

Abstract
Objectives
Dysregulated glycogen synthase kinase-3 (GSK3) may contribute to the pathophysiology of mood disorders and other diseases, and appears to be a target of certain therapeutic drugs. The growing recognition of heightened vulnerability during development to many psychiatric diseases, including mood disorders, led us to test if there are developmental changes in mouse brain GSK3 and its regulation by phosphorylation and by therapeutic drugs.
Methods
GSK3 levels and phosphorylation were measured at seven ages of development in mouse cerebral cortex and hippocampus.
Results
Two periods of rapid transitions in GSK3 levels were identified, a large rise between postnatal day 1 and two to three weeks of age, where GSK3 levels were as high as four-fold adult mouse brain levels, and a rapid decline between two to four and eight weeks of age, when adult levels were reached. Inhibitory serine-phosphorylation of GSK3, particularly GSK3β, was extremely high in one-day postnatal mouse brain, and rapidly declined thereafter. These developmental changes in GSK3 were equivalent in male and female cerebral cortex, and differed from other signaling kinases, including Akt, ERK1/2, JNK, and p38 levels and phosphorylation. In contrast to adult mouse brain, where administration of lithium or fluoxetine rapidly and robustly increased serine-phosphorylation of GSK3, in young mice these responses were blunted or absent.
Conclusions
High brain levels of GSK3 and large fluctuations in its levels and phosphorylation in juvenile and adolescent mouse brain raise the possibility that they may contribute to destabilized mood regulation induced by environmental and genetic factors.
Keywords: brain development, fluoxetine, glycogen synthase kinase-3, lithium, mood disorders
In a landmark study, Kessler and colleagues (1) found that 50% of all lifetime cases of mental disorders begin by the age of 14 years, and 75% by the age of 24 years. This prevalent onset in young people is reflected in the recent large increase in diagnosis and prescriptions of psychotropic medications given to juveniles and adolescents (2, 3). These medications particularly include antidepressants that increase serotonergic signaling, and stimulants that increase dopaminergic signaling. The National Institute of Mental Health (NIMH) has concluded that interventions targeting young, high-risk populations may have remarkable benefits in reducing the risk for psychopathology over the lifespan, which currently afflicts approximately 25% of the population of the United States. However, it is unclear what predisposes young people during development to many major psychiatric diseases, such as mood disorders, schizophrenia, and attention-deficit hyperactivity disorder, and if administered psychotropic medications during development have equivalent effects as in adults (4).
This study was designed to examine glycogen synthase kinase-3 (GSK3) in juvenile, adolescent, and adult mice as a possible risk factor for increased vulnerability to psychiatric diseases during development and as a possible contributor to the potential for differential medication responses in young people and adults. GSK3, comprising GSK3α and GSK3β isoforms, is an important modulator of many cellular functions, such as regulating gene expression, neural plasticity, cellular architecture, and cell survival (5-8). Unlike most protein kinases, GSK3 is constitutively partially active, and most signaling pathways converging on GSK3 decrease its activity. Phosphorylation is a major mechanism regulating GSK3 activity, whereas the expression levels of GSK3 are generally stable. The activity of GSK3 is optimal when phosphorylated on the regulatory tyrosine-279-GSK3α and tyrosine-216-GSK3β, but mechanisms regulating GSK3 tyrosine-phosphorylation remain unclear. In contrast, GSK3 is inhibited by serine-phosphorylation on serine-9 of GSK3β and serine-21 of GSK3α. Many signaling pathways regulate GSK3 activity by this modification, which can be catalyzed by several kinases, particularly Akt/protein kinase B (9, 10).
GSK3 was first linked to mood disorders by the discovery that the mood stabilizer lithium selectively inhibits GSK3 by directly binding the enzyme (11, 12). This direct inhibition by lithium appears to be amplified in vivo at therapeutic lithium levels by an indirect mechanism that increases the inhibitory serine-phosphorylation of GSK3 (13, 14). Antidepressants and antipsychotics also increase inhibitory serine-phosphorylation of GSK3 in vivo (15-20). There is no evidence addressing whether these regulatory effects of therapeutic drugs on GSK3 differ during development from adults. In mice, inhibitors of GSK3 modify several behaviors that may model mood, such as reducing immobility in the forced swim test (indicative of antidepressant-like effects) and reducing amphetamine-induced hyperactivity (a model of manic-like behavior) (21-25). These and many additional findings in animals and humans support the hypothesis that dysregulated GSK3 contributes to susceptibility to mood disorders (26-28).
