

Abstract
Mitochondrial dysfunction is commonly associated with neurodegeneration in the aging brain. In addition, the importance of mitochondrial function during brain development is illustrated by the neurological deficits observed in infants with mitochondrial complex deficiencies. However, the extent to which abnormalities in mitochondrial function might impact neurogenesis during brain development is not well understood. Previously, we demonstrated that adult harlequin (Hq) mutant mice, which have an 80% reduction in the mitochondrial protein apoptosis-inducing factor (AIF), exhibited signs of oxidative stress and progressive loss of adult cerebellar and retinal neurons. To assess whether in addition to its role in postmitotic neuron survival Aif is also necessary for cerebellar development, we analyzed embryos in which Aif was deleted in the prospective midbrain and cerebellum at a very early stage of development using an En1 (engrailed 1) promoter-driven cre recombinase gene. These mutant mice, which died at birth, had midbrain defects and dramatic deficits in cerebellar Purkinje and granule cell precursors. Additional analysis revealed that Aif-null Purkinje cell precursors prematurely entered S-phase, but most failed to undergo mitosis and ultimately died via apoptosis. In contrast, proliferation of mutant granule cell precursors was blocked before S-phase. Mice in which Aif was deleted later in embryogenesis using a nestin promoter-driven cre gene survive for several days after birth, and postnatal granule cell precursors in these mice also failed to enter S-phase. Our results indicate that the loss of Aif results in cell cycle abnormalities in a neuron-specific manner during cerebellar development.
Keywords: conditional knock-out, Purkinje cell precursor, cell cycle arrest, G1-phase, granule cell precursor, S-phase, apoptosis, cerebellum
Introduction
Mitochondrial dysfunction has been suggested to be a common denominator in many neuronal degenerative disorders, acute neuronal injury, and aging in general (Ames, 2004; Barja, 2004; Chan, 2004; Lin and Beal, 2006; McFarland et al., 2007). Although the immature brain is also highly energy dependent and susceptible to excessive reactive oxygen species (ROS), less is known about the in vivo effects of mitochondrial dysfunction on brain development, in particular on neurogenesis (Blomgren and Hagberg, 2006).
Previously, we reported a novel genetic mouse model of mitochondrial dysfunction (Klein et al., 2002). Harlequin (Hq) mice have a proviral insertion in the X-linked apoptosis-inducing factor gene (Aif; also known as Aifm1 and Pcdc8) that results in an 80–90% reduction in AIF expression. AIF is a ubiquitously expressed 57 kDa flavoprotein with similarity in the large C-terminal domain to several bacterial nicotinamide adenine dinucleotide phosphate (NADH)-dependent ferrodoxin oxidoreductases (Susin et al., 1999; Miramar et al., 2001). Normally, AIF is localized in the mitochondrial intermembrane space; however, it translocates to the cytoplasm and nucleus under conditions that induce apoptosis (Susin et al., 1999; Otera et al., 2005). Extramitochondrial targeting of AIF or microinjection of AIF into the cytoplasm of normal cells induces caspase-independent apoptosis, suggesting that AIF has proapoptotic functions (Loeffler et al., 2001; Cande et al., 2004). However, AIF is not necessary for programmed cell death during normal early embryonic development, and indeed, loss of Aif function in embryos causes, rather than prevents, programmed cell death (Brown et al., 2006).
AIF was demonstrated to have a role in bioenergetic and/or redox metabolism by analysis of the Hq mutant mice (Klein et al., 2002; Vahsen et al., 2004). Mice homozygous or hemizygous for the Hq mutation have progressive ataxia beginning at 4–5 months of age, concomitant with the loss of cerebellar neurons (Klein et al., 2002). Progressive loss of retinal neurons also occurs in mutant mice. Biomarker analysis suggests that neuron death in the Hq mutant cerebellum is preceded by oxidative stress. In agreement, an increase in ROS was detected in Aif-deficient cells (Apostolova et al., 2005). This increase was accompanied by the loss of Complex I function of the electron transport chain, which was reversible after treatment with antioxidants. Loss of expression of Complex I proteins has also been reported in Aif-deficient cells and embryos and cerebellar and retinal extracts from the Hq mutant mouse (Vahsen et al., 2004; Apostolova et al., 2005; Brown et al., 2006). Interestingly, terminally differentiated granule cells and retinal neurons in Hq mutant mice undergo aberrant cell cycle re-entry into S-phase before apoptosis in the late S- or G2-phase of the cell cycle (Klein et al., 2002). Furthermore, cell cycle re-entry of these neurons is coincident with the presence of oxidatively damaged DNA, providing direct evidence that oxidative stress stimulates quiescent neurons to leave the G0 phase and re-enter the cell cycle.
In this study, we demonstrate that complete Aif deficiency disrupts both embryonic and postnatal cerebellar neurogenesis in a cell type-specific manner. Our data demonstrate that loss of Aif function prevents entry into S-phase of the cell cycle in both embryonic and postnatal granule cell precursors but results in premature S-phase entry of Purkinje cell precursors, which leads to apoptosis. Together with our previous results on Hq mutant neurons, these data demonstrate that loss of Aif function differentially affects cell cycle regulation in neurons of the embryonic and adult CNS.
Materials and Methods
Mice.
Mice carrying the loxP-flanked Aif (Aifm1tm2Pngr) allele and the En1cre (En1tm6(cre)Alj) allele were genotyped as described previously (Kimmel et al., 2000; Joza et al., 2005). B6.Cg-Tg(Nes-cre)1Kln/J (hereafter referred to as Nes-cre) and the B6.129S4-Gt(ROSA)26tm1Sor (R26R; cre-reporter) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Mutant embryos (Aifflox/Y carrying either the En1cre or Nes-cre gene) and control Aifflox/Y or Aifflox/+ littermates were generated by timed matings of Aifflox/flox mice to mice hemizygous or heterozygous for the cre recombinase genes. The day on which presence of a vaginal plug was detected was designated as embryonic day 0.5 (E0.5). The Animal Care and Use Committee of The Jackson Laboratory approved all animal protocols.
Immunostaining.
Immunohistochemistry was performed as described previously using antibodies to calbindin D-28 (1:1500; Swant, Bellinzona, Switzerland) and colorimetric detection with DAB (Ackerman et al., 1997). For immunofluorescence studies, embryos were fixed in Bouin's fixative or 4% paraformaldehyde (PFA) before paraffin embedding. Sections were incubated with rabbit polyclonal antibodies to cleaved caspase-3 (1:100; Cell Signaling Technology, Danvers, MA), Ki67 (1:1000; Novocastra, Newcastle, UK), phosphorylated histone H3 (1:1000; Millipore, Billerica, MA), phosphorylated retinoblastoma (RB) (1:100; Cell Signaling Technology), cyclin D1 (1:200; Neomarkers, Fremont, CA), and p27Kip (1:100; Santa Cruz Biotechnology, Santa Cruz, CA). Rabbit polyclonal antibodies to AIF (1:500; Epitomics, Burlingame, CA), Gli1 (1:100; Santa Cruz Biotechnology), and Lhx1/5 (1:200; a gift from Dr. Tom Jessell, Columbia University, New York, NY) were used on frozen sections of 4% PFA-fixed/sucrose-impregnated tissues. Expression was visualized with Cy3- or AF488-labeled donkey or goat secondary antibodies.
X-gal staining of tissue sections.
For X-gal staining, embryos were fixed in 2% PFA in PBS, pH 7.0, for 2.5 h. Cryosections were stained as described previously (Goldowitz et al., 2000).
