

Abstract
BACKGROUND AND PURPOSE
Autophagic cell death is considered a self-destructive process that results from large amounts of autophagic flux. In our previous study, GMI, a recombinant fungal immunomodulatory protein cloned from Ganoderma microsporum, induced autophagic cell death in lung cancer cells. The aim of this study was to examine the role of autophagosome accumulation in GMI-mediated cell death.
EXPERIMENTAL APPROACH
Western blot analysis, flow cytometry and confocal microscopy were used to evaluate the effects of different treatments, including silencing of ATP6V0A1 by use of short hairpin RNAi, on GMI-mediated cell death, lung cancer cell viability and autophagosome accumulation in vitro.
KEY RESULTS
Lysosome inhibitors bafilomycin-A1 and chloroquine increased GMI-mediated autophagic cell death. GMI and bafilomycin-A1 co-treatment induced the accumulation of large amounts of autophagosomes, but did not significantly induce apoptosis. GMI elicited autophagy through the PKB (Akt)/mammalian target of rapamycin signalling pathway. Silencing of ATP6V0A1, one subunit of vesicular H+-ATPases (V-ATPases) that mediates lysosome acidification, spontaneously induced autophagosome accumulation, but did not affect lysosome acidity. GMI-mediated autophagosome accumulation and cytotoxicity was increased in shATP6V0A1 lung cancer cells. Furthermore, ATP6V0A1 silencing decreased autophagosome and lysosome fusion in GMI-treated CaLu-1/GFP-LC3 lung cancer cells.
CONCLUSION AND IMPLICATIONS
We demonstrated that autophagosome accumulation induces autophagic cell death in a GMI treatment model, and ATP6V0A1 plays an important role in mediating autophagosome-lysosome fusion. Our findings provide new insights into the mechanisms involved in the induction of autophagic cell death.
Keywords: PKB, ATP6V0A1, autophagy, autophagic cell death, autophagic flux, GMI, immunomodulatory protein, lysosome, mTOR, V-ATPases
Introduction
Autophagy is a lysosomal degradation pathway that controls the quality and balance of cytoplasmic constituents, involving protein and organelles (Rosenfeldt and Ryan, 2011). The three major steps of the autophagy process are (i) initiation with phagophore induction; (ii) autophagosome formation; and (iii) maturation with autolysosome formation. The role of autophagy in pro-death and pro-survival is still elusive due to its dependence on the type and degree of stress and cell type (Janku et al., 2011). It has been suggested that autophagic flux and steady-state autophagy are two different autophagy monitoring methods (Klionsky et al., 2008a). In the canonical definition, autophagic cell death is an eat-to-death process mediated by self-destruction. This suggests that autophagic flux is the critical mechanism in autophagic cell death (Maiuri et al., 2007; Shen and Codogno, 2011). However, it has been demonstrated that steady-state autophagy leads to a physiological dysfunction (Klionsky et al., 2008a). From a comprehensive survey, the level of autophagy induction may be the key to deciding cell survival or death (Levine, 2007). Therefore, a feasible direction for the development of anti-cancer drugs is the killing of cancer cells through induction of an autophagy overload.
The vesicular H+-ATPases (V-ATPases) are a family of ATP-dependent proton pumps. V-ATPases play a critical role in mediating vesicular acidification. V-ATPase binds to the membrane of organelles, including lysosomes, and transfers protons from the cytoplasm to the lumen of bound organelles by ATP hydrolysis (Paroutis et al., 2004). V-ATPase is a multi-subunit complex composed of two functional domains: V0 and V1. V-ATPase is anchored to the membrane through the transmembrane V0 domain. The V0 domain also plays a role in mediating the translocation of protons across the membrane. The major function of the V1 domain is hydrolysis of ATP to supply the energy for inducing proton translocation. ATP6V0A1, the a1 subunit of the V0 sector, has multiple functions, including proton transport, assembly and targeting (Forgac, 1999). It has been documented that the V0 domain and ATP6V0A1 participate in the membrane fusion of vesicles (Bayer et al., 2003; Strasser et al., 2011). Francesca et al. suggested that ATP6V0A1 is required to mediate phagosome–lysosome fusion (Peri and Nusslein-Volhard, 2008).
Fungal immunomodulatory proteins (FIPs) are one family of Lingzhi components with therapeutic effects (Jinn et al., 2006; Boh et al., 2007). In our previous studies, FIPs have been shown to possess anti-cancer abilities and have the potential to act as chemopreventive agents (Liao et al., 2006; 2007; 2008). GMI (Figure S3), an immunomodulatory protein with amino acid sequences highly homologous to those of FIP-gts, inhibits EGF- and TNF-α-mediated migration and invasion in A549 cells (Lin et al., 2010a,b). In our recent study, GMI induced lung cancer cell death via activation of calcium-dependent autophagy in vitro and in vivo (Hsin et al., 2011).
In autophagy research, much attention has been focused on the issues of induction and degradation mechanisms. LC3-II expression is a widely investigated autophagy marker. However, LC3-II does not significantly increase, as assessed by Western blot analysis, in a balanced situation of autophagic flux (Tanida et al., 2005). This suggests that many autophagy inducers, including GMI, elicit an extremely imbalanced autophagic flux in cells, leading to autophagosome accumulation. The aim of this study was to examine the role of autophagosome accumulation in GMI-mediated cell death. We demonstrated that lysosome inhibitors increase GMI-induced autophagosome accumulation and autophagic cell death. Furthermore, ATP6V0A1 was found to be important in mediating autophagosome-lysosome fusion. ATP6V0A1 silencing enhanced GMI-induced cytotoxicity and autophagosome accumulation in lung cancer cells.
Methods
Cell lines and chemicals
A549 human lung adenocarcinoma cells (ATCC, CCL-185) and CaLu-1 human lung squamous carcinoma cells (ATCC, HTB-54) were maintained as described previously (Hsin et al., 2011). Bafilomycin-A1 (B 1793) and chloroquine diphosphate salt (C 6628) were purchased from Sigma (St. Louis, MO, USA). Pan-caspase inhibitor (550377) was purchased from BD Pharmingen (Franklin Lakes, NJ, USA).
Statistical analyses
The data were analysed using Student's t test. P values of <0.05 were considered significant. The data are presented as the mean ± SD.
Results
Lysosome inhibitors enhance the effects of GMI on cell death
To clarify the role of autophagosome accumulation on GMI-mediated cytotoxicity, bafilomycin-A1 and chloroquine were used to prevent autolysosome maturation. Chloroquine inhibits lysozyme activity by increasing pH (Mizushima et al., 2010). Bafilomycin-A1 is a V-ATPase inhibitor that can block autophagosome–lysosome fusion and prevent lysosome acidification (Yamamoto et al., 1998; Klionsky et al., 2008b). A549 and CaLu-1 cells were concurrently treated with GMI and bafilomycin-A1 or chloroquine for 48 h and analysed using the MTT assay. Both lysosome inhibitors significantly enhanced GMI-induced cell death (Figure 1A). In our previous study, several assays were performed to prove that GMI does not predominantly induce apoptosis in lung cancer cells (Hsin et al., 2011). Some studies have reported that combination treatment with bafilomycin-A1 and an autophagy inducer leads to apoptosis (Smith et al., 2010; Yang et al., 2010). To assess the effects of apoptosis on cell death by bafilomycin-A1 and GMI, the pan-caspase inhibitor Z-VAD-FMK was used to block apoptosis. As shown in Figure 1B, Z-VAD-FMK did not reverse the cell death in both cells treated with bafilomycin-A1 and GMI. To further investigate the effect of bafilomycin-A1 on GMI-induced cell death, a clonogenic assay was performed to determine the long-term cytotoxic effects. Co-treatment with GMI and bafilomycin-A1 significantly inhibited colony formation of A549 cells when compared with treatment with GMI alone (Figure 1C). The colonies were counted and their numbers are shown in Figure 1D. To clarify the role of autophagy in cell death induced by co-treatment with GMI and bafilomycin-A1, a VSV-G pseudotyped lentivirus-shRNA system was carried out to knockdown the LC3 expression in A549 cells (Figure 1E). LC3 silencing significantly reversed the cell death induced by co-treatment with GMI and bafilomycin-A1 (Figure 1F). Our results indicate that suppressing autophagosome clearance increases GMI-elicited autophagic cell death.