Little is known about GSK3 during development. Leroy and Brion (29) reported that in whole rat brain the level of GSK3β from birth to 10 days of age was 50–60% higher than in adults. Since GSK3 may contribute to the onset of mood disorders and other psychiatric diseases that commonly occur in young people, we considered that if GSK3 is elevated at susceptible stages of development it may contribute to heightened developmental susceptibility to psychiatric diseases. Therefore, we analyzed developmental changes in the levels of both isoforms of GSK3 in two brain regions of mice, its regulatory phosphorylation, and the regulation of GSK3 by lithium and fluoxetine administered in vivo during development.
Materials and methods
Mice
C57Bl/6 mice (Frederick Cancer Research, Frederick, MD, USA) were housed in a light- and temperature-controlled mouse room. Mice were housed and treated in accordance with National Institutes of Health and the University of Miami and University of Alabama at Birmingham Institutional Animal Care and Use Committee guidelines. Where indicated, mice were injected intraperitoneally (i.p.) with 4 mg/kg lithium chloride (Sigma Chemicals) or 20 mg/kg fluoxetine (from the NIMH Chemical Synthesis and Drug Supply Program).
Mouse cerebral cortex and hippocampus were rapidly dissected in ice-cold phosphate-buffered saline. Brain regions were homogenized in ice-cold lysis buffer containing 20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 10% glycerol, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml pepstatin, 1 mM phenylmethanesulfonyl fluoride, 50 mM NaF, 1 mM sodium orthovanadate, and 100 nM okadaic acid. The lysates were centrifuged at 20,800 × g for 10 min. Protein concentrations in the supernatants were determined using the Bradford protein assay (30).
Immunoblotting
Samples were mixed with Laemmli sample buffer (2% SDS) and placed in a boiling water bath for five min. Proteins (5–20 μg protein) were resolved in 10% SDS-polyacrylamide gels, and transferred to nitrocellulose. Blots were probed with antibodies to phospho-Ser9-GSK3β, phospho-Ser21-GSK3α, phospho-Ser473-Akt, phosphor-Thr308-Akt, total Akt, total extracellular-regulated kinases (ERK) 1/2, phospho-Thr202/Tyr204-ERK1/2, total c-Jun N-terminal kinases (JNKs), phospho-Thr183/Tyr185-JNK, phospho-Thr180/Tyr182-p38, total p38 (Cell Signaling Technology, Beverly, MA, USA), phospho-Tyr279-GSK3α, phospho-Tyr216-GSK3β, total GSK3α (Millipore), or total GSK3β (BD Transduction Labs). Immunoblots were developed using horseradish peroxidase-conjugated goat anti-mouse, or goat anti-rabbit IgG, followed by detection with enhanced chemiluminescence. Protein bands were quantitated with a densitometer and results expressed relative to a standard sample obtained from five eight-week-old mice which was run on every GSK3 immunoblot.
Results
Brain region levels of GSK3 and other kinases during development
Developmental changes in GSK3 were analyzed in the cerebral cortex and hippocampus of C57Bl/6 mice at seven ages: postnatal day 1, juveniles at two weeks and three weeks of age, early adolescent at four weeks of age, middle adolescent at six weeks of age, late adolescent at eight weeks of age, and adult at 16 weeks of age. Examination of the total levels of each isoform of GSK3 revealed generally higher levels at postnatal day 1 than in adults (Fig. 1A). Subsequently, except for GSK3α in the hippocampus, there were large increases in GSK3 levels between postnatal day 1 and juvenile ages. In the cerebral cortex maximal levels of GSK3α and GSK3β occurred at two and three weeks of age that were > 3.5-fold higher than adult levels. This was followed by diminishing levels during adolescence that were still ~two-fold higher than adult levels, which were reached by eight weeks of age. These developmental changes are similar to those reported by Leroy and Brion (29) for GSK3β in rat whole brain. Comparison of males and females at three and six weeks of age showed no significant sex-dependent differences in the levels of either isoform of GSK3 in the cerebral cortex (Fig. 1B).
Fig 1.
