

Abstract
Tamoxifen has both cytostatic and cytotoxic properties for breast cancer. Tamoxifen engaged mitochondrial estrogen receptor beta (ERβ) as an antagonist in MCF-7 BK cells, increasing reactive oxygen species (ROS) concentrations from the mitochondria that were required for cytotoxicity. In part this derived from tamoxifen down-regulating manganese superoxide dismutase (MnSOD) activity through nitrosylating tyrosine 34, thereby increasing ROS. ROS activated protein kinase C delta and c-jun N-terminal kinases, resulting in the mitochondrial translocation of Bax and cytochrome C release. Interestingly, tamoxifen failed to cause high ROS levels or induce cell death in MCF7BK-TR cells due to stimulation of MnSOD activity through agonistic effects at mitochondrial ERβ. In several mouse xenograft models, lentiviral shRNA-induced knockdown of MnSOD caused tumors that grew in the presence of tamoxifen to undergo substantial apoptosis. Tumor MnSOD and mitochondrial ERβ are therefore targets for therapeutic intervention to reverse tamoxifen resistance and enhance a cell death response.
Keywords: estrogen receptor, mitochondria, apoptosis, tamoxifen resistance
Introduction
Tamoxifen (TAM) often prevents primary breast tumor formation or causes the regression of established tumors. One mechanism of action is by TAM inducing apoptotic cell death, demonstrated in many cell types including breast cancer.1–3 In this malignancy, adjuvant radiation and chemotherapies are effective because they induce tumor cell death 4,5 and resistance to adjuvant therapies may arise when treatment does not cause apoptosis. Therefore it is important to understand the mechanisms involved to enhance cell death.
Resistance to TAM develops in as many as 50% of women taking this adjuvant endocrine therapy for breast cancer.6 Both primary (de novo) and secondary (acquired) resistance occurs, and several mechanisms have been proposed. One supported mechanism of resistance is that TAM engages plasma membrane ERα as an agonist, trans-activating members of the epidermal growth factor receptor family including ErbB1 and ErbB2.7 As a result, downstream kinases such as PI3K/AKT enhance growth in part by stimulating steroid receptor coactivator-3 expression and function,8 or the down-regulation of the growth inhibitory. PAX-2 nuclear protein.9 Current efforts to overcome TAM resistance are centered on the inhibition of both ER and ErbB family signaling.7
Numerous, interactive pathways mediate the growth and survival of breast cancers and may underlie resistance to TAM-induced cell death. Here we investigated these mechanisms and demonstrated a novel difference in TAM responsive or resistant cells. The key mediator of the differential responses to TAM is the relative generation of mitochondrial reactive oxygen species (mt-ROS) that induces the intrinsic apoptosis program. TAM engages the mitochondrial ERβ receptor as an antagonist or agonist to differentially regulate manganese superoxide dismutase (MnSOD) activity that impacts ROS levels. Thus, MnSOD is a target to ensure TAM induced apoptosis We further propose this ER pool serves as an ROS rheostat to determine cell fate responses to TAM.
Materials and Methods
Cells and reagents
MCF7 BK and BK-TR cells were from Suzanne Fuqua52, Baylor University, continuously maintained in 100 nM TAM for greater than 1 year. T47D TAM sensitive and resistant cells were from V Craig Jordan, Georgetown University. Materials: SP600125 (JNK1 inhibitor), Rottlerin (PKCδ inhibitor) and GSK3 and c-jun peptide substrates for PKC and JNK kinase assays, respectively, were from (CalBiochem), Rotenone was from EMD4 Biosciences and Mito-Q from Dr. Michael Murphy (Cambridge University). From Santa Cruz, antibodies to ERα (c-terminus, MC-20), blocking ERα or ERβ peptides, PKC, JNK, cytochrome C, Flag, MnSOD, Tom 20, 5'NT, NTF2, and total Bax antibodies were obtained. ERβ (c-terminus) (Invitrogen, 51-7700) and Bax antibodies (BD Bioscience Inc, clone 6A7) were used for Western blot. E2 (Steraloids), PPT (ERα agonist) and DPN (ERβ agonist) (Tocris), and Flag-human-WT and Y34F MnSOD plasmids (Origene) were obtained. Two siRNAs to each ER isoform or a control siRNA were from Santa Cruz.
Apoptosis Assays
For apoptosis studies, TAM-sensitive or resistant cells were subjected to TAM ± other chemicals or steroids, sometimes following siRNA transfection with 2.5 µg each of siRNAs using OLIGOfectamine. Experiments were carried out at 48 hours. Apoptosis was determined by TUNEL kit (Promega, Wisconsin) or Annexin-V expression (Becton-Dickinson, NJ.) as described10.
MTT assay
Cell viability was quantified by a kit that measures mitochondrial dehydrogenase activity in viable cells, based on conversion of MTT to MTT–formazan by mitochondrial enzyme. In brief, MCF7 BK and BK-TR cells were exposed to treatments for up to 72 hours, then incubated with medium containing MTT 1 mg/ml for 4 h at 37 °C. MTT/formazan was extracted overnight at 37 °C with 100 µl extraction buffer. Optical densities at 570 nm were measured.
Cell fraction isolation
Mitochondria were isolated from 70% percent confluent cells, collected by gentle scraping, then centrifuged at 1000 rpm for 5 min to pellet the cells as we described 11,53. Cells were homogenized using the pestle of a tight-fitting Dounce homogenizer until about 90% of the cells were broken, pellets suspended in buffer containing 1 mM EDTA and DNAase and protease inhibitor cocktail (Sigma) and layered onto a discontinues sucrose gradient (1.0–2.5 M). Cytosolic fractions were centrifuged at 100,000×g for 1 hour to obtain membrane fractions The mitochondria were suspended in PBS and protein concentration determined (BCA method).
ER detection in cells
MCF7 cells cultured on coverslips were fixed with paraformaldehyde, permeabilized with 0.2% triton-X, then incubated with antibodies to ERα or ERβ used at 1/50 dilution overnight at 4°C. ER antibodies pre-absorbed with purified ERα or ERβ protein were used as additional negative controls and IgG antibody produced no staining. FITC (Green)-conjugated secondary antibodies (Vector lab Inc.) were added at 1/500 dilution and 2 hours at room temperature for fluorescent visualization. Western blot of cell fractions was previously described 10,18.
Mitochondrial cytochrome C release and membrane potential
MCF7 BK and BK-TR cells were transfected with control or ER isoform siRNAs and the mitochondria isolated. Mitochondrial protein was normalized and the organelles were placed in DMEM and exposed to 5 µM TAM ± 10 µM Mito-Q or SP600125 for 4 hours. Mitochondria were processed for Western blotting of cytochrome C, released into incubation media (supernatant) and remaining in mitochondria. In other experiments, MCF7 BK cells were transfected with one of four siRNAs (control, ERα, ERβ, PKCδ or PKCβ), the cells recovered, then exposed to TAM ± E2 for 4 hours. Cytosol was isolated for Western Blot of cytochrome C after SDS-PAGE separation of proteins. Mitochondrial membrane potentials were measured by the Apo Alert mitochondrial membrane kit (Clontech Laboratories) as we described10.
