

Abstract
Objective
Leukocyte count has been associated with blood pressure, hypertension, and hypertensive complications. We hypothesized that polymorphisms in the CXCL5 gene, which encodes the neutrophilic chemokine ENA-78, are associated with blood pressure in cardiovascular disease (CVD)-free adults and that these polymorphisms are functional.
Methods and results
A total of 192 community-dwelling participants without CVD or risk equivalents were enrolled. Two CXCL5 polymorphisms (−156 G > C (rs352046) and 398 G > A (rs425535)) were tested for associations with blood pressure. Allele-specific mRNA expression in leukocytes was also measured to determine whether heterozygosity was associated with allelic expression imbalance. In −156 C variant carriers, systolic blood pressure (SBP) was 7 mmHg higher than in −156 G/G wild-type homozygotes (131 ± 17 vs. 124 ± 14 mmHg; P = 0.008). Similarly, diastolic blood pressure (DBP) was 4 mmHg higher in −156 C variant carriers (78 ± 11 vs. 74 ± 11 mmHg; P = 0.013). In multivariate analysis of SBP, age, sex, body mass index, and the −156 G > C polymorphism were identified as significant variables. Age, sex, and the −156 G > C SNP were further associated with DBP, along with white blood cells. Allelic expression imbalance and significantly higher circulating ENA-78 concentrations were noted for variant carriers.
Conclusion
CXCL5 gene polymorphisms are functional and associated with variable blood pressure in CVD-free individuals. The role of CXCL5 as a hypertension- and CVD-susceptibility gene should be further explored.
Keywords: CXCL5, ENA-78, Blood pressure, Hypertension, Leukocytes
Introduction
The relationship between inflammation and elevated blood pressure is increasingly being evaluated [1,2]. It has been shown that elevated concentrations of prototypical pro-inflammatory markers such as interleukin-6, C-reactive protein (CRP), and tumor necrosis factor-alpha are associated with increased blood pressure, incidence of hypertension, and the likelihood for hypertensive complications [3-14]. It has been further suggested that this inflammatory-hypertensive relationship results from increased number or activity of common cellular mediators such as white blood cells (WBC) [15,16]. For example, studies have demonstrated elevated WBC count to be associated with increased incident hypertension as well as increased blood pressure within the normal to pre-hypertensive range [17-22].
Although the exact mechanistic relationship between leukocytosis and elevated blood pressure is unknown, it is plausible that low-grade inflammation may be a contributing factor. In this regard, WBC count may be a surrogate marker for increased activation of inflammatory pathways that cause leukocyte recruitment and activation. As such, increased activity of leukocytic chemokines could be related to increased blood pressure.
Epithelial neutrophil activator-78 (ENA-78), a key leukocytic chemokine that is both a neutrophil attractor and activator, has been implicated in many diseases with an inflammatory component (e.g., obesity, diabetes, subclinical atherosclerosis, acute coronary syndromes) [23-32]. We have previously reported that two single nucleotide polymorphisms (SNPs), -156 G > C (rs352046) and 398 G > A (rs425535), in the gene encoding ENA-78 (CXCL5) occur in sites important for transcription and exon splicing [33]. In our previous work, a relationship existed between these SNPs and both plasma concentrations and leukocyte production of the ENA-78 chemokine protein [33]. We then went on to show an association between the CXCL5 -156 G > C polymorphism and worse outcomes in patients with acute coronary syndromes [27]. In the present work, to the extent that ENA-78 is important in neutrophil recruitment and degranulation, we hypothesized that one or both of these polymorphisms (−156 G > C and 398 G > A) could be associated with differences in blood pressure in individuals without established cardiovascular disease (CVD). Specifically, we hypothesized that relatively young individuals without known CVD who were carriers of CXCL5 variant alleles would exhibit higher systolic blood pressure (SBP), diastolic blood pressure (DBP), or pulse pressure (PP) than wild-type homozygotes. Furthermore, to assess whether there was a functional role for these polymorphisms, we measured allele-specific mRNA expression of CXCL5 in leukocytes obtained from CVD-free individuals who were heterozygous for the SNPs at both loci.
Materials and methods
Study population
The study population has been previously described [33]. Briefly, participants were recruited from two sites in the USA and had to be at least 18 years of age without known CVD or CVD-risk equivalents (e.g., diabetes, peripheral vascular disease, 10-year Framingham Risk ≥20%) as defined by National Cholesterol Education Program criteria [34]. Other exclusions were pregnancy, malignancy, substance abuse, and routine use of medications known to affect WBC counts such as systemic steroids and other anti-inflammatory agents. Individuals were excluded from analysis if they were taking anti-hypertensive medications for either cardiovascular or non-cardiovascular indications (e.g., migraine). For blood pressure measurement, subjects were seated for at least 5 min in a quiet, temperature-controlled General Clinical Research Center (GCRC) outpatient clinic room, and two blood pressure measurements were taken at least 5 min apart. The average of the duplicate blood pressure measurements was used for this investigation. Blood samples were obtained from participants enrolled in University of Florida- and Colorado Multiple Institutional Review Board (IRB)-approved studies. All subjects provided written informed consent to specimen and data use in genetic association and related studies.