Bromodeoxyuridine labeling and quantitation.
For bromodeoxyuridine (BrdU) pulse-labeling, pregnant mice were injected intraperitoneally with BrdU at 50 μg/g body weight. Embryos were fixed in 4% PFA and paraffin embedded. Before incubation with a mouse anti-BrdU antibody (1:50; Dako UK, Ely, Cambridgeshire, UK), sections were treated with 4N HCl.
BrdU-positive cells were counted in the cerebellar primordia of three E12.5 wild-type and three mutant embryos from three independent litters. Serial sections (7 μm) were taken from midline, through the medial aspects of the cerebellar primordia (identified by serially sectioning from midline), and continuing 100 μm until the lateral aspects of the cerebellum were reached. Images of immunostained cerebellum were generated from every other section, and immunopositive cells were manually counted using Adobe Photoshop software (Adobe Systems, San Jose, CA). Values were statistically analyzed by the Welch t test using JMP 7.0 (SAS Institute, Cary, NC).
BrdU/Ki67 analysis.
To examine the proportion of progenitors in S-phase or which exit the cell cycle, the labeling index was determined as described previously (Chenn and Walsh, 2002). Pregnant females were injected with BrdU at E12.5 as described above, and embryos were collected 30 min (for S-phase pulse labeling) or 24 h (for cell cycle exit studies) later. For BrdU pulse-labeling of S-phase cells in postnatal cerebella, mice were injected at postnatal day 3 (P3) and killed 30 min postinjection. Embryos and brains were paraffin embedded and serially sectioned as described above. For coimmunofluorescence with antibodies to BrdU and Ki67, antigen retrieval was performed in heated 10 mm citrate buffer, and then sections were incubated in DNaseI (5 U/ml) for 30–60 min before incubation with antibodies.
Cells that were reactive with either or both antibodies were counted in matched sections from three embryos of each genotype. These embryos were obtained from at least two independent litters. Because the cerebellum develops in a lateral to medial manner, we used the medial aspects of the cerebellar primordia as a landmark for cell counts. In the Aif-null cerebellum at E12.5, this landmark is located ∼40 μm more laterally than it is in the wild-type embryo. We counted cells in sections 80 μm from this landmark on E12.5 embryos (∼140 and 180 μm lateral from midline of the developing isthmus in wild-type and mutant embryos, respectively) and 160 μm from the medial aspect of the cerebellar primordial in E13.5 embryos (∼160 and 200 μm lateral from the midline of wild-type and mutant embryos, respectively). Positive cells were counted at midline in P3 cerebella. In E12.5 embryos and P3 cerebella, the percentage of Ki67/BrdU double-positive cells of the total Ki67-expressing cells was determined. This number represents the proportion of cycling progenitor in S-phase during the 30 min of BrdU labeling. In E13.5 embryo, the proportion of BrdU/Ki67 double-positive cells of the total number of BrdU-positive cells reflects the ratio of cells generated on E12.5 that remain in cell cycle 24 h later. Values were analyzed by one-way ANOVA with log transformation to ensure a normal distribution.
Iododeoxyuridine/BrdU analysis.
To determine the relative length of S-phase in mutant and wild-type progenitors, pregnant females were injected intraperitoneally (50 μg/g body weight) with iododeoxyuridine (IrdU) and BrdU at E12.5. Three embryos of each genotype from independent litters were paraffin embedded and serially sectioned on the sagittal plane. Coimmunofluorescence was performed on DNaseI-treated sections using a rat antibody (1:100; Serotec, Oxford, UK) that reacts with BrdU, but not IrdU, and a mouse antibody (1:50; Invitrogen, Carlsbad, CA) that recognizes both BrdU and IrdU as described previously (Burns and Kuan, 2005; Tarui et al., 2005). Expression was visualized with Cy3-conjugated goat or AF488-conjugated donkey secondary antibodies. Cells that had exited S-phase after IrdU administration, but before BrdU administration, were identified as IrdU-positive, but BrdU-negative, cells (i.e., positive with the Invitrogen antibody but not with the Serotec antibody). Those cells that had not exited S-phase by the time of BrdU administration (i.e., were both BrdU and IrdU positive) were reactive with both antibodies. Cells were counted in sagittal sections 80 μm from the medial aspect of the cerebellar primordia. Values were statistically analyzed by one-way ANOVA as described above.
Other BrdU immunofluorescence analysis.
For BrdU/Lhx1/5 or BrdU/cleaved caspase-3 studies, pregnant females were injected with BrdU as described above on E12.5, and embryos were collected on E14.5 and/or E15.5. Coimmunofluorescence was performed on DNaseI-treated sections. Expression was visualized as described above.
Results
AIF is necessary for embryonic cerebellar development
Our analysis of mice with the spontaneous mutation harlequin (Hq) demonstrated that reduced levels of Aif result in progressive death of granule cells and Purkinje cells in the adult cerebellum (Klein et al., 2002). Recently, complete deletion of Aif has been shown to result in lethality by E11.5 (Joza et al., 2005; Brown et al., 2006), suggesting that residual Aif expressed in Hq mice is sufficient for normal embryonic development. To determine whether, in addition to its role in survival of terminally differentiated neurons in the adult cerebellum, Aif is also important for development of the cerebellum, we examined the effects of complete loss of Aif function in the embryonic and early postnatal cerebellum.
Aif was deleted in the progenitors of the midbrain and cerebellum by crossing mice with a floxed Aif allele to the En1cre knock-in deletor strain, which expresses Cre in essentially all progenitors of the midbrain and cerebellum from ∼E8.75 (Kimmel et al., 2000; Chi et al., 2003). Cre function in the early cerebellum was confirmed (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) by crossing En1cre mice to animals carrying the ROSA26 (R26R) cre-reporter strain (Soriano, 1999). To confirm deletion of Aif in cre-expressing embryos, AIF immunostaining of E12.5 and E13.5 En1cre/+; Aifflox/Y (hereafter referred to as Aif mutant) and wild-type embryos was performed (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). AIF-expressing cells were observed throughout the developing midbrain and cerebellum of wild-type but not cre-expressing embryos, demonstrating efficient deletion of Aif. Importantly, AIF expression was not detected in the Aif mutant E12.5 ventricular zone or the Aif mutant E13.5 rhombic lip, the regions that generate Purkinje cell and granule cell precursors, respectively.
Aif mutant mice died soon after birth of unknown causes. Histological examination of mutant embryonic and P0 brains revealed severe cerebellar hypoplasia, which was apparent by E14.5 (Fig. 1). In midsagittal sections, we observed a dramatic reduction in cellularity, particularly near the differentiation zone where recently generated Purkinje cell precursors differentiate (Fig. 1A,D). The developing midbrain tectum was also hypoplastic in Aif mutants at E14.5 (Fig. 1A,D), E16.5 (Fig. 1B,E), and in neonates (Fig. 1C,F), indicating that Aif is also necessary for development and/or survival of midbrain neurons. In addition, edema-like morphology was observed in the midbrain tegmentum at E14.5 and E16.5 (Fig. 1A,B,D,E).
Figure 1.

