

Abstract
Choroid plexus epithelial cells (CPECs) have essential developmental and homeostatic roles related to the CSF and blood–CSF barrier they produce. Accordingly, CPEC dysfunction has been implicated in many neurological disorders, such as Alzheimer's disease, and transplant studies have provided proof-of-concept for CPEC-based therapies. However, such therapies have been hindered by the inability to expand or generate CPECs in culture. During development, CPECs differentiate from preneurogenic neuroepithelial cells and require bone morphogenetic protein (BMP) signaling, but whether BMPs suffice for CPEC induction is unknown. Here we provide evidence for BMP4 sufficiency to induce CPEC fate from neural progenitors derived from mouse embryonic stem cells (ESCs). CPEC specification by BMP4 was restricted to an early time period after neural induction in culture, with peak CPEC competency correlating to neuroepithelial cells rather than radial glia. In addition to molecular, cellular, and ultrastructural criteria, derived CPECs (dCPECs) had functions that were indistinguishable from primary CPECs, including self-assembly into secretory vesicles and integration into endogenous choroid plexus epithelium following intraventricular injection. We then used BMP4 to generate dCPECs from human ESC-derived neuroepithelial cells. These findings demonstrate BMP4 sufficiency to instruct CPEC fate, expand the repertoire of stem cell-derived neural derivatives in culture, and herald dCPEC-based therapeutic applications aimed at the unique interface between blood, CSF, and brain governed by CPECs.
Introduction
Choroid plexus epithelial cells (CPECs) comprise the epithelial compartment of the choroid plexus (CP), the papillary tissue that resides in each of the brain's four ventricles. CPECs have unique structural features, such as extensive apical microvilli and tight junctions unlike those of other neural cell types (Emerich et al., 2005b), and provide important functions. First, they protect the CNS from toxins via direct absorption and their tight junctions, which constitute the blood–CSF barrier (Emerich et al., 2005a). Second, CPECs secrete the CSF, 400–600 ml/day in humans (Emerich et al., 2005b), which contains numerous signaling, hormonal, nutritive, and neurotrophic molecules (Serot et al., 2000; Chodobski and Szmydynger-Chodobska, 2001; Lehtinen et al., 2011) vital for normal CNS development, homeostasis, function, and health.
Although not widely appreciated, CPEC dysfunction is associated with many CNS disorders, including schizophrenia, multiple sclerosis, stroke, and especially Alzheimer's disease (AD) (Weller, 1998; Serot et al., 2003; Sousa et al., 2007). For example, CPECs absorb the amyloid β (Aβ) peptide (Emerich et al., 2005b) and produce transthyretin (Ttr), which abrogates Aβ toxicity (Schwarzman et al., 1994; Costa et al., 2008) and reduces AD pathology in mice (Choi et al., 2007; Buxbaum et al., 2008). CPEC dysfunction has even been hypothesized to cause AD (Silverberg et al., 2003; Maurizi, 2010). Together with transplant studies in animal models (Emerich et al., 2005a; Skinner et al., 2006), these attributes of CPECs make a compelling case for developing CPEC replacements, transplants, and drug screens for enhancing CPEC functions or bypassing the blood–CSF barrier. As long-lived postmitotic secretory cells, engineered CPECs could also be ideal stable vehicles to deliver proteins (Johanson et al., 2005).
This clinical potential has been hindered, however, by the inability to generate or expand CPECs in culture. Studies in mammals indicate that adult CPECs rarely turnover in vivo (Chauhan and Lewis, 1979; Kaplan, 1980). Embryonic stem cells (ESCs) represent a potential cell source in vitro, and bone morphogenetic protein (BMP) signaling has been implicated in CPEC induction in vivo. BMPs are expressed at high levels in the roof plate, which induces and directly differentiates into CPECs (Furuta et al., 1997; Currle et al., 2005). The roof plate and BMP receptors are also required for CPEC specification in mice (Currle et al., 2005; Fernandes et al., 2007), and BMP4 alone can partially rescue CPECs in roof plate-ablated explants (Cheng et al., 2006). BMP4 can also upregulate CPEC gene expression in cortical precursors (Hu et al., 2008) and in mouse ESC-derived neural aggregates (Eiraku et al., 2008), although it is unclear whether bona fide CPEC differentiation occurred in these settings.