Genotype and inflammatory biomarker determination
Genomic DNA was isolated from whole blood or buccal cells using previously described methods [35]. CXCL5 genotypes were determined by polymerase chain reaction (PCR) and pyrosequencing (Qiagen, Valencia, CA, USA) as we have previously described [36]. Circulating high-sensitivity CRP (as a non-specific marker of inflammation) was measured by the Shands Hospital Laboratory at the University of Florida and University of Colorado GCRC. ENA-78 concentrations were measured by cytometric fluorescence detection as previously described (Luminex™100 IS system; Luminex Corp., Austin, TX, USA; Fluorokine® MAP Multiplex Human Cytokine Panel A; R&D Systems, Minneapolis, MN, USA) [37]. Samples were stored at −80°C until CRP and ENA-78 detection was performed.
Allele-specific mRNA quantification
To determine whether variant carrier status results in functional changes at the transcriptional level, we quantified allele-specific mRNA transcripts from leukocytes using pyrosequencing-based methodology [38,39]. Specifically, the presence or absence of allelic expression imbalance was determined using leukocytes obtained from 18 individuals who were heterozygotic for both the −156 G > C and 398 G > A polymorphisms. The 398 G > A SNP was chosen as the genetic biomarker in these experiments because it is located in the coding region of CXCL5, while −156 G > C is a promoter polymorphism and as such cannot be quantified at the mRNA level. Because of the near complete linkage of the studied SNPs, we chose individuals who were heterozygotes at both loci so that 398 G > A genotype might serve as a functional surrogate for the upstream promoter locus.
Leukocyte mRNA was prepared from approximately 6 × 106 cells from each individual using the RNeasy mini kit (Qiagen, Valencia, CA, USA). Cells were rinsed, lysed, and homogenized in buffered solutions and subsequently passed through the RNeasy mini column (Qiagen, Valencia, CA, USA). Following a series of washes at room temperature and 15-min incubation with DNase, concentrations were determined by spectrophotometry (NanoDrop Technologies, Wilmington, DE, USA). cDNA was synthesized using approximately 450 ng of cellular RNA from each individual using a High-Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA) per protocol. Conditions for reverse transcription were 25°C for 10 min followed by 37°C for 2 h. cDNA quality was assessed by comparing cDNA and DNA PCR products generated using intron-spanning primers by gel electrophoresis. For allele-specific transcript quantification, subject DNA and cDNA underwent PCR simultaneously using previously described conditions [36]. PCR products obtained for genotype determination (DNA) and transcript quantification (mRNA) were assayed in parallel pyrosequencing reactions to minimize cycle variability. Pyrosequencing analyses were performed in duplicate on three separate PCR amplification products, and the results were pooled for analysis. Peak heights were determined by the pyrosequencing allele quantification algorithm. In genomic DNA, the ratio of 398A:G alleles for DNA in heterozygotes is expected to be approximately 1, whereas significant deviations from this ratio in mRNA would suggest allele expression imbalance associated with the variant allele.
Statistical analyses
Genotype frequencies were determined by allele counting, and departures from Hardy-Weinberg equilibrium were assessed by chi-square analyses. Differences in blood pressure by genotype groups (0, homozygous for common allele; 1, heterozygous or homozygous for variant allele) were compared using one-way ANOVA. Based on the preexisting sample size and prevalence of variant alleles, we had 80% power with a two-sided α of 0.05 to detect a 6-mmHg difference in SBP, 4-mmHg difference in DBP, and 4-mmHg difference in PP between genotype groups. Multiple regression analysis was performed if blood pressure differences were seen across genotype groups. Covariates for multiple regression were chosen through univariate analyses of age, sex, smoking status (0, non-smoker; 1, current smoker), body mass index (BMI), CRP concentration, ENA-78 concentration, and WBC count. Any variable with a P ≤ 0.1 on univariate analysis was entered into the multivariable model. Because of small numbers of individuals within racial groups, analyses could not be performed within racial strata. However, race (0, white; 1, non-white) was included in all multivariable analyses, and a race-by-genotype interaction term was considered in the regression models to avoid spurious associations secondary to racial differences in allele frequency. Multiple regression using step-type selection methods was performed to determine the joint effects of CXCL5 genotypes and clinical variables on SBP, DBP, or PP. All statistical analyses were performed using SPSS (version 11.5, SPSS Inc., Chicago, IL, USA) or SAS (version 9.1, SAS Institute Inc., Cary, NC, USA). A P value < 0.05 was considered statistically significant.
Results
Baseline demographic characteristics are shown in Table 1. Participants were on average 39 ± 12 years old with blood pressures of 126/75 ± 15/11 mm Hg. -156 G > C and 398 G > A genotypes were determined for 189 and 188 of the 192 individuals, respectively. The overall −156 C and 398A minor allele frequencies were both 15%. Variant allele frequencies differed by race whereby the −156 C allele frequency was 14%, 45%, and 11%, and 398A allele frequency was 13%, 46%, and 9% in Caucasians, blacks, and non-black Hispanics, respectively. Genotype distributions satisfied criteria for Hardy-Weinberg equilibrium (data not shown). The two SNPs were in a high degree of linkage disequilibrium with r2 for Caucasian, black, and Hispanic individuals of 0.82, 1.0, and 0.51, respectively, in our study population.
Table 1.
