGTA, 150 mm sucrose, 100 μm NADPH, and NADPH-driven hydrogen peroxide production was measured by using Amplex Red assay kit (A22188, Molecular Probes). Specificity generation of superoxide by NADPH was confirmed by no measurable activity in the absence of NADPH. The amount of hydrogen peroxide generated was determined by measuring the absorbance of the Amplex Red reagent at 565 nm. Superoxide production was expressed as total fluorescence/mg of protein.
Mitochondrial Aconitase Activity and Cytosolic Citrate Determination
Aconitase activity was determined in pure intact mitochondrial fractions employing the aconitase activity kit from Biovision (K716, Mountain View, CA), expressed as the amount of citrate metabolized to isocitrate (in micromoles) in 1 min, and normalized to milligrams of soluble protein. Fresh mitochondria were resuspended in ice-cold assay buffer and sonicated for 20 s prior to analysis. Citrate levels in the cytosolic fractions obtained from mitochondrial purification were determined with the citrate assay kit (Biovision, K655) as per the manufacturer's protocol.
Pound Mouse Experiments
The Animal Care and Use Committee of the University of Florida approved the protocol. The Pound mouse is a recently established prediabetes/metabolic syndrome model (Charles River, Wilmington, MA) with hyperuricemia (32). The liver tissues used in this study are from a previously published study in which the Pound mouse was treated with allopurinol to assess its effect on fat tissues (32). Basically, C57BL/6NCrl-Leprdb-lb/Crl and lean C57BL/6NCrl 6-week-old mice were maintained on a standard Purina 5001 Diet (PMI, Richmond, IN) under a 12-h light/12-h dark cycle with access to food and water ad libitum. To assess the effects of lowering uric acid levels, obese and lean animals (8–10 animals per group) were either administered for 8 weeks with allopurinol (30 mg/kg) or remained untreated. Samples of liver tissue were collected on ice at sacrifice and rinsed in ice-cold PBS.
Transmission Electron Microscopy
HepG2 cells were pelleted in a microcentrifuge tube and perfused with 2% glutaraldehyde and 2% paraformaldehyde in 0.1 m phosphate buffer. After post-fixation of prepared HepG2 cell blocks in 2% osmium tetroxide, cells were dehydrated and embedded in Epoxy resin. Appropriate areas of interest were selected from sections ∼1 μm thick stained with toluidine blue. About 60–70-nm ultra-thin sections were cut by an ultramicrotome using a diamond knife. Thin sections were stained with 1–2% aqueous uranyl acetate, followed by 1% lead citrate. Stained sections were observed and photographed with H-7650 transmission electron microscope (Hitachi, Japan) at an accelerating voltage of 80 kV.
Hemodialysis Population Study
The Ethical Committee of Ankara Training and Research Hospital (Dikimevi-Ankara, Turkey) approved the study, and all patients gave their informed consent. Between January 1 and June 30, 2010, a total number of 546 chronic hemodialysis patients at least for 6 months from six hemodialysis center entered into the study. The patients had standard hemodialysis for 3.5–4 h three times a week using low molecular weight heparin for anticoagulation; dialysis was performed using high flux membranes and bicarbonate-containing dialysis fluid. Blood flow was usually 300 ml/min with a dialysate flow rate of 500 ml/min. To diminish any confounders that may influence patients with hepatosteatosis, patients with established chronic liver disease, viral hepatitis (hepatitis B and C virus), chronic alcoholism, patients with severe congestive heart failure (New York Heart Association class III to IV), acute infection, and unwillingness to participate to the study were not included in the study (n = 63), resulting in 483 subjects included in the final analysis. We measured the height and dry weight of all patients. Body mass index (BMI) was calculated by dividing the weight (kg) by height squared (m2).
Laboratory and Fatty Liver Assessment
Serum uric acid levels were determined in all samples by the central laboratory of the Ankara Training and Research Hospital, with an Olympus AU2700 autoanalyzer (Olympus Diagnostics, Hamburg, Germany). Hepatosteatosis grade of patients was performed by M. K. and M. D. who were uninformed about the patient characteristics at the time of admission before the blood test results appeared. Hepatosteatosis grading was done using a standard protocol (5) as follows: Grade 1 (mild), slight diffused increase in the echoes in hepatic parenchyma and normal visualization of intrahepatic vessel borders and diaphragm; Grade 2 (moderate), moderately diffuse increase in echoes in the hepatic parenchyma and slightly impaired visualization of the intrahepatic vessel borders and diaphragm; Grade 3 (severe), severe increase in echoes in the hepatic parenchyma and poor or no visualization of intrahepatic vessel borders, diaphragm, and posterior portion of the right lobe.
Statistical Method for Human Study
Analyses were performed using SPSS software (Version 15.0, SPSS, Chicago). The Kolmogorov-Smirnov test was used to determine whether the continuous variables were normally distributed. The study population was assigned into quartiles based on the serum uric acid levels. Comparisons of multiple mean values were carried out by Kruskal-Wallis tests or analysis of variance as appropriate. For comparison of two groups, we used the Student's t test for normally distributed parameters and used Mann-Whitney U test for not normally distributed parameters. The χ2 test was used for categorical variables. A p value of <0.05 was considered statistically significant.
Statistics and Data Analysis
All data are presented as the means ± S.E. Data graphics and statistical analysis were performed using Instat (Version 3.0) and Prism 5 (both Graph Pad Software, San Diego). Data were analyzed for normality tests by using one-way analysis of variance. Multiple group corrections were performed using the method of Bartlett. In most cases, experiments were performed three times with independent replicates. Total data points (n) are identified in figure legends. p values < 0.05 were recognized as statistically significant.
RESULTS
Uric Acid Mediates Fructose-induced Fat Accumulation in HepG2 Cells
To characterize the role of uric acid in the lipogenic ability of fructose, we first analyzed the response of HepG2 cells to increasing amounts of fructose. As shown in Fig. 1A, intracellular TG accumulation was significantly increased after exposure of cells for 72 h in a dose-dependent manner (from 0 to 20 mmol/liter). This increment was associated with higher intracellular uric acid levels as the result of hepatic AMPD activation induced by fructose (Fig. 1, B and C). To analyze whether uric acid generation mediated fructose-induced TG accumulation, we then challenged hepatocytes to fructose in the presence of a xanthine oxidase inhibitor, allopurinol (100 μm), to block uric acid production. As shown in Fig. 1D, allopurinol effectively blocked the production of uric acid after exposure of cells to fructose. Of interest, TG accumulation was also inhibited by allopurinol indicating that uric acid may play a key role in the lipogenic ability of fructose (Fig. 1E).
FIGURE 1.

Uric acid mediates fructose-induced fat accumulation in HepG2 cells. A, intracellular TG levels (in mg of normalized to g of soluble protein) in control and fructose-exposed cells (from 0 to 20 mmol/liter) for 72 h. B, intracellular uric acid levels in control and fructose-exposed cells (from 0 to 20 mmol/liter) for 72 h. C, AMPD activity in control and fructose-exposed cells (from 0 to 20 mmol/liter) for 72 h. D, intracellular uric acid levels in fructose (5 mmol/liter) and fructose + allopurinol (100 μmol/liter)-exposed cells for 72 h. E, intracellular TG levels (mg/g of soluble protein) in fructose (5 mmol/liter) and fructose + allopurinol (100 μmol/liter)-exposed cells for 72 h. *, p < 0.05; **, p < 0.01; ***, p < 0.001; §§, p < 0.01 versus 0 h; §§§, p < 0.001 versus 0 h.