ROS formation
Cultured cells were loaded with 10 µM 2'7'-dichlorodihydrofluorescin diacetate (CM-H2DCFDA, Molecular Probes) for 1 h prior to TAM ± other chemical including Rotenone, 2.5 µM, or steroid incubation. After oxidation by ROS, CM-H2DCF is converted to green-fluorescing dichlorofluorescein (DCF). Mitochondrial superoxide generation was detected by using a specific mitochondrial superoxide indicator, MitoSOX red (Molecular Probes, Eugene, OR, USA). MitoSOX red is a cell-permeative dye that reacts with superoxide for fluorescence upon binding to mitochondrial DNA. Cells grown on specialized coverslips were loaded in the dark with MitoSOX red (5 µM; 10 min at 37°C) then exposed to TAM. At 6 hours, cells were washed with PBS and superoxide was quantified in a NovaStar spectrofluorometer (excitation wavelengths, 396 and 510 nm, emission measured at 580 nm). Values were fold increase in fluorescence from TAM exposure over control cells. In addition, cells were grown on glass coverslips and observed using a fluorescence microscope (Nikon Eclipse 800) with rhodamine filter, then photographed. Most studies were carried out three times, and the mean and SEMs were analyzed by ANOVA plus Schefe's test at a p<0.05 level.
Bax Dimerization and translocation
MCF7 BK cells at near confluency were synchronized, then subjected to TAM ± E2, Rottlerin, or SP600125 for 6 hours except control cultures.
Mitochondria and the remaining cytoplasm were then isolated for Bax localization as we described10.
PKC and JNK activity
PKCδ activity at 10 minutes incubation was determined as phosphorylation of the protein isoform at the active site with antibodies to tyrosine 311 or total PKCδ protein. PKC isoform kinase activity against peptide substrate was also determined10. JNK activity (15 minutes incubation) was determined similarly by in-vitro assay using c-Jun protein as substrate.54
Interfering RNA to PKC and MnSOD studies
MCF7 BK or BK-TR cells were transfected with 2.5 µg each of the siRNAs and immuno-blots for the proteins were carried out 24–48 hours after transfection as we described.50
Manganese superoxide dismutase activity assay
Cells were incubated with TAM for 4 hours, sometimes following siRNA transfections 48 hours earlier. MnSOD activities were determined using a superoxide dismutase activity assay kit (Cayman Chemical) as we described10. Results from 3 experiments were combined and reported as mean absorption ± standard error.
MnSOD nitration
For MnSOD nitration immuno-blots, cells were incubated with Tam ± E2 or DPN for 2 hours and washed with PBS and suspended in lysis buffer (10 mM Tris + HCl, pH 7.4). Anti-MnSOD monoclonal antibody (Santa Cruz) at 2 µg/ml was preincubated with 25 µl of protein A sepharose (Sigma Chemical) for 3 h. Cell lysates were normalized for protein and 500 µg in 300 µl Chaps buffer per condition were added to MnSOD antibody/sepharose beads at 4°C overnight, then eluted with sample buffer for Western blotting. Specific primary antibody to nitrotyrosine (Abcam) followed by peroxidase-conjugated secondary antibody allowed immuno-blot visualization with the ECL detection system (Amersham).
Nitric Oxide
Nitric oxide synthase activity was determined by measuring the conversion of [3H]L-arginine to [3H]citrulline in whole cells. Cells were cultured for 5 min and the conversion of labeled arginine to citrulline was stopped with experimental medium containing unlabeled L-arginine, 4 mM EDTA, and 1 mM Lcitrulline.
The cells were lysed in 20 mM Tris buffer/EDTA and homogenized by a Dounce homogenizer. Nuclear pellets from the homogenates were obtained by centrifugation at 1000 g for 10 min. Supernatants were layered onto a sucrose gradient producing a top layer (cytosol) and a mitochondrial layer at the sucrose gradient interface. Mitochondrial fractions were sonicated and 1ml aliquots from cytosol or mitochondria were added to a slurry (1:1) of Dowex AG5OWX8 (Bio-Rad), vortexed, and applied to an Econo column (Bio-Rad. Effluents from washings were mixed with 15 ml scintillation fluid and counted.
In-Vivo Studies
The VA Medical Center, Long Beach Animal Care and Use Committee approved all procedures. 6-week old athymic (nu/nu) female mice received an E2 pellet under the skin for 6 days (3 days prior to cell injection) then removed, and a 90-day release Tamoxifen pellet (Innovative Research of America) was inserted. MCF7-BK-TR cells (log phase) in 200 µl of cells in PBS/growth factor poor matrigel (50/50, 5×106 cells) were injected subcutaneously into each flank. Tumor size was measured with caliper weekly. At 5 weeks, lentiviral control shRNA or lentiviral MnSOD shRNA (~5×107 virus, MOI=10) was injected for 2 days into either left or right flank tumors (400 µl total volume). At 8 weeks, the mice were sacrificed and tumors harvested. Tumors were divided, one portion fixed in RNA later and the other stored in liquid nitrogen. Modeling de novo resistance, BK-TR cells were suspended.in (50/50)PBS/matrigel with control or MnSOD lentiviral shRNA(s) (5×106 cells/200 µl and virus, MOI=10), injected subcutaneously into the flanks of 6 female mice. Tumors were grown for 5 weeks under TAM pellet conditions, and Annexin V and caspase-7 cleavage was determined. Fixed tumors were paraffin embedded, cut in 5-um sections, and stained with hematoxylin and eosin. For immuno-staining, tissue sections were deparaffinized, dehydrated, antigen retrieved, and incubated in 0.3% H2O2 in methanol for 30 min. Nonspecific staining was blocked using normal serum (1:50 dilution), then incubation overnight occurred at 4°C with primary antibodies against Annexin V (goat polyclonal IgG; Santa Cruz). A brown positive reaction was visualized after incubating the slides with 3,3–diaminobenzidine for 5 min. Apoptotic cells were counted under the microscope. MnSOD knockdown was determined by immuno-blot. Caspase-7 was determined from frozen tumors, pulverized by mortar and pestle, and the proteins extracted with buffer containing protease inhibitors (Sigma). Proteins were homogenized by polytron, the solution centrifuged at 14000×G for 10 minutes. Proteins were separated by 10% SDSPAGE, followed by immuno-blot using caspase-7 mouse monoclonal antibody (Cell Signaling) to detect endogenous un-cleaved and 30 and 20 kDa fragments of this caspase.
Statistical Analysis
Most in-vitro studies were carried out three times, and the mean and SEMs were analyzed by ANOVA plus Schefe's test at a p<0.05 level of significance. In-vivo studies were carried out in 6 mice and analyzed as noted.
Results
Tamoxifen differentially induces ROS formation and apoptosis in BK and BK-TR cells
TAM induces apoptotic cell death in many cells.3–5 In breast cancer, this action could be mediated through large amounts of ROS as induced by numerous tumor cell stressors.12–14 We first found that the SERM induced a significant increase in ROS generation in MCF7-BK cells relative to control (Figure 1a). This occurred from TAM engaging ER as an antagonist, since TAM's effect was significantly reversed by the ER agonist, 17-β-estradiol (E2). In contrast, TAM did not stimulate ROS formation in BK-TR cells, or HCC-1569 cells (ERα and β null), the latter indicating TAM regulates ROS via engaging ER.
Figure 1.



