

Abstract
Glycosaminoglycans (GAGs) are a class of highly negatively charged, unbranched, O-linked polysaccharides that are involved in many diseases. Their role as a protein-binding matrix on cell surfaces has long been recognized, but therapeutic approaches to interfere with protein–GAG interactions have been limited due to the complex chemistry of GAGs, on one hand, and due to the lack of specific antibodies against GAGs, on the other hand. We have developed a protein engineering platform (the so-called CellJammer® technology), which enables us to introduce higher GAG-binding affinity into wild-type GAG-binding proteins and to combine this with impaired biological, receptor-binding function. Chemokines are among the prototypic GAG-binding proteins and here we present selected results of our CellJammer technology applied to several of these proinflammatory proteins. An overview is given of our lead decoy protein, PA401, which is a CXCL8-based mutant protein with increased GAG-binding affinity and decreased CXCR1/2 binding and activation. Major results from our CCL2 and CCL5 programmes are also summarized and the potential for clinical application of these decoy proteins is presented.
Keywords: chemokine, glycosaminoglycan, growth factor
Relevance of protein–glycosaminoglycan interaction for protein activity in vivo
Glycosaminoglycans (GAGs) are long, linear, heterogeneous and highly negatively charged (sulfated) polysaccharide chains composed of repeating disaccharide units that are commonly covalently connected (O-linked) to core proteins, thus forming the so-called proteoglycans, which are located at the cell surface of virtually all eukaryotic cells and constitute the glycocalyx. Despite the fact that the presence of a thin layer covering the endothelial surface was first proposed in 1940 by Danielli (Danielli, 1940), it took another 26 years to have it ‘visualized’ by Luft (1966). For a long time, the GAGs were exclusively regarded as chemically inert coating elements of the cell surface required for its protection or as biochemical fuel in energy metabolism (Weinbaum et al., 2007). It was only rather recently that very specific biological functions have been associated to defined glycan structures. Historically, the best characterized interaction between GAGs and a protein is the activation of antithrombin III by heparin, which ultimately leads to an inhibition of the blood-clotting cascade (Petitou et al., 1988; 1991). The binding of fibroblast growth factors (FGFs) 1 and 2 and their receptors (FGFR) to heparan sulfate (HS) has also attracted much attention over the past 15 years (Lindahl et al., 1989; Rahmoune et al., 1998; Robinson et al., 2005; Harmer, 2006). Specifically for FGFR1, the formation of a tripartite molecular complex comprising FGF2, FGFR1 and the HS GAG chain of a proteoglycan is required to induce receptor dimerization, a step that is required for inducing its activation by autophosphorylation at several intracellular tyrosine residues (Yayon et al., 1991). Chemokines have only rather recently caught up with intensified studies on the role of GAGs for their biological activity.
The major classes of GAGs include heparin and HS, chondroitin sulfate (CS), dermatan sulfate and keratan sulfate, which differ in their core disaccharide units as well as in their glycosidic linkage (see Figure 1). Heparan sulfate proteoglycans (HSPGs), in particular, are found to be involved in many pathophysiological processes and, as major component of the endothelial cell glycocalyx, are involved in regulating leukocyte migration from the bloodstream through the vessel walls to the site of tissue damage (Bishop et al., 2007; Lindahl, 2007; Sarrazin et al., 2011). Within this context, an important role is played by GAGs interacting with chemokines, which are a class of proteins that play a pivotal role in mediating directional cell migration, both during embryonic organ development stages as well as in pathological conditions, such as leukocyte recruitment in autoimmune and inflammatory disorders and cancer. Even if recent publications suggest that soluble chemokines may have a role in inducing chemotactic movement of immune cells within tissues, such as CCL19-induced chemotaxis of dendritic cells within lymph nodes (Schumann et al., 2010; chemokine and receptor nomenclature follows Alexander et al., 2011), binding of chemokines to HSPGs is recognized to be essential for immobilizing chemokines and creating a quasi-solid phase haptotactic gradient on the vascular endothelial cells at the site of inflammation, thus preventing chemokines from being released into the bloodstream and inducing leukocyte surface adhesion (Middleton et al., 1997; 2002; Taylor and Gallo, 2006; Celie et al., 2009).
Figure 1.
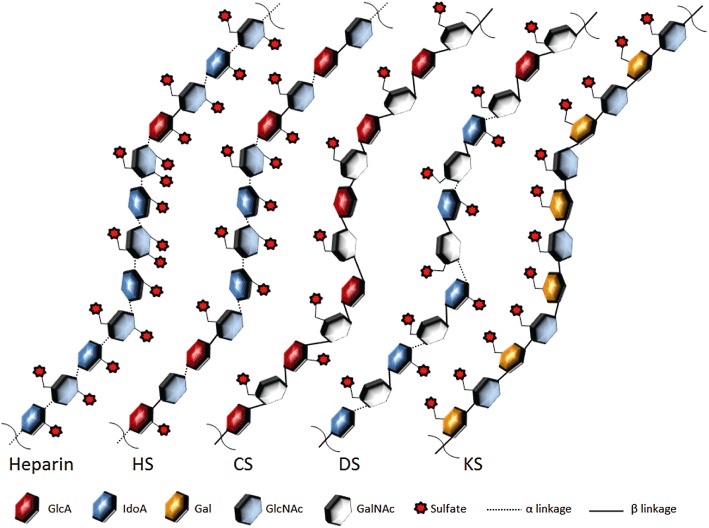
Structures of the main glycosaminoglycans. HS, heparan sulfate; CS, chondroitin sulfate; DS, dermatan sulfate; KS, keratan sulfate.
Originally, saturable in situ binding of various chemokines to post-capillary and small venule endothelial cells was demonstrated by means of an ex vivo autoradiographic approach in the dermis of intact human skin (Rot, 1992; Hub and Rot, 1998). Then, more direct evidence for chemokine presentation on capillary endothelial cells was produced following i.d. injection of CXCL8 (IL-8) in rabbit. In that experiment, CXCL8 could be specifically visualized by using immunoelectron microscopy techniques on luminar endothelial cell membrane of post-capillary venules in the skin, and tissue treatment with heparitinase (an enzyme that hydrolyses HS) markedly reduced CXCL8 immunoreactivity, supporting the role of HS in CXCL8 presentation at the endothelial cell level (Middleton et al., 1997).
Furthermore, in an in vitro model of neutrophil transendothelial migration, CXCL8 was immobilized on the human endothelial cell surface by binding to HS syndecan-1 ectodomains. This bound form of CXCL8 was detached by plasmin, itself generated by endothelial plasminogen activator (Marshall et al., 2003).
Despite all this in vitro and ex vivo evidence, the biological relevance of chemokine/GAG interaction was only relatively recently demonstrated by the generation of a series of engineered chemokine mutants of CCL5 (RANTES), CCL4 (MIP-1β), CCL2 (MCP-1), CXCL12 (SDF-1α) and CCL7 (MCP-3), with impaired GAG-binding properties (Proudfoot et al., 2003; Johnson et al., 2005; Proudfoot, 2006; O'Boyle et al., 2009; Ali et al., 2010). All of these site-directed mutants retained chemotactic activity when tested in vitro, where chemokines were still able to efficiently reach and activate their seven transmembrane GPCRs located on leukocytes in a free-soluble, diffusion gradient and where GAG binding is supposed to play no essential part. However, when these mutants were administered in vivo to rodents, they were unable to induce cell migration even at doses more than 4 logs higher than the corresponding wild-type variants, thus demonstrating that, at least for these chemokines, GAG binding is needed to induce cell migration from the bloodstream to the site of inflammation in vivo.