Uric Acid Induces Fat Accumulation in HepG2 Cells Independently of Fructose
Because our studies suggested that uric acid may contribute to fructose-induced triglyceride accumulation, we next evaluated the effects of uric acid alone on triglyceride accumulation. Human HepG2 cells were exposed to increasing levels of endotoxin-free and crystal-free uric acid from 0 to 750 μmol/liter (4–12 mg/dl), and intracellular TG concentration was measured after 72 h of exposure. Compared with nonexposed cells, TG levels were significantly increased by uric acid in a dose-dependent manner (from 0 to 750 μmol/liter; 250 μmol/liter is considered normouricemia in humans (around 4 mg/dl), 500 μmol/liter is considered hyperuricemia in humans (∼8 mg/dl), and 750 μmol/liter is considered severe hyperuricemia in humans (∼12 mg/dl); see Fig. 2A). The increase in TG content was associated with the accumulation of lipid-containing vacuoles as determined by Oil Red-O staining (Fig. 2B). Importantly, uric acid significantly increased TG accumulation in cells silenced for KHK and AldoB, enzymes involved in fructose metabolism suggesting that uric acid may work independently of direct fructose metabolism (Fig. 2, C and D). In contrast, when fructose was added to the cells, it did not induce TG accumulation in KHK-deficient cells that were unable to generate uric acid, but it significantly did increase intracellular TG levels in AldoB-deficient cells in which the KHK-dependent generation of uric acid during fructose metabolism is intact (Fig. 2, C and D, blue columns). Consistent with this interpretation, decreased uric acid levels were found in KHK-deficient cells treated with fructose, whereas levels were increased in fructose-exposed AldoB-deficient cells (Fig. 2E). Furthermore, KHK up-regulation by fructose was not blocked in AldoB-deficient cells (Fig. 2F).
FIGURE 2.

Uric acid induces fat accumulation in HepG2 cells independently of fructose metabolism. A, concentration of TG (in milligrams normalized to gram of soluble protein) in extracts from cells exposed to different amounts of uric acid for 72 h. B, representative image of Oil Red-O staining of HepG2 cells control or exposed to uric acid (750 μmol/liter). C, concentration of TG (milligrams/g of soluble protein) in control (1st column) or KHK-deficient cells (2nd to 6th columns) exposed to fructose (blue column) or different amounts of uric acid (black columns). D, concentration of TG (milligrams/g of soluble protein) in control (1st column) or AldoB-deficient cells (columns 2–6) exposed to fructose (blue column) or different amounts of uric acid (black columns). E, concentration of uric acid (micrograms/g of soluble protein) in control, KHK-deficient, or AldoB-deficient cells exposed to fructose. F, KHK expression in control or AldoB-deficient cells exposed to fructose. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Uric Acid Induces Mitochondrial Oxidative Stress and Morphology Changes
We next performed studies to identify how uric acid might stimulate fat synthesis. We have previously reported that uric acid induces oxidative stress in vascular cells, tubular cells, and adipocytes, in part by stimulating NADPH oxidase (24, 25, 28, 33). Consistent with this finding, uric acid also stimulated oxidative stress as determined by fluorescence using the oxidative stress marker DCF. Fructose-induced oxidative stress was blocked by allopurinol (Fig. 3, A and B).
FIGURE 3.

Uric acid induces superoxide generation and mitochondrial dysfunction in HepG2 cells. A, cellular oxidative stress as measured with the fluorescent dye DCF in cells exposed to increasing levels of uric acid (from 0 to 750 μmol/liter). B, cellular oxidative stress as measured with the fluorescent dye DCF in cells exposed to 5 mmol/liter fructose in the presence or absence of different amounts of allopurinol (from 0 to 500 μmol/liter) Black columns represent allopurinol concentrations that have a significant effect compared with fructose alone. C, representative image of mitochondrial superoxide generation as measured with the fluorescent dye MitoSOX in control cells and cells exposed to uric acid (750 μmol/liter). D, concentration of MitoSOX in mitochondria from control and cells exposed to uric acid. E, percentage of highly polarized mitochondria as determined with the fluoroprobe JC-1. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
We next asked whether the oxidative stress might be occurring in the mitochondria, because fat metabolism is intricately linked with mitochondrial function. As shown in Fig. 3, C and D, uric acid induced oxidative stress in the mitochondria as determined with the fluorescent compound, MitoSOX, which detects de novo generation of superoxide radicals. Consistent with an increase in mitochondrial oxidative stress, we found that uric acid significantly decreased mitochondrial membrane potential as determined by JC-1 staining (Fig. 3E). Because NADPH oxidases produce superoxide radicals in response to NADPH, and uric acid has been shown to activate the NADPH oxidase subunit NOX4 in other cell types (24), we examined whether the mechanism whereby uric acid promoted mitochondrial oxidative stress could be mediated by the translocation of NOX4 to the mitochondria. To this end, we first determined NOX4 expression in HepG2 cells exposed to uric acid. As shown in Fig. 4, A and B, NOX4 (green) is located in the cytosol of normal HepG2 cells by confocal microscopy. In contrast, upon exposure of cells to uric acid for 24 h, NOX4 gets shifted to the mitochondria where it colocalizes with the specific mitochondrial marker ACAA2 (Fig. 4A, red). NOX4 mitochondrial expressions have been described previously under different stress conditions (31), and immunostaining results were confirmed by Western blot for NOX4 from pure mitochondrial fractions (Fig. 4, C and D). Proof that the NADPH oxidase was functional is shown by measuring H2O2 in isolated mitochondria in response to NADPH substrate (Fig. 4E) and by showing that the increase in mitochondrial oxidants in response to uric acid could be prevented by either apocynin (an antioxidant) or by silencing NOX4 (Fig. 4F). The silencing of NOX4 also partially prevented the rise in intracellular TG in response to fructose or uric acid (Fig. 4G). High levels of uric acid (750 μmol/liter) also induced significant changes in mitochondrial morphology as determined by electron microscopy (Fig. 5). Compared with controls, uric acid-treated HepG2 cells demonstrated mitochondria that were shorter and smaller (Fig. 5, A and B), suggesting mitochondrial fragmentation. Higher magnification (×60,000) revealed a significant decrease in the number of cristae in disarray and a disruption of mitochondrial double membrane in uric acid-treated cells compared with well preserved cristae and double membrane in control cells (Fig. 5, C–E). These studies provide the novel finding that uric acid induces mitochondrial oxidative stress in association with translocation of NADPH oxidase to the mitochondria.
FIGURE 4.

Uric acid induces mitochondrial superoxide generation in HepG2 cells by translocating NOX4 to the mitochondria. A, representative confocal image of NOX4 (green) and the mitochondrial marker ACAA2 (red) in control cells (top) and cells exposed to uric acid (750 μmol/liter, bottom). Colocalization is depicted in yellow. B, confocal analysis of ACAA2 and NOX4 colocalization expressed as percentage of NOX4 pixels colocalizing with ACAA2 pixels in control cells and cells exposed to uric acid. C and D, NOX4 protein expression in isolated pure mitochondrial fraction from cells control and exposed to increasing amounts of uric acid. E, superoxide generation from isolated mitochondria from control cells or incubated with uric acid in response to NADPH addition. F, MitoSOX levels in HepG2 cells exposed to uric acid in the presence of the NOX4 inhibitor, apocynin (100 μmol/liter, left panel) or in cells which NOX4 expression has been stably silenced (right panel). G, concentration of TG (milligrams/g of soluble protein) in control and NOX4-deficient cells exposed to fructose or uric acid. *, p < 0.05; **, p < 0.01.
FIGURE 5.