Tamoxifen (TAM) generates reactive oxygen species (ROS) and apoptosis differentially (a) ROS by DHF fluorescence in TAM sensitive MCF7-BK cells, TAM resistant MCF7-BK-TR cells, and HCC-1569 beast cancer cells (ER null). Bar graph is mean ROS ± SEM from 3 experiments. *p<0.05 for control versus TAM in BK cells; +p<0.05 for TAM versus TAM + E2. (b) TAM differentially induces apoptosis (TUNEL staining) in MCF7-BK cells but not in MCF7BK-TR or ER null cells (HCC-1569). Bar graph shows TUNEL staining, 200 cells in each condition, in three studies. *p<0.05 for control versus TAM; +p<0.05 for TAM versus TAM + E2. (c) TAM-induced ROS is prevented by E2, Mito-Q (10 µM), or Rotenone (2.5 µM). Data are from 3 experiments. *p<0.05 for TAM; +p<0.05 for TAM versus other conditions. Mito Q or Rotenone alone had no effects (not shown). (d) Apoptosis in BK cells from 3 exps; *p<0.05 for TAM, +p<0.05 for TAM versus other conditions. (e) TAM-induces ROS (left) and superoxide generation (right) in the mitochondria of MCF7-BK cells. Data are from 3 exps; *p<0.05 for TAM; +p<0.05 for TAM versus other conditions.
In BK cells, TAM induced a 16-fold increase in apoptosis compared to unexposed(control) cells, significantly reversed by E2 (Figure 1b), and in a concentration-related fashion (Supplemental Figures S1a and S1b). We incubated the cells with 5 µM TAM for many experiments, a concentration that is 1–2 logs less potent than the equivalent of 4-OH TAM that is often used for in vitro studies. No cell death occurred in either BK-TR or HCC-1569 cells exposed to the SERM. ROS generation in normal epithelial cells occurs mainly as a byproduct of oxidative phosphorylation that generates ATP.15 We therefore pretreated BK cells with specific inhibitors of mitochondrial respiratory complexes I (Rotenone) and III (Mito-Q) prior to TAM exposure. Rotenone can induce or inhibit ROS formation,16 but in MCF7 cell culture rotenone inhibits ROS formation.10 The Mito-Q compound is a mitochondrial-targeted derivative of ubiquinone that reduces ROS formation by inhibiting complex III.17
Each inhibitor significantly impaired TAM-generated ROS in BK cells, together reducing ROS by 68% (Figure 1c). The majority of ROS was generated by complex III, as inhibited by Mito-Q, and rotenone and Mito-Q each significantly prevented TAM-induced cell death particularly when combined (Figure 1d). Also, E2 as a receptor agonist reversed both TAM-generated ROS and cell death in BK cells (Figures 1a–d). Thus TAM differentially regulates ROS levels in BK and BK-TR cells underlying cell fate responses. To further support the idea that the majority of ROS originate from mitochondria, we measured total ROS and superoxide levels in BK cells. TAM generated a 4-fold increase in total cellular ROS over control that was substantially inhibited by concomitant Mito-Q or E2 (Figure 1e, left). In the same experiments, TAM stimulated a 3-fold increase specifically in superoxide generation as detected by the Mito-SOX dye, again substantially inhibited by Mito-Q or the sex steroid. These data indicate that the majority of ROS including superoxide originated from the mitochondrial oxidative phosphorylation complexes.
ERβ mediates TAM-induced ROS generation and cell death
We determined the ER isoform involved by knocking down ERα and ERβ in BK and BK-TR cells with specific siRNAs, as previously described 10,18 and shown in Supplemental Figure S2. ERα siRNA had a modest effect on ROS levels and apoptosis induced by TAM in BK cells (Figure 2a). In contrast, ERβ knockdown substantially reversed ROS abundance and cell death in these cells. ROS generation and apoptotic cell death caused by TAM also were antagonized to a much greater extent by 10 nM di-propylnitrile (DPN) (ERβ specific agonist) compared to equimolar propyl-pyrazole-triol (PPT) (ERα specific agonist)19 (Figure 2b).
Figure 2.


ERβ mediates TAM-induced apoptosis in MCF7-BK cells (a) Control, ERα, or ERβ siRNAs were transfected into BK or BK-TR cells for 42 hours, then incubated with TAM for 6 hours. ROS and TUNEL staining were determined at 48 hrs when ER knockdown was maximal. Three experiments (200 cells counted per condition) comprise the bar graph; *p<0.05 for control siRNA versus same plus TAM or TAM + siRNA to ERα; +p<0.05 for TAM + control siRNA versus TAM + ERβ siRNA. (b) DPN but not PPT prevents TAM induced ROS and apoptosis in BK cells after 6 hrs incubation. Bar graphs are three experiments. *p<0.05 for control versus TAM or TAM + PPT; +p<0.05 for TAM versus TAM + DPN. (c) Viability assay in cells incubated ± TAM in 3 exps. (d) Left, BK-TR cells were transfected with siRNAs ± TAM and viability determined at 48 hours. Right, BK and BK-TR cells exposed to TAM ± E2 or E2 alone. *p<0.05 for control versus TAM or versus TAM + ERα siRNA; **p<0.05 for control versus other conditions in BK-TR cells (right); +p<0.05 for TAM versus TAM plus ERβ ± ERαsiRNAs (left) or TAM versus TAM + E2 in BK cells (right); ++p<0.05 for TAM versus E2 in BK cells (right).
These results were confirmed by a second assay, MTT, where TAM induced a progressive loss of viability over three days in BK cells, but promoted the viability of BK-TR cells (Figure 2c). Also, at 48 hours, TAM-enhanced cell viability was significantly prevented only by ERβ but not by ERα siRNA(s) in BK-TR cells (Figure 2d, left). By contrast, TAM-induced loss of viability in BK cells was inhibited by concomitant E2 (Figure 2d, right). However, in BK-TR cells, E2 or TAM comparably improved cell viability, indicating TAM functions here as an agonist.
We carried out similar studies in T47D breast cancer cells sensitive or resistant to TAM.20 TAM-sensitive cells responded with increased ROS production and apoptosis mediated through ERβ but TAM-resistant T47D cells failed to undergo apoptosis to TAM (Supplemental Figure S2).
Distribution of ERα and ERβ in MCF7-BK and BK-TR cells
To further support which ER isoforms and cell pools mediate apoptotic effects, we determined the distribution of ERα and ERβ in MCF7-BK and BK-TR cells. From immuno-fluorescence and Western blot studies, endogenous ERβ is found to be mainly peri-nuclear/mitochondrial (Figures 3a and 3b) but also is present to a lesser extent in nucleus and plasma membrane, comparably in both BK and BK-TR cells. We previously found that in wild type MCF7 cells, ERβ extensively co-localizes with the mitochondrial dye, Mitotracker, and mitochondria are mainly peri-nuclear.10 Thus our finding that ERβ is engaged by TAM as an antagonist to generate mitochondrial-derived ROS that causes apoptosis in BK cells is supported by the localization of this receptor isoform to the mitochondria. However, the differential cell fate response to TAM is unlikely to be due to a large difference in mitochondrial ERβ in the two cell types.
Figure 3.