Baseline characteristics
| Characteristic | N = 192 |
|---|---|
| Age (mean ± SD, years) |
39 ± 12 |
| Women (number (%)) |
124 (65) |
| Race/ethnicity (number (%)) |
|
| White |
148 (77) |
| Black |
12 (6) |
| Hispanic |
19 (10) |
| Other |
13 (7) |
| Family heart disease history (number (%)) |
29 (15.1) |
| Smoking (number (%)) |
35 (18) |
| Body mass index (mean ± SD, kg/m2) |
29.6 ± 7 |
| Blood pressure (mean ± SD, mmHg) |
|
| Systolic |
126 ± 15 |
| Diastolic |
75 ± 11 |
| Pulse pressure (mean ± SD, mmHg) |
51 ± 10 |
| Cholesterol (mean ± SD, mg/dLa) |
|
| Total |
201 ± 43 |
| LDL |
118 ± 36 |
| HDL |
55 ± 17 |
| Triglycerides |
139 ± 107 |
| White blood cell count, (mean ± SD, ×109 cells/L) |
6.3 ± 2.0 |
| C-reactive protein (median (range), mg/Lb) |
1.78 (0.1–16.9) |
| ENA-78 (median (range), pg/mLb) | 362 (32.2–3970) |
aTotal, HDL, and triglycerides available in 94% of subjects; LDL available in 92% of subjects. bCRP and ENA-78 available for 88% and 91% of subjects, respectively.
Genotype association with blood pressure
In −156 C variant carriers, SBP was 7-mmHg higher than in −156 G/G wild-type homozygotes (131 ± 17 vs. 124 ± 14 mmHg; P = 0.008). Similarly, DBP was 4-mmHg higher in −156 C variant carriers (78 ± 11 vs. 74 ± 11 mmHg; P = 0.013). PP did not differ between −156 C variant carriers and wild-type homozygotes (53 ± 11 vs. 51 ± 10; P = 0.22). Because of the high degree of linkage disequilibrium between the 398 G > A and −156 G > C SNPs, blood pressure differences were similar when compared by 398 G > A genotypes. For example, SBP was 130 ± 16 and 125 ± 14 mmHg in 398A variant carriers and 398 G/G homozygotes, respectively (P = 0.033); DBP was 78 ± 11 and 74 ± 11 mmHg, respectively (P = 0.038); and PP was not different between groups (53 ± 11 vs. 51 ± 10 mmHg in 398A carriers and 398 G/G homozygotes, respectively; P = 0.362).
Age (P ≤ 0.001), sex (P ≤ 0.008), and BMI (P ≤ 0.002) were common univariate predictors of SBP, DBP, and PP. Furthermore, WBC count (P = 0.10 for SBP; P = 0.076 for DBP) and both CXCL5 polymorphisms (range P = 0.008 to 0.038) were additional predictors of SBP and DBP, while smoking status was associated with SBP alone (P = 0.038). In terms of circulating CRP and ENA-78 levels, both biomarkers were significant for SBP (P = 0.005 for CRP and P = 0.033 for ENA-78) and PP (P = 0.001 for CRP and P = 0.007 for ENA-78) in univariate analyses. Consistent with our previous report, CXCL5 genotype was associated with ENA-78 protein concentrations in the plasma whereby variant carriers at either SNP locus had higher protein concentrations than wild-type homozygotes (P = 0.003; Figure 1).
Figure 1.
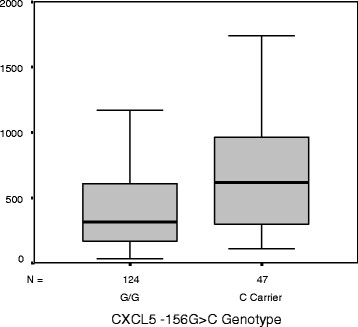
Plasma ENA-78 by CXCL5 -156 G > C genotype. P = 0.003; data were similar for the exon 2 SNP, data not shown (P = 0.001).
In multivariate analysis of SBP, age, sex, BMI, and the CXCL5 -156 G > C promoter polymorphism were identified as significant variables (Table 2). The overall model that included these variables explained 32.5% of the variability in SBP (P < 0.001). Consideration of the 398 G > A polymorphism rather than the −156 G > C promoter SNP resulted in a model in which only age, sex, and BMI were significantly associated with SBP (R2 = 0.301; P < 0.001).
Table 2.
Multivariate predictors of systolic blood pressure in cardiovascular disease-free individuals
| Variable | β | Standard error | Pvalue |
|---|---|---|---|
| Constant |
100 |
4.86 |
<0.0001 |
| Age |
0.313 |
0.094 |
0.001 |
| Sex |
−9.84 |
2.12 |
<0.0001 |
| BMI |
0.637 |
0.160 |
<0.0001 |
| −156 C carrier | 4.93 | 2.30 | 0.034 |
R2 = 0.325; P < 0.0001.
Age, sex, and the −156 G > C SNP were further associated with DBP, along with WBC (Table 3). Consideration of this promoter SNP (model R2 = 0.168; P < 0.0001) was slightly more informative than consideration of the 398 G > A SNP (P = 0.067) in which case age (P < 0.0001), sex (P = 0.001), and WBC (P = 0.02) still remained significant (model R2 = 0.145; P < 0.0001). In multivariable models of PP, only sex (P < 0.004) and BMI (P < 0.0001) were significant (model R2 = 0.247; P < 0.0001).
Table 3.
Multivariate predictors of diastolic blood pressure in cardiovascular disease-free individuals
| Variable | β | Standard error | Pvalue |
|---|---|---|---|
| Constant |
63.13 |
3.42 |
<0.0001 |
| Age |
0.247 |
0.063 |
<0.0001 |
| Sex |
−5.801 |
1.549 |
<0.0001 |
| −156 C carrier |
3.735 |
1.630 |
0.023 |
| WBC | 0.768 | 0.374 | 0.041 |
R2 = 0.168; P < 0.0001.