Uric acid induces mitochondrial morphology changes in HepG2 cells. Transmission electron microscopy images at original magnifications of ×15,000 (A and B) and ×60,000 (C and D). Electron microscopy representative images demonstrating marked alterations in mitochondrial morphology in HepG2 cells incubated with 750 μmol/liter uric acid for 24 h. Short and smaller mitochondria was noted in uric acid-treated cells (B, thick arrow) compared with control (A). Higher magnification (×60,000) revealed a significant decrease in number of cristae in disarray and a disruption of mitochondrial double membrane in uric acid-treated cells (D) compared with well preserved cristae (arrowhead, C) and double membrane (thin arrow, C) in control cells. E, quantization of mitochondrial size in control and uric acid-exposed cells (n >100 mitochondria analyzed). **, p < 0.01.
Mitochondrial Oxidative Stress Stimulates Fat Accumulation
We next examined whether mitochondrial oxidative stress might cause TG accumulation in HepG2 cells. Aconitase, a key enzyme in the citric acid cycle, is inhibited by oxidants (34). Consistent with this, we found that mitochondrial aconitase activity was reduced in uric acid-exposed cells (77.4% decrease, p < 0.01, Fig. 6A). Furthermore, fructose-exposed cells also possessed reduced mitochondrial aconitase activity (68.9% reduction, p < 0.01) that was partially prevented by allopurinol (Fig. 6B). Inhibition of aconitase activity was associated with a significant accumulation of its substrate, citrate, in the cytosol (0.046 versus 0.069 nmol/mg, p < 0.01, Fig. 6C). Citrate is also a substrate for ATP-citrate lyase (ACL), which converts it into acetyl-CoA. Active ACL (determined by Western blotting for phosphorylation at serine 455, Ser(P)455ACL) was increased in the presence of either fructose or uric acid (Fig. 6, D–F). Because citrate release to the mitochondria may necessitate activation of anaplerotic routes to support the TCA cycle, we analyzed intracellular levels of anaplerotic compounds aspartate, pyruvate, glutamate, and hydroxybutyrate (a marker of fat oxidation). Cytosolic levels of pyruvate and glutamate were significantly increased in uric acid (750 μmol/liter)-exposed cells compared with control cells (156.1 versus 166.6 mmol/g for glutamate, p < 0.05; 23.8 versus 27.3 mmol/g for pyruvate, p < 0.05). In contrast, aspartate and hydroxybutyrate levels were found to be significantly lower, the latter perhaps as a consequence of increased lipogenesis (5.8 versus 5.2 mmol/g for aspartate, p < 0.01; 157.7 versus 142.2 mmol/g for hydroxybutyrate, p < 0.05).
FIGURE 6.

Uric acid stimulates de novo lipogenesis by releasing citrate to the cytosol and activating ACL and FAS. A, mitochondrial activity levels of aconitase in control cells and cells exposed to uric acid. B, mitochondrial activity levels of aconitase in control cells and cells exposed to fructose in the presence or absence of allopurinol. C, cytoplasmic citrate levels (nanomoles/mg of soluble protein) in control cells and cells exposed to uric acid. D–F, expression of activated ACL as determined by its phosphorylation at serine 455 in control cells and cells exposed to uric acid at different time points or fructose in the presence or absence of allopurinol. G, cytoplasmic nonmitochondrial citrate levels (nanomoles/mg of soluble protein) in control and cells exposed to uric acid in the presence or absence of radicicol (10 μmol/liter). H, concentration of TG (milligrams/g of soluble protein) in control and fructose or uric acid-exposed cells in the presence or absence of the ACL inhibitor radicicol. I, concentration of TG (milligrams/g of soluble protein) in control and fructose or uric acid-exposed cells in the presence or absence of the FAS inhibitor C75. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To determine whether uric acid-induced TG accumulation was dependent on ACL, we inhibited ACL activity with radicicol (10 μmol/liter) and analyzed the TG response of hepatocytes. In the presence of radicicol, cytoplasmic citrate levels significantly increased in response to uric acid indicating that citrate was being used by ACL (Fig. 6G). TG accumulation was also largely prevented by radicicol in response to either fructose or uric acid (85.3 and 95.5% prevention from fructose and uric acid, respectively, p < 0.01, see Fig. 6H) indicating that citrate-stimulated ACL is involved in fructose or uric acid-mediated TG accumulation. In addition, we found that inhibiting the activity of fatty-acid synthase (FAS) with Cys75 (10 μmol/liter), which ultimately converts acetyl-CoA and malonyl-CoA to long chain saturated fatty acids (palmitate) that will be esterified with glycerol to produce TG, could prevent TG accumulation in response to fructose or uric acid (80.3 and 89.5% prevention from fructose and uric acid, respectively, p < 0.01; see Fig. 6I). Thus, uric acid increases fat in hepatocytes by stimulating mitochondrial oxidative stress that blocks aconitase and increases citrate, which then stimulates fat synthesis. Although allopurinol may have off-target effects considering its antioxidant properties, the prevention of fructose-induced lipogenesis seems to be dependent on its ability to reduce uric acid levels because adding back uric acid restored the metabolic effects of fructose (Fig. 7, A–C).
FIGURE 7.
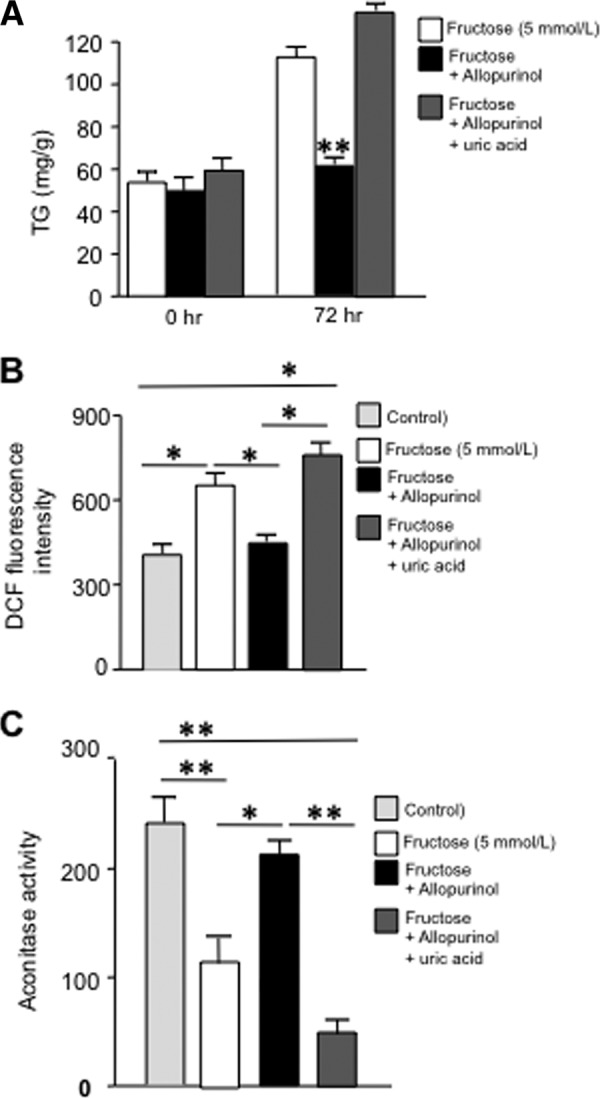
Adding back uric acid (750 μmol/liter) blocks the inhibitory effects of allopurinol (100 μmol/liter) in fructose-induced mitochondrial dysfunction and TG accumulation. A, intracellular TG (milligrams/g of soluble protein) levels in HepG2 cells exposed to fructose alone or in the presence of allopurinol only or along with uric acid. B, DCF fluorescent units in HepG2 cells exposed to fructose alone or in the presence of allopurinol only or along with uric acid. C, mitochondrial aconitase activity in HepG2 cells exposed to fructose alone or in the presence of allopurinol only or along with uric acid. *, p < 0.05; **, p < 0.01.