Loss of Aif function results in hypoplasia of the embryonic cerebellum. A–F, Near midline, sagittal sections of wild-type (A–C) and En1Cre/+; Aifflox/Y (D–F) cerebella at E14.5 (A, D), E16.5 (B, E), and P0 (C, F) were stained with hematoxylin and eosin. Edema-like morphology (arrowheads) was observed in the mutant midbrain. Note also the deficiency of cells in the differentiating zone, where Purkinje cell precursors normally reside, and the paucity of granule cell precursors in the external granule cell layer in mutant cerebella. DZ, Differentiating zone; PCL, Purkinje cell layer; Te, midbrain tectum; Tg, midbrain tegmentum, VZ, ventricular zone. Scale bar, 100 μm
Examination of parasagittal sections of Aif mutant and wild-type cerebella at E14.5 revealed that the number of granule cell precursors in the external granule cell layer (EGL) was also markedly reduced in the absence of Aif (Fig. 2 A–D). In contrast, the area between the ventricular zone and the differentiation zone in the Aif mutant cerebellum appeared hypercellular (Fig. 2E,F). This later observation, combined with the observed decrease in cell number in the differentiation zone, suggested that Purkinje cells do not develop properly in the absence of Aif. To examine this possibility more closely, we performed immunohistochemistry with antibodies to the Purkinje cell marker calbindin D-28. As shown in Figure 2 (G,H) there is a marked reduction in the number of calbindin-positive cells in the Aif null cerebellum, confirming that Purkinje cell development is disrupted.
Figure 2.

Neuronal precursors are decreased in the Aif-null cerebellum. A–H, Parasagittal sections of E14.5 wild-type (A, C, E, G) and En1Cre/+; Aifflox/Y (B, D, F, H) cerebella are shown. Sections were stained with hematoxylin and eosin (A–F) or immunostained with calbindin D-28 (Calb) antibody (G, H). The boxes in A and B demarcate the regions that are shown at higher magnification in C, E and D, F, respectively. Note the reduction in cell number in the region normally occupied by the external granule layer in the mutant cerebellum, delineated by a dashed line in C and D. The bracket in E delineates the ventricular zone. Note the hypercellularity in the corresponding region in En1Cre/+; Aifflox/Y cerebellum (F, asterisks) and lack of calbindin-positive Purkinje cell precursors in the mutant cerebellum. VZ, Ventricular zone. Scale bars, 100 μm.
Loss of Aif function increases the number of cells in S-phase during Purkinje cell neurogenesis
The results described above suggested that Aif may be necessary for generation, differentiation, and/or survival of both Purkinje cell and granule cell precursors. To examine a potential role for Aif in Purkinje cell neurogenesis, BrdU pulse labeling was performed. Purkinje cell precursors arise between E11 and E13 from the ventricular zone adjacent to the fourth ventricle in a lateral to medial gradient and subsequently migrate to the differentiating zone in the cerebellar primordium (Altman and Bayer, 1997; Goldowitz and Hamre, 1998). E12.5 embryos were pulse-labeled with BrdU in utero for 30 min. In wild-type embryos, BrdU-positive nuclei were found scattered through the ventricular zone, whereas in Aif mutants, BrdU-positive nuclei extended more deeply into the developing cerebellum (Fig. 3 A–D). Quantification of BrdU-positive cells in sections cut just lateral to the midline (i.e., through the prospective vermis) (n = 3 for both wild-type and for Aif mutant embryos) demonstrated an increase in the number of cells in S-phase in the Aif mutant cerebellum over the number in the wild-type cerebellum (1743 ± 28 and 1093 ± 57, respectively; p = 0.002). Significantly, the total number of cells was approximately the same in the ventricular zone at this stage in Aif mutant as in wild-type embryos, thus demonstrating that a higher proportion of the mutant cells were in S-phase. In agreement with the apparent increase in cells in S-phase, the increase in BrdU-positive cells in the Aif mutant embryos was accompanied by an increase in the number of cells expressing the phosphorylated (serine807/811) form of RB, which is associated with progression through the G1 restriction point into S-phase (Fig. 3E,F), and the G1 cyclin, cyclin D1 (data not shown). Consistent with RB phosphorylation, reduced levels of p27Kip, an inhibitor of cyclin-dependent kinase 2 (CDK2) that blocks cell cycle progression in G0 or early G1, were detected in Aif mutant Purkinje cell precursors (Fig. 3G,H). However, immunofluorescence with an antibody to the mitosis marker, phosphorylated histone H3 (pH3), revealed fewer cells in M-phase in Aif mutant ventricular zone than in wild-type at E12.5 (Fig. 3I,J), suggesting that most Aif mutant progenitors that enter S-phase do not undergo mitosis.
Figure 3.
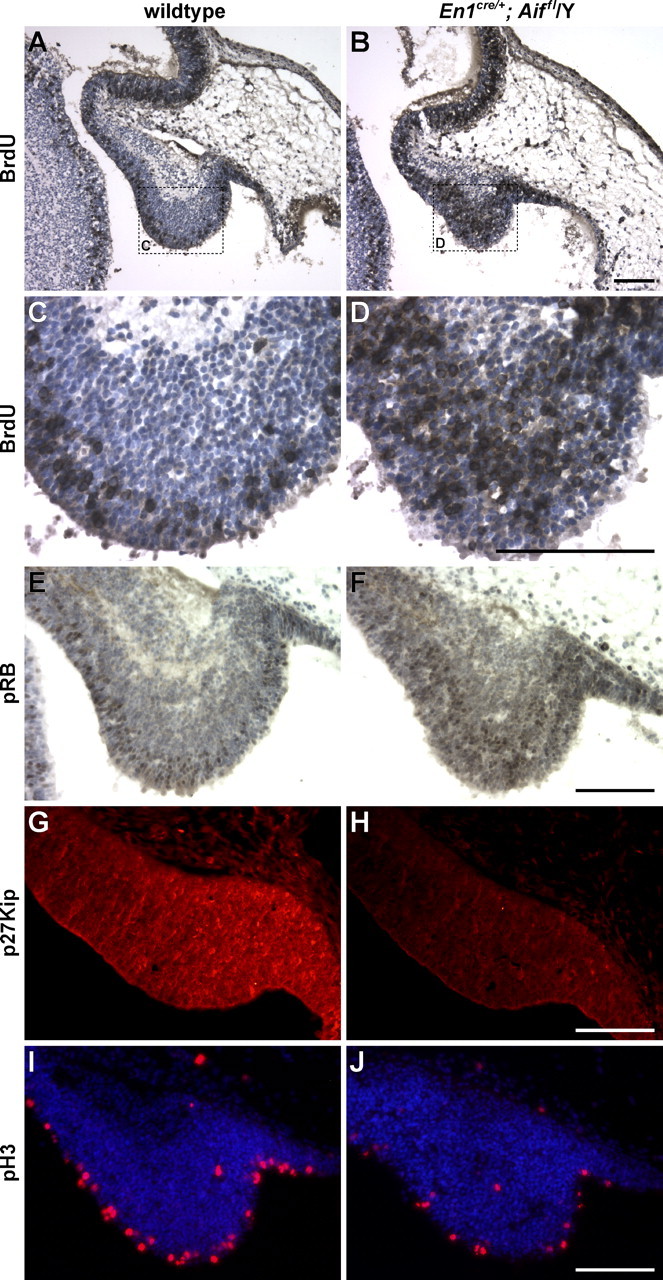
Loss of Aif function causes an increase in cells in S-phase of the cell cycle. A–J, Parasagittal sections are shown of cerebella from E12.5 wild-type (A, C, E, G, I) and En1Cre/+; Aifflox/Y (B, D, F, H, J) embryos pulse-labeled with BrdU in utero. The boxes in A and B delineate the regions shown at higher magnification in C and D, respectively. The sections were immunostained with antibodies to BrdU (A–D), phosphorylated RB (pRB; E, F), p27Kip (G, H), or pH3 (I, J). Sections were counterstained with hematoxylin (A–F) or Hoechst 33342 (I, J). Note that the increase in the number of cells in S-phase in the mutants is not accompanied by an increase in pH3-positive cells, suggesting that these cells do not undergo mitosis. Scale bars, 100 μm.