Here we provide evidence for BMP4 sufficiency to induce CPEC differentiation from mouse and human ESC-derived neural progenitors. This induction, which occurs with low to moderate efficiency in the presence of BMP4 alone, is restricted to critical periods enriched for early neuroepithelial cells (NECs). In addition, derived CPECs (dCPECs) induced from mouse ESCs had self-assembly, secretory, and engraftment properties indistinguishable from those of primary CPECs, and dCPECs could be similarly induced from human ESC-derived NECs using BMP4.
Materials and Methods
Mouse ESC line derivation.
The M1 mouse ESC line was generated by Robin Wesselschmidt (Primogenix) from blastocysts derived from R26CreER/CreER;Lhx2cKO/cKO × R26+/+;Lhx2cKO/cKO matings (C57BL/6J with minor CD-1 background) (Mangale et al., 2008). The M1 line genotype is R26CreER/+Lhx2cKO/cKO (for the studies reported here, Lhx2 inactivations were not performed; M1 was used solely as a control line). Lines were expanded by the University of California Irvine (UCI) Transgenic Mouse Facility on mitomycin C-treated mouse embryonic fibroblasts (MEFs; Millipore) in 0.1% gelatin-coated plates in Low Osmo DMEM (Hyclone) with 15% ESC-qualified fetal bovine serum (Hyclone), 5% serum replacement (Hyclone), 2 mm l-glutamine (Life Technologies), 1000 U/ml leukemia inhibitory factor (LIF) (Millipore), 0.1 mm β-mercaptoethanol (Sigma-Aldrich), and penicillin-streptomycin (Life Technologies). The M2 line (TTR::RFP hemizygous) and seven other lines (1 wild-type, 2 CAG::H2B-GFP hemizygous, and 4 TTR::RFP;CAG::H2B-GFP compound hemizygous) were derived from TTR::RFP × CAG::H2B-GFP blastocysts (mostly CD1 background with C57BL/6J and ICR) (Hadjantonakis and Papaioannou, 2004; Kwon and Hadjantonakis, 2009) by the UCI Transgenic Mouse Facility using the 2i method (Li et al., 2008) with 0.1% gelatin-coated plates in ESGRO complete basal media (Millipore), 0.1% embryonic stem cell-qualified FBS, glycogen synthase kinase inhibitor 3 beta (CHIR99021, Stemgent), MEK inhibitor (PD0325901, Stemgent), and 1000 U/ml LIF. Before all experiments, mouse ESCs were cultured for at least two passages after thawing. Mouse ESCs were expanded and maintained at 5% CO2 with daily media changes and every other day splits. Experiments were conducted on cells between passage numbers 11 and 40. The M1 and M2 lines were confirmed for pluripotency (Oct4, Sox2, and alkaline phosphatase staining approaching 100%), normal chromosome numbers, and mycoplasma negativity.
Mouse ESC culture and differentiation.
Neural differentiation in “SFEBq” (serum-free floating culture of embryoid body-like aggregate with quick reaggregation) aggregates was performed as described previously (Eiraku et al., 2008). Briefly, mouse ESCs were dissociated to single cells using TrypLE Express (Life Technologies), placed onto gelatin-coated plates for 2 h to reduce MEF load, then plated and spun at varying concentrations onto ultralow attachment U-bottom 96-well plates (#7007, Corning) to form aggregates. Differentiation media (Eiraku et al., 2008) was replaced with fresh media containing 0.5–50 ng/ml BMP4 (R&D Systems) for another 5–7 days in vitro (DIV) with (M1) or without every other day BMP4 replacement (M2). (Note: We found that one-time BMP4 addition was equally effective to every other day replacement at inducing CPEC gene expression in M1 cells; data not shown.) Neural differentiations via neural rosette (NR) formation were also attempted (Elkabetz et al., 2008), but these took longer (11 DIV total), were more variable, and yielded lower neural induction efficiency in our hands (∼50% Nestin positivity; data not shown). A combined SFEBq/NR method, in which 5-day SFEBq aggregates were transferred into adherent NR conditions for another 2 DIV resulted in consistent and efficient neural induction (∼95% Nestin positivity; data not shown), but involved more manipulations than the SFEBq method alone.