Lowering Uric Acid Reduces Hepatic Steatosis in the Pound Mouse Model of Metabolic Syndrome
Because lowering intracellular uric acid levels could prevent fat accumulation in fructose-exposed HepG2 cells, we wanted to know if lowering uric acid might also prevent fatty liver in an animal model of hyperuricemia and metabolic syndrome. The pound mouse has a leptin receptor defect and develops obesity, insulin resistance, increased visceral fat, hyperuricemia, and fatty liver. We had previously reported that the lowering of uric acid with allopurinol (30 mg/kg in drinking water) in these mice can improve insulin resistance, lower blood pressure, and reduce the inflammatory response in the visceral fat (32). Livers from that original study were analyzed for their fat content and the effect of allopurinol treatment. Allopurinol effectively reduced intrahepatic uric acid levels (p < 0.001; Fig. 8A), TG content (p < 0.01; Fig. 8B), and hepatic cholesterol content (p < 0.01; Fig. 8C). These findings were paralleled with significantly decreased activity of NOX4 in the mitochondria of pound mice receiving allopurinol (Fig. 8D) with concomitant higher mitochondrial aconitase activity (Fig. 8E) and lower cytoplasmic citrate release (Fig. 8F).
FIGURE 8.
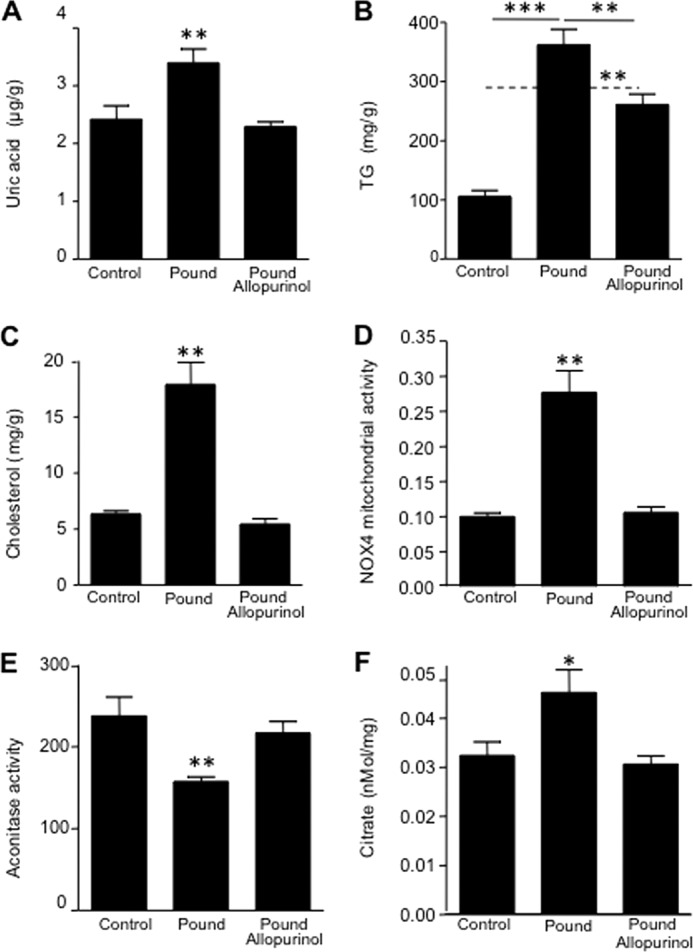
Lowering uric acid prevents hepatic steatosis in the pound mouse. A, quantity of uric acid in liver extracts from lean, pound, and pound with allopurinol mice. B, concentration of TG (milligrams/g of soluble protein) in liver extracts from lean, pound, and pound with allopurinol mice. C, concentration of cholesterol (total and sterol esters) in liver extracts from lean, pound, and pound with allopurinol mice. D, NOX4 mitochondrial activity in liver extracts from lean, pound, and pound with allopurinol mice. E, mitochondrial aconitase activity in liver extracts from lean, pound, and pound with allopurinol mice. F, cytoplasmic citrate in levels liver extracts from lean, pound, and pound with allopurinol mice. (n = 5 for each group.) *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Serum Uric Acid Levels Positively Correlate with Hepatic Steatosis in Humans Independent of Obesity
As mentioned in the Introduction, NAFLD is strongly associated with hyperuricemia (13–22), but one potential explanation could be because many patients with hyperuricemia have obesity and metabolic syndrome. One potential way to determine whether hyperuricemia may predict NAFLD independent of obesity is to examine the hemodialysis population, as hyperuricemia is common even when obesity is absent. We therefore evaluated the relationship of hyperuricemia with ultrasound-diagnosed fatty liver in a hemodialysis population. General and serum parameters are shown in Table 1. Average serum uric acid levels in patients on hemodialysis were 6.3 ± 1.6 mg/dl, and patients were divided into four quartiles as follows: Q1, 4.28 ± 0.7; Q2, 5.7 ± 0.3; Q3, 6.7 ± 0.3; and Q4, 8.4 ± 0.9 mg/dl. Serum uric acid levels were associated with increasing prevalence of hepatic steatosis (Table 2), although this association was only observed in nondiabetic subjects (Table 3). For each increasing quartile of serum uric acid, there was a significant trend for increasing prevalence of steatosis severity. Although NAFLD is primarily observed in obese subjects, in hemodialysis patients NAFLD can occasionally be seen in lean subjects (BMI <20). As shown in Table 4, those subjects with NAFLD whose BMI was <20 also had significantly higher uric acid levels than those with a BMI less than 20 who did not have steatosis. These studies show an important association of uric acid with hepatic steatosis independent of obesity.
TABLE 1.
Overall and serum parameters in each uric acid quartile
The following abbreviations are used: T-cholesterol, total cholesterol; Kt/V, dialyzer clearance of urea·dialysis time/volume of body water; ALT, alanine transaminase; ALP, alkaline phosphatase; GGT, γ-glutamyl transpeptidase; ARB-ACEi, angiotensin receptor blockers-angiotensin-converting enzyme inhibitors.