To examine whether the cells in S-phase in Aif-null cerebellum were indeed Purkinje cell precursors, we examined expression of early markers of Purkinje cell progenitors. Pregnant dams were injected with BrdU at E12.5, and embryos were harvested at E14.5. Coimmunostaining was performed with antibodies to BrdU and Lhx1/5, transcription factors that are first expressed in Purkinje cell precursors as they leave the ventricular zone (Morales and Hatten, 2006; Zhao et al., 2007). Some mutant BrdU-positive, Lhx1/5-negative cells remained in the ventral aspects of the cerebellar primordia in, or near, the ventricular zone (Fig. 4 A–D). This subpopulation was not observed in the wild-type cerebellum (data not shown) and likely represents progenitors that failed to migrate from the ventricular zone and begin the differentiation process. However, most of the BrdU+, Aif-null cells located more medially expressed Lhx1/5, confirming that these cells were indeed Purkinje cell precursors. In addition, the Lhx1/5-negative ventricular zone was grossly the same thickness in wild-type and mutant cerebella (data not shown), indicating large changes in numbers of nondifferentiated cells in the mutant ventricular zone are not apparent at E14.5.
Figure 4.

Aif-null Purkinje cell precursors prematurely enter S-phase. A–D, En1Cre/+; Aifflox/Y embryos were pulse-labeled with BrdU in utero at E12.5, the time of Purkinje cell precursor generation, and collected at E14.5. Parasagittal sections were immunostained with antibodies to BrdU (A) and Lhx1/5 (B) and were counterstained with Hoechst 33342 to visualize nuclei (C). Merges of BrdU and Lhx1/5 immunostained images are shown in D. Note that most of the BrdU-positive cells in the medial cerebellum are positive for Lhx1/5, suggesting that these cells are Purkinje cell precursors. Scale bar, 100 μm.
Loss of Aif function in Purkinje cell precursors causes premature entry into S-phase
To determine whether the increase in cells in S-phase in Aif-null embryos is attributable to an increase in proliferating progenitor cells or to an increase in the percentage of progenitor cells that are in S-phase, we determined the proportion of proliferating cells that were in S-phase. We pulse-labeled E12.5 embryos for 30 min and then immunostained their cerebella with antibodies to BrdU and Ki67, a protein expressed in all proliferating cells, and thus all progenitors. We found that a higher percentage of Ki67+ cells were also BrdU positive in the Aif-null cerebellum (Fig. 5). This result demonstrates that the increase in the number of S-phase cells is caused by changes in cell cycle length.
Figure 5.
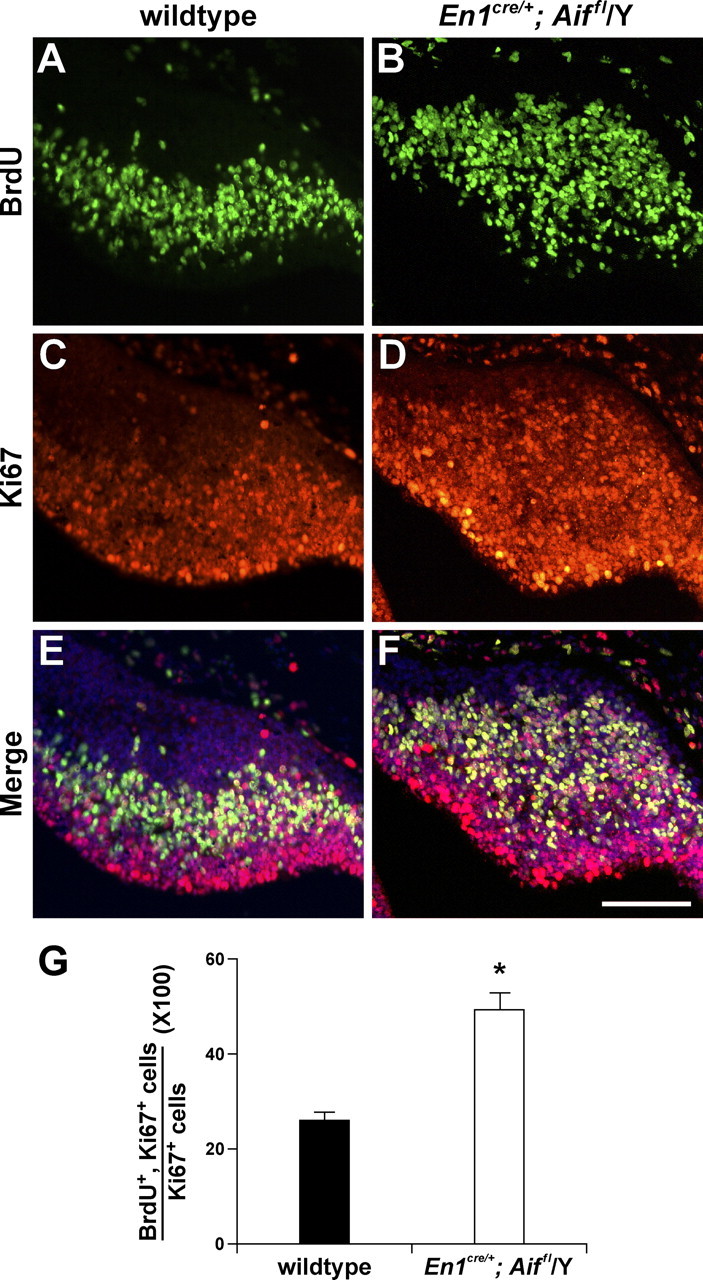
Cell cycle length is shortened in Aif-null Purkinje cell precursors. A–F, Parasagittal sections of cerebella from E12.5 wild-type (A, C, E) and En1Cre/+; Aifflox/Y (B, D, F) embryos were pulse-labeled with BrdU in utero and immunostained with antibodies to BrdU (A, B) and Ki67 (C, D). Images of BrdU- and Ki67-immunostained sections, counterstained with Hoechst 33342, are merged in E and F. G, The bar graphs indicate the percentages of proliferating cells that were in the S-phase of the cell cycle in wild-type and mutant embryos. Using Ki67 as a marker for proliferating cells, these percentages were calculated by determining the ratio of BrdU+, Ki67+ cells to the total number of Ki67+ cells. Values are the means ± SEM of three independent experiments. *p < 0.002. Scale bar, 100 μm.
To determine what aspect of the cell cycle is affected by Aif deficiency, we performed IrdU/BrdU incorporation experiments in mutant and wild-type embryos. DNA undergoing replication was labeled in E12.5 embryos in utero by first injecting pregnant dams with IrdU and then 2 h later with BrdU, and embryos were collected 30 min post-BrdU injection (Fig. 6). We determined the fraction of cells that had exited S-phase between the two injections by counting the number of cerebellar ventricular cells that were labeled with only IrdU (IrdU+/BrdU− cells, which exited S-phase before BrdU was injected) and the number of cells that were labeled with both thymidine analogs (IrdU+/BrdU+ cells, which were still in S-phase at the time of BrdU injection) (Burns and Kuan, 2005; Tarui et al., 2005). We found that the percentage of cells that had exited S-phase during the 2 h labeling period was similar in wild-type and mutant cerebellum (wild type, 24.5 ± 1.3; mutant, 22.6 ± 1.9; p > 0.05), suggesting that loss of Aif does not change the length of S-phase. In addition, these data demonstrate that mutant cells exit S-phase and proceed normally into G2. Together, our data suggest that G1 of the cell cycle is shorter than normal in proliferating Aif-null cells during the time of Purkinje cell generation, resulting in premature entry into S-phase.