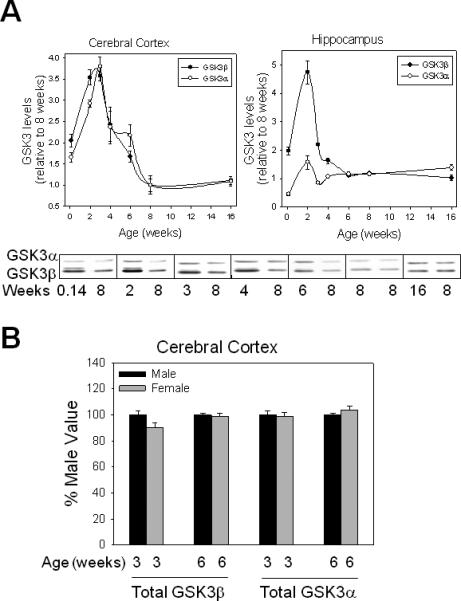
Developmental changes in total levels of GSK3α and GSK3β in mouse brain. (A) Representative immunoblots and quantitation of total GSK3α and GSK3β in mouse cerebral cortex and hippocampus obtained at postnatal day 1, juveniles at two and three weeks of age, early adolescent at four weeks of age, middle adolescent at six weeks of age, late adolescent at eight weeks of age, and adult at 16 weeks of age. Values shown are relative to values from eight-week-old mice. Means ± standard error (SEM) (n = 4–5). (B) Comparison of male and female levels of GSK3 isoforms in mouse cerebral cortex. Tissue was obtained from male and female mice at three and six weeks of age. Values from female mice are shown relative to those from male mice of the same age. Means ± SEM (n = 4). GSK3 = glycogen synthase kinase-3.
These large biphasic changes in the levels of GSK3 during early development led us to examine other intracellular signaling kinases to determine if this pattern of expression of GSK3 was common or distinct. Kinases examined include ERK1/2, JNK, p38, and Akt. ERK1/2 in the cerebral cortex, JNK in the cerebral cortex and hippocampus, and p38 in the cerebral cortex displayed increases between postnatal day 1 and juvenile age (Fig. 2). In contrast to the other kinases, the level of Akt was high at postnatal day 1 and it remained high through juvenile ages. Like GSK3, levels of JNK and Akt diminished between juvenile and adult ages, whereas ERK1/2 and p38 levels remained relatively stable. Collectively, the levels of each of these intracellular signaling kinases displayed different changes during development, and the developmental changes for each were similar in the cerebral cortex and the hippocampus with the notable exception of the initial levels of ERK1/2.
Fig 2.
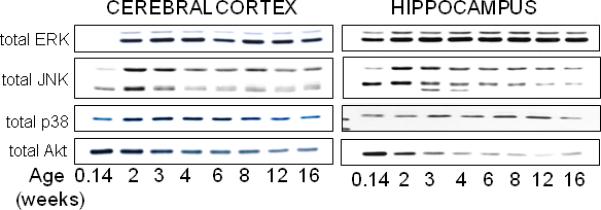
Developmental changes in total levels of extracellular-regulated kinases (ERK1/2), c-Jun N-terminal kinases (JNK), p38, and Akt. Immunoblots of total levels of ERK1/2, JNK, p38, and Akt in mouse cerebral cortex and hippocampus obtained at postnatal day 1, juveniles at two and three weeks of age, early adolescent at four weeks of age, middle adolescent at six weeks of age, late adolescent at eight weeks of age, and adult at 12 and 16 weeks of age. Immunoblots are representative of two independent experiments.
Brain region phosphorylation of GSK3 and other kinases during development
We also examined the phosphorylation status of GSK3 during development. The extent of serine-phosphorylation of GSK3, especially GSK3β, was remarkably high on postnatal day 1 (Fig. 3A). This was followed by a drastic decline in GSK3 phosphorylation during juvenile ages. Phospho-Ser9-GSK3β levels remained higher in juvenile-aged mice than in adults, followed by a gradual decline until adult levels were attained by eight weeks of age. The developmental pattern of tyrosine-phosphorylation of GSK3 was very different from its serine-phosphorylation (Fig. 3B). Tyrosine-phosphorylation levels of GSK3β in the cerebral cortex and of GSK3α in the hippocampus remained relatively constant with age. In contrast, hippocampal tyrosine-phosphorylation of GSK3β and cortical tyrosine-phosphorylation of GSK3α were low at postnatal day 1 and through early development, followed by an increase to adult phosphorylation levels. Comparison of males and females at three and six weeks of age showed no significant sex-dependent differences in the phosphorylation status of either isoform of GSK3 in the cerebral cortex (Fig. 4).
Fig 3.