| Entire group (n = 483) | 1st quartile (n = 118) | 2nd quartile (n = 127) | 3rd quartile (n = 116) | 4th quartile (n = 122) | p for trend | |
|---|---|---|---|---|---|---|
| Uric acid (mg/dl) | 6.3 ± 1.6 | 4.28 ± 0.7 | 5.7 ± 0.3 | 6.7 ± 0.3 | 8.4 ± 0.9 | 0.001 |
| Steatosis (n, %) | 111, 23% | 18, 15.3% | 22, 17.3% | 26, 22.4% | 45, 36.9% | 0.001 |
| Age (year) | 56.3 ± 16.2 | 56.7 ± 17.3 | 56.6 ± 16.2 | 56.4 ± 15.2 | 55.5 ± 17 | 0.94 |
| Gender, male (n, %) | 261, 54%% | 51, 43.2% | 79, 62.2% | 58, 50% | 73, 59.8% | 0.069 |
| Diabetes mellitus (n, %) | 158, 32.7% | 33, 28% | 38, 30% | 35, 30.2% | 52, 42.6% | 0.02 |
| Hemodialysis duration (month) | 42.9 ± 36.1 | 43.5 ± 37.4 | 46.5 ± 39.5 | 42 ± 32.8 | 39.3 ± 33.9 | 0.35 |
| T-cholesterol (mg/dl) | 178.5 ± 50.1 | 178.5 ± 52.3 | 176.2 ± 48.8 | 176.9 ± 50.9 | 182.2 ± 48.8 | 0. 80 |
| LDL cholesterol (mg/dl) | 105.6 ± 37.3 | 102.7 ± 37.8 | 105.4 ± 37.5 | 103.3 ± 37.1 | 108.4 ± 38.8 | 0.65 |
| HDL cholesterol (mg/dl) | 39.6 ± 11.9 | 39.7 ± 10.9 | 38.9 ± 11.4 | 39.5 ± 12.8 | 40.3 ± 12.6 | 0.77 |
| Triglyceride (mg/dl) | 169.3 ± 95.0 | 172.4 ± 116.6 | 156.2 ± 86.4 | 173.2 ± 86.7 | 176.1 ± 86.1 | 0.12 |
| Kt/V | 1.45 ± 0.35 | 1.5 ± 0.38 | 1.5 ± 0.25 | 1.51 ± 0.39 | 1.39 ± 0.27 | 0.15 |
| Albumin | 3.6 ± 0.6 | 3.6 ± 0.5 | 3.5 ± 0.7 | 3.6 ± 0.7 | 3.7 ± 0.5 | 0.45 |
| ALT | 18.3 ± 15.0 | 17.7 ± 15.1 | 16.7 ± 9.4 | 19.6 ± 13.0 | 19.3 ± 20.5 | 0.30 |
| ALP | 115.8 ± 106.5 | 110.9 ± 108.2 | 99.2 ± 64.3 | 118.6 ± 101 | 135 ± 138.2 | 0.15 |
| GGT | 48.9 ± 68.4 | 42.6 ± 39.1 | 58.8 ± 64 | 45 ± 36 | 48 ± 37 | 0.09 |
| Body mass index (kg/m2) | 23.9 ± 4.3 | 24.2 ± 5.1 | 23.9 ± 4.0 | 23.6 ± 4.2 | 24.1 ± 4.1 | 0.48 |
| Medications | ||||||
| Vitamin D (n, %) | 127, 26.3% | 32, 27.1% | 41, 32.3% | 21, 18.1% | 33, 27% | 0.424 |
| Statin (n, %) | 94, 19.6% | 19, 16.2% | 19, 15.1% | 23, 19.8% | 33, 27.3% | 0.019 |
| ARB-ACEi (n, %) | 223, 46.4% | 47, 39.8% | 61, 48% | 57, 49.6% | 58, 47.9% | 0.21 |
| Beta-blockers (n, %) | 181, 37.5% | 41, 34.7% | 49, 38.6% | 48, 41.4% | 43, 35.2% | 0.84 |
| Calcium channel blockers (n, %) | 123, 25.6% | 32, 27.4% | 30, 23.8% | 27, 23.3% | 34, 27.9% | 0.93 |
TABLE 2.
The prevalence and severity of hepatosteatosis for each uric acid quartile
| Entire group (n = 483) | 1st quartile (n = 118) | 2nd quartile (n = 127) | 3rd quartile (n = 116) | 4th quartile (n = 122) | p for trend | |
|---|---|---|---|---|---|---|
| Uric acid (mg/dl) | 6.3 ± 1.6 | 4.28 ± 0.7 | 5.7 ± 0.3 | 6.7 ± 0.3 | 8.4 ± 0.9 | <0.001 |
| Steatosis (n, %) | 111, 23% | 18, 15.3% | 22, 17.3% | 26, 22.4% | 45, 36.9% | <0.001 |
| Without steatosis (n, %) | 372, 77% | 100, 84.7% | 105, 82.7% | 90, 77.6% | 77, 63.1% | <0.001 |
| Mild steatosis (n, %) | 70, 14.5% | 9, 7.6% | 20, 15.7% | 18, 15.5% | 23, 18.9% | 0.021 |
| Moderate steatosis (n, %) | 26, 5.4% | 7, 5.9% | 1, 0.8% | 7, 6% | 11, 9% | 0.15 |
| Severe steatosis (n, %) | 15, 3.1% | 2, 1.7% | 1, 0.8% | 1, 0.9% | 11, 9% | 0.002 |
TABLE 3.
The prevalence of steatosis in lower and higher uric acid group in diabetic and nondiabetic patients
| Diabetic group |
Nondiabetic group |
|||||
|---|---|---|---|---|---|---|
| Lower group (n = 77) | Higher group (n = 81) | p value | Lower group (n = 163) | Higher group (n = 162) | p value | |
| Uric acid (mg/dl) | 5.23 ± 0.8 | 7.8 ± 0.9 | <0.001 | 4.9 ± 0.9 | 7.4 ± 1.1 | <0.001 |
| Steatosis (n, %) | 27, 35% | 29, 36% | 0.92 | 13, 8% | 42, 25.9% | <0.001 |
| Age (year) | 61.5 ± 13.8 | 60 ± 13.6 | 0.44 | 54.5 ± 17 | 53.7 ± 16.7 | 0.72 |
| Gender, male (n, %) | 31, 40.3% | 38, 47% | 0.40 | 94, 57.7% | 98, 60.5% | 0.60 |
| BMI (kg/m2) | 24.7 ± 4.3 | 24.6 ± 4.1 | 0.81 | 23.6 ± 4.5 | 23.6 ± 4.3 | 0.62 |
TABLE 4.
Comparison of serum uric acid level in patients with and without hepatosteatosis in cachectic group (BMI <20) patients without diabetes mellitus
| Patients with hepatosteatosis (n = 12) | Patients without hepatosteatosis (n = 54) | p value | |
|---|---|---|---|
| Serum uric acid level (mg/dl) | 7.17 ± 1.84 | 5.85 ± 1.34 | 0.023 |
| Gender (male) (n, %) | 8, 64.8% | 35, 66.7% | 0.90 |
| Age (years) | 49.6 ± 18.8 | 43.0 ± 17.4 | 0.25 |
Our results are summarized in Fig. 9, in which uric acid in hepatocytes leads to an increase in the lipogenic response of hepatocytes to fructose, which further leads to even greater intracellular uric acid levels. This effect results in mitochondrial oxidative stress and mitochondrial aconitase inhibition, with cytoplasmic citrate release and activation of lipogenesis through ACL and FAS.
FIGURE 9.
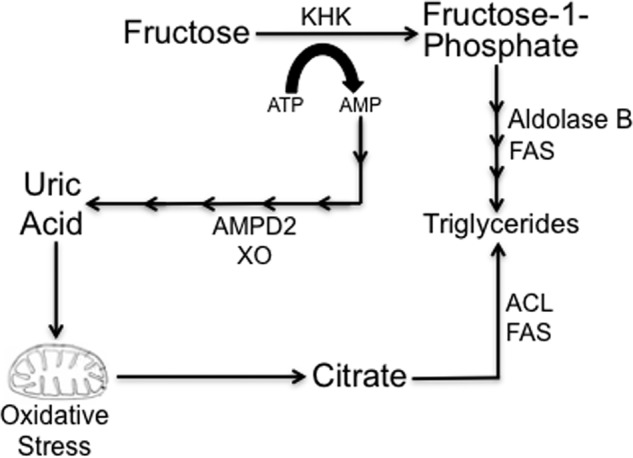
Potential mechanisms whereby uric acid mediates fat accumulation in hepatocytes. Uric acid is a by-product of fructose metabolism generated from the ATP depletion induced by KHK in fructose phosphorylation. AMP is converted to uric acid by the action of several enzymes, including AMP deaminase (AMPD2) and xanthine oxidase (XO). Direct fructose metabolism results in increased TG synthesis by AldoB and fatty-acid synthase (FAS). Similarly, uric acid induces mitochondrial oxidative stress and citrate release to the cytosol for de novo triglyceride synthesis from citrate and acetyl-CoA via ATP citrate lyase (ACL) and FAS.