Allelic expression imbalance
Allele-specific mRNA quantification was performed to determine whether there is a functional basis for the differences seen in blood pressure based on CXCL5 genotypes (see ‘Materials and methods’ section for rationale of 398 G > A as marker SNP). Importantly, there was consistently higher expression of CXCL5 mRNA from the 398A allele compared to the 398 G allele in heterozygous individuals (Figure 2A). For example, individual heterozygotes displayed anywhere from 2.2-fold to 3.4-fold higher expression of 398A variant transcripts compared to the 398 G allele, with a mean ratio of 2.9 (Figure 2B; P = 7.4E-15).
Figure 2.
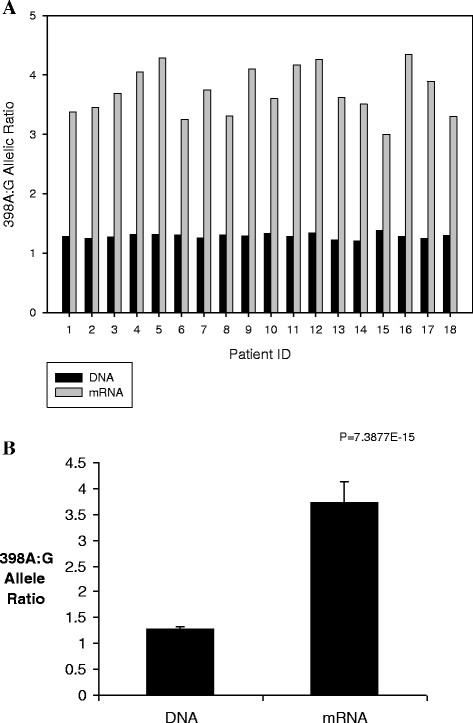
Allele-specific CXCL5 mRNA expression in leukocytes. (A) Allelic mRNA and DNA ratios were measured in 18 cardiovascular disease-free individuals heterozygous for the 398 G > A SNP. The A/G ratios in DNA were close to 1 suggesting equal abundance of both alleles, whereas there was consistently higher expression of mRNA from the 398A allele compared to the 398 G allele. (B) Pooled 398A/G ratios from 18 heterozygous individuals. The sample displayed 2.9-fold higher expression of 398A variant transcripts compared to the 398 G allele (P = 7.4E-15). Data are presented as mean ± SD
Discussion
Accumulating evidence points to a relationship between inflammation and blood pressure. Data suggest that WBC counts are associated with incident hypertension and correlated with blood pressure concentrations. We hypothesized that WBC count is a surrogate for leukocytic chemokine activity and that the CXCL5 gene, which encodes the neutrophil attractor ENA-78, may be an important determinant of blood pressure. We demonstrated a significant, independent relationship between CXCL5 polymorphisms and SBP and DBP in the overall population of CVD-free individuals. Variant carriers of the −156 G > C promoter SNP had 7-mmHg and 4-mmHg higher SBP and DBP, respectively, than those with the wild-type −156 G/G genotype. Because of the epidemiologically significant difference in CVD risk conferred by blood pressure differences of this magnitude, and since variant carriers represent approximately 30% of the population studied, CXCL5 polymorphisms should be considered as a potential novel biomarker of pre-hypertension, hypertension, and CVD risk requiring future study. However, it is important to emphasize that genetic associations are preliminary and will require confirmation in additional populations.
Of particular interest, WBC count (along with traditional variables such as age, sex, smoking status, and BMI) was significantly associated with SBP and DBP in univariate analysis among CVD-free individuals. This finding supports the report by Orakzai et al. that demonstrated a relationship between WBC counts and SBP among nearly 3,500 white individuals without CVD and with SBP < 140 mmHg on entry [20]. It also supports data from other clinical cohorts showing an association between WBC count, major WBC components (e.g., neutrophils), and blood pressure [21,22,40,41]. However, in our analysis WBC count was no longer a significant predictor of SBP when CXCL5 genotype was included in multivariable analysis, suggesting genotype may capture the contribution of inflammation to SBP more effectively than WBC count. WBC did, however, remain a significant predictor of DBP in multivariate analysis, along with age, sex, and CXCL5 -156 G > C genotype.
To determine whether there is any functional basis for an observed association between CXCL5 variant alleles and blood pressure, we performed allele expression imbalance experiments in a subset of participants. The exonic 398 G > A allele was chosen as the genetic marker given its location in the coding region of the mRNA. However, the 398 G/A heterozygous individuals (N = 18) were also heterozygous for the promoter polymorphism, which minimizes confounding of an association by differing genotypes at the upstream locus. It was noted that variant carriers displayed nearly threefold higher expression of variant CXCL5 mRNA transcripts from the 398A allele. This novel finding is consistent with our previous observation that variant carriers exhibited higher plasma and leukocyte-produced ENA-78 than wild-type homozygotes and that the promoter and exonic SNPs occur in transcription factor binding and splicing enhancer sites, respectively [33]. Given that the −156 G > C and 398 G > A SNPs are in near perfect linkage disequilibrium, it is unclear which polymorphism is the causal variant and functionally contributes to the blood pressure phenotype. However, the −156 G > C promoter SNP was more significantly correlated with blood pressure in our study. Further functional studies of these SNPs are warranted.