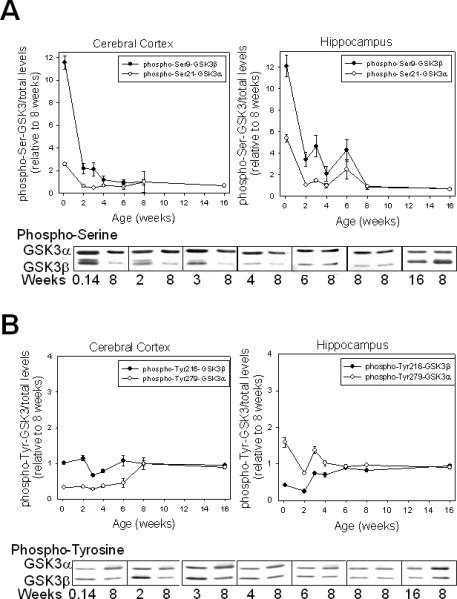
Developmental changes in serine-phosphorylation and tyrosine-phosphorylation of GSK3α and GSK3β in mouse brain. (A) Representative immunoblots and quantitation of phospho-Ser21-GSK3α and phospho-Ser9-GSK3β in mouse cerebral cortex and hippocampus obtained from seven ages of mice. Values shown are ratios of phospho-serine to total GSK3 and are relative to values from eight-week-old mice. Means ± standard error (SEM) (n = 4–5). (B) Representative immunoblots and quantitation of phospho-Tyr279-GSK3α and phospho-Tyr216-GSK3β in mouse cerebral cortex and hippocampus obtained from seven ages of mice. Values shown are ratios of phospho-tyrosine to total GSK3 and are relative to values from eight-week-old mice. Means ± SEM (n = 4–5). GSK3 = glycogen synthase kinase-3.
Fig 4.
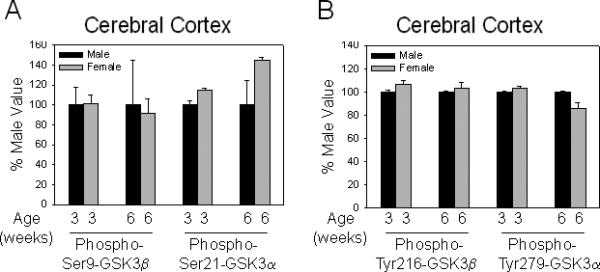
Comparison of phosphorylation of GSK3 isoforms in male and female mice. (A) Comparison of male and female mouse serine-phosphorylation of GSK3 isoforms in the cerebral cortex at three and six weeks of age. Means ± standard error (SEM) (n = 4). (B) Comparison of male and female mouse tyrosine-phosphorylation of GSK3 isoforms in the cerebral cortex at three and six weeks of age. Values from female mice are shown relative to those from male mice of the same age. Means ± SEM (n = 4). GSK3 = glycogen synthase kinase-3.
Developmental changes in the phosphorylation of other signaling kinases were measured to compare with GSK3. In both the cerebral cortex and hippocampus the phosphorylation of ERK1/2 and JNK were low at postnatal day 1 and increased to adult levels by two weeks of age (Fig. 5). A low mobility, highly phosphorylated form of p38 was detected in the cerebral cortex at postnatal day 1, and after diminishing early, the phosphorylation of p38 gradually increased after three to four weeks of age. The phosphorylation of Akt on both Ser-473 and Thr-308 was high at postnatal day 1 and juvenile ages, followed by a decline to adult levels.
Fig 5.
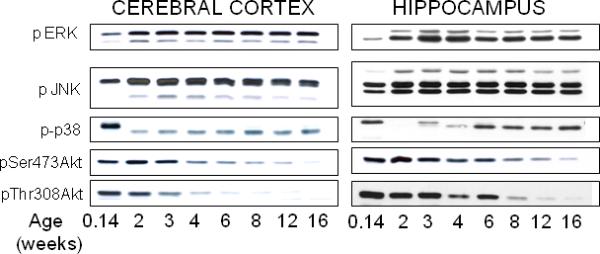
Developmental changes in phosphorylation of extracellular-regulated kinases (ERK1/2), c-Jun N-terminal kinases (JNK), p38, and Akt. Representative immunoblots of phosphorylated ERK1/2, JNK, p38, and Akt in mouse cerebral cortex and hippocampus obtained at postnatal day 1, juveniles at two and three weeks of age, early adolescent at four weeks of age, middle adolescent at six weeks of age, late adolescent at eight weeks of age, and adult at 12 and 16 weeks of age. Immunoblots are representative of two independent experiments.