Figure 1.
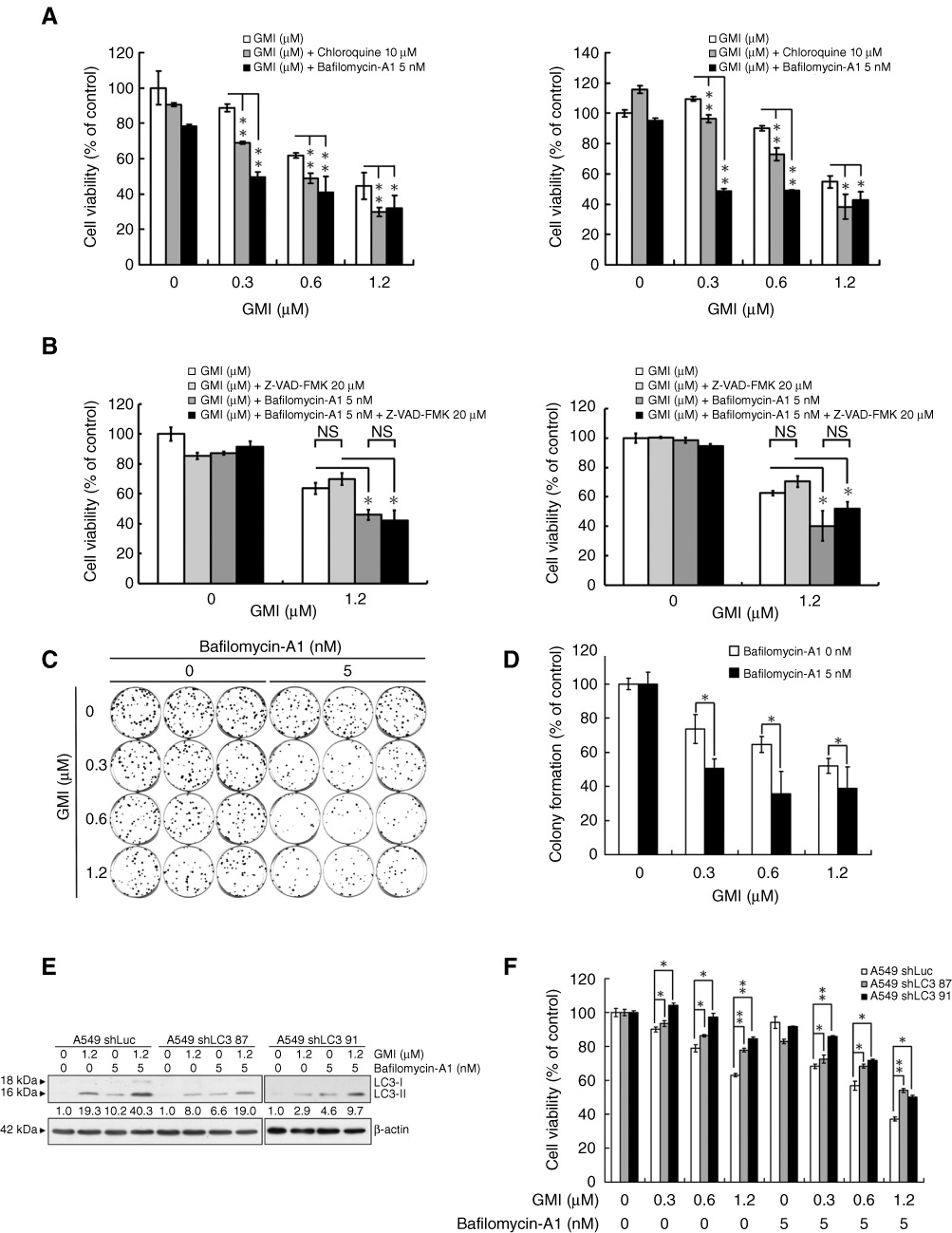
Effect of lysosome inhibitors on GMI-induced autophagic cell death in non-small cell lung cancer cells. (A) A549 and CaLu-1 cells (5 × 103 cells per well of 96-well plate) were co-treated with various concentrations of GMI (0, 0.3, 0.6 and 1.2 µM) and chloroquine (0, 10 µM) or bafilomycin-A1 (0, 5 nM) for 48 h. Cell viability was analysed on MTT assay. The data are presented as mean ± SD of triplicate experiments. (B) A549 and CaLu-1 cells (5 × 103 cells per well of 96-well plate) were pretreated with 20 µM pan-caspase inhibitor Z-VAD-FMK for 1 h, followed by exposure to GMI (0, 1.2 µM) and bafilomycin-A1 (0, 5 nM) for 48 h. MTT assay was performed to measure the cell viability. The data are presented as mean ± SD of triplicate experiments. (C) Equal numbers of cells from the A549 cell pools co-treated with GMI and bafilomycin-A1 were plated and stained as described in Appendix S1. Shown are three dishes from each pool. (D) The number of colonies was counted under a dissecting microscope. The number of cells in each colony had to be larger than 50. Shown are the relative colony numbers. The number of A549 cells without GMI treatment was set at 100%. The original numbers of colonies of A549 cells without or with bafilomycin-A1 treatment are as follows: bafilomycin-A1 0 nM, 88.3 ± 3.5; bafilomycin-A1 5 nM, 74.7 ± 6.7. (E) Equal amounts of total cell lysates from A549 shLuc and A549 shLC3 cells (5 × 105 cells per 60 mm dish) after 1.2 µM GMI treatment were analysed by Western blot. (F) After co-treatment with bafilomycin-A1 (0, 5 nM) and GMI (0, 0.3, 0.6 and 1.2 µM) for 48 h, the cell viabilities of A549 shLuc and A549 shLC3 cells (5 × 103 cells per well of 96-well plate) were measured on MTT assay. The data are presented as mean ± SD of triplicate experiments. The symbols (*) and (**) indicate P < 0.05 and P < 0.0001 using Student's t-test. NS, not significant.
Bafilomycin-A1 enhances GMI-induced autophagosome accumulation
To investigate the effect of bafilomycin-A1 on GMI-induced autophagosome accumulation, we established the stable expression of GFP-LC3 in CaLu-1 cells (CaLu-1/GFP-LC3). To rule out the influence of GFP-LC3 expression in CaLu-1 cells, confirmation experiments were performed to prove that stable expression of GFP-LC3 did not change the response of CaLu-1 cells to GMI and bafilomycin-A1 treatment (Figure S1). On Western blot, bafilomycin-A1 increased GMI-mediated endogenous LC3-II and GFP-LC3-II accumulation (Figure 2A,B). We found cleaved GFP expression in different GFP-LC3 stable CaLu-1 cell clones without stimulation (Figure 2B and data not shown). It is suggested that GFP-LC3 was degraded by endogenous autophagic flux or protease in CaLu-1 cells. Thus, inhibition of cleaved GFP expression by bafilomycin-A1 was not obvious, even though GFP-LC3-II degradation was blocked. To confirm the effects of apoptosis on cell death induced by bafilomycin-A1 and GMI, PARP and caspase 3 were investigated by Western blot analysis. Bafilomycin-A1 and GMI co-treatment did not significantly increase PARP and caspase 3 cleavage in A549 cells (Figure 2A). Interestingly, GMI blocked bafilomycin-A1-induced apoptosis (Figure 2A). It has been reported that autophagy can counteract apoptosis activation (Maiuri et al., 2007). This suggested that GMI predominantly induces autophagy in A549 cells. In CaLu-1 cells, bafilomycin-A1 and 0.3 µM GMI slightly increased apoptosis when compared with cells exposed to bafilomycin-A1 or GMI alone (Figure 2B).
Figure 2.