In addition to genotype and traditional covariates, we included plasma CRP and ENA-78 protein concentrations in our analyses. While CRP and ENA-78 were significantly associated with SBP (and PP) in univariate analyses, they fell out of the models when CXCL5 genotype was included. This suggests that in our analyses, genotype is more significantly associated with the blood pressure phenotype than systemically circulating concentrations of the non-specific inflammatory mediator CRP and the CXCL5 protein product ENA-78. While this observation may appear somewhat contradictory, it can be postulated that CXCL5 gene polymorphisms may be better indicators of chemokine activity at the target organ (e.g., endothelium) level than a measurement in the circulation. Because of trans-acting influences on systemic biomarker expression, polymorphisms in CXCL5 may be more robustly associated with blood pressure. In fact, we have shown a similar finding in a different population for the endothelial nitric oxide synthase gene where NOS3 gene polymorphisms, but not measures of circulating NO activity, were associated with arterial stiffness in children with type 1 diabetes [42,43]. Further support for this observation can be found in a case–control study of the role of ENA-78 in patients with ischemic stroke. Zaremba et al. demonstrated that serum ENA-78 protein concentrations were not different between stroke patients and controls; contrarily, it was demonstrated that ENA-78 concentrations were significantly higher (twofold) in the cerebrospinal fluid of stroke patients compared with controls [44]. Taken in sum, it is possible that genotype more effectively captures the likelihood for local preponderance of chemokine activity than plasma protein level.
In general, there is biological plausibility for the role of CXCL5 in CVD. For example, the protein product of CXCL5, ENA-78, belongs to the same class of chemokines as IL-8, IP-10, and I-TAC, which have been previously implicated in atherosclerotic inflammation [23,45]. ENA-78 has been shown to be chemotactic for neutrophils and stimulate neutrophilic degranulation causing release of myeloperoxidase and generating reactive oxygen species [24,25]. In addition, ENA-78 is involved in platelet-dependent activation of monocytes, displays angiogenic properties, and has been implicated in diseases such as obesity, diabetes, subclinical atherosclerosis, acute coronary syndromes, ischemic stroke, abdominal aortic aneurysm, and thrombosis [27,28,32,44,46-51]. Hypertension is a risk factor for adverse events such as atherosclerosis, stroke, and abdominal aortic aneurysm, and ENA-78 is overexpressed in these situations. We have shown CXCL5 polymorphisms to be associated with ENA-78 concentrations, blood pressure, and prognosis following acute coronary syndromes [27,33]. Thus, the role of CXCL5 in CVD should be further explored. As final hypothesis-generating evidence of a link between the CXCL5 pathway and blood pressure, statins have been hypothesized to have mild antihypertensive effects, and we have shown that atorvastatin reduces ENA-78 production from human endothelial cells in a dose-dependent fashion [52,53]. Our findings, along with existing data, support the need for future investigation of CXCL5 as a hypertension- and CVD-susceptibility gene.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ALB performed statistical analyses and drafted the manuscript. CLA enrolled the study subjects and drafted the manuscript. HA assisted in the molecular genetic studies and provided critical revision of the manuscript. TYL assisted in the molecular genetic studies. GJW assisted in the molecular genetic studies. RSS assisted with the clinical study. IZ conceived the manuscript, enrolled the study subjects, and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Amber L Beitelshees, Email: abeitels@medicine.umaryland.edu.
Christina L Aquilante, Email: christina.aquilante@ucdenver.edu.
Hooman Allayee, Email: hallayee@usc.edu.
Taimour Y Langaee, Email: langaee@cop.ufl.edu.
Gregory J Welder, Email: gw21stunna@gmail.com.
Richard S Schofield, Email: richard.schofield@medicine.ufl.edu.
Issam Zineh, Email: Issam.Zineh@fda.hhs.gov.
Acknowledgments
We thank Dr. Julie A. Johnson for her thoughtful comments regarding the manuscript. We thank Lauren Burt and Lynda Stauffer for their laboratory assistance. This work was supported by American Heart Association Florida/Puerto Rico Affiliate Scientist Development Grant 0435278B, American College of Clinical Pharmacy Kos Dyslipidemia Research and Pharmacotherapy New Investigator Awards, American Association of Colleges of Pharmacy New Investigator Program Award, the University of Colorado Denver General Clinical Research Center (RR00051), and NIH C06 Grant RR17568. ALB is supported by K23 HL091120.