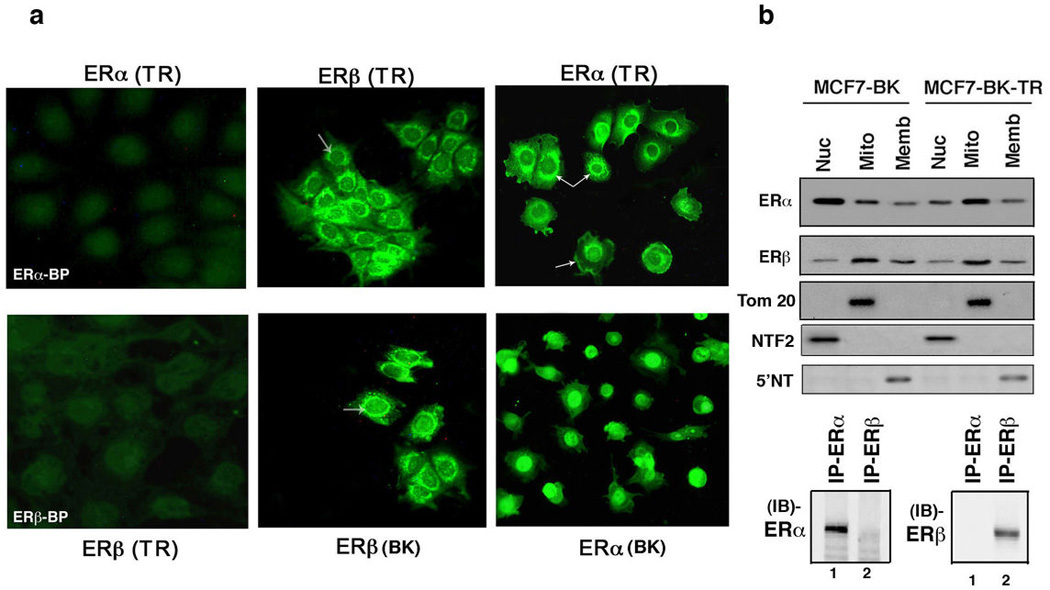
ER isoform distribution in MCF7 cells (a) Imuno-fluorescent microscopy of ERα and ERβ distribution in MCF7-BK and BK-TR cells. Arrows indicate peri-nuclear ERβ (middle panels) and peri-nuclear and membrane ERα (right panels) in each cell type. ERα or ERβ blocking peptides prevented ER isoform detection (left panels). (b), Representative Western blots of the endogenous ER protein isoforms in fractions of BK and BK-TR cells. Tom 20 is a mitochondrial protein, NTF2 is a nuclear protein, 5'NT is an integral membrane protein. The study was done twice and the validation of antibody specificity is shown by immunoprecipitation (IP) followed by immunoblot (IB) with specific ER isoform antibodies.
In contrast, ERα is predominantly extra-nuclear/mitochondrial in BK-TR cells (arrows) but mainly nuclear in BK cells. Since ERα does not appear to be important for the differential cell fate response to TAM, we do not believe these results are important in this regard. We previously validated ER antibody specificity in cells11. Here, we first immuno-precipitated ERα or ERβ from whole cell lysates with the appropriate antibody, then carried out immuno-blots (IB) with each ER isoform antibody (Figure 3b). These results validated that each ER isoform antibody was specific. We also immuno-blotted the cell fractions with antibodies to Tom 20 (mitochondrial), NTF2 (nuclear) and 5'NT (membrane) proteins, as we previously published to establish the purity of cell fractions from MCF7 and other cells.10,11
TAM regulates MnSOD activity that modulates cell fate
Oxidative stress stimulates mitochondrial ROS production21 but several scavenger enzymes prevent cell damage and apoptosis. Especially important is MnSOD that exists exclusively in mitochondria and rapidly reduces superoxide to hydrogen peroxide.22 Genetic deletion of MnSOD in mice is early post-natal lethal,23 indicating ROS quenching by this oxidant scavenger is probably essential. E2 rapidly activates MnSOD activity and prevents radiation-induced cell death in MCF7 cells10 and therefore we postulated that the differential regulation of this enzyme's activity by TAM regulates ROS levels that dictate cell fate.
MnSOD activity was significantly reduced in BK cells but increased in BK-TR cells at 2 hours of exposure to TAM (Figure 4a) and was consistent with the different levels of ROS found in the respective cells. Furthermore, TAM-induced inhibition of MnSOD activity in BK cells would be predicted to increase superoxide levels, as we demonstrated in Figure 1e. MnSOD activity regulation depended upon ERβ and the protein levels of MnSOD were not affected (Figure 4a). We therefore determined how TAM differentially regulates MnSOD activity. The peroxynitrite ROS inactivates MnSOD by nitrosylating tyrosine 34.24,25 TAM stimulated the overall tyrosine nitration of endogenous MnSOD in BK cells but inhibited tyrosine nitration in BK-TR cells, consistent with the differential effects on enzyme activity (Figure 4b). Interestingly, E2 and the ERβ agonist DPN inhibited TAM-stimulated MnSOD nitration in BK cells, indicating TAM acts as an ERβ antagonist. In BK-TR cells, the estrogenic compounds acted like TAM and reduced the control MnSOD nitration, consistent with TAM engaging ERβ as agonist.
Figure 4.

TAM differentially modulates manganese superoxide dismutase activity by nitrosylation via ERβ (a) TAM decreases endogenous MnSOD activity in BK cells (left), but increases activity in BK-TR cells (right). The data are three exps combined. *p<0.05 for control siRNA versus same + TAM or TAM + siRNA to ERα; +p<0.05 for TAM versus TAM + siRNA ERβ. (b) Nitration of endogenous MnSOD by TAM was determined by Western blot using an anti-nitrotyrosine antibody. Molecular weight markers are shown and the representative study was repeated. (c) TAM differentially modulates expressed WT but not mutant MnSOD activity in BK and BK-TR cells. MnSOD activity was from protein immuno-precipitated with anti-Flag antibody in 3 exps. *p<0.05 for control versus TAM (d) TAM differentially modulates nitration of only expressed WT MnSOD. Bar graphs are 3 exps, *p<0.05 for control versus TAM.
To implicate tyrosine 34 nitration in the differential effects of TAM on MnSOD activity, we expressed Flag-tagged, human wild type (WT) or Y34F MnSOD: the latter is a mutant that abrogates nitration at this regulatory site. TAM suppressed Flag-WT MnSOD activity in BK-transfected cells but stimulated activity in BK-TR cells (Figure 4c), similar to endogenous MnSOD regulation (Figure 4a). Importantly, TAM failed to significantly modulate the basal activity of the Y34F mutant enzyme in either cell. Additionally, TAM stimulated the overall tyrosine nitration of Flag-WT MnSOD in BK cells and suppressed nitration in BK-TR cells (Figure 4d). In these two cells, nitration inversely correlated to the differential regulation by TAM of endogenous or expressed WT MnSOD activities (Figures 4a and 4c) and ROS levels (Figure 1). By contrast, TAM did not modulate the overall nitration of Y34F MnSOD in either cell type. These results implicate tyrosine 34 as the specific target for differential nitration and regulation of MnSOD activity by TAM.
To link enhanced MnSOD activity to TAM resistance, we knocked down this enzyme with siRNA as previously validated in MCF7 cells.10 MCF7 BK-TR cells expressing control siRNA and exposed to TAM showed few cells undergoing apoptotic cell death (Figure 5a). In contrast, MnSOD knockdown caused a 15-fold increase in apoptosis but only in the presence of TAM. We then further investigated how TAM modulates MnSOD nitrosylation. Nitric oxide (NO) inhibits cytochrome oxidase in respiratory complexes, increasing peroxynitrite generation that nitrosylates tyrosine 34 of MnSOD.25 TAM stimulated NO formation only in the mitochondria but not the cytoplasm of BK cells and reduced NO formation in BK-TR cell mitochondria (Figure 5b). The inhibitor of mitochondrial Complex III, Mito Q, prevented the differential regulation of NO by TAM, indicating Complex III is involved in modulating NO levels/generation.