Figure 6.

Loss of Aif function does not alter S-phase length of Purkinje cell precursors. A–F, E12.5 embryos were sequentially labeled in utero with IrdU and BrdU at E12.5. Parasagittal sections of wild-type (A, C, E) and En1Cre/+; Aifflox/Y (B, D, F) cerebella were immunostained with antibodies that react with both IrdU and BrdU (A, B) or only BrdU (C, D). Images of IrdU- and BrdU-immunostained sections, counterstained with Hoechst 33342, are merged in E and F. Scale bar, 100 μm.
Undifferentiated Aif null Purkinje cell precursors undergo apoptosis at E15.5
To further investigate our observation that Aif null Purkinje cell precursors that exit S-phase fail to progress through the cell cycle into M-phase, we analyzed cell cycle exit in mutant and wild-type cerebella. Pregnant dams were injected with BrdU, and 24 h later, E13.5 embryos were collected. Coimmunofluorescence was performed with antibodies to BrdU and Ki67. Cells that remained in the cell cycle during the 24 h after labeling were positive for both BrdU and Ki67. In contrast, cells that exited the cell cycle during this time were positive for BrdU but did not express Ki67. In the wild-type cerebellum, most BrdU-positive cells did not express Ki67, demonstrating that most cells had exited the cell cycle and begun terminal differentiation within 24 h of having been labeled. In contrast, in Aif mutant cerebella, there were more BrdU-positive, Ki67-positive cells (Fig. 7), providing evidence that mutant Purkinje cell precursors fail to exit the cell cycle. Moreover, because we found that mutant cells do complete S-phase, but do not enter mitosis (Figs. 6, 3I,J), it is likely that they are arrested at the G2 checkpoint.
Figure 7.

Aif-null Purkinje cell precursors fail to exit the cell cycle. A–F, Parasagittal sections of cerebella from E13.5 wild-type (A, C, E) and En1Cre/+; Aifflox/Y (B, D, F) embryos labeled in utero with BrdU at E12.5 were immunostained with antibodies to BrdU (A, B) and Ki67 (C, D). Images of BrdU- and Ki67-immunostained sections, counterstained with Hoechst 33342, are merged in E and F. G, The bars indicate the percentages of cells that were in S-phase ∼24 h earlier but then exited the cell cycle. These percentages were determined by calculating the ratio of BrdU+, Ki67− cells to the total number of BrdU+ cells and multiplying by 100. Values are the means ± SEM of three independent experiments. *p < 0.0001. Scale bar, 100 μm.
To determine whether Aif-deficient null Purkinje cell precursors survive in the cerebellum, we examined En1cre/+; Aifflox/Y cerebella at E13.5–E16.5 by immunostaining with antibodies to cleaved caspase-3, a marker for apoptotic cells. Positive cells were not detected in either wild-type or Aif mutant cerebella at E13.5 or E14.5 (data not shown). However, many cleaved caspase 3-positive cells were observed in the interior of Aif-null cerebella at E15.5 (Fig. 8A,B) and at E16.5, although at the later stage, they were reduced in number (data not shown). Cleaved caspase 3-positive cells were not observed in wild-type cerebella at E15.5 or E16.5 (Fig. 8C) (data not shown), nor were they observed in mutant or wild-type EGL at any of these stages (data not shown).
Figure 8.

Undifferentiated Purkinje cell precursors die at E15.5 in Aif-null cerebellum. A–I, Parasagittal sections of E15.5 En1Cre/+; Aifflox/Y (A, B, D–I) and wild-type (C) cerebella were immunostained with cleaved caspase-3 antibodies (A–C). Sections were counterstained with hematoxylin. The box in A delineates the region shown at higher magnification in B. D–F, Parasagittal sections of the E15.5 mutant cerebella were stained with antibody to Lhx1/5 (D) and counterstained with Hoechst 33342 (E) to visualize nuclei. Note that many pyknotic nuclei are found in Lhx1/5-positive cells (F, arrowheads in merged image). G–I, Embryos at E12.5, the time of Purkinje cell precursor generation, were pulse-labeled with BrdU in utero and collected at E15.5. Coimmunofluorescence was performed with antibodies to BrdU (G) and cleaved caspase-3 and counterstained with Hoechst 33342 to visualize nuclei before confocal analysis (H). Note that most cells that express cleaved caspase-3 are also BrdU positive (I,arrowheads in merged image). Scale bars: A–C, 50 μm; D–I, 20 μm.
Examination of the E15.5 mutant cerebellum immunostained with Lhx1/5 antibodies demonstrated that many apoptotic cells expressed Lhx1/5 (Fig. 8 D–F). β-Tubulin III (Tuj1), a cytoskeletal component found in neuronal but not glial precursors, was also expressed in these cells (data not shown). To confirm that the dying cells represented Purkinje cell precursors generated at E12.5 from abnormally cycling precursors, Aif-null embryos were labeled with BrdU in utero at E12.5, a time when only Purkinje cells are generated from the ventricular zone, and then collected at E15.5. Immunostaining with antibodies to BrdU and cleaved caspase-3 demonstrated many double-positive cells (Fig. 8 G–I). Although other cell types may also die, these data provide evidence that many Purkinje cell precursors undergo apoptosis several days after they are generated.
Loss of Aif function disrupts the G1/S phase transition in granule cell precursors
In addition to loss of Purkinje cell precursors, we observed a dramatic decrease in granule cell precursors in the EGL of the En1cre/+; Aifflox/Y cerebellum (Fig. 2 A–D). Neither caspase-3 immunoreactive cells nor cells with pyknotic nuclei were observed in the mutant EGL at E13.5–P0 (data not shown), suggesting that rather than inducing cell death, as is the case for Purkinje cells, loss of Aif function may instead disrupt the generation of granule cell precursors. To investigate this hypothesis, embryos were BrdU pulse-labeled in utero at E13.5, when granule cells are first born and emanate from the rhombic lip to form the EGL. The number of BrdU-positive cells in the EGL at E13.5 was greatly reduced in En1cre/+; Aifflox/Y embryos compared with wild-type embryos (Fig. 9 A–D), although no overt decrease in the size of the rhombic lip neuroepithelium was observed at E12.5, 1 d before granule cell precursor generation (Fig. 9E,F). These data suggest that loss of Aif function in embryonic granule cell precursors results in cell cycle arrest before entry into S-phase.
Figure 9.