Human ESC culture and differentiation.
Human ESCs (H1 and H9 lines; WiCell Research Institute) were cultured and maintained using standard procedures (Thomson et al., 1998) on 0.1% gelatin-coated plates with mitomycin C-treated MEFs (Millipore) in KO DMEM/F12 (Life Technologies) with 20% Knockout Serum Replacement (Life Technologies), 2 mm GlutaMax (Life Technologies), 4 ng/ml FGF2 (Life Technologies), 0.055 mm β-mercaptoethanol (Life Technologies), and nonessential amino acids (NEAAs, Life Technologies). Human ESCs were maintained at 5% CO2 with daily media changes and were passaged approximately weekly. Human ESC lines were confirmed for pluripotency (Oct4, Sox2, and alkaline phosphatase staining approaching 100%), normal chromosome numbers, and mycoplasma negativity. Experiments were performed on cells between passages 30 and 70.
Neural inductions via NR formation were performed as described previously (Zhang et al., 2001). Human ESC colonies were partially dissociated with 1 mg/ml dispase, then plated in suspension to form embryoid bodies (EBs) for 3 DIV. EBs were then transferred to neural induction media [DMEM/F12 (Life Technologies), N2 (Life Technologies), NEAA, heparin (2 μg/ml), and FGF2 (20 ng/ml)] for another 2 DIV. On day 6, EBs were transferred to adherent poly-d-lysine/laminin-coated plates with neural induction media, which was fully replaced every other day. On day 16, media was fully replaced with neural induction media containing BMP4 (0.5–15 ng/ml), with full media replacement every other day. For neural induction via SFEBq aggregates, human ESC colonies were completely detached from the well with 1 mg/ml dispase, then plated on 0.1%-gelatin-coated wells in culture media with 10 μm ROCK (Rho kinase) inhibitor (Y27632, Reagents Direct) to remove MEF load for 1 h. Colonies were then collected and treated with 0.25% trypsin-EDTA (Life Technologies) for single cell dissociation. Cells were counted and plated in human SFEBq media (Eiraku et al., 2008) onto ultralow attachment U-bottom 96-well plates (Corning) with 10 μm ROCK inhibitor. SFEBq media with ROCK inhibitor was partially replaced (∼⅔) at 3 DIV, with similar media replacement every third day thereafter without ROCK inhibitor. At 21 DIV, SFEBq media with varying BMP4 concentrations was added for 2 weeks with full replacement of BMP4-containing media every third day until cultures were stopped at 35 DIV.
CPEC vesicle culture.
Primary CP cells or mouse SFEBq-derived cells were cultured on Matrigel as previously described (Thomas and Dziadek, 1993; Swetloff et al., 2006). Briefly, Matrigel (BD Biosciences) was mixed 1:1 with DMEM (Life Technologies) and incubated for at least 30 min at 37°C. BMP4-treated and control mouse SFEBq aggregates at 10–12 DIV (5 DIV neural induction followed by 5–7 DIV CPEC induction) were dissociated with TrypLE Express (Life Technologies) and plated at 500,000–600,000 cells/well onto Matrigel-coated six-well plates in MEF media (DMEM with 10% FBS, 2 mm l-glutamine, NEAA, and sodium pyruvate). After 5 (primary CPEC and M1) or 8 DIV (M2), selected cultures were treated with 100 μm acetazolamide (Sigma-Aldrich) and 5 mm ouabain (Sigma-Aldrich) for 24 h, followed by washout 2–3× with fresh media. Times for vesicle recovery following washout were similar to previous descriptions of primary CPEC vesicles (Swetloff et al., 2006).
Stereotaxic injections and analysis.