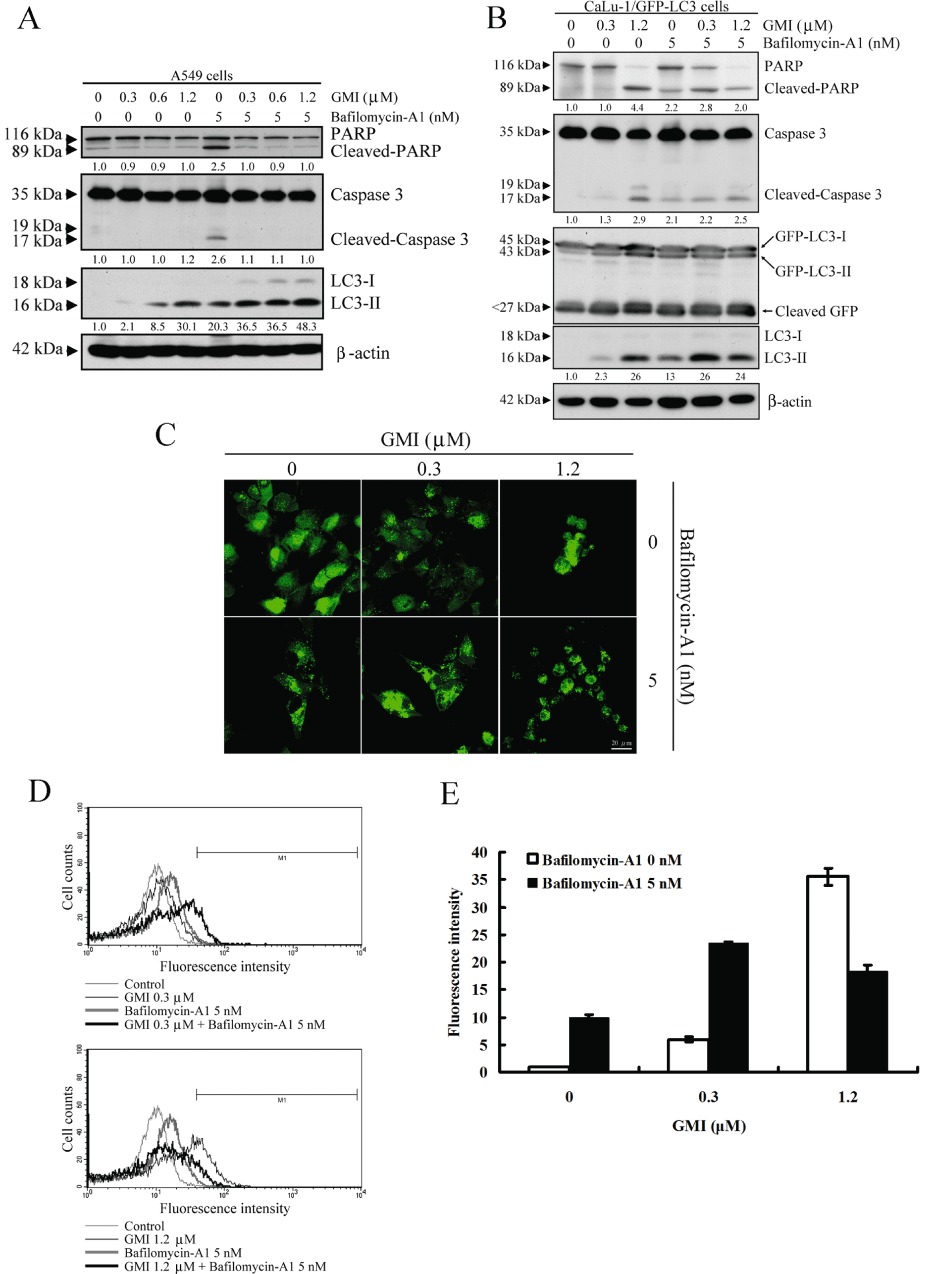
Effect of bafilomycin-A1 and GMI co-treatment on apoptosis and autophagosome accumulation in non-small cell lung cancer NSCLC cells. (A) PARP, caspase 3 cleavage and LC3 conversion were determined on Western blot after A549 cells (2 × 105 cells per 35 mm dish) were treated with 5 nM bafilomycin-A1 and various concentrations of GMI for 48 h. β-Actin is a loading control. (B) Changes in PARP and caspase 3, and conversions of GFP-LC3 and endogenous LC3 in GMI and bafilomycin-A1-treated CaLu-1/GFP-LC3 cells (2 × 105 cells per 35 mm dish) were detected on Western blot. β-Actin is a loading control. The software ImageJ was used to quantify the band intensity of cleaved PARP, cleaved caspase 3 and LC3-II. The data showed the relative expression standardized by the β-actin protein level, and the ratio without GMI and bafilomycin-A1 treatment was set at 1. (C) CaLu-1/GFP-LC3 cells (5 × 104 cells per well of 24-well plate) were treated with 5 nM bafilomycin-A1 and various concentrations of GMI for 48 h. The GFP-LC3 dots induced by GMI and bafilomycin-A1 in CaLu-1/GFP-LC3 cells were observed under confocal microscope. Scale bar indicates 20 µm. (D) After bafilomycin-A1 (0, 5 nM) and GMI (0, 0.3 and 1.2 µM) treatment for 48 h, CaLu-1/GFP-LC3 cells (5 × 105 cells per 60 mm dish) were harvested with trypsin-EDTA. The relative level of GFP-LC3 fluorescence intensity was analysed by flow cytometry. (E) Quantification of cells developing GFP-LC3 dots in CaLu-1/GFP-LC3 cells. The percentages of developed GFP-LC3 dots in GMI- and bafilomycin-A1-treated CaLu-1/GFP-LC3 cells were calculated based on the results of fluorescence-activated cell sorting assay.
Using confocal microscope, autophagosome accumulation was markedly enhanced after co-treatment with bafilomycin-A1 and 0.3 µM GMI (Figure 2C). It was difficult to count the number of autophagosomes in the shrinking cells after GMI treatment. GFP-LC3-labelled autophagosomes can be measured by flow cytometry in living cells (Shvets et al., 2008). Therefore, we quantified the autophagosome fractional volume after bafilomycin-A1 and GMI treatment by flow cytometric analysis. The data indicate that autophagosome accumulation is significantly up-regulated in CaLu-1/GFP-LC3 cells treated with bafilomycin-A1 and 0.3 µM GMI (Figure 2D,E). However, 1.2 µM GMI and bafilomycin-A1 did not increase autophagosome accumulation in CaLu-1/GFP-LC3 cells. This phenomenon is consistent with the LC3-II expression in Figure 2B. We suggest that combined treatment with high-dose GMI and bafilomycin-A1 induces severe cell damage, leading to a partial decrease in cancer cells with autophagosomes (Figure 2D,E). Furthermore, these data indicate that GMI blocks autophagosome clearance in lung cancer cells.
GMI induces autophagy through PKB/mTOR inhibition
The PKB/mTOR signalling cascade is a critical autophagy regulation pathway (Fu et al., 2009; Rubinsztein et al., 2011). As shown in Figure 3A and B, GMI significantly decreased PKB and p70S6K phosphorylation in A549 and CaLu-1 cells. GMI suppressed PKB phosphorylation after 1 h of treatment in the time course experiments in A549 cells (Figure 3C). A small amount of LC3-II expression was elicited after 6 h of GMI treatment (Figure 3C). CA-PKB overexpression attenuated GMI-induced LC3-II expression in A549 cells (Figure 3D). As shown in Figure 3D, CA-PKB obviously restored reduced GSK-3β levels induced by GMI treatment. However, CA-PKB only partially reversed GMI-induced p70S6K phosphorylation inhibition. This indicates that GMI inhibited mTOR not only through the PKB pathway. Moreover, rapamycin, an mTOR inhibitor, enhanced low-dose GMI-mediated autophagy (data not shown). These results demonstrate that GMI-stimulated autophagy is primarily dependent on the PKB pathway in lung cancer cells and that mTOR also plays a role in the GMI-induced autophagy.
Figure 3.