Figure 5.

MnSOD knockdown reverses TAM resistance and nitric oxide regulates MnSOD nitrosylation. (A) MCF7-BK-TR cells underwent substantial apoptosis (TUNEL) from TAM in the presence of MnSOD siRNA. The bar graph is 3 exps, *p<0.05 for Tam versus TAM + siRNA to MnSOD. (B) Nitric oxide was determined as arginine to citrulline conversion from 3 exps. Cytoplasm is the cell fraction after mitochondria were removed. *p<0.05 for control versus TAM in mitochondria. +p<0.05 or TAM versus TAM plus Mito-Q. (C) Flag-H-WT or Y34F MnSOD were expressed, the cells incubated with TAM ± the NO inhibitor, LNMMA, 100 µM, for 2 hours. Flag-WT or mutant MnSOD were immuno-precipitated from cell lysates for Western blot with anti-nitrotyrosine antibody. Total MnSOD protein is also shown. Three studies were combined. *p<0.05 for H-MnSOD (wt) versus H-MnSOD (wt) + TAM in BK cells, +p<0.05 for H-MnSOD (wt) + TAM versus H-MnSOD (wt) + TAM + L-NMMA, **p<0.05 for H-MnSOD (wt) versus H-MnSOD (wt) + TAM in BK-TR cells.
By using an NO inhibitor, L-NMMA, we found that TAM caused an NO dependent nitrosylation of expressed WT MnSOD in BK cells (Figure 5c, lanes 3 versus 5), correlating to decreased MnSOD activity (Figures 4a and 4c). TAM or NO did not affect Y34F MnSOD nitrosylation in BK cells. In BK-TR cells, basal nitrosylation of WT but not Y34F MnSOD was strong and TAM substantially inhibited only WT enzyme nitrosylation (Figure 5c, lanes 1 versus 3). The results in BK-TR cells correlate to increased MnSOD activity, and L-NMMA had no effect when added with TAM. Thus, in BK-TR cells, TAM stimulates MnSOD activity by inhibiting NO generation and its target nitrosylation of tyrosine 34. This leads to reduced ROS and cell survival.
Mediators of ROS-induced cell death
In BK cells, TAM induced a decrease in mitochondrial membrane permeability that correlated to stimulating the intrinsic mitochondrial apoptosis pathway (Supplemental Figure S4). We therefore sought to identify targets of ROS that mediated engagement of this pathway.
Protein kinase Cδ (PKCδ) is implicated in mitochondrial-related cell death, and the hydrogen peroxide ROS promotes PKCδ translocation to the organelle to modify pro-apoptotic BCL-2 family activities.26,27 Additionally, c-Jun, N-terminal kinase (JNK) contributes to apoptosis in breast cancer cells.28,29 We found that TAM stimulated both JNK and PKCδ activities in BK but not BK-TR cells, and through mitochondrial ROS since Mito-Q reversed these actions (Figures 6a and b).
Figure 6.
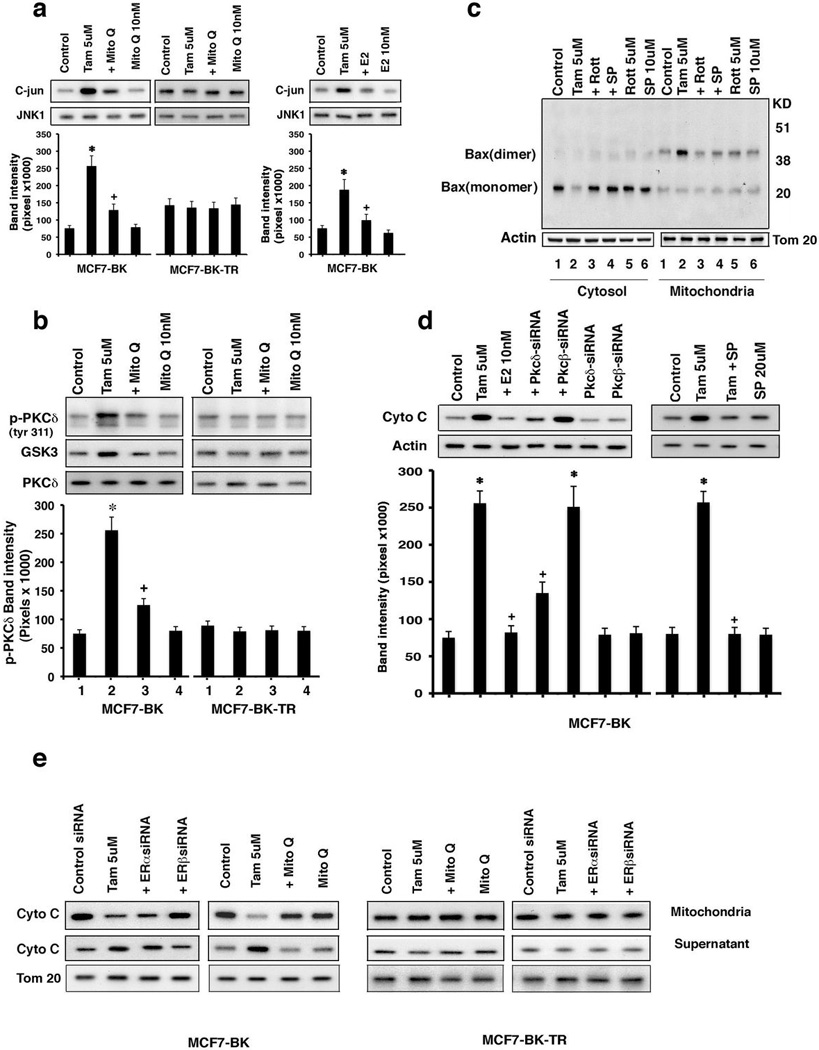
TAM activates JNK and PKCδ in MCF7-BK to induce apoptosis (a) JNK activity or (b) PKCδ kinase assays in cells exposed to TAM ± Mito-Q or E2. Total protein was used as a normalization factor. Bar graphs are JNK activity or PKCδ tyr311 phosphorylation from 3 exps. *p<0.05 for TAM; +p<0.05 for TAM versus TAM + Mito Q or TAM + E2. (c) Bax translocation to mitochondria and dimerization induced by TAM in BK cells but not to TAM ± Rottlerin (PKCδ inhibitor) or SP600125 (JNK inhibitor). The study was repeated. (d) TAM causes cytochrome C release via PKCδ or JNK into BK cell cytosol. Bar graph is 3 exps combined. *p<0.05 for control versus TAM or TAM + PKCβ siRNA; +p<0.05 for TAM versus TAM + E2 or TAM + PKCδ siRNA or TAM + SP. (e) TAM stimulates cytochrome C release through ERβ from isolated mitochondria of BK cells depending upon mitochondrial ROS. Actin serves as a protein normalization control.
A possible target of this signaling is the pro-apoptotic protein Bax. In response to death stimuli, Bax translocates to the mitochondria, undergoes dimerization/activation, and inserts into the outer mitochondrial membrane.30,31 This promotes cytochrome C release into the cytoplasm, stimulating. apoptosome formation.32 Under our control conditions, Bax exists predominantly as a monomer in the cytosol of the TAM-sensitive BK cells (Figure 6c, lanes 1).