For Ara-C treatments, 10–12 day mouse SFEBq aggregates were dissociated and cultured on poly-d-lysine/laminin in media (DMEM with 10% embryonic stem cell-qualified FBS, 2 mm l-glutamine, 1 mm Na Pyruvate, and 0.1 mm NEAA) containing 20 μm cytosine arabinoside (Ara-C) (Matsumoto et al., 2010; Menheniott et al., 2010) for 7 DIV, washed to remove Ara-C, then cultured for an additional 7 DIV. On the day of injection, cells were dissociated with TrypLE Express (Life Technologies), incubated with 8 μm Vybrant carboxyfluorescein diacetate-succinimidyl ester (CFDA-SE) Cell Tracker dye (Life Technologies) for 15 min at 37°C, resuspended in MEF media for 30 min at 37°C, then resuspended in HBSS at 500–1000 cells/μl.
Primary CP cells from lateral and fourth ventricles were prepared from postnatal day 3–5 CAG::H2B-GFP or CD1 mice (Charles River) followed by Vybrant CFDA-SE dye labeling as described above. CP tissue was dissected, digested in type II collagenase (2.5 mg/ml; Life Technologies) in HBSS for 15–20 min followed by TrypLE Express for 15–20 min, then resuspended in HBSS at 12,000–40,000 cells/μl. Five thousand (SFEBq-derived) or 120–200K cells (primary CP) were unilaterally injected via Hamilton syringe with 30-gauge needle into the anterior-midregion of the left lateral ventricle (anteroposterior, −0.1 mm; mediolateral, −1.0 mm; dorsoventral, −2.5 mm from bregma) of 6–8-week-old CD1 female mice. One to two days after injection, recipients were killed by cardiac perfusion with 4% PFA/PBS, brains were postfixed 1 h in 4% PFA/PBS on ice, then transferred to 30% sucrose/PBS overnight at 4°C. Serial 30 μm coronal cryosections of the 2–2.5 mm region spanning the injection site were processed and analyzed. All procedures involving live mice were performed according to approved Institutional Animal Care and Use Committee protocols and guidelines at UCI.
qRT-PCR, histochemistry, immunostaining, in situ hybridization, and electron microscopy.
qRT-PCR was performed as described previously (Currle et al., 2005; Hu et al., 2008) on a StepOne Plus (Applied Biosystems) using 18S normalization, Excel for statistical tests (two-tailed t tests assuming equal variance), and KaleidaGraph for graphing. All primers and amplicons were validated as described previously (Currle et al., 2005; Hu et al., 2008). Sequences will be provided upon request. All qRT-PCR studies are reported as mean ± SEM for at least two biological replicates representing cultures initiated on different days. Histochemistry, immunostaining, and in situ hybridization (ISH) were performed as described previously (Currle et al., 2005; Cheng et al., 2006; Hu et al., 2008; Mangale et al., 2008). SFEBq aggregates were processed similarly to embryonic mouse brain, using 4% PFA/PBS fixation for 1 h on ice, 30% sucrose/PBS infusion overnight at 4°C, flash-freezing in Tissue Tek O.C.T. compound (Sakura Finetek USA) with a heat extractor, immediate 20 μm cryosectioning, and immunostaining performed on the same day. For nuclear antigens, aggregates were occasionally fixed in 4% PFA/0.1% saponin/0.1 m sucrose. Human cell ISH on coverslips was performed without dehydration steps. Toluidine blue stains and electron microscopy were performed by the UCI Pathology Services Core. Antibodies used were as follows: Aqp1 (1:500; AB2219, Millipore), Nestin [1:500; Rat-401, Developmental Studies Hybridoma Bank (DSHB)], RC2 (1:200; DSHB), Pax6 (1:2000; DSHB), ZO1 (1:100; 61-7300, Life Technologies, Sox1 (1:50; sc17318, Santa Cruz Biotechnology), Ttr (1:500; ab9015, Abcam), Alexa 488-conjugated and 555-conjugated secondary antibodies (1:100; Life Technologies). Additional reagents comprised the following: Hoechst 33342 (Life Technologies), phalloidin-TRITC (1:1000, added to secondary antibody solution; Sigma-Aldrich), Ttr ISH template [IMAGE (Integrated Molecular Analysis of Genomes and Their Expression Consortium) clone 1078224].