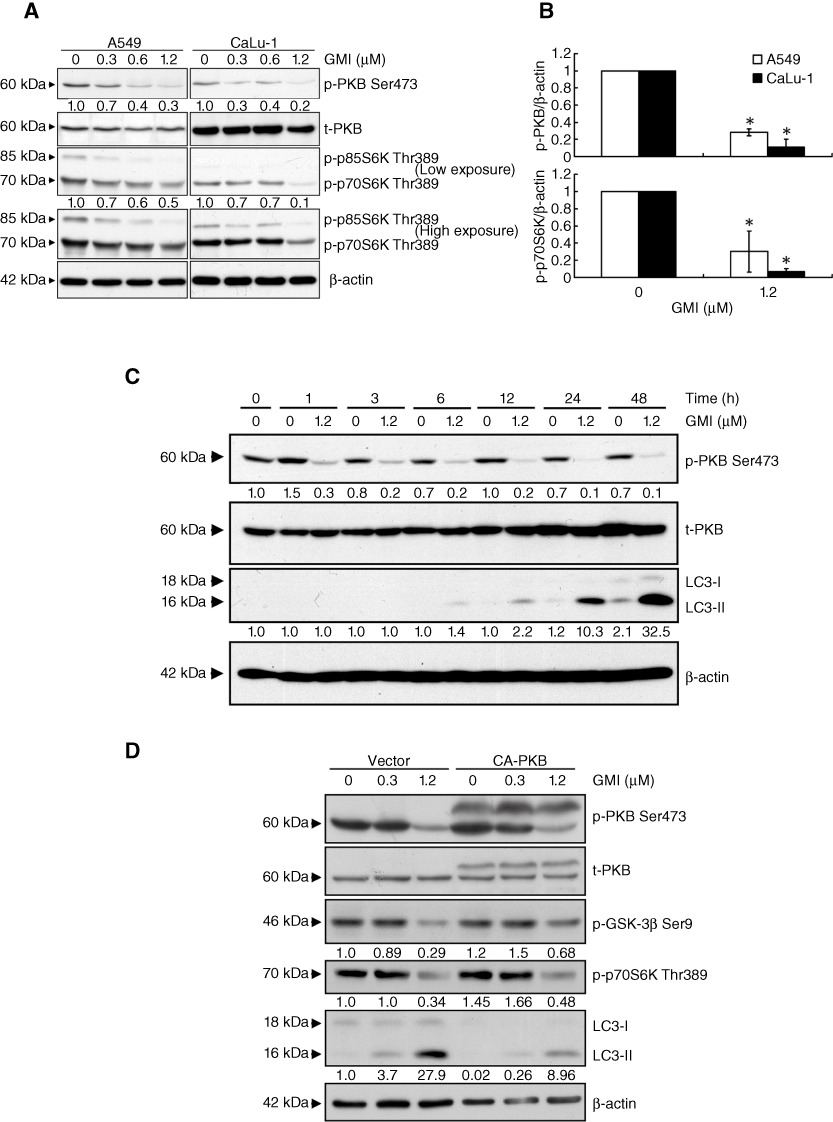
Effect of the PKB/mTOR signalling pathway in GMI-mediated autophagy in non-small cell lung cancer cells. (A) A549 and CaLu-1 cells (2 × 105 cells per 35 mm dish) were treated with GMI (0, 0.3, 0.6 and 1.2 µM) for 48 h. Equal amounts of total cell lysates were subject to immunoblotting with anti-phospho-PKB, anti-PKB and anti-phospho-p70S6K. β-Actin is a loading control. (B) Statistical analysis of Western blotting. The protein levels of p-PKB and p-p70S6K were standardized by the β-actin protein level, and each value is expressed as an intensity showing the ratio of the expression in A549 and CaLu-1 cells treated with GMI to that in untreated cells. The ratio of cells without GMI treatment was set at 1. The symbol (*) indicates P < 0.05 by Student's t-test. (C) A549 cells (2 × 105 cells per 35 mm dish) were treated without or with 1.2 µM GMI for various time periods (0, 1, 3, 6, 12, 24 and 48 h). Protein loading was determined by Western blot against β-actin. (D) A549 cells (1 × 105 cells per 35 mm dish) transfected with empty vector or constitutively active PKB (CA-PKB) expression vector for 24 h were treated with various concentrations of GMI (0, 0.3 and 1.2 µM) for 48 h. Phospho-PKB, PKB, phospho-GSK-3β, phospho-p70S6K expression and LC3 conversion were determined on Western blot. The software ImageJ was used to quantify the band intensity.
ATP6V0A1 silencing does not affect lysosomal acidity
In Figures 1 and 2, we demonstrated that bafilomycin-A1 enhances GMI-mediated autophagosome accumulation and autophagic cell death. Moreover, the combination of bafilomycin-A1 and GMI treatment did not markedly increase apoptosis. This prompted us to examine whether GMI and bafilomycin-A1 induce autophagic cell death through autophagosome accumulation in lung cancer cells. Recently, mTOR complex 1 has been shown to regulate the expression of several genes encoding V-ATPase subunits, including ATP6V0A1 (Pena-Llopis et al., 2011). Thus, we investigated the effects of GMI on ATP6V0A1 expression. Using real-time (RT)-PCR, GMI down-regulated ATP6V0A1 mRNA expression in lung cancer cells (Figure 4A). Two bands of ATP6V0A1 RT-PCR products were produced for ATP6V0A1 isoforms (NP_001123492.1, NP_001123493.1 and NP_005168.2). To further investigate the function of ATP6V0A1 in lysosome acidity, VSV-G pseudotyped lentivirus-shRNA system was used to knockdown the ATP6V0A1 expression. As shown in Figure 4B, the different silencing effects of two shRNA clones against ATP6V0A1 were investigated in both lung cancer cell lines. To detect the effect of ATP6V0A1 knockdown on lysosomal acidity, A549 and CaLu-1/GFP-LC3 cells untreated and treated with GMI were stained with acridine orange and visualized using a fluorescence microscope (Figure 4C). Flow cytometric analysis was performed to quantify the acidic vesicular organelle (AVO) fractional volume after GMI treatment (Figure 4D,E). A549 shATP6V0A1 33 cells possessed flattened morphology with elevated lysosomal mass-like cellular senescence (Figure 4E and data not shown). High-dose GMI induced severe cell damage, leading to a decrease in cancer cells with AVOs (Figure 4D,E). These data suggest that ATP6V0A1 silencing did not affect the lysosomal acidity.
Figure 4.
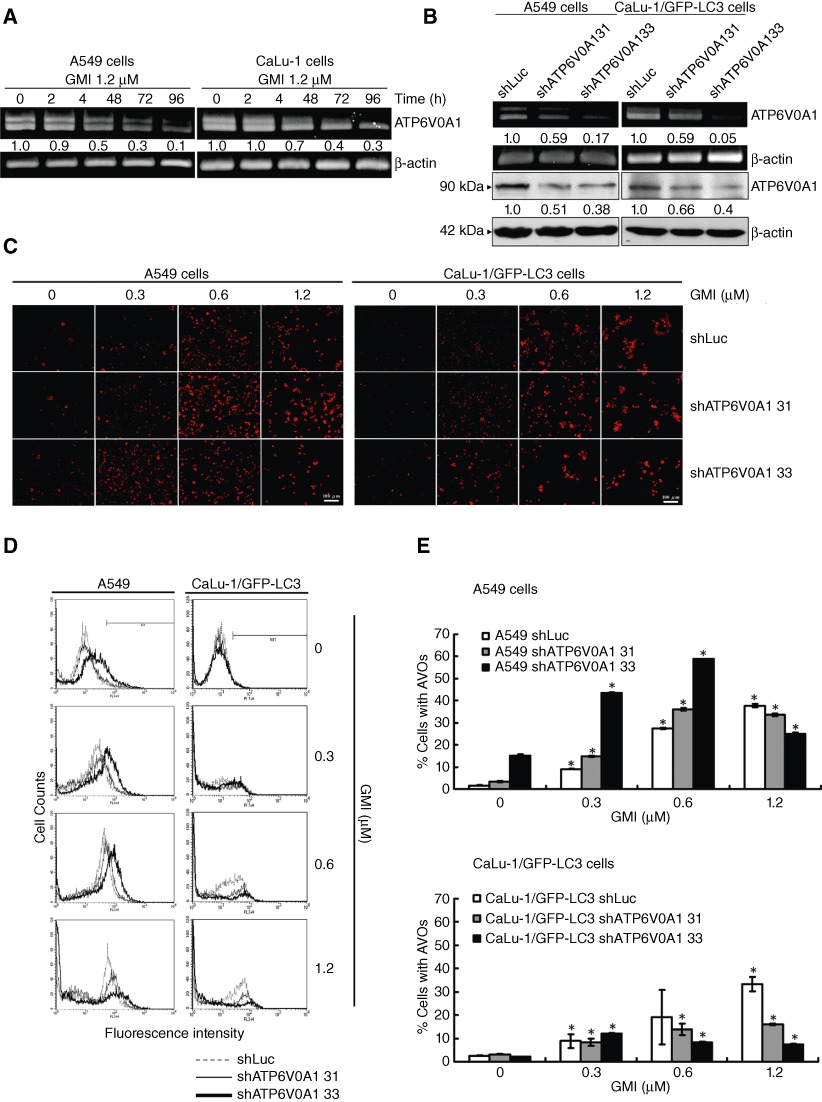
Effect of ATP6V0A1 silencing on GMI-induced acidic vesicular organelle (AVO) development in non-small cell lung cancer cells. (A) A549 and CaLu-1 cells (5 × 105 cells per 60 mm dish) were treated without or with 1.2 µM GMI for various time periods (0, 24, 48, 72 and 96 h). The total RNA was reverse-transcribed into cDNA, and the expressions of ATP6V0A1 and β-actin were analysed by PCR. (B) Total RNA and cell lysate were collected from A549 shLuc, A549 shATP6V0A1, CaLu-1/GFP-LC3 shLuc and CaLu-1/GFP-LC3 shATP6V0A1 cells. ATP6V0A1 expression was observed by RT-PCR and Western blot. β-Actin served as an internal control. The software ImageJ was used to quantify the band intensities of ATP6V0A1. (C) Acridine orange was used to stain AVOs in A549 shLuc, A549 shATP6V0A1, CaLu-1/GFP-LC3 shLuc and CaLu-1/GFP-LC3 shATP6V0A1 cells (5 × 104 cells per well of 24-well plate) untreated or treated with GMI (0.3, 0.6 and 1.2 µM) for 48 h. The cells were investigated under a red filter fluorescence microscope. Scale bar indicates 100 µm. (D) The indicated cell lines (5 × 105 cells per 60 mm dish) were treated with GMI for 48 h, followed by staining with acridine orange. The fluorescence-activated cells were analysed by flow cytometry. (E) Quantification of cells with AVO development in indicated cell lines. The percentages of developed AVOs in GMI-treated cells were calculated based on the results of fluorescence-activated cell sorting assay. The symbol (*) indicates P < 0.05 by Student's t-test.