TAM caused both the translocation of Bax to the mitochondrial cell fraction and dimerization of this protein (lanes 2) but Rottlerin (PKCδ inhibitor) and SP 600125 (JNK1 inhibitor) significantly prevented both actions (Figure 6c, lanes 2–4). Thus, mtROS-induced kinase activation likely results in BAX participating in the intrinsic (mitochondrial) apoptosis pathway in these cells.
To link PKC to apoptosome formation, we used siRNAs as previously validated.10 Specifically, PKCδ knockdown significantly prevented TAM-induced cytochrome C release into the cytoplasm of BK cells and this action of TAM also depended upon JNK activity (Figure 6d).
At what sub-cellular site does TAM act since ERβ is predominantly in mitochondria (Figure 3)? BK and BK-TR cells were transfected with ERα, ERβ, or control siRNAs, the cells recovered, and mitochondria were isolated and placed into culture. TAM caused secretion of cytochrome C protein from isolated BK mitochondria into the culture media (supernatant) (Figure 6e, left 2 panels), inhibited by only ERβ siRNA. Mito-Q also substantially reversed cytochrome C secretion from BK mitochondria linking TAM-generated ROS in this organelle. In contrast, there were no effects from any treatment on BK-TR cells. (Figure 6e, right 2 panels).
MnSOD serves as an in-vivo target to restore TAM sensitivity
Tamoxifen resistance in women develops after initial tumor response of varying duration or as a primary/de novo event and we modeled both situations in-vivo. At two separate flank sites, BK-TR cells were injected into mice where 90-day, Tam release pellets were inserted and the tumor cells were grown for 8 weeks. At week 5, comparable size tumors were seen in each flank (Figure 7a) and either control or MnSOD lentiviral shRNAs were injected into the palpable tumors on each of 2 successive days. The lentiviral shRNA(s) were first evaluated in cultured BK-TR cells, and the MnSOD shRNA induced substantial apoptosis only in the presence of TAM (Supplemental Figure S5).
Figure 7.


MnSOD knockdown disrupts the in-vivo growth of TAM resistant breast tumors (a), Left, MnSOD knockdown restores sensitivity to the tumor-repressing effects of TAM. Pictures are representative in-vivo tumors from 3 mice. Arrows indicate the tumors. Right, line graphs of measured in-vivo tumor volumes (n=6 mice). *p<0.05 for control versus MnSOD lenti-viral shRNA injected tumors. (b), Ex-vivo tumor volumes (n=6) (left) and pictures of extracted tumors from 3 mice. *p<0.05 for tumors injected with control shRNA versus MnSOD shRNA. (c) MnSOD protein immuno-blots from 3 tumors in each group. Actin is protein control (d) Annexin-V expression in tissue sections of representative tumors. Bar graph is from 300 cells in each of three tissue sections from all tumors, results combined. *p<0.05 for control versus MnSOD shRNA-injected tumors. (e) Cleaved caspase-7 in protein homogenates of representative tumors.
Tumors injected with lentiviral control shRNA showed continued growth at 8 weeks (1690 ± 146 (SEM) mm3)(Fig. 7a). In contrast, lentiviral MnSOD shRNA resulted in substantial tumor regression (480 ± 62 mm3), compared both to 5 weeks growth and to control shRNA-injected tumors at 8 weeks (*p<0.05, n=6) (Figure 7a). The removed tumors showed significantly different weights (Figure 7b), expression of MnSOD protein (Figure 7c), staining for Annexin V (Figure 7d), and cleaved caspase-7 (Figure 7e). Thus in-vivo TAM resistance can be reversed by inhibiting MnSOD, restoring a significant apoptotic response in mature tumors. We also injected BK-TR cells in matrigel into both flanks of nude mice, one site co-injected with lentiviral control shRNA and the other with lentiviral MnSOD shRNA. Control shRNA tumor cells rapidly grew into palpable tumors while MnSOD shRNA tumors very slowly progressed over 5 weeks (Supplemental Figures S6a). MnSOD protein was significantly reduced by the lentiviral shRNA against this protein, tumor weights and size were significantly diminished, and Annexin V expression and caspase-7 cleavage were demonstrated only in MnSOD shRNA tumors (Supplemental Figures S6b–d). We believe that the incomplete knockdown of MnSOD in some tumor cells allowed expansion into the small tumors seen at 5 weeks.
Discussion
In breast cancer, adjuvant therapies generate large amounts of ROS that cause oxidative damage and induce apoptosis.33 By contrast, moderate ROS formation is often required for rapid signaling by growth factor receptors 21,34 including estrogen actions in breast cancer.35 Also, mitochondrial gene mutations that result in moderate ROS levels promote cancer development.36,37 Thus, the tight regulation of ROS underlies cancer fate.
TAM resistance may arise from several mechanisms. One mechanism is that increased membrane ERα and the resulting rapid signaling promotes TAM resistance 7, 38. Interestingly, we found enhanced expression of ERα at the membrane of the BK-TR cells and not in BK cells. However, ERα does not seem to play a role in the adjuvant resistance of BK-TR cells investigated here. From our models, TAM acts in sensitive breast cancer cells as an ERβ antagonist increasing mitochondrial ROS concentration due to diminished MnSOD activity, and activating the intrinsic apoptosis pathway. Although TAM also has cytostatic properties, cytotoxicity is an important mechanism in the effectiveness of this SERM. 3,29 Resistance to TAM occurs from the SERM acting as an ERβ agonist, up-regulating MnSOD activity that diminishes ROS to result in cell survival and growth. Although we don't know why cancer cells become TAM resistant or why TAM engages ERβ as an agonist in this setting, we have uncovered a mechanism that differentiates sensitivity versus resistance and a potential target to reverse the latter. Importantly, knocking down MnSOD impressively changed BK-TR SERM resistance to sensitivity, resulting in a 15-fold increase in TAM induced apoptotic cell death. High MnSOD expression in ER-positive breast tumors was identified as a significant negative prognostic factor for patient survival, associated with the development of distant metastases.39 Insufficient numbers of recurring tumors however did not allow analysis of contributing factors.
In many cell types including breast cancer, others and we have localized a significant expression of endogenous ERβ to mitochondria10,40–42 where in several models, ERβ mediates cell survival.10,43,44 However, the role of ERβ in breast cancer is unclear. In women with this malignancy, ERβ is expressed at relatively low amounts but correlates to TAM resistance45 and the cytoplasmic expression of the endogenous ERβ2 isoform correlates to poor prognosis from this cancer.46
In sensitive cells, TAM caused substantial cell death through mtROS dependent kinase activation that induced the intrinsic (mitochondrial) apoptosis program. Importantly, TAM directly stimulated the release of cytochrome C from isolated mitochondria of sensitive but not resistant cells in both ERβ and ROS dependent fashions. Decreased ROS abundance in TAM-resistant cells resulted primarily from ROS quenching by enhanced MnSOD activity.
We provide the novel insight that TAM differentially regulates MnSOD nitration at tyrosine 34 and hence enzyme activity in BK versus BK-TR cells through the differential generation of NO in mitochondria. TAM/ERβ was found to activate a mitochondrial NO synthase (blocked by L-NAMME) to produce NO that is known to inhibit cytochrome oxidase in respiratory complexes, increasing peroxynitrite generation that nitrosylates tyrosine 34 of MnSOD.47 TAM stimulated NO formation only in the mitochondria of BK cells and reduced NO formation in BKTR cell mitochondria. The inhibitor of mitochondrial Complex III, Mito Q, prevented the differential regulation of NO by TAM, indicating that Complex III is likely also involved in TAM modulating NO levels/generation in some way. In mice, genetic deletion of MnSOD leads to neonatal death due to cell apoptosis.48 Thus, other oxidant scavenging proteins can't fully compensate for the loss of MnSOD, establishing its importance.