In the same period, generation of mutant mice with inactivated N-acetylglucosamine N-deacetylase-N-sulfotransferase-1 (the enzyme responsible for addition of sulfate to the heparin sulfate chains), specifically in endothelial cells and leukocytes, resulted in impaired neutrophil infiltration in several inflammatory models in vivo. These effects were due to the changes in HS in the endothelial cells and to a reduction of transcytosis and apical presentation of chemokines, further highlighting the importance of HSPGs–chemokine interaction in inflammation in vivo (Wang et al., 2005).
Finally, it has been recently reported that an artificially created chemokine gradient, generated by means of an implant of mCXCL2-containing gel into the cremaster muscle in mice, resulted in endothelial chemokine sequestration, exclusively in venules, that was HS-dependent and was responsible for directional migration of leukocytes to the site of implant in vivo (Massena et al., 2010).
Binding of chemokines to GAGs may also induce protein homo-oligomerization as well as formation of hetero-oligomers with other chemokines; for example, the in vitro formation of CCL8-CCL11 and CCL2-CCL11 heterodimers in the presence of the heparin pentasaccharide Arixtra® (Hoogewerf et al., 1997; Crown et al., 2006; Salanga and Handel, 2011). This type of GAG ligand-induced homo-/hetero-oligomerization is assumed to amplify the bioactivity of chemokines by concentrating the proteins at their hot spot of activity.
The formation of chemokine oligomers may appear to be of minimal importance, considering that for most of the chemokines, it is the monomeric form that is believed to bind to the GPCRs on leukocytes to induce cell migration in vitro[e.g. Paavola et al. (1998) for CCL2]. However, oligomerization seems to be required for the in vivo functioning of at least some chemokines. This was shown by the observation that engineered obligate monomers of CCL2, CCL5, CCL4 and CXCL10 were not functional in vivo (Proudfoot et al., 2003; Handel et al., 2008; Campanella et al., 2006, and reviewed in Salanga and Handel, 2011).
To further increase the complexity of this scenario is the recent report that engineered obligate monomer and obligate dimer of CXCL8 were both capable to induce cell recruitment when instilled into the lung in mice, even if the two forms had distinct in vivo recruitment profile, and wild type had intermediate characteristics, suggesting it exists as natural equilibrium between monomer and dimer (Das et al., 2010). The same group has just reported that these same CXCL8 mutants were also able to induce neutrophil recruitment in mouse peritoneum, but the relative potency of monomer and dimer was quite different compared to what was observed in the lungs (Gangavarapu et al., 2012). By additionally generating various mutants of the GAG-binding residues, they concluded that a GAG-mediated tissue-specific difference in chemokine gradient was responsible for the observed difference in in vivo neutrophil recruitment between the lungs and peritoneum.
Another important consequence of chemokine binding to GAGs is protection of the protein against proteolytic degradation, by this means increasing the natural lifetime of the chemokine in complex with GAGs and therefore its duration of action (Wagner et al., 1998; Sadir et al., 2004; Johnson et al., 2005; Rot, 2010). The chemokine–GAG interaction seems therefore not only a means for marking the point of highest chemokine concentration as the finish line for cell migration, possibly in a tissue-specific manner, but also a means for structurally activating and for protecting chemokines in order to develop their full biological activity for a prolonged time at their site of release.
It is now almost generally agreed that, in inflammatory conditions, the major site of chemokine–GAG interactions for mediating leukocyte recruitment from the bloodstream is at the level of the vascular endothelium of the inflamed tissues (Hoogewerf et al., 1997; Middleton et al., 2002; Constantinescu et al., 2003). However, GAGs and HSPGs, in particular, are expressed not only on the endothelial cells but also on the cell surface of leukocytes (e.g. Manakil et al., 2001; Campbell and Owen, 2007). This may further contribute to presenting chemokines to their high-affinity GPCRs in a tri-molecular complex, GAG–chemokine–GPCR, in this case, on the leukocyte cell surface (i.e. referring to a cis interaction compared with the traditionally assumed trans interaction between chemokines and GAGs/HSPGs located on the endothelium and the respective GPCR being located on the leukocyte). However, as already pointed out by Celie et al. (2009), the fact that at least in an in vitro setting chemokines modified for reduced or no GAG binding at all are still able, by simple diffusion, to efficiently bind/signal via the receptor(s) on leukocytes and induce chemotaxis argues against a major importance of the HSPGs–chemokine cis interaction. It is, however, possible that, as already suggested by Ali et al. (2000), the cis interaction may allow lower concentrations of the chemokine to activate the receptor, possibly through a mechanism that involves the chemokine sequestration on the cell surface. In this case, the cis interaction would play quite an important and underestimated role in the (early) in vivo inflammatory processes.
Similarly, binding of chemokines to GAGs can also ‘protect’ them from agents other than enzymes, affecting the success of the development of therapeutic antibodies if these were raised against the soluble protein. Structural rearrangements of the protein upon GAG binding as well as the change of overall/surface charge can influence or mask the antibody binding epitope, rendering the chemokine un-accessible to the antibody, or simply interfering with the antibody binding due to the high charge of the GAG ligand. This was most probably the reason for the lack of activity in phase II clinical trials of the anti-hCXCL-8 antibody, ABX-IL8 from Abgenix Inc. because the antibody was specifically raised to recognize only soluble CXCL-8, and not CXCL-8 localized on endothelial cells, that is, bound to GAGs (Yang et al., 1999).
Are GAGs suitable targets for therapeutic intervention?
From this brief description of the general importance of GAG–protein binding and the HSPGs–chemokine interaction, in particular, it seems that HSPGs should be very interesting therapeutic targets for the development of anti-inflammatory strategies, especially in the context of chronic inflammation, autoimmune diseases and oncology/angiogenesis (Rek et al., 2009b). In general, protein–GAG interactions are considered as molecular encounters with low affinity and low specificity, that is, (i) their dissociation constants (KD values) are found in the low micromolar range, although higher affinities (KD in the nanomolar range) especially for growth factors have been observed (Mohammadi et al., 2005), and (ii) their target glycan sequence is either not known at all (as for most chemokines) or can be estimated only from screening with animal-derived oligosaccharides. Contradicting this general observation, the high-affinity interaction of anti-thrombin III with its specific heparin pentasaccharide seems to be an exception to this rule. By any means, there seems to be sufficient potential and need to interfere with protein–GAG interactions.