Imaging.
Epifluorescence and brightfield imaging and processing were performed as described previously (Hu et al., 2008; Mangale et al., 2008). Confocal images and z-stacks were acquired using a Zeiss LSM510 or LSM710 confocal microscope and Zen LE software. Aqp1-stained vesicles were imaged on a Nikon Eclipse Ti (optics for imaging through plastic) and captured using Nikon NIS-Elements AR3.00 software. Phase-contrast images were taken on a NikonTS100 or EVOS microscope (Advanced Microscopy Group). All images were compiled in Adobe Photoshop, with image adjustments restricted to brightness, contrast, and levels. Any images shown in figures as comparisons were acquired and processed in parallel using identical settings.
Flow cytometry.
M2-derived aggregates treated with or without BMP4 were dissociated with TrypLE Express (Life Technologies), filtered (40 μm mesh strainer, BD Biosciences), then stained with the green LIVE/DEAD Fixable Stain Kit (Life Technologies) according to manufacturer protocol in sorting buffer [1× Dulbecco's PBS without Ca2+/Mg2+ (Life Technologies), 1% FBS (Hyclone), penicillin-streptomycin (Life Technologies), and 10 μg/ml DNase I (Worthington Biochemical)]. Undifferentiated M2 mouse ESCs were processed in parallel to gate for RFP using a FACSAria II Cell Sorter (BD Biosciences) and FlowJo software (Tree Star). Cells were gated by side-scatter area and forward-scatter area (debris and dead cell exclusion), by 488 nm emission (dead cell exclusion), and by forward-scatter height and forward-scatter area (single cell selection) before analyzing for RFP expression. Reported values reflect two independent runs for each condition (0, 5, and 50 ng/ml BMP4; 10,000 events per run).
Cell quantification.
Nestin, Pax6, and Lhx2 images were manually counted in Photoshop from two biological replicates and two different confocal planes per replicate. Denominators for Nestin studies included all Hoechst-stained cells, including those in central cavitating regions of SFEBq aggregates. The reported Nestin-positive percentages therefore represent conservative estimates for the viable cell population. Pax6 and Lhx2 counts were restricted to viable peripheral regions of aggregates. Positivity figures were as follows: Nestin, 96.9 ± 3.13% (n = 4890 cells); Lhx2, 36.8 ± 1.58% (n = 4036 cells); Pax6, 50.8 ± 5.12% (n = 5387 cells). For blinded scoring of Sox1 and Nestin immunostains, matched unprocessed confocal images (Zeiss LSM 510) were scored as follows: 2+, 1+, and 0 corresponded to >50%, <50%, and undetectable Sox1 positivity, respectively; 3+, 2+, 1+, and 0 corresponded to >50%, 10–50%, <10%, and undetectable Nestin positivity, respectively. Total aggregates counted were n = 28 (5-day aggregates) and n = 31 (7-day aggregates). For quantification of stereotaxically injected cells, only those cells that were CFDA-SE/Hoechst colabeled and had intact Hoecsht-stained nuclei (indicative of cell viability) were counted.
Results
Efficient neural induction from mouse ESC lines
Since CPECs differentiate in vivo from neuroepithelium, we first compared protocols for deriving neuroepithelial cells from mouse ESCs. After comparing three such protocols (see Materials and Methods), we adopted the “SFEBq” aggregate method (Eiraku et al., 2008) (Fig. 1A), then optimized this method for cell numbers/well in 96-well U-bottom plates using nine newly derived mouse ESC lines (see Materials and Methods). While aggregate diameters scaled positively with cells/well for each of nine lines (data not shown), qRT-PCR screens for generic (Nestin, Sox1) and forebrain-restricted (Foxg1, Lhx2) neural progenitor markers after 5 DIV yielded optima ranging from 1000 to 9000 cells/well (data not shown). For the two main mouse ESC lines used in our studies (M1 and M2), optima were 2000 and 6000 cells/well, respectively (Fig. 1B).
Figure 1.