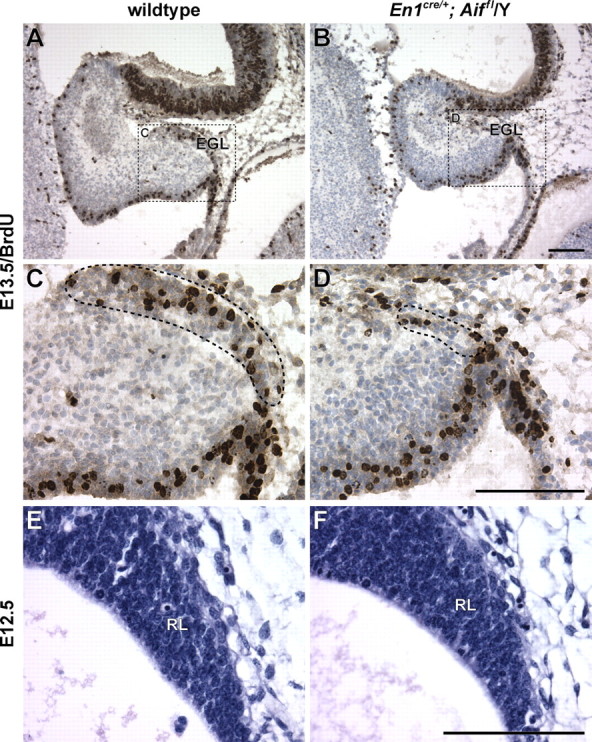
Proliferation of granule cell precursors is decreased in the embryonic Aif-null cerebellum. E13.5 embryos were pulse-labeled in utero with BrdU for 1 h. A–D, BrdU immunostaining of parasagittal sections of wild-type (A, C) and En1Cre/+; Aifflox/Y (B, D) cerebella. Sections were counterstained with hematoxylin. The dashed boxes in A and B demarcate the regions shown at higher magnification in C and D, respectively. The EGL is demarcated by a dashed line (C, D). E, F, Hematoxylin- and eosin-stained sections of the E12.5 rhombic lip (RL) in wild-type (E) and mutant (F) embryos. Scale bars, 100 μm.
Much of granule cell neurogenesis occurs postnatally between P3 and P15 when granule cell precursors in the EGL undergo clonal expansion, before their radial migration past Purkinje cells to form the inner granule cell layer. To determine the effect of loss of Aif function on postnatal granule cell neurogenesis, we deleted Aif using a cre transgene driven by the nestin (Nes) promoter (Tronche et al., 1999), which functions at a later stage than En1cre. Aif-deficient mice generated using this Nes-cre deleter line were born in normal Mendelian ratios and were indistinguishable from wild-type mice until P3, after which the mutant pups grew poorly. Death of Nes-cre; Aifflox/Y mice occurred between P5 and P8.
As evidenced by examination of embryos from crosses of the Nes-cre mice to the R26R reporter strain, cre-mediated deletion occurs at a much later stage in cerebellar development than when En1cre was used. Little expression of β-gal was detected in the cerebellum of cre-positive reporter mice at or before E12.5, but the vast majority of cells were β-gal-positive in the E14.5 cerebellum (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). In agreement, AIF expression was still detected in the E12.5 ventricular zone in Nes-cre; Aifflox/Y embryos (data not shown). Consistent with the delayed function of cre in the Nes-cre strain, early cerebellar development proceeded normally in Nes-cre; Aifflox/Y embryos. BrdU pulse labeling at E13.5 did not reveal abnormalities in early granule cell neurogenesis in mutant embryos (supplemental Fig. 4, available at www.jneurosci.org as supplemental material).
Although early embryonic granule cell development appeared normal in Nes-cre; Aifflox/Y mice, a failure of later granule cell neurogenesis was suggested by the observation that these mice displayed mild cerebellar hypoplasia accompanied by delayed fissure formation at P1 (Fig. 10A,C), which was more pronounced at P3 and was accompanied by a markedly thinner EGL. Because Purkinje cell production of Sonic hedgehog (SHH) promotes postnatal granule cell proliferation (Wallace, 1999; Wechsler-Reya and Scott, 1999; Lewis et al., 2004), we stained P1 cerebella with calbindin antibodies and found no obvious defects in Purkinje cell generation in the Nes-cre; Aifflox/Y cerebellum (Fig. 10B,D). In addition, granule cell precursors in the P3 mutant EGL expressed apparently normal levels of Gli1, a transcriptional activator of SHH that is upregulated in the EGL by SHH signaling (Corrales et al., 2004), and cyclin D1, a G1 cyclin, the expression of which is promoted by SHH (Kenney and Rowitch, 2000; Kenney et al., 2003; Oliver et al., 2003) (Fig. 10 E–L).
Figure 10.

Purkinje cell development is normal in the Nes-cre; Aifflox/Y mutant early postnatal cerebellum. A–D, P1 midline sagittal sections of wild-type (A, B) and Nes-cre; Aifflox/Y mutant (C, D) cerebellum were stained with hematoxylin and eosin (H+E; A, C), or immunostained with antibody to calbindin D-28 (Calb; B, D). E–L, P3 midline sagittal sections of wild-type (E, F, I, J) and Nes-cre; Aifflox/Y (G, H, K, L) cerebella are immunostained with antibodies to Gli1 (E–H) or cyclin D1 (CycD1; I–L). Merged sections counterstained with Hoescht 33342 to visualize the dense nuclei of the EGL are shown in F, H and J, L. PCL, Purkinje cell layer. Scale bars: A–D, 100 μm; E–L, 40 μm.
To further investigate granule cell proliferation, we determined the proportion of these progenitors that were in S-phase in mice at P3. Pups were injected with BrdU and killed 30 min later. Coimmunofluorescence staining with antibodies to BrdU and Ki67 revealed that, as expected during early postnatal cerebellar development, the majority of cells in both the wild-type and mutant EGL were cycling, as evidenced by their expression of Ki67 (Fig. 11 A–D). However, a significantly lower percentage of granule cell progenitors in the mutant EGL entered S-phase, as evidenced by the decreased number of Ki67-positive progenitors that had incorporated BrdU (Fig. 11 E–I). These data suggest that as in embryonic granule cell precursors, loss of Aif function in postnatal granule cell precursors also results in cell cycle arrest before S-phase.
Figure 11.

Proliferation of granule cell progenitors is decreased in the postnatal Aif-null cerebellum. P3 mice were pulse-labeled with BrdU for 30 min. A–H, Midline sagittal sections of the cerebella from these wild-type (A, C, E, G) and Nes-cre; Aifflox/Y (B, D, F, H) mice are shown. The dashed boxes in A and B demarcate the regions shown at higher magnification in C, E, G and D, F, H, respectively. The sections were immunostained with antibodies to Ki67 (A–D) and BrdU (E, F). Images of Ki67- and BrdU-immunostained sections, counterstained with Hoechst 33342, are merged in G and H. I, The bar graphs show the percentages of proliferating cells that were in the S-phase of the cycle. These percentages were determined by calculating the ratio of BrdU+ and Ki67+ cells to the total number of Ki67+ cells. Values are the means ± SEM of three independent experiments. *p < 0.002. Scale bars, 100 μm.
Discussion
The production of terminally differentiated neurons from neuroepithelial stem cells in the developing CNS requires a precise balance between cell proliferation, cell cycle exit, and subsequent differentiation. Although cellular stress, including mitochondrial dysfunction, impacts the survival of terminally differentiated neurons, little is known regarding the effects of mitochondrial deficiency on neurogenesis in the developing brain. Here, we demonstrate that absence of the mitochondrial oxidoreductase, AIF, in the developing cerebellum results in cell cycle defects, which lead to a reduction in the generation and survival of neuronal precursors. These cell cycle defects occur in both Aif-null granule cell and Purkinje cell precursors; however, the nature of these defects differs dramatically. Aif-null granule cell progenitors arrest at the G1/S restriction point. In contrast, Aif-null Purkinje cell precursors rapidly progress through this restriction point but subsequently arrest at the G2/M checkpoint, thus failing to complete mitosis and differentiate.