Phosphorylation of GSK3 induced by lithium or fluoxetine
Lithium is the prototypical mood stabilizer used to treat bipolar disorder (26, 31). Lithium is also an inhibitor of GSK3 (11, 12) and part of its therapeutic effect as a mood stabilizer may result from its stimulation of increased inhibitory serine-phosphorylation of GSK3 (13). Although the mechanisms by which lithium increases phospho-Ser-GSK3 remain unresolved (14), since they may contribute to its therapeutic actions, we examined if lithium administration was capable of increasing phospho-Ser-GSK3 similarly in young mice as in adults. For this purpose, we measured the time-dependent increases in phospho-Ser-GSK3 in cerebral cortex and hippocampus following administration of an acute dose of lithium to mice of three ages; three, five, and 12 weeks, ages that express maximal, intermediate, and adult levels of GSK3. In adults, acute lithium administration caused a rapid, transient increase in the serine-phosphorylation of both GSK3 isoforms in both brain regions (Fig. 6). The lithium-induced increase in serine-phosphorylation of both GSK3 isoforms was blunted in three- and five-week-old mice in both the cerebral cortex and hippocampus.
Fig 6.
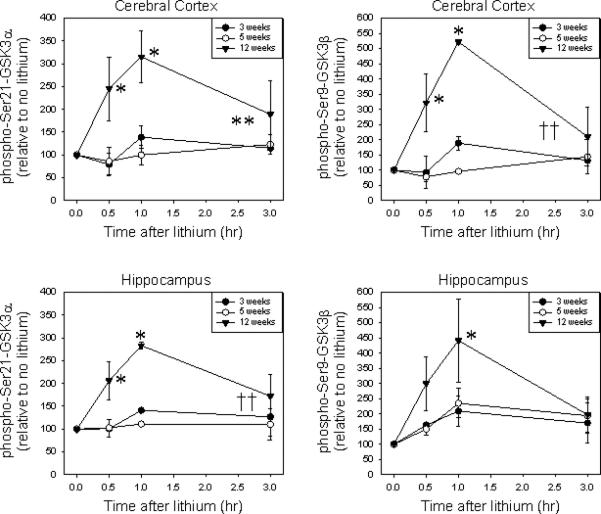
Effects of lithium administration on serine-phosphorylation of GSK3 in mouse brain. Serine-phosphorylation and total levels of GSK3 were measured in three-week-old, five-week-old, and 12-week-old mouse brain regions 0.5, 1.5, and 3.0 hours after administration of 4 mg/kg lithium chloride. Values are presented as percent of untreated mice of the same age. Means ± standard error (SEM) (n = 3) per time point for each age. GSK3 = glycogen synthase kinase-3.
*p < 0.05 compared with lithium-free controls of the same age (two-way ANOVA, Bonferroni post-test).
**p < 0.05.
††p < 0.07 response to lithium in adult mice compared with young mice (one-way ANOVA).
Fluoxetine is a widely used antidepressant that increases serine-phosphorylation of GSK3 in mouse brain in vivo, presumably by increasing serotonergic activity, which may contribute to its therapeutic actions (15). Therefore, we tested if there are differences in the capacity of fluoxetine to increase serine-phosphorylation of GSK3 in the brains of three-, five-, and twelve-week-old mice. As observed with lithium, fluoxetine-induced increases in serine-phosphorylation of GSK3 were blunted in the brains of three- and five-week-old mice compared with adult mice (Fig. 7).
Fig 7.
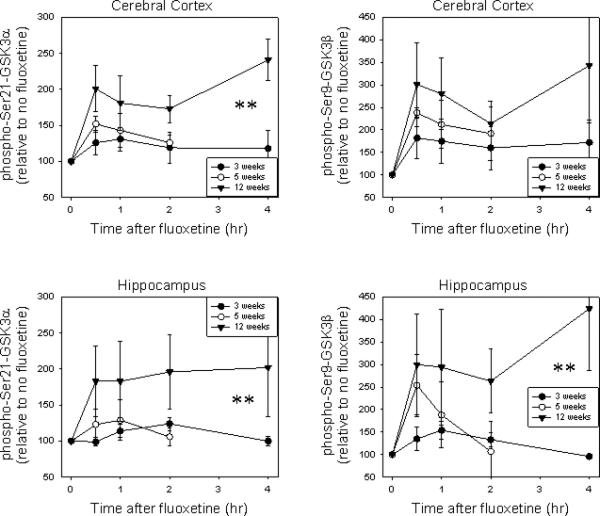
Effects of fluoxetine administration on serine-phosphorylation of GSK3 in mouse brain. Serine-phosphorylation and total levels of GSK3 were measured in three-week-old, five-week-old, and 12-week-old mouse brain regions 0.5, one, two, and four hours after administration of 20 mg/kg fluoxetine. Values are presented as percent of untreated mice of the same age. Means ± standard error (SEM) (n = 3). GSK3 = glycogen synthase kinase-3.