ATP6V0A1 silencing sensitizes lung cancer cells to GMI-induced cell death
In spite of the lack of change in lysosome acidification, cell density decreased in shATP6V0A1 lung cancer cells without or with GMI treatment when compared with shLuc cells (Figure 5A). To confirm this observation, cell viability was analysed using the MTT assay. As shown in Figure 5B and C, ATP6V0A1 silencing slowed down lung cancer cell proliferation. In comparison with A549 shLuc cells, GMI significantly increased cell death in A549 shATP6V0A1 cells (Figure 5B). ATP6V0A1 silencing elevated GMI-mediated cell death more markedly in CaLu-1/GFP-LC3 cells (Figure 5C). To further analyse the cell death induced by GMI in ATP6V0A1-silenced cells, propidium iodide staining was performed and cells were analysed by flow cytometry. As shown in Figure 5D, ATP6V0A1 silencing increased the cell death in GMI-treated A549 cells.
Figure 5.
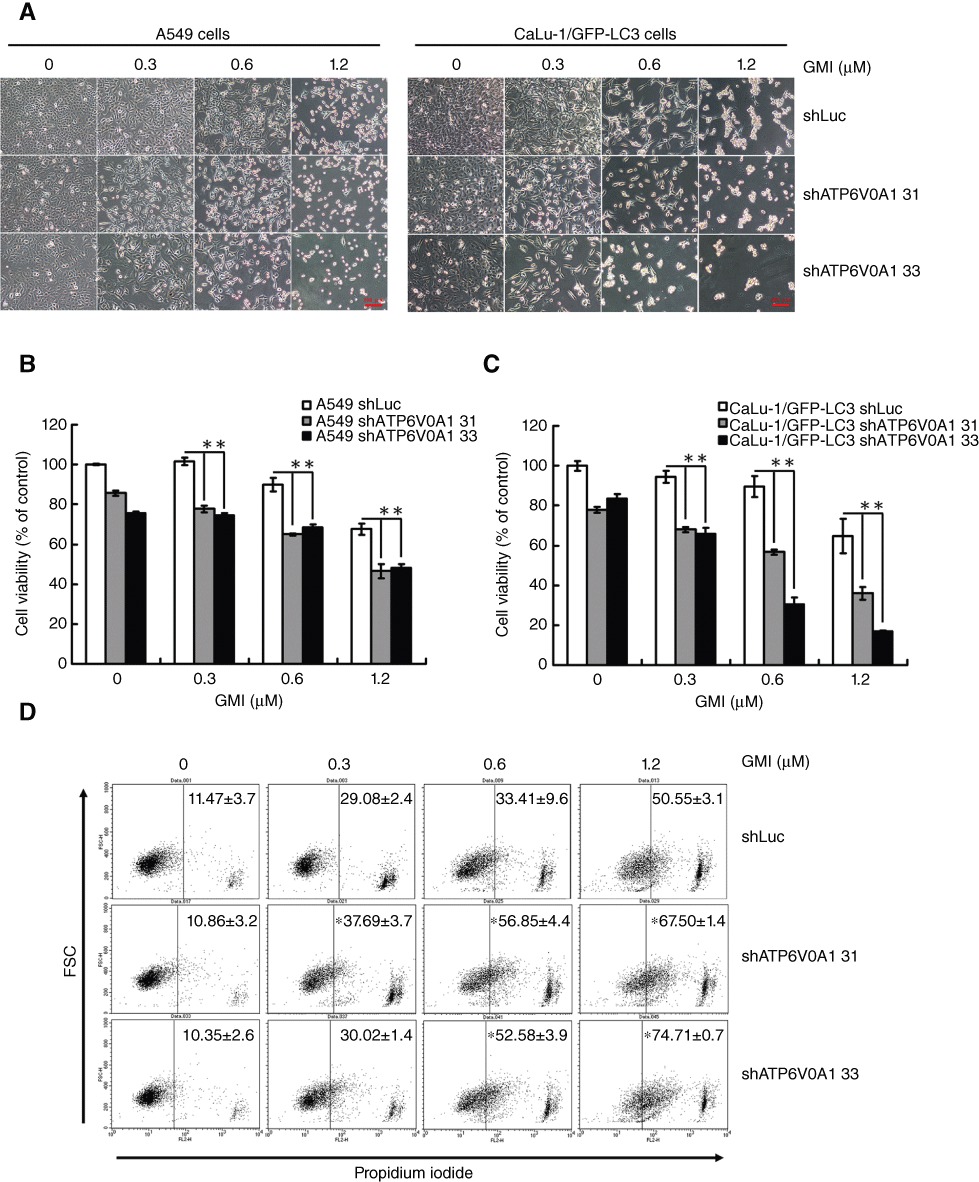
Effect of ATP6V0A1 silencing on GMI-induced cell death in non-small cell lung cancer cells. (A) A549 shLuc, A549 shATP6V0A1, CaLu-1/GFP-LC3 shLuc and CaLu-1/GFP-LC3 shATP6V0A1 cells were treated with GMI for 48 h. Cell morphology was observed under an inverted microscope. Scale bar indicates 100 µm. (B) A549 shLuc, A549 shATP6V0A1, (C) CaLu-1/GFP-LC3 shLuc and CaLu-1/GFP-LC3 shATP6V0A1 cells (5 × 103 cells per well of 96-well plate) were treated with GMI (0, 0.3, 0.6 and 1.2 µM) for 48 h. MTT assay was used to estimate the cell viability. The symbol (**) indicates P < 0.0001 by Student's t-test. (D) A549 shLuc, A549 shATP6V0A1 cells (2 × 105 cells per well of 6-well plate) were treated with GMI (0, 0.3, 0.6 and 1.2 µM) for 48 h, followed by staining with propidium iodide. The fluorescence-activated cells were analysed by flow cytometry. The symbol (*) indicates P < 0.05 by Student's t-test.