The ‘classical’ medicinal chemistry approach would be to synthetically and/or enzymically generate a target protein-specific glycan structure (or a mimetic thereof) and to test its inhibitory activity for interfering with binding. Despite the initial success of Sanofi and Organon in co-developing several forms of AT-III-specific heparin pentasaccharide as anti-thrombotic agents, resulting in heparin-like pentasaccharide Fondaparinux (Arixtra®), an a priori GAG synthesizing approach seems currently less pursued, although some biotech companies such as Momenta Pharmaceuticals Inc. and Endotis Pharma are still active in this area. This may be due to (i) the difficulty to identify unique, disease- and protein-specific GAG epitope(s) and (ii) the considerable synthetic effort required to synthesize even short GAG oligosaccharides. We have recently shown that human microvascular endothelial cells change their GAG sulfation pattern after exposure to an inflammatory trigger (TNF-α; Krenn et al., 2008), and it has also been postulated that these changes are cell-/tissue- and time-specific. In particular, different sulfation patterns in the HS chains can favour binding of some chemokine versus others (Esko and Selleck, 2002; de Paz et al., 2007), dictating a certain degree of selectivity and timing of the attraction of different leukocyte population. There is a very complex enzymic machinery responsible for the synthesis and post-synthesis modifications of GAGs (Prydz and Dalen, 2000; Rek et al., 2009b) that allows for the formation of a wide range of polysaccharide sequences and modifications (i.e. N- and 2-,3-,6-O- sulfations, epimerization, deacetylation and desulfation) that cannot consequently be easily predicted. As an example, when only sulfation is considered, a HS tetrasaccharide can exhibit up to 576 different sulfation patterns. When the other possible chain modifications are also considered, 20 different saccharide building blocks (i.e. different with respect to sulfation, acetylation and epimerization) are able to form up to 1.44 × 1015 different hexasaccharides (Gesslbauer and Kungl, 2006). Unfortunately, there are currently no experimental methods available that allow sequencing of GAGs and thus relating their structure to function.
From a biopharmaceutical point of view, it proved very difficult to raise high-affinity monoclonal antibodies against protein-specific GAG sequences. Although a few groups were successful in generating antibodies against various tissue-specific GAGs by phage display (see Lensen et al., 2005), no therapeutic antibody that inhibits well-defined protein–GAG interaction has reached a clinical phase. Antibodies raised against the entire proteoglycans (core protein plus glycan chains) have been available for some time (Fjeldstad and Kolset, 2005); however, their potential in treating human diseases is not clearly predictable. An anti-glypican 3 antibody (GC33) is now being evaluated in two clinical trials (Chugai Pharmaceutical Co. Ltd.; Nakano et al., 2010) in monotherapy or in combination with the multiple-kinase inhibitor, sorafenib (Nexavar®), for safety and tolerability in patients with advanced metastatic hepatocellular carcinoma. Preliminary anti-tumour activity will also be evaluated. Results from these clinical trials will shed light on safety and therapeutic potential of this type of pharmacological intervention.
For all reasons mentioned earlier, we believe that there is still a need and the opportunity to develop new strategies to interfere with disease-specific GAG–protein interactions.
Changing the paradigm: the CellJammer® approach
The key information on how a protein specifically interacts with its ligand is not only contained in the structure and conformation of the ligand, but also in the structure and conformation of the protein. With this in mind and with a growing literature on amino acids involved in specific protein–GAG interactions, we turned the common paradigm around and used the structural and ligand-specific information contained in naturally GAG-binding proteins to engineer them to become better glycan binders and to be therefore able to antagonize defined protein–GAG interactions (see Figure 2). For this purpose, we have developed a protein-based technology platform that enables us, in principle, to mutagenize the entire class of GAG-binding proteins and so to obtain biopharmaceuticals that circumvent the problems of ill-defined GAG ligands and their complexity for therapeutic purposes (Potzinger et al., 2006; Rek et al., 2009b). These ‘dominant’ mutations (increasing GAG-binding affinity) of a naturally GAG-binding protein, such as a chemokine, need to be complemented by ‘negative’ (or knock-out) mutations, which inhibit the wild type protein's natural bioactivity, i.e. GPCR-binding/activation, in the case of chemokines.
Figure 2.
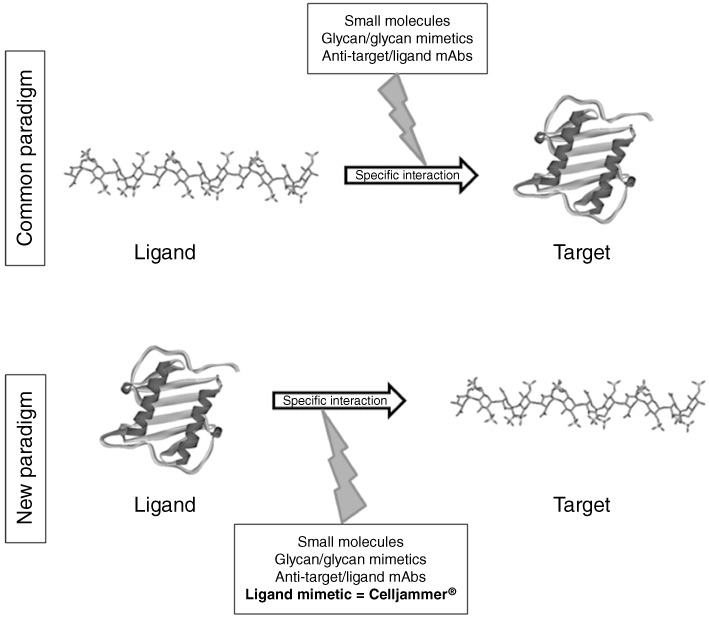
Schematic representation of the CellJammer® approach for interfering with protein–glycosaminoglycan interactions.
We have called our technology the CellJammer® platform as it was originally developed to use engineered chemokines for preventing cell traffic from the lumen of a vessel into the inflamed tissue. The platform takes advantage of the protein-intrinsic recognition potential for its specific, or better its selective, GAG co-receptor and to improve it to create a protein-based antagonist for the wild-type protein/GAG binding. This is achieved by replacing non-crucial amino acids in the GAG-binding domain of the selected wild-type protein with basic amino acids, thereby increasing the electrostatic component of the protein–GAG interaction and therefore its affinity. Side-chain contacts of the protein with its GAG ligand, which represent hydrogen bonding or van der Vaals interactions, are meant to be conserved during the engineering process as these interactions are responsible for recognizing a specific GAG oligosaccharide sequence. On top of this ‘affinity maturation’, the side chains responsible for natural protein–protein contacts, such as chemokine–GPCR interactions, and thus for the wild-type protein's bioactivity are either deleted or replaced by alanine residues. As chemokines need to adopt a certain conformation in order to interact not only with GAG ligands but also with other chemokines (see Weber and Koenen, 2006) and GPCRs in the case of native chemokines – thereby creating an interlinked multiple chemokine–GAG network – our decoys are derived by a structure-conserving approach, which is initially tested by computational energy minimization and by molecular dynamics simulation of the engineered mutant under investigation. Using this approach, the knowledge of the precise structural nature of the glycan co-receptor is not needed, as long as the positioning of the specific/selective GAG recognition domain is retained, and is extended by site-directed replacements against basic amino acids to increase the electrostatic binding strength of the engineered decoy protein for its GAG ligand.