References
- Watson T, Goon PK, Lip GY. Endothelial progenitor cells, endothelial dysfunction, inflammation, and oxidative stress in hypertension. Antioxid Redox Signal. 2008;10:1079–1088. doi: 10.1089/ars.2007.1998. [DOI] [PubMed] [Google Scholar]
- Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, Vinh A, Weyand CM. Inflammation, immunity, and hypertension. Hypertension. 2011;57:132–140. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae CU, Lee RT, Rifai N, Ridker PM. Blood pressure and inflammation in apparently healthy men. Hypertension. 2001;38:399–403. doi: 10.1161/01.HYP.38.3.399. [DOI] [PubMed] [Google Scholar]
- Abramson JL, Weintraub WS, Vaccarino V. Association between pulse pressure and C-reactive protein among apparently healthy US adults. Hypertension. 2002;39:197–202. doi: 10.1161/hy0202.104270. [DOI] [PubMed] [Google Scholar]
- Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM. C-reactive protein and the risk of developing hypertension. JAMA. 2003;290:2945–2951. doi: 10.1001/jama.290.22.2945. [DOI] [PubMed] [Google Scholar]
- Sung KC, Suh JY, Kim BS, Kang JH, Kim H, Lee MH, Park JR, Kim SW. High sensitivity C-reactive protein as an independent risk factor for essential hypertension. Am J Hypertens. 2003;16:429–433. doi: 10.1016/S0895-7061(03)00566-1. [DOI] [PubMed] [Google Scholar]
- Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens. 2005;19:149–154. doi: 10.1038/sj.jhh.1001785. [DOI] [PubMed] [Google Scholar]
- Margolis KL, Manson JE, Greenland P, Rodabough RJ, Bray PF, Safford M, Grimm RH Jr, Howard BV, Assaf AR, Prentice R. Women’s Health Initiative Research Group. Leukocyte count as a predictor of cardiovascular events and mortality in postmenopausal women: the Women’s Health Initiative Observational Study. Arch Intern Med. 2005;165:500–508. doi: 10.1001/archinte.165.5.500. [DOI] [PubMed] [Google Scholar]
- Vazquez-Oliva G, Fernandez-Real JM, Zamora A, Vilaseca M, Badimon L. Lowering of blood pressure leads to decreased circulating interleukin-6 in hypertensive subjects. J Hum Hypertens. 2005;19:457–462. doi: 10.1038/sj.jhh.1001845. [DOI] [PubMed] [Google Scholar]
- Horne BD, Anderson JL, John JM, Weaver A, Bair TL, Jensen KR, Renlund DG, Muhlestein JB. Intermountain Heart Collaborative Study Group. Which white blood cell subtypes predict increased cardiovascular risk? J Am Coll Cardiol. 2005;45:1638–1643. doi: 10.1016/j.jacc.2005.02.054. [DOI] [PubMed] [Google Scholar]
- Lakoski SG, Herrington DM, Siscovick DM, Hulley SB. C-reactive protein concentration and incident hypertension in young adults: the CARDIA study. Arch Intern Med. 2006;166:345–349. doi: 10.1001/archinte.166.3.345. [DOI] [PubMed] [Google Scholar]
- Sesso HD, Wang L, Buring JE, Ridker PM, Gaziano JM. Comparison of interleukin-6 and C-reactive protein for the risk of developing hypertension in women. Hypertension. 2007;49:304–310. doi: 10.1161/01.HYP.0000252664.24294.ff. [DOI] [PubMed] [Google Scholar]
- Wang TJ, Gona P, Larson MG, Levy D, Benjamin EJ, Tofler GH, Jacques PF, Meigs JB, Rifai N, Selhub J, Robins SJ, Newton-Cheh C, Vasan RS. Multiple biomarkers and the risk of incident hypertension. Hypertension. 2007;49:432–438. doi: 10.1161/01.HYP.0000256956.61872.aa. [DOI] [PubMed] [Google Scholar]
- Lakoski SG, Cushman M, Siscovick DS, Blumenthal RS, Palmas W, Burke G, Herrington DM. The relationship between inflammation, obesity and risk for hypertension in the Multi-Ethnic Study of Atherosclerosis (MESA) J Hum Hypertens. 2011;25:73–79. doi: 10.1038/jhh.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman M, Blum A, Baruch R, Kaplan E, Benjamin M. Leukocytes and coronary heart disease. Atherosclerosis. 2004. pp. 1–6. [DOI] [PubMed]
- Karthikeyan VJ, Lip GY. White blood cell count and hypertension. J Hum Hypertens. 2006;20:310–312. doi: 10.1038/sj.jhh.1001980. [DOI] [PubMed] [Google Scholar]
- Friedman GD, Selby JV, Quesenberry CP Jr. The leukocyte count: a predictor of hypertension. J Clin Epidemiol. 1990;43:907–911. doi: 10.1016/0895-4356(90)90074-Y. [DOI] [PubMed] [Google Scholar]
- Shankar A, Klein BE, Klein R. Relationship between white blood cell count and incident hypertension. Am J Hypertens. 2004;17:233–239. doi: 10.1016/j.amjhyper.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Gillum RF, Mussolino ME. White blood cell count and hypertension incidence. The NHANES I Epidemiologic Follow-up Study. J Clin Epidemiol. 1994;47:911–919. doi: 10.1016/0895-4356(94)90195-3. [DOI] [PubMed] [Google Scholar]
- Orakzai RH, Orakzai SH, Nasir K, Santos RD, Rana JS, Pimentel I, Carvalho JA, Meneghello R, Blumenthal RS. Association of white blood cell count with systolic blood pressure within the normotensive range. J Hum Hypertens. 2006;20:341–347. doi: 10.1038/sj.jhh.1001992. [DOI] [PubMed] [Google Scholar]