ATP6V0A1 silencing enhances GMI-mediated autophagosome accumulation
It has been reported that ATP6V0A1 mediates phagosome and lysosome fusion without affecting lysosomal acidity (Peri and Nusslein-Volhard, 2008). ATP6V0A1 may play an important role in autolysosome formation. Using Western blot analysis, ATP6V0A1 silencing was shown to induce LC3-II expression in lung cancer cells without treatment, especially in CaLu-1/GFP-LC3 cells (Figure 6A,B). In comparison with A549 shLuc cells, GMI-induced LC3-II expression increased in A549 shATP6V0A1 31 cells, but not in A549 shATP6V0A1 33 cells (Figure 6A). This may be due to the senescence-like phenomenon in A549 shATP6V0A1 33 cells. Furthermore, the phenomenon was observed in both CaLu-1/GFP-LC3 shATP6V0A1 cells, especially following 0.3 µM GMI treatment (Figure 6B). GFP-LC3-II expression was consistent with that of endogenous LC3-II expression in CaLu-1/GFP-LC3 cells (Figure 6B). As with the bafilomycin-A1 treatment (Figure 2A,B), ATP6V0A1 silencing did not significantly increase apoptosis after GMI treatment (Figure 6C,D).
Figure 6.

Effect of ATP6V0A1 silencing on GMI-induced autophagosome accumulation in non-small cell lung cancer cells. Equal amounts of total cell lysates from GMI-treated (A) A549 and (B) CaLu-1/GFP-LC3 cells were analysed on Western blot. PARP expression and change in the (C) A549 and (D) CaLu-1/GFP-LC3 cells treated with GMI were analysed on immunoblotting assay. Band intensity was quantified by the software Image J.
Autophagosome accumulation and autophagosome–lysosome interaction were investigated in CaLu-1/GFP-LC3 shLuc and shATP6V0A1 cells by confocal microscopy. As shown in Figure 7A, endogenous and 0.3 µM GMI-induced autophagosomes fused with lysosomes; 1.2 µM GMI induced autophagosomes partly fused with lysosomes (Figure 7A). Large green puncta spontaneously accumulated in CaLu-1/GFP-LC3 shATP6V0A1 cells (Figure 7B,C). Larger numbers of large green puncta were induced by GMI in CaLu-1/GFP-LC3 shATP6V0A1 cells when compared with CaLu-1/GFP-LC3 shLuc cells (Figure 7B,C). Similar to the results shown in Figure 4D and E, high-dose GMI treatment resulted in a decrease in CaLu-1/GFP-LC3 shATP6V0A1 33 cells with AVOs (Figure 7C). Using Image software (Nikon, Tokyo, Japan), shATP6V0A1 was shown to decrease autophagosome and lysosome co-localization in CaLu-1/GFP-LC3 cells with or without GMI treatment (Figure 7). The quantification analysis was performed to confirm the effects of GMI and ATP6V0A1 silencing on the fusion of autophagosome and lysosome (Figure 7D). These effects were also investigated in anti-LAMP2 antibody-stained CaLu-1/GFP-LC3 shATP6V0A1 cells (Figure S2). These results suggest that ATP6V0A1 mediates autophagosome–lysosome fusion.
Figure 7.
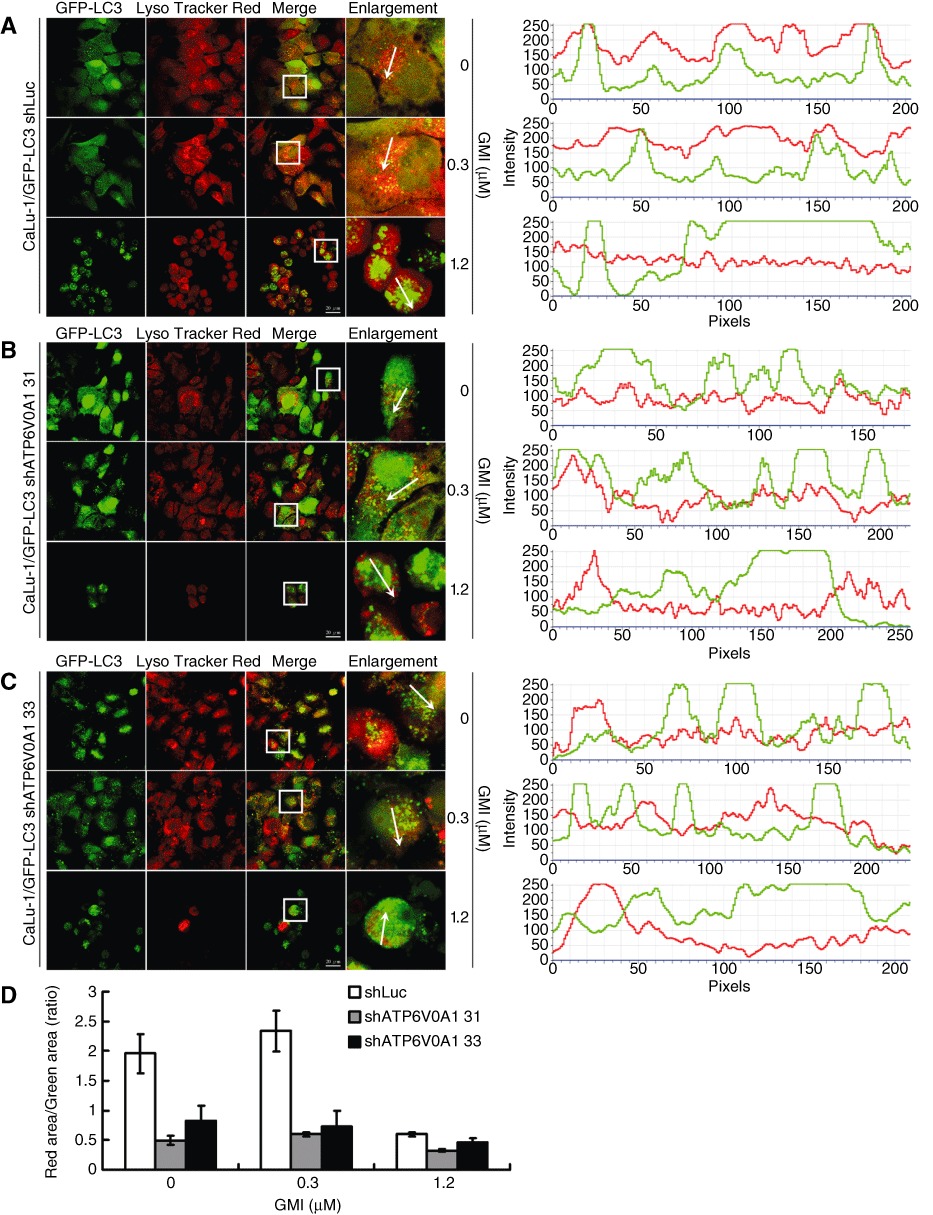
Effect of ATP6V0A1 silencing on GMI-induced autolysosome formation in non-small cell lung cancer cells. After GMI treatment for 48 h, CaLu-1/GFP-LC3 (A; right panel) shLuc and (B, C; right panel) shATP6V0A1 cells (5 × 104 cells per well of 24-well plate) were stained with LysoTracker. GFP-LC3 dots and lysosomes were visualized on confocal microscope. Box indicates enlarged area. Scale bar indicates 20 µm. The co-localization was indicated by the orientation of the arrow and analysed by software NIS Elements D 3.0 (A, B and C; left panel). (D) Autophagosome-lysosome co-localization quantification. The quantification method was described in Appendix S1.
Discussion
In our previous study, MTT assay, clonogenic assay and cell cycle analysis were performed to prove that autophagy activation by GMI induces lung cancer cell death, not inhibition of cell proliferation. We also demonstrated that GMI induced autophagic flux in lung cancer cells (Hsin et al., 2011). In this study, we demonstrated that increasing autophagosome accumulation by lysosome inhibitors and ATP6V0A1 silencing enhances GMI-mediated autophagic cell death. Chloroquine has been found to induce autophagic cell death (Ramser et al., 2009; Geng et al., 2010). PM02734 (Elisidepsin) induces autophagy by compromising the clearance of autophagosome and kills lung cancer cells (Ling et al., 2011). These findings accord with our hypothesis that an abundant accumulation of autophagosomes mediates autophagic cell death. Furthermore, according to the LC3 flux measurement in our previous study (Ling et al., 2011), GMI decreases LC3 flux in lung cancer cells (Figure 2A,B). These findings indicate that GMI not only activates autophagosome formation but also interferes with autolysosome maturation. We believe that autophagosome accumulation is the primary mechanism by which GMI kills cancer cells. However, we could not rule out the effects of autophagic flux on the cytotoxicity elicited by GMI. Unfortunately, to date, there is no appropriate method for increasing the flux rate of autophagy. Therefore, it is difficult to clarify the effects of autophagosome accumulation and autophagic flux on autophagic cell death.