DISCUSSION
Increased fatty acid synthesis, through the lipogenic pathway in liver, results in the development of hepatic steatosis (NAFLD) that contributes to the development of chronic hepatic inflammation and insulin resistance. The fructose component of added sugars has been particularly implicated in the etiology of these syndromes as the administration of fructose (or sucrose) to rats results in fatty liver as well as all features of the metabolic syndrome (6, 36–38).
Although the lipogenic ability of fructose has been known for years, the molecular mechanism whereby fructose potentiates fat accumulation in the liver remains poorly understood. It is classically accepted that fructose can be directly metabolized to triglycerides in a mechanism involving the action of three major enzymes as follows: fructokinase (KHK), which phosphorylates fructose to fructose 1-phosphate; AldoB, which breaks fructose 1-phosphate into dihydroxyacetone phosphate and glyceraldehyde; and FAS, which metabolizes acetyl-CoA produced from dihydroxyacetone phosphate and glyceraldehydes into triglycerides. Nevertheless, studies in which fructose has been labeled have shown that only 3% of the fructose is incorporated into triglycerides (39, 40), and this cannot explain why fructose is so lipogenic (41).
In this study, we have identified an additional pathway whereby fructose induces its lipogenic effects. We show that uric acid, either alone or as a by-product generated by the initial phosphorylation of fructose by KHK, directly regulates hepatic lipogenesis through the generation of mitochondrial oxidative stress. First, we found that exposure of HepG2 cells to uric acid at levels considered to be hyperuricemic in humans led to the development of hepatic steatosis characterized by an increase in de novo lipogenesis and triglyceride accumulation. This was due to enhanced mitochondrial translocation of the NADPH oxidase isoform, NOX4, which increased the generation of superoxide by the mitochondria. We found that aconitase, which is present in the mitochondrial matrix and is sensitive to changes in the mitochondrial oxidative state, was reduced in activity, and this led to citrate accumulation in the mitochondria with release into the cytosol where it acted as the substrate for de novo lipogenesis. Specifically, increased cytoplasmic citrate levels activated the ATP-sensitive enzyme, ACL by phosphorylation at Ser455, which converted it to acetyl-CoA for de novo lipogenesis through FAS. These findings suggest that uric acid-dependent regulation of mitochondrial function could be critical for the modulation of lipid homeostasis in fatty liver and that the lipogenic ability of fructose may be mediated at least partially by the generation of uric acid. In the settings of fructose, a condition associated with elevated uric acid production, we confirmed that blockade of uric acid generation with allopurinol prevented fructose-induced lipogenesis.
Although the direct metabolism of fructose to triglycerides partially explains why fructose is lipogenic, the uric acid-dependent pathway may explain two paradoxes in the lipogenic effects of fructose. First, when the fructose molecule is radiolabeled, only 1–3% of the fructose is directly converted into triglycerides (41). Second, people with hereditary fructose intolerance, a rare disorder in which AldoB is nonfunctional, develop fatty liver in response to dietary fructose even though it cannot be fully metabolized to triglycerides (42). Because fructose is distinct from glucose in its ability to increase intracellular uric acid, our studies provide an explanation for why fructose (or sucrose) can induce fatty liver much easier than glucose (or starch) in animals (7, 38).
Hyperuricemia is also a prevalent finding in patients presenting with NAFLD and metabolic syndrome, although its clinical meaning is still controversial and often underestimated. Numerous studies have consistently demonstrated that the presence of metabolic syndrome and its individual components is associated with serum uric acid levels (43–45), but the importance of hyperuricemia still remains a matter of debate. In this context, our group and others have reported that lowering uric acid can block both fructose-induced metabolic syndrome as well as improve features of metabolic syndrome in other animal models (32, 46). Xu et al. (23) also reported that lowering uric acid can improve fatty liver in the Mongolian gerbil. Pilot clinical studies have also reported benefits of lowering uric acid on inflammation, endothelial function, blood pressure, and insulin resistance (47–50). These studies are reawakening the possibility that uric acid may have a causal role in metabolic syndrome and cardiovascular disease (51).
The novel finding in this study is that uric acid can directly stimulate hepatic fat accumulation. Consistent with an independent deleterious role of uric acid in hepatic steatosis, we show that lowering hepatic uric acid could reduce fatty liver both in the fructose-exposed hepatocyte and in the pound mouse, an animal model of metabolic syndrome and hyperuricemia. These studies suggest that allopurinol could be helpful in the treatment of fatty liver in humans. However, the doses used in these animal studies are severalfold higher than we typically use in patients; the justification for doing this is because in rodents the xanthine oxidase activity is about 100 times higher compared with humans (52).
In conclusion, this study provides the first direct evidence that uric acid can stimulate fat synthesis in the hepatocyte. We identify a mechanism that involves the translocation of NADPH oxidase to the mitochondria with inactivation of aconitase, accumulation of citrate, and a stimulation of fat synthesis. We also show that this accounts for an important mechanism by which fructose induces hepatic fat accumulation. Together, our study may provide a mechanism for why soft drink intake is strongly linked with fatty liver (2, 3, 53, 54) and why uric acid is such a potent predictor of NAFLD (13–22). Further studies investigating the role of purine-degrading pathways and metabolites in metabolic syndrome seem warranted.
This work was supported, in whole or in part, by National Institutes of Health Grants HL-68607 and RC4 DK090869-01 (to R. J. J.). This work was also supported by Amway, Cardero, Danone, and Questcor (to R. J. .J.) and by startup funds from the University of Colorado (to R. J. J.) and Korea Healthcare Technology R&D Project, Ministry for Health, Welfare, and Family Affairs, Republic of Korea Grant A101742 (to D.-H. K.). M. A. L., T. I., and R. J. J. are listed as inventors on a patent application from the University of Colorado related to developing isoform-specific fructokinase inhibitors in the treatment of disorders associated with obesity and insulin resistance. T. N. and R. J. J. are listed as inventors on several patent applications related to lowering uric acid as a means to prevent or treat metabolic syndrome. Dr. Johnson also has a patent with the University of Washington and Merck for the use of allopurinol to treat hypertension. Dr. Johnson also discloses that he has consulted for Ardea, Astellas, Danone, and Novartis, that he is on the scientific board of Amway. R. J. J. and T. N. have patent applications related to lowering uric acid as a means to prevent fatty liver and metabolic syndrome. R. J. J., T. I., and M. L. also have patent applications related to blocking fructose metabolism as a means for preventing or treating metabolic syndrome. Dr. Johnson has also consulted for Novartis, Danone, Ardea, Mitsubishi Tanabe, and Astellas. Dr. Johnson also has two lay books on sugar, including the Sugar Fix (Rodale, 2008) and the Fat Switch (Mercola.com, 2012).
- NAFLD
- nonalcoholic fatty liver disease
- KHK
- Ketohexokinase
- TG
- triglyceride
- AMPD
- AMP deaminase
- FAS
- fatty-acid synthase
- ACL
- ATP-citrate lyase
- DCF
- 2′,7′-dichlorofluorescein
- AldoB
- aldolase B
- ACL
- ATP-citrate lyase
- BMI
- body mass index
- FAS
- fatty-acid synthase.