**p < 0.05 response to fluoxetine in adult mice compared with young mice (one-way ANOVA).
Discussion
The causes of the heightened susceptibility of young people to several prevalent psychiatric diseases, such as mood disorders, remain largely unknown (4). This problem is of increasing importance because of the rapidly growing number of young people diagnosed with psychiatric disorders and the associated rising numbers of psychotropic medications prescribed for them (2, 3). This study examined GSK3 in developing mouse brain to address the possibility that developmental changes in GSK3 may be linked to heightened susceptibility to psychiatric diseases in the young. This hypothesis was based on much evidence from pre-clinical, clinical, and genetic studies that dysregulated GSK3 likely increases susceptibility to mood disorders as well as other psychiatric diseases (26-28), and that GSK3 is inhibited by mood stabilizers, antidepressants, and atypical antipsychotics (11, 12, 15-20). The results show that the brain levels and regulation of GSK3 undergo large fluctuations during development and that lithium- and fluoxetine-induced inhibitory serine-phosphorylation of GSK3 are blunted in young compared with adult mouse brain.
GSK3 levels were generally higher in the brains of young mice than adult mice until approximately six to eight weeks of age. One day after birth, the levels of GSK3α and GSK3β were ~1.8-fold higher than adult levels in the cerebral cortex, and hippocampal GSK3β, but not GSK3α, was two-fold adult levels. With the exception of hippocampal GSK3α, the brain region levels of GSK3 subsequently increased, reaching peak levels in juveniles two to three weeks after birth which were nearly four-fold higher than adult levels. These results extend the report by Leroy and Brion (29) that GSK3β levels are higher in whole brain of young mice than adults.
High brain levels of GSK3 in young mice raise the possibility that mood regulation during development may be more sensitive to disruption than adult brain in response to impairments in the inhibitory control of GSK3. For example, serotonin and brain-derived neurotrophic factor (BDNF) normally contribute to maintaining inhibitory control of GSK3 in the brain (15, 32), and each may be deficient in depression (33, 34). Disruptions in the inhibition of GSK3 due to depression-associated deficits in BDNF or serotonin may contribute to dysregulated GSK3 and susceptibility to mood disorders (28), which may be accentuated in developing brains. Thus, a moderate 25% increase in GSK3 activity in adults due to these deficiencies would be four-fold greater in young mice that express four times greater brain levels of GSK3. Previous studies have indicated that elevated GSK3 is not alone sufficient to cause depression, but heightens vulnerability to stress-induced depression-like behavior and is a major target of lithium's effects in mice (25, 35), These findings raise the possibility that high GSK3 levels during development may contribute to the heightened susceptibility of young people to mood disorders and other psychiatric diseases that involve dysregulated GSK3.
The regulatory serine and tyrosine phosphorylation of GSK3 were also examined in mouse brain regions during development. The inhibitory serine-phosphorylation of GSK3, particularly GSK3β, was remarkably high in one day postnatal mouse brain. This modification inhibits GSK3 in all known signaling pathways except for the Wnt pathway that regulates β-catenin, which functions independently of GSK3 serine-phosphorylation (6, 36). Thus, the high serine-phosphorylation of GSK3 at one day postnatal age may serve to allow normal Wnt signaling, which is critical for brain development (36, 37), while inhibiting other actions of GSK3. The signal causing the early high serine-phosphorylation of GSK3 remains unidentified. Although Akt, a common mediator of GSK3 serine-phosphorylation, exhibited high levels in young mouse brain and was highly phosphorylated on its activating residues at one day postnatal compared with adult brain, it remained highly active at two and three weeks of age; ages when the serine-phosphorylation of GSK3 fell precipitously. In comparison, the signaling kinases ERK1/2, p38, and JNK displayed different developmental patterns of change in levels and phosphorylation. Compared with GSK3 serine-phosphorylation, the tyrosine phosphorylation of GSK3 was relatively stable during development, although it was notably low for one isoform of GSK3 in each brain region examined in young mice. Differences between GSK3α and GSK3β in both serine- and tyrosine-phosphorylation during development highlight growing evidence for differences in the regulation and functions of these two isoforms of GSK3 (6).