In a recent study, Fan et al. suggested an interesting mechanism involving PKB, autophagy and apoptosis in glioma (Fan et al., 2010). PI-101, a dual inhibitor of PI3K and mTOR, induces apoptosis in U373 cells after bafilomycin-A1 treatment or LAMP2 silencing (inhibition of autophagy maturation). However, PI-101 does not trigger apoptosis in the same glioma cells after 3-MA treatment or VPS34 silencing (inhibition of autophagy initiation). These results are similar to our findings. Furthermore, combined treatment with GMI and bafilomycin-A1 only induced apoptosis in CaLu-1 cells. Taken together, these data indicate that the effects of an autophagy inducer and bafilomycin-A1 treatment on apoptotic cell death may depend on cell type.
It is important to quantify the cells undergoing autophagy and autophagosome accumulation in autophagy research. The direct and precise method involves counting green dots in GFP-LC3 transfected cells. A recent study indicated that larger GFP-LC3 dots are formed by several autophagosomes (Ganley et al., 2011). However, it is difficult to count the autophagosomes in the shrinking cells following treatment with GMI. To overcome this problem, flow cytometric analysis was performed to quantify the GFP-LC3 dot fractional volume in living CaLu-1/GFP-LC3 cells. The fluorescence intensity following 1.2 µM GMI treatment was much higher than that following bafilomycin-A1 and 1.2 µM GMI co-treatment (Figure 2D,E). Using Western blots, 1.2 µM GMI was shown to increase GFP-LC3-I expression in CaLu-1/GFP-LC3 cells (Figure 2D). This highlights the defect of investigating GFP-LC3 puncta in living cells directly by flow cytometry. We suggest that Western blots should be used to assess the expression of GFP-LC3-I.
mTOR is a well-documented autophagy regulator. PKB and AMPK have been reported to regulate mTOR/autophagy pathway (Yang and Klionsky, 2010). However, interestingly, Mammucari et al. demonstrated that FoxO3 controls autophagy through a PKB-dependent and mTOR-independent pathway (Mammucari et al., 2007). In the present study, GMI inhibited the PKB/mTOR signalling pathway in lung cancer cells (Figure 3A). To further prove the role of the PKB/mTOR pathway in GMI-induced autophagy, a CA-PKB expression plasmid was used to reverse the inhibition of PKB by GMI. CA-PKB could not markedly activate the PKB signalling pathway because of the strong PKB activity in A549 cells. After GMI treatment, CA-PKB obviously reversed the inhibition of GSK-3β, a downstream target of PKB. This suggests that CA-PKB works in A549 cells. Furthermore, GMI-mediated LC3 conversion was blocked by CA-PKB. However, CA-PKB only partially prevented the GMI-mediated p70S6K inhibition. This result indicates that GMI is a new dual PKB/mTOR inhibitor and inhibits mTOR activity through a PKB-independent pathway. It has been reported that p53 inhibition and calcium inhibit mTOR and induce autophagy through the AMPK pathway (Hoyer-Hansen et al., 2007; Tasdemir et al., 2008). In our previous study, we demonstrated that GMI induced autophagy by increasing calcium and decreasing p53 expression. This implies that GMI suppresses mTOR through the AMPK pathway. According to the present results, PKB plays a major role in GMI-induced autophagy. In our unpublished data, we found that rapamycin can enhance GMI-mediated autophagy. This suggests that mTOR inhibition also plays a role in autophagy induction by GMI. Taken together, these results suggest that GMI-induced autophagy is primarily dependent on PKB inhibition and partially dependent on mTOR inhibition.
A decade ago, the V0 sector of V-ATPase was implicated to participate in membrane fusion. However, the lethality of V-ATPase disruption caused difficulties in a further study (Scott et al., 2011). ATP6V0A1 is an important subunit in the V-ATPase complex assembly, proton transport and membrane fusion. It has been reported that ATP6V0A1 knockdown blocks phagosome–lysosome fusion without changing the lysosome acidity (Peri and Nusslein-Volhard, 2008). At least one similar gene can replace ATP6V0A1 and perform its function in the acidification of lysosomes in zebrafish (Peri and Nusslein-Volhard, 2008). Consistent with previous findings, ATP6V0A1 silencing did not affect lysosome acidity in lung cancer cells in this study. However, ATP6V0A1 silencing decreased the cell viability, which is a limitation for further investigations, such as animal experiments.
LC3-II spontaneously accumulated in shATP6V0A1 lung cancer cells, especially in CaLu-1 cells (Figure 6A,B). Endogenous autophagy in CaLu-1 cells was higher than in A549 cells in terms of LC3-II expression (data not shown). As shown in Figure 6B, cleaved GFP, an autophagic flux marker, increased in CaLu-1/GFP-LC3 cells in a silencing efficiency-dependent manner. This indicates that ATP6V0A1 silencing delays trafficking between the autophagosome and lysosome. We found that GMI significantly inhibited ATP6V0A1 mRNA expression in lung cancer cells (Figure 4A). This may explain why autophagosomes do not co-localize with lysosomes in CaLu-1/GFP-LC3 cells after 1.2 µM GMI treatment (Figure 7A). To demonstrate that shATP6V0A1-enhanced autophagosome accumulation does not only occur in the GMI model, thapsigargin was used to stimulate autophagy in A549 shATP6V0A1 31 cells. Thapsigargin is thought to induce autophagy through inhibition of autophagosome–lysosome fusion (Ganley et al., 2011). The mechanism by which thapsigargin induces autophagy, triggering autophagosome formation or blocking autophagosome turnover, remains highly controversial (Decuypere et al., 2011). From our data, shATP6V0A1 significantly increased LC3-II expression induced by various concentrations of thapsigargin (data not shown). These data suggest that ATP6V0A1 plays an important role in mediating autophagosome–lysosome fusion and GMI is an efficient autophagosome accumulator, increasing the formation of and decreasing the degradation of autophagosomes in lung cancer cells.
In conclusion, we provide new insights into autophagosome accumulation-induced autophagic cell death and ATP6V0A1-mediated autophagosome–lysosome fusion. Our results strongly support the anti-cancer function of GMI through accumulation of autophagosomes. Furthermore, chloroquine is an approved drug in clinical use (Janku et al., 2011). We found that chloroquine elevates the GMI-mediated cell death. We expect more clinical drugs that block autophagosome–lysosome fusion and lysosome function, such as chloroquine, to be developed in the future.
Acknowledgments
The authors would like to thank MycoMagic Biotechnology Co., Ltd., for supplying the GMI sample. We would like to thank Dr. Tamotsu Yoshimori and Dr. Noboru Mizushima for providing the LC3 cDNA. Confocal microscopy, flow cytometry and use of microplate reader were performed at the Instrument Center of Chung Shan Medical University, which is partly supported by the National Science Council (NSC 100-2320-B-040-002-MY3) and Chung Shan Medical University Hospital (CSH-2010-D-003, CSH-2011-D-001).
Glossary
- ATP6V0A1
ATPase, H+ transporting, lysosomal V0 subunit a1
- AVOs
acidic vesicular organelles
- CA-PKB:
constitutively active PKB
- MTOR
mammalian target of rapamycin
- PKB
protein kinase B
- ShRNA
short hairpin RNA
- V-ATPases
vesicular H+-ATPases
Conflicts of interest
The authors do not have any conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 The responses of CaLu-1/GFP-LC3 cells were similar to those of parental. CaLu-1 cells after GMI and bafilomycin-A1 treatment. (A) CaLu-1/GFP-LC3 cells (5 ×103 cells per well of 96-well plate) were co-treated with GMI (0, 1.2 μM) and bafilomycin-A1 (0, 5 nM) for 48 h. Cellviability was analysed on MTT assay. The data are presented as mean± SD of triplicate experiments. (B) CaLu-1 cells (2 ×105 cells per 35 mm dish) transfected with GFP expression vector for 24 h were treated with various concentrations of GMI (0, 0.3 and 1.2 μM) and bafilomycin-A1 (0, 5 nM) for 48h. Cleaved-PARP, caspase 3, GFP and LC3 conversion were determined on immunoblotting assay.