REFERENCES
- 1. Adams L. A., Lymp J. F., St Sauver J., Sanderson S. O., Lindor K. D., Feldstein A., Angulo P. (2005) The natural history of nonalcoholic fatty liver disease. A population-based cohort study. Gastroenterology 129, 113–121 [DOI] [PubMed] [Google Scholar]
- 2. Ouyang X., Cirillo P., Sautin Y., McCall S., Bruchette J. L., Diehl A. M., Johnson R. J., Abdelmalek M. F. (2008) Fructose consumption as a risk factor for nonalcoholic fatty liver disease. J. Hepatol. 48, 993–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lim J. S., Mietus-Snyder M., Valente A., Schwarz J. M., Lustig R. H. (2010) The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 7, 251–264 [DOI] [PubMed] [Google Scholar]
- 4. Abdelmalek M. F., Lazo M., Horska A., Bonekamp S., Lipkin E. W., Balasubramanyan A., Bantle J. P., Johnson R. J., Diehl A. M., Clark J. M. (2012) Higher dietary fructose is associated with impaired hepatic ATP homeostasis in patients with nonalcoholic fatty liver disease. Hepatology 56, 952–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abdelmalek M. F., Suzuki A., Guy C., Johnson R. J., Diehl A. M. (2007) Fructose-induced hyperuricemia as a causal mechanism for nonalcoholic fatty liver disease. Hepatology 46, 293A (Abstr. 128) [Google Scholar]
- 6. Havel P. J. (2005) Dietary fructose: implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutr. Rev. 63, 133–157 [DOI] [PubMed] [Google Scholar]
- 7. Ackerman Z., Oron-Herman M., Grozovski M., Rosenthal T., Pappo O., Link G., Sela B. A. (2005) Fructose-induced fatty liver disease. Hepatic effects of blood pressure and plasma triglyceride reduction. Hypertension 45, 1012–1018 [DOI] [PubMed] [Google Scholar]
- 8. van den Berghe G., Bronfman M., Vanneste R., Hers H. G. (1977) The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. Biochem. J. 162, 601–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Perheentupa J., Raivio K. (1967) Fructose-induced hyperuricaemia. Lancet 2, 528–531 [DOI] [PubMed] [Google Scholar]
- 10. Reiser S., Powell A. S., Scholfield D. J., Panda P., Ellwood K. C., Canary J. J. (1989) Blood lipids, lipoproteins, apoproteins, and uric acid in men fed diets containing fructose or high amylose cornstarch. Am. J. Clin. Nutr. 49, 832–839 [DOI] [PubMed] [Google Scholar]
- 11. Cox C. L., Stanhope K. L., Schwarz J. M., Graham J. L., Hatcher B., Griffen S. C., Bremer A. A., Berglund L., McGahan J. P., Keim N. L., Havel P. J. (2012) Consumption of fructose- but not glucose-sweetened beverages for 10 weeks increases circulating concentrations of uric acid, retinol-binding protein-4, and γ-glutamyl transferase activity in overweight/obese humans. Nutr. Metab. (Lond) 24, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Choi J. W., Ford E. S., Gao X., Choi H. K. (2008) Sugar-sweetened soft drinks, diet soft drinks, and serum uric acid level. The third national health and nutrition examination survey. Arthritis Rheum. 59, 109–116 [DOI] [PubMed] [Google Scholar]
- 13. Xu C., Yu C., Xu L., Miao M., Li Y. (2010) High serum uric acid increases the risk for nonalcoholic fatty liver disease. A prospective observational study. PLoS ONE 5, e11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuo C. F., Yu K. H., Luo S. F., Chiu C. T., Ko Y. S., Hwang J. S., Tseng W. Y., Chang H. C., Chen H. W., See L. C. (2010) Gout and risk of nonalcoholic fatty liver disease. Scand. J. Rheumatol. 39, 466–471 [DOI] [PubMed] [Google Scholar]
- 15. Li Y., Xu C., Yu C., Xu L., Miao M. (2009) Association of serum uric acid level with nonalcoholic fatty liver disease. A cross-sectional study. J. Hepatol. 50, 1029–1034 [DOI] [PubMed] [Google Scholar]
- 16. Lee K. (2009) Relationship between uric acid and hepatic steatosis among Koreans. Diabetes Metab. 35, 447–451 [DOI] [PubMed] [Google Scholar]
- 17. Yamada T., Suzuki S., Fukatsu M., Wada T., Yoshida T., Joh T. (2010) Elevated serum uric acid is an independent risk factor for nonalcoholic fatty liver disease in Japanese undergoing a health checkup. Acta Gastroenterol. Belg 73, 12–17 [PubMed] [Google Scholar]
- 18. Petta S., Cammà C., Cabibi D., Di Marco V., Craxì A. (2011) Hyperuricemia is associated with histological liver damage in patients with nonalcoholic fatty liver disease. Aliment. Pharmacol. Ther. 34, 757–766 [DOI] [PubMed] [Google Scholar]
- 19. Lee J. W., Cho Y. K., Ryan M., Kim H., Lee S. W., Chang E., Joo K. J., Kim J. T., Kim B. S., Sung K. C. (2010) Serum uric acid as a predictor for the development of nonalcoholic fatty liver disease in apparently healthy subjects. A 5-year retrospective cohort study. Gut Liver 4, 378–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lonardo A., Loria P., Leonardi F., Borsatti A., Neri P., Pulvirenti M., Verrone A. M., Bagni A., Bertolotti M., Ganazzi D., Carulli N. (2002) Fasting insulin and uric acid levels but not indices of iron metabolism are independent predictors of nonalcoholic fatty liver disease. A case-control study. Dig. Liver Dis. 34, 204–211 [DOI] [PubMed] [Google Scholar]
- 21. Ryu S., Chang Y., Kim S. G., Cho J., Guallar E. (2011) Serum uric acid levels predict incident nonalcoholic fatty liver disease in healthy Korean men. Metab. Clin. Exp. 60, 860–866 [DOI] [PubMed] [Google Scholar]
- 22. Sirota J. C., McFann K., Targher G., Johnson R. J., Chonchol M., Jalal D. (2012) Association between serum uric acid levels and ultrasound-diagnosed nonalcoholic fatty liver disease in the National Health and Nutrition Examination Survey. Metabolism S0026-0495(12)0031 7-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu C. F., Yu C. H., Xu L., Sa X. Y., Li Y. M. (2010) Hypouricemic therapy. A novel potential therapeutic option for nonalcoholic fatty liver disease. Hepatology 52, 1865–1866 [DOI] [PubMed] [Google Scholar]
- 24. Sautin Y. Y., Nakagawa T., Zharikov S., Johnson R. J. (2007) Adverse effects of the classic antioxidant uric acid in adipocytes. NADPH oxidase-mediated oxidative/nitrosative stress. Am. J. Physiol. Cell Physiol 293, C584–C596 [DOI] [PubMed] [Google Scholar]
- 25. Yu M. A., Sánchez-Lozada L. G., Johnson R. J., Kang D. H. (2010) Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J. Hypertens. 28, 1234–1242 [PubMed] [Google Scholar]
- 26. Kang D. H., Park S. K., Lee I. K., Johnson R. J. (2005) Uric acid-induced C-reactive protein expression. Implication on cell proliferation and nitric oxide production of human vascular cells. J. Am. Soc. Nephrol. 16, 3553–3562 [DOI] [PubMed] [Google Scholar]
- 27. Kanellis J., Watanabe S., Li J. H., Kang D. H., Li P., Nakagawa T., Wamsley A., Sheikh-Hamad D., Lan H. Y., Feng L., Johnson R. J. (2003) Uric acid stimulates monocyte chemoattractant protein-1 production in vascular smooth muscle cells via mitogen-activated protein kinase and cyclooxygenase-2. Hypertension 41, 1287–1293 [DOI] [PubMed] [Google Scholar]