Since increasing numbers of young people are being treated with mood stabilizers and antidepressants, it is important to determine if the actions of these drugs differ with age. Both lithium and fluoxetine cause large increases in the inhibitory serine-phosphorylation of GSK3 in adult mouse brain (13, 15), actions that may contribute to their therapeutic actions as a mood stabilizer and antidepressant, respectively. However, as opposed to adult mice, administration of each of these drugs to young mice had very blunted effects on the serine-phosphorylation of GSK3. Although examining specific ages of mice necessitated studying acute responses—rather than the effects of chronic treatments that are required for therapeutic effects— and these findings suggest that the signaling mechanisms mediating increased serine-phosphorylation of GSK3 induced by lithium and fluoxetine differ in young and adult mouse brain.
GSK3 regulates many crucial neuronal processes, acting as an integrator of several major signaling pathways linked to neurotransmitter receptors, hormones, and growth factors (6, 10). Impairments in these signaling systems that converge on GSK3, such as deficient serotonergic activity, reduce the inhibitory control of GSK3. In the young, the high levels of GSK3 and the age-related large fluctuations in levels and regulatory phosphorylation of GSK3 may create an unstable condition that is especially susceptible to imbalance by environmental and genetic influences. Furthermore, the diminished capacities of lithium and fluoxetine to increase the inhibitory serine-phosphorylation of GSK3 raise the possibility that in the young mechanisms controlling GSK3 and pharmacological control of GSK3 may differ from adults.
Acknowledgements
We thank Anna Zmijewska for excellent technical assistance and the National Institute of Mental Health (NIMH) Chemical Synthesis and Drug Supply Program for fluoxetine. This research was supported by grants from the NIMH (MH038752, MH090236, MH092970).
Footnotes
Disclosure
The authors of this paper do not have any commercial associations that might pose a conflict of interest in connection with this manuscript.
References
- 1.Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- 2.Moreno C, Laje G, Blanco C, Jiang H, Schmidt AB, Olfson M. National trends in the outpatient diagnosis and treatment of bipolar disorder in youth. Arch Gen Psychiatry. 2007;64:1032–1039. doi: 10.1001/archpsyc.64.9.1032. [DOI] [PubMed] [Google Scholar]
- 3.Rohde P, Silva SG, Tonev ST, et al. Achievement and maintenance of sustained response during the Treatment for Adolescents With Depression Study continuation and maintenance therapy. Arch Gen Psychiatry. 2008;65:447–455. doi: 10.1001/archpsyc.65.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andersen SL, Teicher MH. Stress, sensitive periods and maturational events in adolescent depression. Trends Neurosci. 2008;31:183–191. doi: 10.1016/j.tins.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Grimes CA, Jope RS. The multi-faceted roles of glycogen synthase kinase-3β in cellular signaling. Prog Neurobiology. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 6.Kaidanovich-Beilin O, Woodgett JR. GSK-3: functional insights from cell biology and animal models. Frontiers Mol Neurosci. 2011;4:40–49. doi: 10.3389/fnmol.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chuang DM, Wang Z, Chiu CT. GSK-3 as a target for lithium-induced neuroprotection against excitotoxicity in neuronal cultures and animal models of ischemic stroke. Front Mol Neurosci. 2011;4:15. doi: 10.3389/fnmol.2011.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Progress Neurobiol. 2006;79:173–189. doi: 10.1016/j.pneurobio.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woodgett JR. Physiological roles of glycogen synthase kinase-3: potential as a therapeutic target for diabetes and other disorders. Curr Drug Targets Immune Endocr Metabol Disord. 2003;3:281–290. doi: 10.2174/1568008033340153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jope RS, Johnson GVW. The glamour and gloom of glycogen synthase kinase-3 (GSK3). Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 11.Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- 13.De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3β phosphorylation by sodium valproate and lithium. Neuropharmacol. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- 14.Jope RS. Lithium and GSK3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol Sci. 2003;24:441–443. doi: 10.1016/S0165-6147(03)00206-2. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Zhu W, Roh M-S, Friedman AB, Rosborough, Jope RS. In vivo regulation of glycogen synthase kinase-3β (GSK3β) by serotonergic activity in mouse brain. Neuropsychopharmacology. 2004;29:1426–1431. doi: 10.1038/sj.npp.1300439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Rosborough K, Friedman AB, Zhu W, Roth KA. Regulation of mouse brain glycogen synthase kinase-3 by atypical antipsychotics. Int J Neuropsychopharmacology. 2007;10:7–19. doi: 10.1017/S1461145706006547. [DOI] [PubMed] [Google Scholar]