- Schillaci G, Pirro M, Pucci G, Ronti T, Vaudo G, Mannarino MR, Porcellati C, Mannarino E. Prognostic value of elevated white blood cell count in hypertension. Am J Hypertens. 2007;20:364–369. doi: 10.1016/j.amjhyper.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Tatsukawa Y, Hsu WL, Yamada M, Cologne JB, Suzuki G, Yamamoto H, Yamane K, Akahoshi M, Fujiwara S, Kohno N. White blood cell count, especially neutrophil count, as a predictor of hypertension in a Japanese population. Hypertens Res. 2008;31:1391–1397. doi: 10.1291/hypres.31.1391. [DOI] [PubMed] [Google Scholar]
- Walz A, Burgener R, Car B, Baggiolini M, Kunkel SL, Strieter RM. Structure and neutrophil-activating properties of a novel inflammatory peptide (ENA-78) with homology to interleukin 8. J Exp Med. 1991;174:1355–1362. doi: 10.1084/jem.174.6.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz A, Schmutz P, Mueller C, Schnyder-Candrian S. Regulation and function of the CXC chemokine ENA-78 in monocytes and its role in disease. J Leukoc Biol. 1997;62:604–611. doi: 10.1002/jlb.62.5.604. [DOI] [PubMed] [Google Scholar]
- Walz A, Strieter RM, Schnyder S. Neutrophil-activating peptide ENA-78. Adv Exp Med Biol. 1993;351:129–137. doi: 10.1007/978-1-4615-2952-1_14. [DOI] [PubMed] [Google Scholar]
- Wislez M, Philippe C, Antoine M, Rabbe N, Moreau J, Bellocq A, Mayaud C, Milleron B, Soler P, Cadranel J. Upregulation of bronchioloalveolar carcinoma-derived C-X-C chemokines by tumor infiltrating inflammatory cells. Inflamm Res. 2004;53:4–12. doi: 10.1007/s00011-003-1215-3. [DOI] [PubMed] [Google Scholar]
- Zineh I, Beitelshees AL, Welder GJ, Hou W, Chegini N, Wu J, Cresci S, Province MA, Spertus JA. Epithelial neutrophil-activating peptide (ENA-78), acute coronary syndrome prognosis, and modulatory effect of statins. PLoS One. 2008;3:e3117. doi: 10.1371/journal.pone.0003117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavey C, Lazennec G, Lagarrigue S, Clape C, Iankova I, Teyssier J, Annicotte JS, Schmidt J, Mataki C, Yamamoto H, Sanches R, Guma A, Stich V, Vitkova M, Jardin-Watelet B, Renard E, Strieter R, Tuthill A, Hotamisligil GS, Vidal-Puig A, Zorzano A, Langin D, Fajas L. CXC ligand 5 is an adipose-tissue derived factor that links obesity to insulin resistance. Cell Metab. 2009;9:339–349. doi: 10.1016/j.cmet.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez M, Miquel R, Colmenero J, Moreno M, Garcia-Pagan JC, Bosch J, Arroyo V, Ginès P, Caballería J, Bataller R. Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology. 2009;136:1639–1650. doi: 10.1053/j.gastro.2009.01.056. [DOI] [PubMed] [Google Scholar]
- Yang Z, Zhang Z, Wen J, Wang X, Lu B, Zhang W, Wang M, Feng X, Ling C, Wu S, Hu R. Elevated serum chemokine CXC ligand 5 levels are associated with hypercholesterolemia but not a worsening of insulin resistance in Chinese people. J Clin Endocrinol Metab. 2010;95:3926–3932. doi: 10.1210/jc.2009-2194. [DOI] [PubMed] [Google Scholar]
- Keeley EC, Moorman JR, Liu L, Gimple LW, Lipson LC, Ragosta M, Taylor AM, Lake DE, Burdick MD, Mehrad B, Strieter RM. Plasma chemokine levels are associated with the presence and extent of angiographic coronary collaterals in chronic ischemic heart disease. PLoS One. 2011;6:e21174. doi: 10.1371/journal.pone.0021174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Yang Z, Lu B, Li Q, Ye Z, He M, Huang Y, Wang X, Zhang Z, Wen J, Liu C, Qu S, Hu R. Serum CXC ligand 5 is a new marker of subclinical atherosclerosis in type 2 diabetes. Clin Endocrinol (Oxf) 2011;75(6):766–770. doi: 10.1111/j.1365-2265.2011.04119.x. [DOI] [PubMed] [Google Scholar]
- Zineh I, Aquilante CL, Langaee TY, Beitelshees AL, Arant CB, Wessel TR, Schofield RS. CXCL5 gene polymorphisms are related to systemic concentrations and leukocyte production of epithelial neutrophil-activating peptide (ENA-78) Cytokine. 2006;33:258–263. doi: 10.1016/j.cyto.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III) JAMA. 2001;285:2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- Andrisin TE, Humma LM, Johnson JA. Collection of genomic DNA by the noninvasive mouthwash method for use in pharmacogenetic studies. Pharmacotherapy. 2002;22:954–960. doi: 10.1592/phco.22.12.954.33598. [DOI] [PubMed] [Google Scholar]
- Zineh I, Welder GJ, Langaee TY. Development and cross-validation of sequencing-based assays for genotyping common polymorphisms of the CXCL5 gene. Clin Chim Acta. 2006;370:72–75. doi: 10.1016/j.cca.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Zineh I, Welder GJ, DeBella AE, Arant CB, Wessel TR, Schofield RS. Atorvastatin effect on circulating and leukocyte-produced CD40 ligand concentrations in people with normal cholesterol levels: a pilot study. Pharmacotherapy. 2006;26:1572–1577. doi: 10.1592/phco.26.11.1572. [DOI] [PubMed] [Google Scholar]