The CellJammer® approach applied to chemokines
Considering the extremely important role played by chemokines in driving a plethora of human pathologies, as well as the essential and well-studied role of GAG binding for chemokine in vivo functions, the first focus of the CellJammer® technology was directed towards engineering this class of proteins. A CellJammer® chemokine is supposed to displace the corresponding wild-type protein from its GAG ligand, but not to engage with the GPCR on the leukocytes. As a result, a reduction of the inflammatory processes will occur. Critical steps in the inflammation-driven leukocyte vascular extravasation and CellJammer® (chemokine) mutant actions are illustrated in Figure 3.
Figure 3.
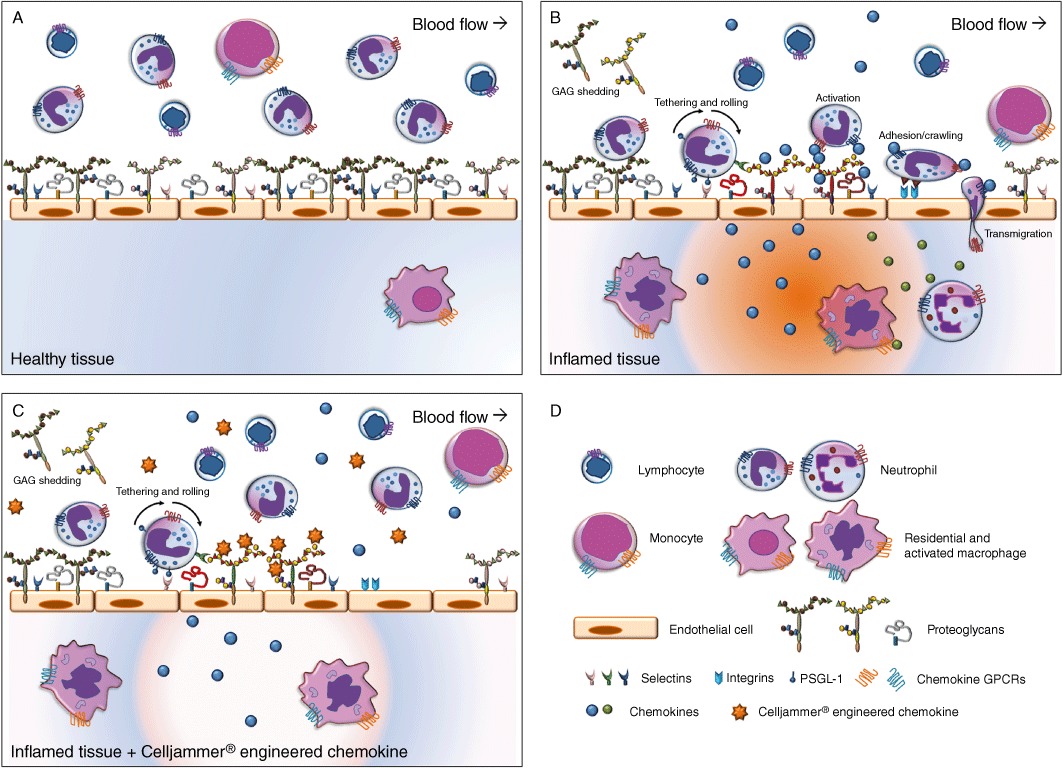
Schematic representation of the presumed mode of action of CellJammer® proteins.
PA401: a CXCL8-based decoy protein
The binding of CXCL8 to HSPGs on the surface of endothelial cells is crucial for the recruitment of neutrophils from the bloodstream to the site of inflammation, and this chemokine is one of the main players responsible for the acute infiltration of neutrophils into inflamed tissues in humans (Mukaida, 2003; Cowburn et al., 2008). High levels of CXCL8 are found in several severe human inflammatory pathologies, which are characterized by high levels of neutrophilic infiltrates, including lung neutrophilic inflammatory disease such as chronic obstructive pulmonary disease, neutrophilic asthma and cystic fibrosis, as well as in autoimmune diseases such as rheumatoid arthritis, psoriasis and Crohn's disease. Based on the predicted structure of the CXCL8/GAG complex (Krieger et al., 2004), we applied our engineering approach and generated a series of CXCL8 mutants by replacing either single or combinations of non-crucial amino acids in the CXCL8 GAG-binding site with basic amino acid residues. Computational affinity maturation, combined with in silico structural analysis, was used to select between energetically stable and less stable CXCL8 mutants and to categorize GAG-binding energies, on order to predict relative GAG-binding affinities of the various CXCL8 mutants. In addition to knocking-in high GAG-binding affinity, the first six amino acids of human CXCL8, including the so-called ELR motif, were deleted to knock-out the chemokine's interaction with its specific GPCRs, CXCR1 and CXCR2, on leukocytes. After expression of the mutant proteins in Escherichia coli, the CXCL8 mutant with the highest affinity for HS was selected by in vitro screening. By this means, the mutant CXCL8[Δ6,F17K,F21K,E70K,N71K] turned out as our lead compound PA401 for the treatment of CXCL8-related diseases. Knock-out of GPCR activation and impaired binding to human CXCR1 and CXCR2 receptors was confirmed in the modified Boyden chamber assay and by in vitro displacement of [125I]-CXCL8 by PA401 from human recombinant receptors, expressed on HEK-293 cells.
In vivo anti-inflammatory activity of PA401 was then assessed in experimental models of kidney ischaemia/reperfusion and acute renal allograft damage in rats, models where the early inflammatory response is responsible for allograft rejection. In line with its mode of action, in the ischaemia/reperfusion model PA401 was able to limit proximal tubular damage and reduce neutrophil and monocyte infiltration that is associated with acute damage, resulting in an overall better conserved renal tubular architecture. In the Fisher344 to Lewis rat transplant model, at 7 days post-transplant, PA401 treatment was able to dose-dependently reduce tubulointerstitial and glomeruli cell infiltration, especially of ED-1 positive macrophages and CD8 positive T cells, as well as tubulitis a sign of acute allograft rejection. In addition, at the highest dose tested, it improved glomerular and vascular rejection (Bedke et al., 2010). The activity observed in these models suggests that early treatment of allograft with PA401 may contribute to reduce acute allograft inflammatory damage and preserve renal morphology, limiting mid- to long-term chronic dysfunction. These positive data were the basis of an Orphan Medicinal Product designation for PA401 in delayed graft function after solid organ transplantation, by the EMA and the FDA.
More recently, strong evidence of PA401's activity in murine models for lung inflammation has been obtained. In acute LPS, as well as in chronic smoke-induced murine lung inflammation, PA401 strongly reduced cell infiltrates in bronchoalveolar lavage as well as in lung tissue [preliminary data presented at the European Respiratory Society Congress and at the American Thoracic Society International Conference (Adage et al., 2010; 2011b)]. These data support the clinical development of PA401 for those human lung diseases, which are characterized by acute and chronic neutrophilic infiltration.