Recent studies have linked lysine 122 acetylation of MnSOD to the downregulation of enzyme activity.49 Sirtuin 3 is a de-acetylase that is mainly mitochondrial in location and prevents lysine122 acetylation that results from radiation and other stresses. As a result, sirtuin 3 maintains MnSOD activity to reduce superoxide levels and genomic stress. Interestingly, female sirtuin 3 knockout mice develop ERα and progesterone receptor positive mammary tumors, correlating to the reduced sirtuin 3 levels described in human breast cancer.50 Whether mitochondrial ERβ in breast tumor epithelial cells affects sirtuin 3 expression and function is unknown.
We found that TAM resistant tumors significantly grew in-vivo in the presence of this SERM but underwent substantial regression/apoptosis upon MnSOD knockdown. As shown in-vitro, TAM exposure was required for extensive cell death of MnSOD knockdown cells. This model indicates a possible target to reestablish sensitivity in women whose tumors were initially inhibited by TAM but subsequently acquired resistance. We also found that our model of de novo TAM resistant tumor development was substantially prevented by MnSOD knockdown. This is not accomplished by deleting ERβ since TAM-induced mitochondrial ROS generation and cell death requires engaging this steroid receptor, as seen in sensitive cells. Rather we inhibited the downstream mediator of TAM-resistance, MnSOD, to allow sufficient ROS levels to cause apoptotic cell death. Recently reported, cancer stem cells produce lower levels of ROS than non-tumor initiating, transformed epithelial cells, attributed to increased expression of ROS scavenging systems that also confer radiation resistance.51 Thus augmenting ROS levels may be important in several therapeutic contexts.
In summary, down-regulating mitochondrial MnSOD in ER positive breast cancers may prevent and reverse tumor resistance to adjuvant TAM therapy. The results also support developing a selective ERβ antagonist and delivering this reagent into breast tumors to stimulate the important cytotoxic mechanisms delineated here, perhaps providing therapeutic benefit.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
The work was supported by grants from the Research Service of the Department of Veteran's Affairs, and NIH CA-10036 to ERL and SPORE in Breast Cancer (#P50CA89018), Department of Defense Center of Excellence Grant (#W81XWH-06-1-0590) (VCJ) and Georgetown Lombardi Comprehensive Cancer Spore Grant (#P30CA051008).
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
Supplementary information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Han P, Kang JH, Li HL, Hu SX, Lian HH, Qiu PP, et al. Antiproliferation and apoptosis induced by tamoxifen in human bile duct carcinoma QBC939 cells via upregulated p53 expression. Biochem Biophys Res Commun. 2009;385:251–256. doi: 10.1016/j.bbrc.2009.05.059. [DOI] [PubMed] [Google Scholar]
- 2.Huang CC, Cheng HH, Lin KL, Cheng JS, Tsai JY, Liao WC, et al. Tamoxifen-induced [Ca2+]I rise and apoptosis in corneal epithelial cells. Toxicology. 2009;255:58–64. doi: 10.1016/j.tox.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Moriai R, Tsuji N, Moriai M, Kobayashi D, Watanabe N. Survivin plays as a resistant factor against taxoxifen-induced apoptosis in human breast cancer cells. Breast Cancer Res Treat. 2009;117:261–271. doi: 10.1007/s10549-008-0164-5. [DOI] [PubMed] [Google Scholar]
- 4.Luce A, Courtin A, Levalois C, Altmeyer-Morel S, Romeo PH, Chevillard S, et al. Death receptor pathways mediate targeted and non-targeted effects of ionizing radiations in breast cancer cells. Carcinogenesis. 2009;30:432–439. doi: 10.1093/carcin/bgp008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chalut KJ, Ostrander JH, Giacomelli MG, Wax A. Light scattering measurements of subcellular structure provide noninvasive early detection of chemotherapy-induced apoptosis. Cancer Res. 2009;69:1199–1204. doi: 10.1158/0008-5472.CAN-08-3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Early Breast Cancer Trialists' Collaborative Group. Tamoxifen for early breast cancer: an overview of the randomized trials. Lancet. 1998;351:1451–1467. [PubMed] [Google Scholar]
- 7.Arpino G, Weichmann L, Osborne CK, Schiff R. Cross talk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endo Rev. 2008;29:217–233. doi: 10.1210/er.2006-0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osborne CK, Bardou V, Hopp TA, Chamnes GC, Hilsenbeck SG, Fuqua SA, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and Her2/neu in tamoxifen resistance in breast cancer. J Natl Canc Inst. 2003;95:353–361. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 9.Hurtado A, Holmes KA, Geistlinger TR, Hutcheson IR, Nicholson RI, Brown M, et al. Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature. 2008;456:663–666. doi: 10.1038/nature07483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pedram A, Razandi M, Wallace DC, Levin ER. Functional estrogen receptors in the mitochondria of breast cancer cells. Mol Biol Cell. 2006;17:2125–2137. doi: 10.1091/mbc.E05-11-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pedram A, Razandi M, O'Mahony F, Lubahn D, Levin ER. Estrogen receptor beta inhibits cardiac fibrosis. Mol Endocrinol. 2010;24:2152–2165. doi: 10.1210/me.2010-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao D, Powolny AA, Singh SV. Benzyl isothiocyanate targets mitochondrial respiratory chain to trigger reactive oxygen species dependent apoptosis in human breast cancer cells. J Biol Chem. 2008;283:30151–30163. doi: 10.1074/jbc.M802529200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di X, Shiu RP, Newsham IF, Gewirtz DA. Apoptosis, autophagy, accelerated senescence and reactive oxygen in the response of human breast tumor cells to adriamycin. Biochem Pharmacol. 2009;77:1139–1150. doi: 10.1016/j.bcp.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 14.Chua PJ, Yip GW, Bay BH. Cell cycle arrest induced by hydrogen peroxide is associated with modulation of oxidative stress related genes in breast cancer cells. Exp Biol Med. 2009;234:1086–1094. doi: 10.3181/0903-RM-98. [DOI] [PubMed] [Google Scholar]
- 15.Poyton RO, Ball KA, Castello PR. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol Metab. 2009;20:332–340. doi: 10.1016/j.tem.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Chapman KE, Sinclair SE, Zhuang D, Hassid A, Desai L, Waters CM. Cyclic mechanical strain increases reactive oxygen species production in pulmonary epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2005;289:L834–L841. doi: 10.1152/ajplung.00069.2005. [DOI] [PubMed] [Google Scholar]
- 17.James AM, Cocheme HM, Smith RA, Murphy MP. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J Biol Chem. 2005;280:21295–21312. doi: 10.1074/jbc.M501527200. [DOI] [PubMed] [Google Scholar]
- 18.Pedram A, Razandi M, Levin ER. Nature of functional estrogen receptors at the plasma membrane. Mol Endocrinol. 2006;20:1996–2009. doi: 10.1210/me.2005-0525. [DOI] [PubMed] [Google Scholar]