- 28. Corry D. B., Eslami P., Yamamoto K., Nyby M. D., Makino H., Tuck M. L. (2008) Uric acid stimulates vascular smooth muscle cell proliferation and oxidative stress via the vascular renin-angiotensin system. J. Hypertens. 26, 269–275 [DOI] [PubMed] [Google Scholar]
- 29. Baldini P. M., De Vito P., Antenucci D., Vismara D., D'Aquilio F., Luly P., Zalfa F., Bagni C., Di Nardo P. (2004) Atrial natriuretic peptide induces cell death in human hepatoblastoma (HepG2) through the involvement of NADPH oxidase. Cell Death Differ. 11, S210–S212 [DOI] [PubMed] [Google Scholar]
- 30. Chaney A. L., Marbach E. P. (1962) Modified reagents for determination of urea and ammonia. Clin. Chem. 8, 130–132 [PubMed] [Google Scholar]
- 31. Block K., Gorin Y., Abboud H. E. (2009) Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. U.S.A. 106, 14385–14390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baldwin W., McRae S., Marek G., Wymer D., Pannu V., Baylis C., Johnson R. J., Sautin Y. Y. (2011) Hyperuricemia as a mediator of the proinflammatory endocrine imbalance in the adipose tissue in a murine model of the metabolic syndrome. Diabetes 60, 1258–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cirillo P., Gersch M. S., Mu W., Scherer P. M., Kim K. M., Gesualdo L., Henderson G. N., Johnson R. J., Sautin Y. Y. (2009) Ketohexokinase-dependent metabolism of fructose induces proinflammatory mediators in proximal tubular cells. J. Am. Soc. Nephrol. 20, 545–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hausladen A., Fridovich I. (1994) Superoxide and peroxynitrite inactivate aconitases but nitric oxide does not. J. Biol. Chem. 269, 29405–29408 [PubMed] [Google Scholar]
- 35.Deleted in proof
- 36. Johnson R. J., Perez-Pozo S. E., Sautin Y. Y., Manitius J., Sanchez-Lozada L. G., Feig D. I., Shafiu M., Segal M., Glassock R. J., Shimada M., Roncal C., Nakagawa T. (2009) Hypothesis. Could excessive fructose intake and uric acid cause type 2 diabetes? Endocr. Rev. 30, 96–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tappy L., Lê K. A. (2010) Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 90, 23–46 [DOI] [PubMed] [Google Scholar]
- 38. Roncal-Jimenez C. A., Lanaspa M. A., Rivard C. J., Nakagawa T., Sanchez-Lozada L. G., Jalal D., Andres-Hernando A., Tanabe K., Madero M., Li N., Cicerchi C., Mc Fann K., Sautin Y. Y., Johnson R. J. (2011) Sucrose induces fatty liver and pancreatic inflammation in male breeder rats independent of excess energy intake. Metab. Clin. Exp. 60, 1259–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nikkilä E. A. (1969) Control of plasma and liver triglyceride kinetics by carbohydrate metabolism and insulin. Adv. Lipid Res. 7, 63–134 [DOI] [PubMed] [Google Scholar]
- 40. Nikkilä E. A., Kekki M. (1972) Effects of dietary fructose and sucrose on plasma triglyceride metabolism in patients with endogenous hypertriglyceridemia. Acta Med. Scand. Suppl. 542, 221–227 [DOI] [PubMed] [Google Scholar]
- 41. Van den Berghe G. (1986) Fructose. Metabolism and short term effects on carbohydrate and purine metabolic pathways. Prog. Biochem. Pharmacol. 21, 1–32 [PubMed] [Google Scholar]
- 42. Odièvre M., Gentil C., Gautier M., Alagille D. (1978) Hereditary fructose intolerance in childhood. Diagnosis, management, and course in 55 patients. Am. J. Dis. Child. 132, 605–608 [DOI] [PubMed] [Google Scholar]
- 43. Kodama S., Saito K., Yachi Y., Asumi M., Sugawara A., Totsuka K., Saito A., Sone H. (2009) Association between serum uric acid and development of type 2 diabetes. Diabetes Care 32, 1737–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Masuo K., Kawaguchi H., Mikami H., Ogihara T., Tuck M. L. (2003) Serum uric acid and plasma norepinephrine concentrations predict subsequent weight gain and blood pressure elevation. Hypertension 42, 474–480 [DOI] [PubMed] [Google Scholar]
- 45. Dehghan A., van Hoek M., Sijbrands E. J., Hofman A., Witteman J. C. (2008) High serum uric acid as a novel risk factor for type 2 diabetes. Diabetes Care 31, 361–362 [DOI] [PubMed] [Google Scholar]
- 46. Nakagawa T., Hu H., Zharikov S., Tuttle K. R., Short R. A., Glushakova O., Ouyang X., Feig D. I., Block E. R., Herrera-Acosta J., Patel J. M., Johnson R. J. (2006) A causal role for uric acid in fructose-induced metabolic syndrome. Am. J. Physiol. Renal Physiol. 290, F625–F631 [DOI] [PubMed] [Google Scholar]
- 47. Feig D. I., Soletsky B., Johnson R. J. (2008) Effect of allopurinol on the blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA 300, 922–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kanbay M., Huddam B., Azak A., Solak Y., Kadioglu G. K., Kirbas I., Duranay M., Covic A., Johnson R. J. (2011) A randomized study of allopurinol on endothelial function and estimated glomerular filtration rate in asymptomatic hyperuricemic subjects with normal renal function. Clin. J. Am. Soc. Nephrol. 6, 1887–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ogino K., Kato M., Furuse Y., Kinugasa Y., Ishida K., Osaki S., Kinugawa T., Igawa O., Hisatome I., Shigemasa C., Anker S. D., Doehner W. (2010) Uric acid-lowering treatment with benzbromarone in patients with heart failure. A double-blind placebo-controlled crossover preliminary study. Circ. Heart Fail. 3, 73–81 [DOI] [PubMed] [Google Scholar]
- 50. Kanbay M., Ozkara A., Selcoki Y., Isik B., Turgut F., Bavbek N., Uz E., Akcay A., Yigitoglu R., Covic A. (2007) Effect of treatment of hyperuricemia with allopurinol on blood pressure, creatinine clearance, and proteinuria in patients with normal renal functions. Int. Urol. Nephrol. 39, 1227–1233 [DOI] [PubMed] [Google Scholar]
- 51. Feig D. I., Kang D. H., Johnson R. J. (2008) Uric acid and cardiovascular risk. N. Engl. J. Med. 359, 1811–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xu P., LaVallee P., Hoidal J. R. (2000) Repressed expression of the human xanthine oxidoreductase gene. E-box and TATA-like elements restrict ground state transcriptional activity. J. Biol. Chem. 275, 5918–5926 [DOI] [PubMed] [Google Scholar]
- 53. Abdelmalak M., Sukzuki A., Guy C., Unalp-Arida A., Colvin R., Johnson R. J., Diehl A. M. (2010) Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 51, 1961–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Argalious M. Y., Dalton J. E., Cywinski J. B., Seif J., Abdelmalak M., Sessler D. I. (2012) Association between preoperative statin therapy and postoperative change in glomerular filtration rate in endovascular aortic surgery. Br. J. Anaesth. 109, 161–167 [DOI] [PubMed] [Google Scholar]