PA508: a CCL2-based decoy protein
Another chemokine for which the GAG-binding motif has been considerably well described and characterized (Lau et al., 2004), and for which the in vivo activity has been shown to be dependent on GAG binding (see above), is CCL2. This proinflammatory chemokine binds to the GPCR CCR2, which is expressed on several leukocyte populations, including T and NK cells, monocytes, dendritic cells and basophils, and governs their directional trafficking via the interaction with cell-surface GAGs. High levels of CCL2 have been found in a variety of diseases that feature a monocyte-rich inflammatory component, such as atherosclerosis, rheumatoid arthritis, inflammatory bowel disease, multiple sclerosis, type 2 diabetes, obesity and congestive heart failure, making this chemokine a central player in a broad variety of human pathologies.
We have recently reported on the generation of CCL2-based CellJammer® mutants. The results of in vitro and cell-based characterization of four CCL2 mutants have been described (Piccinini et al., 2010). As a result of this screening, CCL2[Met,Y13A,S21K,Q23R] has been selected as the lead compound, PA508, of ProtAffin's MCP-1 programme. For CCL2, the amino terminus and the residue Y13 are essential for protein signalling via the CCR2 receptor. In PA508, retention of the N-terminal Met and the mutation of Tyr13 toAla were effective for significantly reducing GPCR binding, as proven by in vitro agonistic radio-binding of [125I]-CCL2 on human recombinant CCR2 receptor expressed in HEK-293 cells (Liehn et al., 2010) and by lack of induction of intracellular calcium release and of chemotactic capacity (Liehn et al., 2010; Piccinini et al., 2010). Regarding the CCL2/–GAG interactions, the residues conferring affinity to GAGs are Arg and Lys at positions 18, 19, 24, 49 and 59, and His66 (Lau et al., 2004). Ser21 and Glu23 are solvent-accessible amino acids proximal to those in the GAG-binding site. In Figure 4, the structures of wild-type CCL2 and PA508 are compared and the amino acids responsible for increased GAG binding in PA508 are highlighted (Piccinini et al., 2010).
Figure 4.
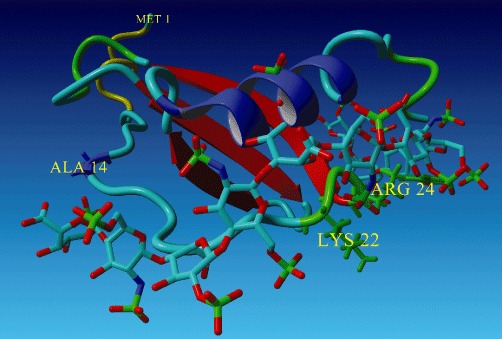
Model structure of PA508 docked to a heparin octasaccharide. Explicitly shown are the amino acid residues responsible for increased GAG-binding (green) and knocked-out CCR2 activation (blue). The structure for 1DOK.pdb (cf. P13500 UniProt) was used as the template for the model of the PA508 mutant. Heparin was placed in proximity to 21K and 23R of the PA508 model, using 1HPN.pdb as a model of heparin. The protein models were held rigid and the heparin polymer was minimized and adjusted to fit to the protein models. Using each individual protein model, further minimization accommodating side-chain relaxation was also performed. MMFF94 force field was used.
In view of the important role played by CCL2 in leading the inflammatory response occurring during the development of the restenotic changes that follow coronary intervention and lead to myocardial infarction (Frangogiannis et al., 2002; Weber et al., 2008), PA508 activity was initially tested in a mechanistic setting by evaluating ex vivo effects on monocytic cell adhesion to injured murine carotid arteries excised 1 day post injury. PA508 significantly and dose-dependently reduced the luminal cell adhesion (Liehn et al., 2010), confirming its proposed mode of action. Next, PA508 was tested in ApoE-/- mice, animals that are characterized by impaired clearing of plasma lipoproteins and prone to development of atherosclerosis, in a model of wire-induced endothelial denudation injury of the carotid artery. Three weeks post injury, the animals receiving daily treatment with PA508 had significantly reduced neointima formation, associated with a reduction in macrophage infiltrates in the plaque (Liehn et al., 2010).
These initial positive results prompted further investigation in a model of myocardial ischaemia/reperfusion, as it is recognized that a major complication in case of coronary stenosis combined with unstable plaque is myocardial infarction characterized by tissue damage associated with inflammatory monocyte infiltration. Injury was induced by coronary occlusion and reperfusion in ApoE-/- mice, followed by daily treatment with PA508 for 1 week. Ex vivo functional parameters, analysed by studies in isolated hearts (Langendorff perfusion), were improved by PA508 treatment. In particular, PA508 treatment resulted in a significant normalization of the left ventricle developed pressure and cardiac output both in the absence or presence of the inotropic agent, dobutamine. A dose-dependent significant decrease in the infarct volume, as measured by histomorphometric analysis performed on the heart after functional parameters measurements, was also observed, with reduction of the number of infiltrating monocytes at all treatment doses. Significant normalization of myofibroblast and collagen content in the infarcted area was observed only at the higher dose tested. Finally, in vivo measurement in another groups of animals confirmed significantly preserved heart functions in animals treated with PA508 (Liehn et al., 2010).
PA508 also showed significant activity in a rat model of experimental autoimmune uveitis induced by a peptide of the retinal soluble antigen (PDSAg), which is also recognized as auto-antigen for the human disease (Diedrichs-Moehring et al., 2005). The effects of daily treatment with PA508, as amelioration of the severity of the disease pathology, were evaluated by clinical score assessment using an ophthalmoscope during the study and confirmed at histological level at study completion (Piccinini et al., 2010).
Finally, significant activity of PA508 in a MOG35–55-induced chronic experimental autoimmune encephalomyelitis model of multiple sclerosis in mice has been recently reported (Adage et al., 2011a), which further broadens the therapeutic potential of PA508.
CCL5-based decoy proteins: initial engineering
Another important chemokine for early inflammatory responses, as well as for chronic diseases characterized by delayed type sensitivity reactions, rheumatoid arthritis and progressive glomerulonephritis, is CCL5. While originally considered a T cell-specific chemokine, it is now known to be expressed by a number of other cell types, including epithelial cells and platelets. CCL5 acts as a potent chemoattractant for many cell types, such as monocytes, NK cells, memory T-cells, eosinophils and dendritic cells. Moreover, CCL5 has been shown to play a role in immune responses to viral infections, with one of its receptors, CCR5, being used by HIV to enter the cell. This receptor is a major target for anti-HIV drugs that are based on blocking viral entry, such as the CCR5-blocking agent maraviroc, which is in the market for CCR5-tropic HIV-1.
We have initially characterized the importance of CCL5–GAG interactions, particularly with respect to size-defined heparin and HS oligosaccharides, and binding isotherms were obtained by isothermal fluorescence titration experiments. We then confirmed the importance of chemokine oligomerization to allow proper presentation of CCL5 to the GPCRs, by generating oligomerization-deficient CCL5 mutants, which showed impaired chemotactic activity in vitro (Rek et al., 2009a). Based on these premises, we have then generated a series of 10 CCL5 mutants, engineered to increase GAG-binding affinity based on specific amino acid replacement in the region next to the CCL5 GAG-binding epitope 44RKNR47. The amino acids to be replaced were identified as Thr43, Asn46, Gln48 and Val49, and in addition, an extension of this linear epitope was attempted by further engineering of Ala22 and His23 in the N-proximal loop (Brandner et al., 2009). In order to affect the GPCR binding, the N-terminus Met residue was retained. This change is known to lead to a functional receptor antagonist (Proudfoot et al., 1996). The mutants were thoroughly characterized in vitro for their biophysical properties, and two of them, CCL5[Met,A22K] and CCL5[Met,H23K], were initially evaluated in vivo in a model of experimental autoimmune uveitis (Brandner et al., 2009). In this model, CCL5[Met,H23K] exhibited transient beneficial activity by reducing the clinical scores in uveitis. Our CCL5/RANTES programme is currently being evaluated with respect to further potential clinical indications.