Although the exact function of AIF remains unclear, absence of this protein results in complex I deficiency and oxidative damage (Klein et al., 2002; Vahsen et al., 2004; Apostolova et al., 2005; Brown et al., 2006). Therefore, the differential response of cells to the loss of Aif function may be reflective of variation in the oxidative state among cell types, as shown previously for granule cells and astrocytes (Harrison et al., 2005). In addition, precursors of different neuronal cell types may have different energy requirements or differ in their antioxidant capacity or their response to ROS-induced damage. For example, granule cell neurogenesis may require more ATP than Purkinje cell generation, thus leading to the cell cycle arrest of granule cell precursors at the G1/S checkpoint, previously shown to be a sensor of the cellular metabolic status and a common checkpoint for arrest in cells with DNA damage caused by acute oxidative stress (Mandal et al., 2005).
In contrast to the arrest at the G1/S restriction point in granule cell progenitors, loss of Aif function during Purkinje cell generation causes an increase in the number of cells that enter S-phase in the ventricular zone neuroepithelium. However, unlike the aberrant DNA synthesis observed in postmitotic granule cells in the aging Hq mutant cerebellum (Klein et al., 2002), which is caused by cell cycle reentry, Aif-null Purkinje cell precursors have a shortening of the G1 phase of the cell cycle. Cell cycle length dramatically increases during cortical neurogenesis caused by an increase in G1 length but not S-, G2-, or M-phase (Caviness and Takahashi, 1995). This increase in G1 length occurs in neurogenic precursors and not in proliferating neuroepithelial cells (Calegari et al., 2005). Furthermore, extension of G1 length has been shown to lead to an increase in neurogenesis, without a concomitant increase in proliferation of neuroepithelial cells (Malatesta et al., 2000; Canzoniere et al., 2004). Therefore, the G1 shortening observed in the absence of Aif might itself prevent neurogenic divisions or differentiation of Purkinje cell precursors. The failure of most mutant cells to progress through later stages of the cell cycle may also be attributable to altered length of G1 and/or cellular stress.
During the G1-phase, numerous intracellular and extracellular signals are integrated and determine whether a cell will replicate its DNA or begin the differentiation process (Sherr and Roberts, 1999; Massague, 2004). Although the molecular pathways that control G1 progression vary substantially between cell types, entry into S-phase in all cells is achieved by activation of CDKs, which ultimately phosphorylate Rb leading to E2F transcription factor activation. In agreement with our finding that loss of Aif function leads to an increase in the number of cells that enter S-phase, we observed both a decrease in levels of the CDK inhibitor p27Kip and an increase in phosphorylated Rb in the Aif-null ventricular zone during the time of Purkinje cell neurogenesis. Although not reported previously in the nervous system, experimental findings in other systems have demonstrated that increases in ROS are linked, in a cell type-specific manner, to aberrant entry into S-phase and abnormal cell cycle progression. Oxidative stress-induced cytostatis has been shown to be mediated in part via the Forkhead box subgroup O (FOXO) transcription factors that control transcription of ROS regulatory genes (Tothova et al., 2007). Loss of FOXO transcription factors in hematopoetic stem cells resulted in an increase in ROS and inappropriate progression into S/G2/M, both of which were rescued by administration of antioxidants. Similarly, G1 was shortened in HeLa cells overexpressing a dominant-negative form of thioredoxin reductase 2, a mitochondrial antioxidant (Kim et al., 2003).
In contrast to previous studies suggesting Aif is necessary for the survival, but not the generation of early postmitotic neurons in developing telencephalon (Cheung et al., 2006), our results demonstrate that in the developing cerebellum, Aif is clearly necessary for granule cell and Purkinje cell neurogenesis. Purkinje cell defects were not observed in Nes-cre; Aifflox/Y mice in which Aif was deleted only after Purkinje cell precursor generation has occurred. Similarly, degeneration of Purkinje cells or granule cells does not occur for several months after cre-mediated deletion of Aif in postnatal neurons (R.I. and S.L.A., unpublished data), suggesting that survival of postmitotic neurons in the early postnatal or young adult cerebellum does not require Aif function. These results, combined with our previous studies on the Hq mutant mouse, demonstrate that Aif is required in neurons and their progenitors at two different times: during neurogenesis for completion of the cell cycle and again in aging terminally differentiated neurons for their continued survival.
Footnotes
This work was supported by National Institutes of Health Grants AG19358 (S.L.A.) and HD25331 (G.R.M.) and CORE Grant CA34196 (The Jackson Laboratory). S.L.A. is an investigator of the Howard Hughes Medical Institute. We thank the histology service at The Jackson Laboratory for embedding tissues, Dr. Jim Denegre in the Bioimaging Service for assistance with confocal analysis, and Weidong Zhang for statistical analysis. We are also grateful to Drs. Doris Brown and Salvador Martinez, who produced and analyzed the first En1Cre/+; Aifflox/Y embryos that were studied; Yusuke Kageyama for technical assistance; Dr. Tom Jessell for the Lhx1/5 antibody; and Drs. Rob Burgess, Kevin Mills, and Kyuson Yun for comments on this manuscript.
References
- Ackerman SL, Kozak LP, Przyborski SA, Rund LA, Boyer BB, Knowles BB. The mouse rostral cerebellar malformation gene encodes an UNC-5-like protein. Nature. 1997;386:838–842. doi: 10.1038/386838a0. [DOI] [PubMed] [Google Scholar]
- Altman J, Bayer SA. Development of the cerebellar system: in relation to its evolution, structure and functions. New York: CRC; 1997. [Google Scholar]
- Ames BN. Delaying the mitochondrial decay of aging. Ann NY Acad Sci. 2004;1019:406–411. doi: 10.1196/annals.1297.073. [DOI] [PubMed] [Google Scholar]
- Apostolova N, Cervera AM, Victor VM, Cadenas S, Sanjuan-Pla A, Alvarez-Barrientos A, Esplugues JV, McCreath KJ. Loss of apoptosis-inducing factor leads to an increase in reactive oxygen species, and an impairment of respiration that can be reversed by antioxidants. Cell Death Differ. 2005;13:354–357. doi: 10.1038/sj.cdd.4401776. [DOI] [PubMed] [Google Scholar]
- Barja G. Free radicals and aging. Trends Neurosci. 2004;27:595–600. doi: 10.1016/j.tins.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Blomgren K, Hagberg H. Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic Biol Med. 2006;40:388–397. doi: 10.1016/j.freeradbiomed.2005.08.040. [DOI] [PubMed] [Google Scholar]
- Brown D, Yu BD, Joza N, Benit P, Meneses J, Firpo M, Rustin P, Penninger JM, Martin GR. Loss of Aif function causes cell death in the mouse embryo, but the temporal progression of patterning is normal. Proc Natl Acad Sci USA. 2006;103:9918–9923. doi: 10.1073/pnas.0603950103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns KA, Kuan CY. Low doses of bromo- and iododeoxyuridine produce near-saturation labeling of adult proliferative populations in the dentate gyrus. Eur J Neurosci. 2005;21:803–807. doi: 10.1111/j.1460-9568.2005.03907.x. [DOI] [PubMed] [Google Scholar]