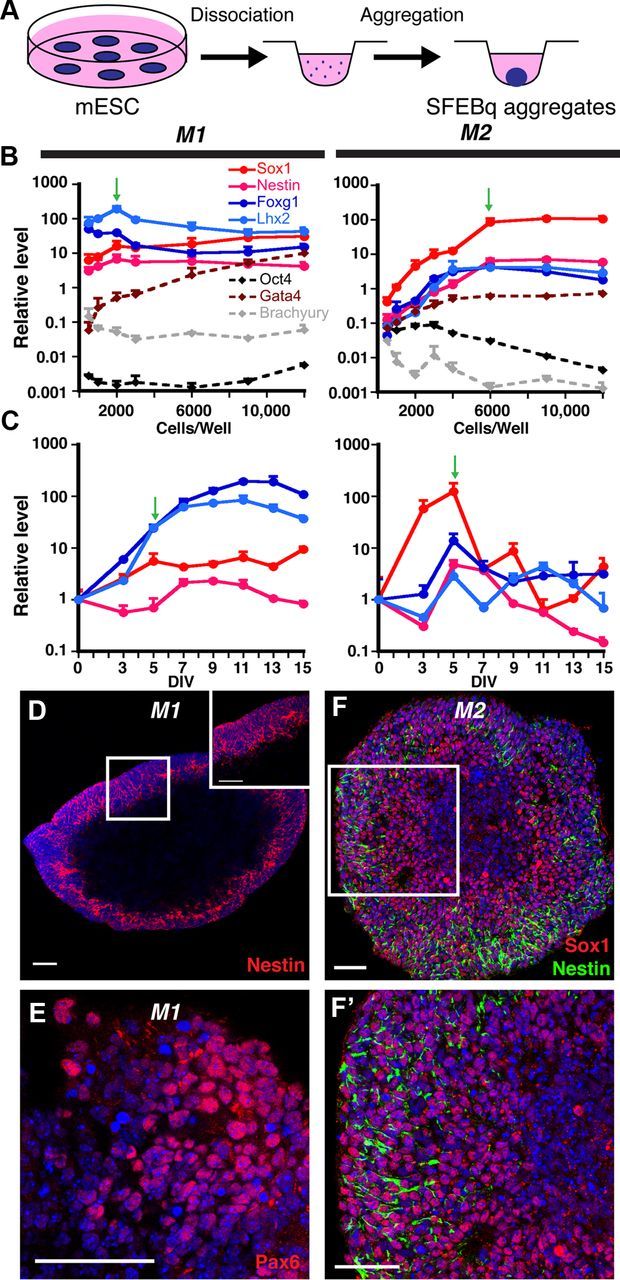
Efficient neural induction from mouse ESCs. A, Schematic of neural differentiation using the SFEBq method (Eiraku et al., 2008). B, Cells/well optimization for neural induction (qRT-PCR, normalized to starting mouse ESCs); 2000 and 6000 cells/well (green arrows) yield optimal gene expression profiles for the M1 and M2 mouse ESC lines, respectively. C, Temporal analysis of 2000-cell (M1) or 6000-cell (M2) aggregates (qRT-PCR, normalized to mouse ESCs). Plateaus for the four NEC markers are seen in M1 aggregates, whereas they peak, then decrease in M2 aggregates. Green arrows designate the time point at which most CPEC differentiations were initiated. D–F', Nestin, Pax6, and Sox1 ICC of 7-day M1 aggregates and 5-day M2 aggregates reveal efficient neural induction. Scale bars, 50 μm.