Conclusions and future perspectives
From the data mentioned above, it is clear that GAGs are therapeutically amenable drug targets with a strong involvement in many disease areas. The three examples presented here show the high potential for the development of more glycan-targeting biopharmaceuticals. Future work will address the specificity of the different decoy proteins with respect to certain GAG epitopes. Currently, we are establishing an analytical method by which an IC50 value for the competition of several pre-bound GAG-binding proteins by individual decoy proteins is derived. A comparative IC50 analysis of various decoy proteins on the same GAG ligand against a panel of GAG-binding proteins will allow prediction of the selectivity or the range of activity of a certain decoy protein. Another major challenge facing us is the first clinical trial with our lead compound, PA401, to see whether the therapeutic concept presented here is safe and has potential medical value for humans. Furthermore, the increase of bioavailability by extending the serum half life of our decoy proteins using standard technologies such as PEGylation is being intensively addressed. Finally, in our in-house database, we have collected all the experimental and theoretical information needed for engineering any GAG-binding protein according to our platform technology. This should open doors to development programmes for for further decoy proteins in a variety of disease indications.
Glossary
- CS
chondroitin sulfate
- FGF
fibroblast growth factor
- GAG
glycosaminoglycan
- HS
heparan sulfate
- HSPG
heparan sulfate proteoglycans
Conflict of interest
T. Adage, A. Falsone, M. Trinker, J. Robinson, B. Gesslbauer and A.J. Kungl are employees of ProtAffin Biotechnologie AG. A.J. Kungl is also a shareholder of ProtAffin Biotechnologie AG.
References
- Adage T, Rek A, Kungl AJ. 2010. A decoy CXCL8 with increased glycosaminoglycan binding has potent anti-inflammatory activity in lung inflammation models. European Respiratory Society Annual Meeting. Barcelona, September 18–22. Abstract 5151.
- Adage T, Piccinini A-M, Knebl K, Rek A, Kungl AJ. 2011a. A novel CCL2- based decoy with increased glycosaminoglycan binding is active in the mouse MOG35-55 EAE model of Multiple Sclerosis. 5th Joint Triennial Congress of the European and Americas Committees for Treatment and Research in Multiple Sclerosis. Amsterdam, 19–22 October. Poster topic 21- Immunomodulation 1. Poster 474.
- Adage T, Rek A, Kungl AJ. Pharmacological profile of a novel decoy CXCL8- based biologic therapeutic in murine models of lung inflammation. Am J Respir Crit Care Med. 2011b;183:A4071. [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali S, Palmer AC, Banerjee B, Fritchley SJ, Kirby JA. Examination of the function of RANTES, MIP-1-alpha, and MIP-1beta following interaction with heparin-like glycosaminoglycans. J Biol Chem. 2000;275:11721–11727. doi: 10.1074/jbc.275.16.11721. [DOI] [PubMed] [Google Scholar]
- Ali S, O'Boyle G, Hepplewhite P, Tyler JR, Robertson H, Kirby JA. Therapy with nonglycosaminoglycan-binding mutant CCL7: a novel strategy to limit allograft inflammation. Am J Transplant. 2010;10:47–58. doi: 10.1111/j.1600-6143.2009.02868.x. [DOI] [PubMed] [Google Scholar]
- Bedke J, Nelson PJ, Kiss E, Muenchmeier N, Rek A, Behnes C-L, et al. A novel CXCL8 protein-based antagonist in acute experimental renal allograft damage. Mol Immunol. 2010;47:1047–1057. doi: 10.1016/j.molimm.2009.11.012. [DOI] [PubMed] [Google Scholar]
- Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- Brandner B, Rek A, Diedrichs-Möhring M, Wildner G, Kungl AJ. Engineering the glycosaminoglycan-binding affinity, kinetics and oligomerization behaviour of RANTES: a tool for generating chemokine-based glycosaminoglycan antagonists. Protein Eng Des Sel. 2009;22:367–373. doi: 10.1093/protein/gzp013. [DOI] [PubMed] [Google Scholar]
- Campanella GS, Grimm J, Manice LA, Colvin RA, Medoff BD, Wojtkiewicz GR, et al. Oligomerization of CXCL10 is necessary for endothelial cell presentation and in vivo activity. J Immunol. 2006;177:6991–6998. doi: 10.4049/jimmunol.177.10.6991. [DOI] [PubMed] [Google Scholar]
- Campbell EJ, Owen CA. The sulphate groups of chondroitin sulphate- and heparan sulphate- containing proteoglycans in neutrophil plasma membranes are novel binding sites for human leukocyte elastase and cathepsin G. J Biol Chem. 2007;282:14645–14654. doi: 10.1074/jbc.M608346200. [DOI] [PubMed] [Google Scholar]
- Celie JWAM, Beelen RHJ, van den Born J. Heparan sulfate proteoglycans in extravasation: assisting leukocyte guidance. Front Biosci. 2009;14:4932–4949. doi: 10.2741/3578. [DOI] [PubMed] [Google Scholar]
- Constantinescu AA, Vink JA, Spaan JA. Endothelial cell glycocalix modulates immobilization of leukocytes at the endothelial surface. Arterioscler Thromb Vasc Biol. 2003;23:1541–1547. doi: 10.1161/01.ATV.0000085630.24353.3D. [DOI] [PubMed] [Google Scholar]
- Cowburn SC, Condliffe AM, Farhai N, Summers C, Chilvers ER. Advances in neutrophil biology. Clinical implications. Chest. 2008;134:606–612. doi: 10.1378/chest.08-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crown SE, Yu Y, Sweeney MD, Leary JA, Handel TM. Heterodimerization of CCR2 chemokines and regulation by glycosaminoglycan binding. J Biol Chem. 2006;281:25438–25446. doi: 10.1074/jbc.M601518200. [DOI] [PubMed] [Google Scholar]
- Danielli JF. Capillary permeability and edema in the perfused frog. J Cell Physiol. 1940;98:109–129. doi: 10.1113/jphysiol.1940.sp003837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das ST, Rajagopalan L, Guerrero-Plata A, Sai J, Richmond A, Garofalo RP, et al. Monomeric and dimeric CXCL8 are both essential for in vivo neutrophil recruitment. Plos ONE. 2010;5:e11754. doi: 10.1371/journal.pone.0011754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paz JL, Moseman EA, Noti C, Polito L, von Andrian UH, Seeberger PH. Profiling heparin-chemokine interactions using synthetic tools. ACS Chem Biol. 2007;2:735–744. doi: 10.1021/cb700159m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diedrichs-Moehring M, Nelson PJ, Proudfoot AE, Thurau SR, Wilder G. Role of CC chemokine receptor antagonist Met-RANTES in experimental autoimmune uveitis and oral tolerance induction. J Neuroimmunol. 2005;164:22–30. doi: 10.1016/j.jneuroim.2005.02.023. [DOI] [PubMed] [Google Scholar]
- Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in Heparan Sulfate. Annu Rev Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- Fjeldstad K, Kolset SO. Decreasing the metastatic potential in cancers targeting the heparan sulphate proteoglycans. Curr Drug Targets. 2005;6:665–682. doi: 10.2174/1389450054863662. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- Gangavarapu P, Rajagopalan L, Kolli D, Guerrero-Plata A, Garofalo RP, Rajarathnam K. The monomer-dimer equilibrium and glycosaminoglycan interaction of chemokine CXCL8 regulate tissue-specific neutrophil recruitment. J Leukoc Biol. 2012;91:259–265. doi: 10.1189/jlb.0511239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesslbauer B, Kungl AJ. Glycomic approaches toward drug development: therapeutically exploring the glycosaminoglycome. Curr Opin Mol Ther. 2006;8:521–528. [PubMed] [Google Scholar]
- Handel TM, Johnson Z, Rodrigues DH, Dos Santos AC, Cirillo R, Muzio V, et al. An engineered monomer of CCL2 has anti-inflammatory properties emphasizing the importance of oligomerization for chemokine activity in vivo. J Leukoc Biol. 2008;84:1101–1108. doi: 10.1189/jlb.0108061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmer NJ. Insights into the role of heparan sulphate in fibroblast growth factor signalling. Biochem Soc Trans. 2006;34:442–445. doi: 10.1042/BST0340442. [DOI] [PubMed] [Google Scholar]
- Hoogewerf AJ, Kuschert GSV, Proudfoot AEI, Borlat F, Clark-Lewis I, Power CA, et al. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry. 1997;36:13570–13578. doi: 10.1021/bi971125s. [DOI] [PubMed] [Google Scholar]
- Hub E, Rot A. Binding of RANTES, MCP-1, MCP-3 and MIP-1 alpha to cells in human skin. Am J Pathol. 1998;152:749–757. [PMC free article] [PubMed] [Google Scholar]
- Johnson Z, Proudfoot AE, Handel TM. Interaction of chemokines and glycosaminoglycans: a new twist in the regulation of chemokine function with opportunities for therapeutic intervention. Cytokine Growth Factor Rev. 2005;16:625–636. doi: 10.1016/j.cytogfr.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Krenn EC, Wille I, Gesslbauer B, Poteser M, van Kuppevelt TH, Kungl AJ. Glycanogenomics: a qPCR-approach to investigate biological glycan function. Biochem Biophys Res Commun. 2008;375:297–302. doi: 10.1016/j.bbrc.2008.07.144. [DOI] [PubMed] [Google Scholar]
- Krieger E, Geretti E, Brandner B, Goger B, Wells TN, Kungl AJ. A structural and dynamic model for the interaction of interleukin-8 and glycosaminoglycans: support from isothermal fluorescence titrations. Proteins. 2004;54:768–775. doi: 10.1002/prot.10590. [DOI] [PubMed] [Google Scholar]
- Lau EK, Paavola CD, Johnson Z, Gaudry J-P, Geretti E, Borlat F, et al. Identification of the glycosaminoglycan binding site of the CC chemokine, MCP-1. J Biol Chem. 2004;279:22294–22305. doi: 10.1074/jbc.M311224200. [DOI] [PubMed] [Google Scholar]
- Lensen JF, Rops AL, Wijnhoven TJ, Hafmans T, Feitz WF, Oosterwijk E, et al. Localization and functional characterization of glycosaminoglycan domains in the normal human kidney as revealed by phage display-derived single chain antibodies. J Am Soc Nephrol. 2005;16:1279–1288. doi: 10.1681/ASN.2004050413. [DOI] [PubMed] [Google Scholar]
- Liehn EA, Piccinini A-M, Koenen RR, Soehnlein O, Adage T, Fatu R, et al. A new monocyte chemotactic protein-1/chemokine CC motif ligand-2 competitor limiting neointima formation and myocardial ischemia/reperfusion injury in mice. J Am Coll Cardiol. 2010;56:1857–1852. doi: 10.1016/j.jacc.2010.04.066. [DOI] [PubMed] [Google Scholar]
- Lindahl U. Heparan sulfate-protein interactions – a concept for drug design? Thromb Haemost. 2007;98:109–115. [PubMed] [Google Scholar]
- Lindahl U, Backstrom G, Thunberg L, Leder IG. Evidence for a 3-O-sulfated d-glucosamine residue in the antithrombin-binding sequence of heparin. Proc Natl Acad Sci USA. 1989;77:6551–6555. doi: 10.1073/pnas.77.11.6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luft JH. Fine structures of capillary layer as revealed by ruthenium red. Fed Proc. 1966;25:1773–1783. [PubMed] [Google Scholar]
- Manakil JF, Surgerman PB, Li H, Seymour GJ, Bartold PM. Cell-surface proteoglycan expression by lymphocytes from peripheral blood and gingival in health and periodontal disease. J Dent Res. 2001;80:1704–1710. doi: 10.1177/00220345010800080501. [DOI] [PubMed] [Google Scholar]
- Marshall LJ, Ramdin LSP, Brooks T, Charlton P, Shute JK. Plasminogen activator inhibitor-1 supports IL-8-mediated neutrophil transendothelial migration by inhibition of the constitutive shedding of endothelial IL-8/hparan sulfate/syndecan-1 complexes. J Immunol. 2003;171:2057–2065. doi: 10.4049/jimmunol.171.4.2057. [DOI] [PubMed] [Google Scholar]
- Massena S, Christoffersson G, Hjertström E, Zcharia E, Vlodavsky I, Ausmees N, et al. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional crawling of neutrophils. Blood. 2010;116:1924–1931. doi: 10.1182/blood-2010-01-266072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton J, Neil S, Wintle J, Clark-Lewis I, Moore H, Lam C, et al. Trancytosis and surface presentation of IL-8 by venular endothelial cells. Cell. 1997;91:385–395. doi: 10.1016/s0092-8674(00)80422-5. [DOI] [PubMed] [Google Scholar]
- Middleton J, Patterson AM, Gardner L, Schmutz C, Ashton BA. Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood. 2002;100:3853–3860. doi: 10.1182/blood.V100.12.3853. [DOI] [PubMed] [Google Scholar]
- Mohammadi M, Olsen SK, Goetz R. A protein canyon in the FGF–FGF receptor dimer selects from an a' la carte menu of heparan sulfate motifs. Curr Opin Struct Biol. 2005;15:1–11. doi: 10.1016/j.sbi.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Mukaida N. Pathophysiological roles of interleukin-8/CXCL8 in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol. 2003;284:L566–L577. doi: 10.1152/ajplung.00233.2002. [DOI] [PubMed] [Google Scholar]
- Nakano K, Ishiguro T, Konishi H, Tanaka M, Sugimoto M, Sugo I, et al. Generation of a humanized anti-glypican 3 antibody by CDR grafting and stability optimization. Anticancer Drugs. 2010;21:907–916. doi: 10.1097/CAD.0b013e32833f5d68. [DOI] [PubMed] [Google Scholar]