Figure S2 Autophagosome and lysosome distribution in ATP6V0A1 silenced CaLu-1/GFP-LC3 cells. (A)CaLu-1/GFP-LC3 shLuc, shATP6V0A1 31 cells (4×105 cells per well of 60 mm plate) were treated with GMI (0, 1.2 μM) for 48 h, followed by staining with anti-LAMP2 antibody. The box shows an enlarged area. Scale bar represents 20 μm. (B) Software NIS Elements D 3.0 was used to analyse the co-localization data.
Figure S3 GMI protein sequence.
Appendix S1 Expression and purification of GMI protein.
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author of the article.
References
- Bayer MJ, Reese C, Buhler S, Peters C, Mayer A. Vacuole membrane fusion: V0 functions after trans-SNARE pairing and is coupled to the Ca2+-releasing channel. J Cell Biol. 2003;162:211–222. doi: 10.1083/jcb.200212004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boh B, Berovic M, Zhang J, Zhi-Bin L. Ganoderma lucidum and its pharmaceutically active compounds. Biotechnol Annu Rev. 2007;13:265–301. doi: 10.1016/S1387-2656(07)13010-6. [DOI] [PubMed] [Google Scholar]
- Decuypere JP, Bultynck G, Parys JB. A dual role for Ca(2+) in autophagy regulation. Cell Calcium. 2011;50:242–250. doi: 10.1016/j.ceca.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Fan QW, Cheng C, Hackett C, Feldman M, Houseman BT, Nicolaides T, et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal. 2010;3:ra81. doi: 10.1126/scisignal.2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgac M. Structure and properties of the vacuolar (H+)-ATPases. J Biol Chem. 1999;274:12951–12954. doi: 10.1074/jbc.274.19.12951. [DOI] [PubMed] [Google Scholar]
- Fu L, Kim YA, Wang X, Wu X, Yue P, Lonial S, et al. Perifosine inhibits mammalian target of rapamycin signaling through facilitating degradation of major components in the mTOR axis and induces autophagy. Cancer Res. 2009;69:8967–8976. doi: 10.1158/0008-5472.CAN-09-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley IG, Wong PM, Gammoh N, Jiang X. Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell. 2011;42:731–743. doi: 10.1016/j.molcel.2011.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Kohli L, Klocke BJ, Roth KA. Chloroquine-induced autophagic vacuole accumulation and cell death in glioma cells is p53 independent. Neuro Oncol. 2010;12:473–481. doi: 10.1093/neuonc/nop048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Hsin IL, Ou CC, Wu TC, Jan MS, Wu MF, Chiu LY, et al. GMI, an immunomodulatory protein from Ganoderma microsporum, induces autophagy in non-small cell lung cancer cells. Autophagy. 2011;7:873–882. doi: 10.4161/auto.7.8.15698. [DOI] [PubMed] [Google Scholar]
- Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8:528–539. doi: 10.1038/nrclinonc.2011.71. [DOI] [PubMed] [Google Scholar]
- Jinn TR, Wu CM, Tu WC, Ko JL, Tzen JT. Functional expression of FIP-gts, a fungal immunomodulatory protein from Ganoderma tsugae in Sf21 insect cells. Biosci Biotechnol Biochem. 2006;70:2627–2634. doi: 10.1271/bbb.60232. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008a;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy. 2008b;4:849–950. doi: 10.4161/auto.6845. [DOI] [PubMed] [Google Scholar]
- Levine B. Cell biology: autophagy and cancer. Nature. 2007;446:745–747. doi: 10.1038/446745a. [DOI] [PubMed] [Google Scholar]
- Liao CH, Hsiao YM, Hsu CP, Lin MY, Wang JC, Huang YL, et al. Transcriptionally mediated inhibition of telomerase of fungal immunomodulatory protein from Ganoderma tsugae in A549 human lung adenocarcinoma cell line. Mol Carcinog. 2006;45:220–229. doi: 10.1002/mc.20161. [DOI] [PubMed] [Google Scholar]
- Liao CH, Hsiao YM, Sheu GT, Chang JT, Wang PH, Wu MF, et al. Nuclear translocation of telomerase reverse transcriptase and calcium signaling in repression of telomerase activity in human lung cancer cells by fungal immunomodulatory protein from Ganoderma tsugae. Biochem Pharmacol. 2007;74:1541–1554. doi: 10.1016/j.bcp.2007.07.025. [DOI] [PubMed] [Google Scholar]
- Liao CH, Hsiao YM, Lin CH, Yeh CS, Wang JC, Ni CH, et al. Induction of premature senescence in human lung cancer by fungal immunomodulatory protein from Ganoderma tsugae. Food Chem Toxicol. 2008;46:1851–1859. doi: 10.1016/j.fct.2008.01.044. [DOI] [PubMed] [Google Scholar]
- Lin CH, Hsiao YM, Ou CC, Lin YW, Chiu YL, Lue KH, et al. GMI, a Ganoderma immunomodulatory protein, down-regulates tumor necrosis factor alpha-induced expression of matrix metalloproteinase 9 via NF-kappaB pathway in human alveolar epithelial A549 cells. J Agric Food Chem. 2010a;58:12014–12021. doi: 10.1021/jf103068w. [DOI] [PubMed] [Google Scholar]
- Lin CH, Sheu GT, Lin YW, Yeh CS, Huang YH, Lai YC, et al. A new immunomodulatory protein from Ganoderma microsporum inhibits epidermal growth factor mediated migration and invasion in A549 lung cancer cells. Process Biochem. 2010b;45:1537–1542. [Google Scholar]
- Ling YH, Aracil M, Zou Y, Yuan Z, Lu B, Jimeno J, et al. PM02734 (elisidepsin) induces caspase-independent cell death associated with features of autophagy, inhibition of the Akt/mTOR signaling pathway, and activation of death-associated protein kinase. Clin Cancer Res. 2011;17:5353–5366. doi: 10.1158/1078-0432.CCR-10-1948. [DOI] [PubMed] [Google Scholar]
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paroutis P, Touret N, Grinstein S. The pH of the secretory pathway: measurement, determinants, and regulation. Physiology (Bethesda) 2004;19:207–215. doi: 10.1152/physiol.00005.2004. [DOI] [PubMed] [Google Scholar]
- Pena-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, Wolff NC, Tran TA, Zou L, et al. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011;30:3242–3258. doi: 10.1038/emboj.2011.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri F, Nusslein-Volhard C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. 2008;133:916–927. doi: 10.1016/j.cell.2008.04.037. [DOI] [PubMed] [Google Scholar]
- Ramser B, Kokot A, Metze D, Weiss N, Luger TA, Bohm M. Hydroxychloroquine modulates metabolic activity and proliferation and induces autophagic cell death of human dermal fibroblasts. J Invest Dermatol. 2009;129:2419–2426. doi: 10.1038/jid.2009.80. [DOI] [PubMed] [Google Scholar]
- Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis. 2011;32:955–963. doi: 10.1093/carcin/bgr031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- Scott CC, Bissig C, Gruenberg J. Duelling functions of the V-ATPase. EMBO J. 2011;30:4113–4115. doi: 10.1038/emboj.2011.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HM, Codogno P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy. 2011;7:457–465. doi: 10.4161/auto.7.5.14226. [DOI] [PubMed] [Google Scholar]
- Shvets E, Fass E, Elazar Z. Utilizing flow cytometry to monitor autophagy in living mammalian cells. Autophagy. 2008;4:621–628. doi: 10.4161/auto.5939. [DOI] [PubMed] [Google Scholar]
- Smith DM, Patel S, Raffoul F, Haller E, Mills GB, Nanjundan M. Arsenic trioxide induces a beclin-1-independent autophagic pathway via modulation of SnoN/SkiL expression in ovarian carcinoma cells. Cell Death Differ. 2010;17:1867–1881. doi: 10.1038/cdd.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser B, Iwaszkiewicz J, Michielin O, Mayer A. The V-ATPase proteolipid cylinder promotes the lipid-mixing stage of SNARE-dependent fusion of yeast vacuoles. EMBO J. 2011;30:4126–4141. doi: 10.1038/emboj.2011.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- Yang PM, Liu YL, Lin YC, Shun CT, Wu MS, Chen CC. Inhibition of autophagy enhances anticancer effects of atorvastatin in digestive malignancies. Cancer Res. 2010;70:7699–7709. doi: 10.1158/0008-5472.CAN-10-1626. [DOI] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.