- 19.Harrington WR, Sheng S, Barnett DH, Petz LN, Katzenellenbogen JA, Katzenellenbogen BS. Activities of estrogen receptor alpha- and beta selective ligands at diverse estrogen responsive gene sites mediating transactivation or transrepression. Mol Cell Endocrinol. 2003;206:13–22. doi: 10.1016/s0303-7207(03)00255-7. [DOI] [PubMed] [Google Scholar]
- 20.Park WC, Liu H, Macgregor-Schafer J, Jordan VC. Deregulation of estrogen induced telomerase activity in tamoxifen-resistant breast cancer cells. Int J Oncol. 2005;27:1459–1466. [PubMed] [Google Scholar]
- 21.Halliwell B. Antioxidant defense mechanisms: from the beginning to the end (of the beginning) Free Radic Res. 1999;31:261–272. doi: 10.1080/10715769900300841. [DOI] [PubMed] [Google Scholar]
- 22.Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature. 2000;407:390–395. doi: 10.1038/35030140. [DOI] [PubMed] [Google Scholar]
- 23.Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr, Dionne L, Lu N, et al. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacMillan-Crow LA, Crow JP, Thompson JA. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry. 1998;37:1613–1622. doi: 10.1021/bi971894b. [DOI] [PubMed] [Google Scholar]
- 25.Yamakura F, Taka H, Fujimura T, Murayama K. Inactivation of human manganese-superoxide dismutase by peroxynitrite is caused by exclusive nitration of tyrosine 34 to 3-nitrotyrosine. J Biol Chem. 1998;273:14085–14089. doi: 10.1074/jbc.273.23.14085. [DOI] [PubMed] [Google Scholar]
- 26.Majumder PK, Mishra NC, Sun X, Bharti A, Kharbanda S, Saxena S, et al. Targeting of protein kinase C delta to mitochondria in the oxidative stress response. Cell Growth Differ. 2001;12:465–470. [PubMed] [Google Scholar]
- 27.Brodie C, Blumberg PM. Regulation of cell apoptosis by protein kinase C delta. Apoptosis. 2003;8:19–27. doi: 10.1023/a:1021640817208. [DOI] [PubMed] [Google Scholar]
- 28.Mingo-Sion AM, Marietta PM, Koller E, Wolf DM, Van Den Berg CL. Inhibition of JNK reduces G2/M transit independent of p53, leading to endoreduplication, decreased proliferation, and apoptosis in breast cancer cells. Oncogene. 2004;23:596–604. doi: 10.1038/sj.onc.1207147. [DOI] [PubMed] [Google Scholar]
- 29.Mandlekar S, Kong AN. Mechanisms of tamoxifen-induced apoptosis. Apoptosis. 2001;6:469–477. doi: 10.1023/a:1012437607881. [DOI] [PubMed] [Google Scholar]
- 30.Gross A, Jockel J, Wei MC, Korsmeyer SJ. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 1998;17:3878–3885. doi: 10.1093/emboj/17.14.3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dejean LM, Martinez-Caballero S, Guo L, Hughes C, Teijido O, Ducret T, et al. Oligomeric Bax is a component of the putative cytochrome c release channel MAC, mitochondrial apoptosis-induced channel. Mol Biol Cell. 2005;16:2424–2432. doi: 10.1091/mbc.E04-12-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jurgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benhar M, Engelberg D, Levitzki A. ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep. 2002;3:420–425. doi: 10.1093/embo-reports/kvf094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aslan M, Ozben T. Oxidants in receptor tyrosine kinase signal transduction pathways. Antioxid Redox Signal. 2003;5:781–788. doi: 10.1089/152308603770380089. [DOI] [PubMed] [Google Scholar]
- 35.Felty Q, Singh KP, Roy D. Estrogen-induced G(1)/S transition of G(0)-arrested estrogen-dependent breast cancer cells is regulated by mitochondrial oxidant signaling. Oncogene. 2005;24:4883–4893. doi: 10.1038/sj.onc.1208667. [DOI] [PubMed] [Google Scholar]
- 36.Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci USA. 2005;102:719–724. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanisahi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–664. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- 38.Fan P, Wang J, Santen RJ, Yue W. Long-term treatment with tamoxifen facilitates translocation of estrogen receptor alpha out of the nucleus and enhances its interaction with EGFR in MVF-7 breast cancer cells. Can Res. 2007;67:1352–1360. doi: 10.1158/0008-5472.CAN-06-1020. [DOI] [PubMed] [Google Scholar]
- 39.Van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 40.Milanesi L, Vasconsuelo A, de Boland AR, Bland R. Expression and subcellular distribution of native estrogen receptor beta in murine C2C12 cells and skeletal muscle tissue. Steroids. 2009;74:489–497. doi: 10.1016/j.steroids.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 41.Chen JQ, Delannoy M, Cooke C, Yager JD. Mitochondrial localization of ERalpha and ERbeta in human MCF7 cells. Am J Physiol Endocrinol Metab. 2004;286:E1011–E1022. doi: 10.1152/ajpendo.00508.2003. [DOI] [PubMed] [Google Scholar]
- 42.Yang SH, Liu R, Perez EJ, Wen Y, Stevens SM, Jr, Valencia T, et al. Mitochondrial localization of estrogen receptor beta. Proc Natl Acad Sci USA. 2004;101:4130–4135. doi: 10.1073/pnas.0306948101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flynn JM, Dimitrijevich SD, Younes M, Skliris G, Murphy LC, Cammarata PR. Role of wild-type estrogen receptor-beta in mitochondrial cytoprotection of cultured normal male and female human lens epithelial cells. Am J Physiol Endocrinol Metab. 2008;295:E637–E647. doi: 10.1152/ajpendo.90407.2008. [DOI] [PubMed] [Google Scholar]
- 44.Hsieh YC, Yu HP, Suzuki T, Choudhry MA, Schwacha MG, Bland KI, et al. Upregulation of mitochondrial respiratory complex IV by estrogen receptorbeta is critical for inhibiting mitochondrial apoptotic signaling and restoring cardiac functions following trauma-hemorrhage. J Mol Cell Cardiol. 2006;41:511–521. doi: 10.1016/j.yjmcc.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 45.Fuqua SA, Cui Y. Estrogen and progesterone receptor isoforms: clinical significance in breast cancer. Breast Cancer Res Treat. 2004;87:S3–S10. doi: 10.1007/s10549-004-1577-4. [DOI] [PubMed] [Google Scholar]
- 46.Speirs V. The evolving role of oestrogen receptor beta in clinical breast cancer. Breast Cancer Res. 2008;10:111. doi: 10.1186/bcr2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamakura F, Taka H, Fujimura T, Murayama K. Inactivation of human manganese-superoxide dismutase by peroxynitrite is caused by exclusive nitration of tyrosine 34 to 3-nitrotyrosine. J Biol Chem. 1998;273:14085–14089. doi: 10.1074/jbc.273.23.14085. [DOI] [PubMed] [Google Scholar]
- 48.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 49.Kim H-S, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010;17:41–51. doi: 10.1016/j.ccr.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tao R, Coleman MC, Pennington JD, Ozden O, Park S-H, Jiang H, et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Molecular Cell. 2010;40:893–903. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barone I, Brusco L, Gu G, Selever J, Beyer A, Covington KR, et al. Loss of Rho GDIα resistance to tamoxifen via effects on estrogen receptor α. J Natl Cancer Inst. 2011;103:538–552. doi: 10.1093/jnci/djr058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trounce I, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 54.Razandi M, Pedram A, Levin ER. Plasma membrane estrogen receptors signal to anti-apoptosis in breast cancer. Mol Endocrinol. 2000;14:1434–1447. doi: 10.1210/mend.14.9.0526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.