- Calegari F, Haubensak W, Haffner C, Huttner WB. Selective lengthening of the cell cycle in the neurogenic subpopulation of neural progenitor cells during mouse brain development. J Neurosci. 2005;25:6533–6538. doi: 10.1523/JNEUROSCI.0778-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cande C, Vahsen N, Kouranti I, Schmitt E, Daugas E, Spahr C, Luban J, Kroemer RT, Giordanetto F, Garrido C, Penninger JM, Kroemer G. AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene. 2004;23:1514–1521. doi: 10.1038/sj.onc.1207279. [DOI] [PubMed] [Google Scholar]
- Canzoniere D, Farioli-Vecchioli S, Conti F, Ciotti MT, Tata AM, Augusti-Tocco G, Mattei E, Lakshmana MK, Krizhanovsky V, Reeves SA, Giovannoni R, Castano F, Servadio A, Ben-Arie N, Tirone F. Dual control of neurogenesis by PC3 through cell cycle inhibition and induction of Math1. J Neurosci. 2004;24:3355–3369. doi: 10.1523/JNEUROSCI.3860-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviness VS, Jr, Takahashi T. Proliferative events in the cerebral ventricular zone. Brain Dev. 1995;17:159–163. doi: 10.1016/0387-7604(95)00029-b. [DOI] [PubMed] [Google Scholar]
- Chan PH. Mitochondria and neuronal death/survival signaling pathways in cerebral ischemia. Neurochem Res. 2004;29:1943–1949. doi: 10.1007/s11064-004-6869-x. [DOI] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- Cheung EC, Joza N, Steenaart NA, McClellan KA, Neuspiel M, McNamara S, MacLaurin JG, Rippstein P, Park DS, Shore GC, McBride HM, Penninger JM, Slack RS. Dissociating the dual roles of apoptosis-inducing factor in maintaining mitochondrial structure and apoptosis. EMBO J. 2006;25:4061–4073. doi: 10.1038/sj.emboj.7601276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi CL, Martinez S, Wurst W, Martin GR. The isthmic organizer signal FGF8 is required for cell survival in the prospective midbrain and cerebellum. Development. 2003;130:2633–2644. doi: 10.1242/dev.00487. [DOI] [PubMed] [Google Scholar]
- Corrales JD, Rocco GL, Blaess S, Guo Q, Joyner AL. Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellum development. Development. 2004;131:5581–5590. doi: 10.1242/dev.01438. [DOI] [PubMed] [Google Scholar]
- Goldowitz D, Hamre K. The cells and molecules that make a cerebellum. Trends Neurosci. 1998;21:375–382. doi: 10.1016/s0166-2236(98)01313-7. [DOI] [PubMed] [Google Scholar]
- Goldowitz D, Hamre KM, Przyborski SA, Ackerman SL. Granule cells and cerebellar boundaries: analysis of Unc5h3 mutant chimeras. J Neurosci. 2000;20:4129–4137. doi: 10.1523/JNEUROSCI.20-11-04129.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JF, Hollensworth SB, Spitz DR, Copeland WC, Wilson GL, LeDoux SP. Oxidative stress-induced apoptosis in neurons correlates with mitochondrial DNA base excision repair pathway imbalance. Nucleic Acids Res. 2005;33:4660–4671. doi: 10.1093/nar/gki759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joza N, Oudit GY, Brown D, Benit P, Kassiri Z, Vahsen N, Benoit L, Patel MM, Nowikovsky K, Vassault A, Backx PH, Wada T, Kroemer G, Rustin P, Penninger JM. Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol Cell Biol. 2005;25:10261–10272. doi: 10.1128/MCB.25.23.10261-10272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney AM, Rowitch DH. Sonic hedgehog promotes G1 cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol. 2000;20:9055–9067. doi: 10.1128/mcb.20.23.9055-9067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003;130:15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]
- Kim MR, Chang HS, Kim BH, Kim S, Baek SH, Kim JH, Lee SR, Kim JR. Involvements of mitochondrial thioredoxin reductase (TrxR2) in cell proliferation. Biochem Biophys Res Commun. 2003;304:119–124. doi: 10.1016/s0006-291x(03)00547-3. [DOI] [PubMed] [Google Scholar]
- Kimmel RA, Turnbull DH, Blanquet V, Wurst W, Loomis CA, Joyner AL. Two lineage boundaries coordinate vertebrate apical ectodermal ridge formation. Genes Dev. 2000;14:1377–1389. [PMC free article] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- Lewis PM, Gritli-Linde A, Smeyne R, Kottmann A, McMahon AP. Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev Biol. 2004;270:393–410. doi: 10.1016/j.ydbio.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Loeffler M, Daugas E, Susin SA, Zamzami N, Metivier D, Nieminen A-L, Brothers G, Penninger JM, Kroemer G. Dominant cell death induction by extramitochondrially targeted apoptosis-inducing factor. FASEB J. 2001;15:758–767. doi: 10.1096/fj.00-0388com. [DOI] [PubMed] [Google Scholar]
- Malatesta P, Gotz M, Barsacchi G, Price J, Zoncu R, Cremisi F. PC3 overexpression affects the pattern of cell division of rat cortical precursors. Mech Dev. 2000;90:17–28. doi: 10.1016/s0925-4773(99)00224-5. [DOI] [PubMed] [Google Scholar]
- Mandal S, Guptan P, Owusu-Ansah E, Banerjee U. Mitochondrial regulation of cell cycle progression during development as revealed by the tenured mutation in Drosophila. Dev Cell. 2005;9:843–854. doi: 10.1016/j.devcel.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Massague J. G1 cell-cycle control and cancer. Nature. 2004;432:298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- McFarland R, Taylor RW, Turnbull DM. Mitochondrial disease–its impact, etiology, and pathology. Curr Top Dev Biol. 2007;77:113–155. doi: 10.1016/S0070-2153(06)77005-3. [DOI] [PubMed] [Google Scholar]
- Miramar MD, Costantini P, Ravagnan L, Saraiva LM, Haouzi D, Brothers G, Penninger JM, Peleato ML, Kroemer G, Susin SA. NADH oxidase activity of mitochondrial apoptosis-inducing factor. J Biol Chem. 2001;276:16391–16398. doi: 10.1074/jbc.M010498200. [DOI] [PubMed] [Google Scholar]
- Morales D, Hatten ME. Molecular markers of neuronal progenitors in the embryonic cerebellar anlage. J Neurosci. 2006;26:12226–12236. doi: 10.1523/JNEUROSCI.3493-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM, Wickramasinghe R, Scott MP, Wechsler-Reya RJ. Transcriptional profiling of the Sonic hedgehog response: a critical role for N-myc in proliferation of neuronal precursors. Proc Natl Acad Sci USA. 2003;100:7331–7336. doi: 10.1073/pnas.0832317100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Ohsakaya S, Nagaura Z, Ishihara N, Mihara K. Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J. 2005;24:1375–1386. doi: 10.1038/sj.emboj.7600614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Tarui T, Takahashi T, Nowakowski RS, Hayes NL, Bhide PG, Caviness VS., Jr Overexpression of p27Kip1, probability of cell cycle exit, and laminar destination of neocortical neurons. Cereb Cortex. 2005;15:1343–1355. doi: 10.1093/cercor/bhi017. [DOI] [PubMed] [Google Scholar]
- Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, Armstrong SA, Passegue E, DePinho RA, Gilliland DG. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Vahsen N, Candé C, Brière JJ, Bénit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, Feraud O, Debili N, Wissing S, Engelhardt S, Madeo F, Piacentini M, Penninger JM, Schägger H, Rustin P, Kroemer G. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace VA. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol. 1999;9:445–448. doi: 10.1016/s0960-9822(99)80195-x. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Kwan KM, Mailloux CM, Lee WK, Grinberg A, Wurst W, Behringer RR, Westphal H. LIM-homeodomain proteins Lhx1 and Lhx5, and their cofactor Ldb1, control Purkinje cell differentiation in the developing cerebellum. Proc Natl Acad Sci USA. 2007;104:13182–13186. doi: 10.1073/pnas.0705464104. [DOI] [PMC free article] [PubMed] [Google Scholar]