- O'Boyle G, Mellor P, Kirby JA, Ali S. Anti-inflammatory therapy by intravenous delivery of non-heparan sulfate-binding CXCL12. FASEB J. 2009;23:3906–3916. doi: 10.1096/fj.09-134643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paavola C, Hemmerich S, Grunberger D, Polsky I, Bloom A, Freedman R, et al. Monomeric monocyte chemoattractant protein-1 (MCP-1) binds and activates the MCP-1 receptor CCR2B. J Biol Chem. 1998;273:33157–33165. doi: 10.1074/jbc.273.50.33157. [DOI] [PubMed] [Google Scholar]
- Petitou M, Lormeau JC, Choay J. Interaction of heparin and antithrombin III. The role of O-sulfate groups. Eur J Biochem. 1988;176:637–640. doi: 10.1111/j.1432-1033.1988.tb14324.x. [DOI] [PubMed] [Google Scholar]
- Petitou M, Lormeau JC, Choay J. A new synthetic pentasaccharide with increased anti-factor Xa activity: possible role for anionic clusters in the interaction of heparin and antithrombin III. Semin Thromb Hemost. 1991;7(Suppl. 2):143–146. [PubMed] [Google Scholar]
- Piccinini A-M, Knebl K, Rek A, Wildner G, Diedrichs-Möhring M, Kungl AJ. Rationally evolving MCP-1/CCL2 into a decoy protein with potent anti-inflammatory activity in vivo. J Biol Chem. 2010;285:8782–8792. doi: 10.1074/jbc.M109.043299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potzinger H, Geretti E, Brandner B, Wabitsch V, Piccinini AM, Rek A, et al. Developing chemokine mutants with improved proteoglycan affinity and knocked-out GPCR activity as anti-inflammatory recombinant drugs. Biochem Soc Trans. 2006;34:435–437. doi: 10.1042/BST0340435. [DOI] [PubMed] [Google Scholar]
- Proudfoot AE. The biological relevance of chemokine-proteoglycan interactions. Biochem Soc Trans. 2006;34:422–426. doi: 10.1042/BST0340422. [DOI] [PubMed] [Google Scholar]
- Proudfoot AE, Power CA, Hoogewerf AJ, Montjovent MO, Borlat F, Offord RE, et al. Extension of recombinant human RANTES by the retention of the initiating methionine produces a potent antagonist. J Biol Chem. 1996;271:2599–2603. doi: 10.1074/jbc.271.5.2599. [DOI] [PubMed] [Google Scholar]
- Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, et al. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci USA. 2003;100:1885–1890. doi: 10.1073/pnas.0334864100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prydz K, Dalen KT. Synthesis and sorting of proteoglycans. J Cell Sci. 2000;113:193–205. doi: 10.1242/jcs.113.2.193. [DOI] [PubMed] [Google Scholar]
- Rahmoune H, Chen HL, Gallagher JT, Rudland PS, Fernig DG. Interaction of heparan sulfate from mammary cells with acidic fibroblast growth factor (FGF) and basic FGF. Regulation of the activity of basic FGF by high and low affinity binding sites in heparan sulfate. J Biol Chem. 1998;273:7303–7310. doi: 10.1074/jbc.273.13.7303. [DOI] [PubMed] [Google Scholar]
- Rek A, Brandner B, Geretti E, Kungl AJ. A biophysical insight into the RANTES-glycosaminoglycan interaction. Biochim Biophys Acta. 2009a;1794:577–582. doi: 10.1016/j.bbapap.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Rek A, Krenn E, Kungl AJ. Therapeutically targeting protein-glycan interactions. Br J Pharmacol. 2009b;157:686–694. doi: 10.1111/j.1476-5381.2009.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson CJ, Harmer NJ, Goodger SJ, Blundell TL, Gallagher JT. Cooperative dimerization of fibroblast growth factor 1 (FGF1) upon a single heparin saccharide may drive the formation of 2:2:1 FGF1.FGFR2c.heparin ternary complexes. J Biol Chem. 2005;280:42274–42282. doi: 10.1074/jbc.M505720200. [DOI] [PubMed] [Google Scholar]
- Rot A. Binding of neutrophil attractant/activation protein-1 (interleukin 8) to resident dermal cells. Cytokine. 1992;4:347–352. doi: 10.1016/1043-4666(92)90077-5. [DOI] [PubMed] [Google Scholar]
- Rot A. Chemokine patterning by glycosaminoglycans and interceptors. Front Biosci. 2010;15:645–660. doi: 10.2741/3638. [DOI] [PubMed] [Google Scholar]
- Sadir RA, Imberty A, Baleux F. Heparan sulphate/heparin oligosaccharides protect stromal cell-derived factor-1 (SDF-1)/CXCL12 against proteolysis induced cy CD26/dipeptidyl peptidase IV. J Biol Chem. 2004;279:43854–43860. doi: 10.1074/jbc.M405392200. [DOI] [PubMed] [Google Scholar]
- Salanga CL, Handel TM. Chemokine oligomerization and interaction with receptors and glycosaminoglycans: the role of structural dynamics in function. Exp Cell Res. 2011;317:590–601. doi: 10.1016/j.yexcr.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol. 2011;3:1–33. doi: 10.1101/cshperspect.a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann K, Lämmermann T, Bruckner M, Legler DF, Polleux J, Spatz JP, et al. Immobilized chemokine fields and soluble chemokine gradients cooperatively shape migration patterns of dendritic cells. Immunity. 2010;32:703–713. doi: 10.1016/j.immuni.2010.04.017. [DOI] [PubMed] [Google Scholar]
- Taylor KR, Gallo RL. Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. FASEB J. 2006;20:9–22. doi: 10.1096/fj.05-4682rev. [DOI] [PubMed] [Google Scholar]
- Wagner L, Yang OO, Garcia-Zepeda EA, Ge Y, Kalams SA, Walker BD, et al. Beta-chemokines are releases from HIV-1-specific citolytic T-cell granules complexed to proteoglycans. Nature. 1998;391:908–911. doi: 10.1038/36129. [DOI] [PubMed] [Google Scholar]
- Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. 2005;6:902–910. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- Weber C, Koenen RR. Fine-tuning leukocyte responses: towards a chemokine ‘interactome’. Trends Immunol. 2006;26:265–273. doi: 10.1016/j.it.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Weber C, Zerneke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol. 2008;8:810–815. doi: 10.1038/nri2415. [DOI] [PubMed] [Google Scholar]
- Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng. 2007;9:121–167. doi: 10.1146/annurev.bioeng.9.060906.151959. [DOI] [PubMed] [Google Scholar]
- Yang XD, Corvalan JRF, Wang P, Roy CMN, Davis CG. Fully human anti-interleukin-8 monoclonal antibodies: potential therapeutics for the treatment of inflammatory disease states. J Leukoc Biol. 1999;66:401–410. doi: 10.1002/jlb.66.3.401. [DOI] [PubMed] [Google Scholar]
- Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64:841–848. doi: 10.1016/0092-8674(91)90512-w. [DOI] [PubMed] [Google Scholar]