- 17.Alimohamad H, Rajakumar N, Seah YH, Rushlow W. Antipsychotics alter the protein expression levels of beta-catenin and GSK-3 in the rat medial prefrontal cortex and striatum. Biol Psychiatry. 2005;57:533–542. doi: 10.1016/j.biopsych.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 18.Roh MS, Seo MS, Kim Y, et al. Haloperidol and clozapine differentially regulate signals upstream of glycogen synthase kinase 3 in the rat frontal cortex. Exp Mol Med. 2005;39:353–360. doi: 10.1038/emm.2007.39. [DOI] [PubMed] [Google Scholar]
- 19.Okamoto H, Voleti B, Banasr M, et al. Wnt2 expression and signaling is increased by different classes of antidepressant treatments. Biol Psychiatry. 2010;68:521–527. doi: 10.1016/j.biopsych.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beaulieu JM, Zhang X, Rodriguiz RM, et al. Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc Natl Acad Sci USA. 2008;105:1333–1338. doi: 10.1073/pnas.0711496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beaulieu JM, Sotnikova TD, Yao WD, et al. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci USA. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gould TD, Einat H, Bhat R, Manji HK. AR-A014418, a selective GSK-3 inhibitor, produces antidepressant-like effects in the forced swim test. Int J Neuropsychopharmacology. 2004;7:387–390. doi: 10.1017/S1461145704004535. [DOI] [PubMed] [Google Scholar]
- 23.Kaidanovich-Beilin O, Milman A, Weizman A, Pick CG, Eldar-Finkelman H. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on β-catenin in mouse hippocampus. Biol Psychiatry. 2004;55:781–784. doi: 10.1016/j.biopsych.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 24.O'Brien WT, Harper AD, Jove F, et al. Glycogen synthase kinase-3β haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci. 2004;24:6791–6798. doi: 10.1523/JNEUROSCI.4753-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Brien WT, Huang J, Buccafusca R, et al. Glycogen synthase kinase-3 is essential for β-arrestin-2 complex formation and lithium-sensitive behaviors in mice. J Clin Invest. 2011;121:3756–3762. doi: 10.1172/JCI45194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phiel CJ, Klein PS. Molecular targets of lithium action. Annu Rev Pharmacol Toxicol. 2001;41:789–813. doi: 10.1146/annurev.pharmtox.41.1.789. [DOI] [PubMed] [Google Scholar]
- 27.Gould TD, Picchini AM, Einat H, Manji HK. Targeting glycogen synthase kinase-3 in the CNS: implications for the development of new treatments for mood disorders. Curr Drug Targets. 2006;7:1399–1409. doi: 10.2174/1389450110607011399. [DOI] [PubMed] [Google Scholar]
- 28.Jope RS. Glycogen synthase kinase-3 in the etiology and treatment of mood disorders. Frontiers Mol Neurosci. 2011;4:16–26. doi: 10.3389/fnmol.2011.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leroy K, Brion JP. Developmental expression and localization of glycogen synthase kinase-3β in rat brain. J Chem Neuroanatomy. 1999;16:279–293. doi: 10.1016/s0891-0618(99)00012-5. [DOI] [PubMed] [Google Scholar]
- 30.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 31.Jope RS. Anti-bipolar therapy: mechanism of action of lithium. Mol Psychiatry. 1999;4:117–128. doi: 10.1038/sj.mp.4000494. [DOI] [PubMed] [Google Scholar]
- 32.Mai L, Jope RS, Li X. BDNF-mediated signal transduction is modulated by GSK3β and mood stabilizing agents. J Neurochem. 2002;82:75–83. doi: 10.1046/j.1471-4159.2002.00939.x. [DOI] [PubMed] [Google Scholar]
- 33.Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- 34.Pittenger C, Duman RS. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology. 2008;33:88–109. doi: 10.1038/sj.npp.1301574. [DOI] [PubMed] [Google Scholar]
- 35.Polter A, Beurel E, Yang S, et al. Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology. 2010;35:1761–1774. doi: 10.1038/npp.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McNeill H, Woodgett JR. When pathways collide: collaboration and connivance among signalling proteins in development. Nat Rev Mol Cell Biol. 2010;11:404–413. doi: 10.1038/nrm2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yanfeng W, Saint-Jeannet JP, Klein PS. Wnt-frizzled signaling in the induction and differentiation of the neural crest. Bioessays. 2003;25:317–325. doi: 10.1002/bies.10255. [DOI] [PubMed] [Google Scholar]