- Shiao YH, Crawford EB, Anderson LM, Patel P, Ko K. Allele-specific germ cell epimutation in the spacer promoter of the 45 S ribosomal RNA gene after Cr(III) exposure. Toxicol Appl Pharmacol. 2005;205:290–296. doi: 10.1016/j.taap.2004.10.017. [DOI] [PubMed] [Google Scholar]
- Sun A, Ge J, Siffert W, Frey UH. Quantification of allele-specific G-protein beta3 subunit mRNA transcripts in different human cells and tissues by Pyrosequencing. Eur J Hum Genet. 2005;13:361–369. doi: 10.1038/sj.ejhg.5201334. [DOI] [PubMed] [Google Scholar]
- Tian N, Penman AD, Mawson AR, Manning RD Jr, Flessner MF. Association between circulating specific leukocyte types and blood pressure: the atherosclerosis risk in communities (ARIC) study. J Am Soc Hypertens. 2010;4:272–283. doi: 10.1016/j.jash.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeli F, Angeli E, Ambrosio G, Mazzotta G, Cavallini C, Reboldi G, Verdecchia P. Neutrophil count and ambulatory pulse pressure as predictors of cardiovascular adverse events in postmenopausal women with hypertension. Am J Hypertens. 2011;24:591–598. doi: 10.1038/ajh.2011.18. [DOI] [PubMed] [Google Scholar]
- Haller MJ, Pierce GL, Braith RW, Silverstein JH. Serum superoxide dismutase activity and nitric oxide do not correlate with arterial stiffness in children with type 1 diabetes mellitus. J Pediatr Endocrinol Metab. 2006;19:267–269. doi: 10.1515/jpem.2006.19.3.267. [DOI] [PubMed] [Google Scholar]
- Zineh I, Beitelshees AL, Haller MJ. NOS3 polymorphisms are associated with arterial stiffness in children with type 1 diabetes. Diabetes Care. 2007;30:689–693. doi: 10.2337/dc06-1697. [DOI] [PubMed] [Google Scholar]
- Zaremba J, Skrobanski P, Losy J. The level of chemokine CXCL5 in the cerebrospinal fluid is increased during the first 24 hours of ischaemic stroke and correlates with the size of early brain damage. Folia Morphol (Warsz) 2006;65:1–5. [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2002. pp. 868–874. [DOI] [PubMed]
- Damas JK, Gullestad L, Ueland T, Solum NO, Simonsen S, Froland SS, Aukrust P. CXC-chemokines, a new group of cytokines in congestive heart failure–possible role of platelets and monocytes. Cardiovasc Res. 2000;45:428–436. doi: 10.1016/S0008-6363(99)00262-X. [DOI] [PubMed] [Google Scholar]
- Hamid C, Norgate K, D’Cruz DP, Khamashta MA, Arno M, Pearson JD, Frampton G, Murphy JJ. Anti-beta2GPI-antibody-induced endothelial cell gene expression profiling reveals induction of novel pro-inflammatory genes potentially involved in primary antiphospholipid syndrome. Ann Rheum Dis. 2007;66:1000–1007. doi: 10.1136/ard.2006.063909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm T, Damas JK, Holven K, Nordoy I, Brosstad FR, Ueland T, Währe T, Kjekshus J, Frøland SS, Eiken HG, Solum NO, Gullestad L, Nenseter M, Aukrust P. CXC-chemokines in coronary artery disease: possible pathogenic role of interactions between oxidized low-density lipoprotein, platelets and peripheral blood mononuclear cells. J Thromb Haemost. 2003;1:257–262. doi: 10.1046/j.1538-7836.2003.00065.x. [DOI] [PubMed] [Google Scholar]
- Middleton RK, Lloyd GM, Bown MJ, Cooper NJ, London NJ, Sayers RD. The pro-inflammatory and chemotactic cytokine microenvironment of the abdominal aortic aneurysm wall: a protein array study. J Vasc Surg. 2007;45:574–580. doi: 10.1016/j.jvs.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Hasani Ranjbar S, Amiri P, Zineh I, Langaee TY, Namakchian M, Heshmet R, Sajadi M, Mirzaee M, Rezazadeh E, Balaei P, Tavakkoly Bazzaz J, Gonzalez-Gay MA, Larijani B, Amoli MM. CXCL5 gene polymorphism association with diabetes mellitus. Mol Diagn Ther. 2008;12:391–394. doi: 10.1007/BF03256304. [DOI] [PubMed] [Google Scholar]
- Turner NA, Das A, O’Regan DJ, Ball SG, Porter KE. Human cardiac fibroblasts express ICAM-1, E-selectin and CXC chemokines in response to proinflammatory cytokine stimulation. Int J Biochem Cell Biol. 2011;43:1450–1458. doi: 10.1016/j.biocel.2011.06.008. [DOI] [PubMed] [Google Scholar]
- Milionis HJ, Liberopoulos EN, Achimastos A, Elisaf MS, Mikhailidis DP. Statins: another class of antihypertensive agents? J Hum Hypertens. 2006;20:320–335. doi: 10.1038/sj.jhh.1002001. [DOI] [PubMed] [Google Scholar]
- Zineh I, Luo X, Welder GJ, Debella AE, Wessel TR, Arant CB, Schofield RS, Chegini N. Modulatory effects of atorvastatin on endothelial cell-derived chemokines cytokines, and angiogenic factors. Pharmacotherapy. 2006;26:333–340. doi: 10.1592/phco.26.3.333. [DOI] [PubMed] [Google Scholar]