M1-derived and M2-derived aggregates were then examined for temporal expression of neural markers. qRT-PCR analyses of 2000-cell (M1) or 6000-cell aggregates (M2) revealed significant expression of Sox1, Foxg1, and Lhx2 by 5 DIV (Fig. 1C). Interestingly, after 5 DIV, neural marker levels were maintained in M1 aggregates, while they decreased markedly in M2 aggregates (Fig. 1C). Similar maintenance patterns were seen with two other mouse ESC lines (one with each pattern; data not shown). By 5 DIV, M1 and M2 aggregates often showed nuclear fragmentation or frank cavitation centrally, with neural progenitor antigen expression peripherally (Fig. 1D–F), as described by others (Eiraku et al., 2008). Based on conservative counts (i.e., counting all nuclei, including in central regions), Nestin immunopositivity approached 95% in M1 aggregates (Fig. 1D), which is comparable to previous reports (Watanabe et al., 2005; Eiraku et al., 2008). Essentially all peripheral cells demonstrated positivity for either Nestin or Sox1 (Fig. 1D,F,F'). The dorsal telencephalic markers Pax6 and Lhx2 were also expressed (51 and 38% positive, respectively) (Fig. 1C; see Material and Methods). In contrast, markers for ESCs (Oct4), endoderm (Gata4), and mesoderm (Brachyury) decreased or remained near their basal levels seen in ESCs (Fig. 1B). The SFEBq aggregate method therefore led to efficient neural induction by 5 DIV, although maintenance of neural progenitor gene expression varied across mouse ESC lines.
BMP4 sufficiency and dose-dependency to induce CPEC differentiation
Since BMP4 can rescue CPEC fate following roof plate ablation (Cheng et al., 2006) and upregulate CPEC genes in cortical progenitors (Hu et al., 2008), we applied BMP4 to the SFEBq aggregates (Fig. 2A). When given to 5-day M1 neural aggregates for 5–7 DIV (0.5–50 ng/ml supplemented every other day), BMP4 dramatically upregulated multiple CPEC markers (Ttr, Msx1, Aqp1, Cldn1, and Lmx1a) in a concentration-dependent fashion. CPEC marker induction occurred at the expense of the neural progenitor markers Sox1 and Foxg1. Similar results were obtained using 5-day M2 neural aggregates (Fig. 2B).
Figure 2.
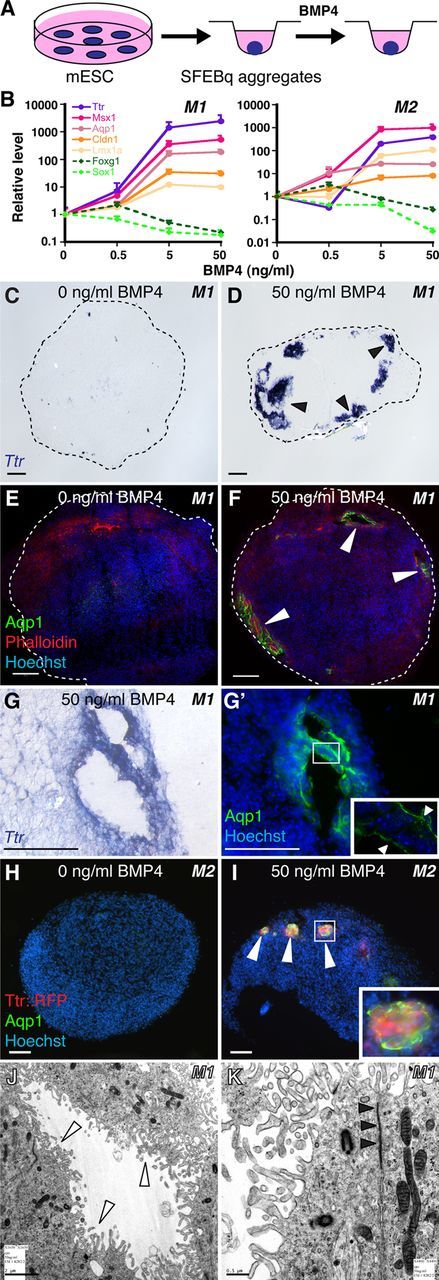
BMP4 sufficiency and concentration dependence for CPEC differentiation from mouse ESC-derived neural progenitors. A, Schematic of the differentiation method. Neural aggregates are treated for 5–7 DIV with BMP4. B, qRT-PCR of 5-day M1 and M2 aggregates treated with BMP4 (normalized to no BMP4 controls). CPEC markers are strongly induced, while neural progenitor markers Sox1 and Foxg1 are downregulated in a BMP4 concentration-dependent manner. C–G', ISH and ICC of sectioned M1 aggregates. Ttr and Aqp1 expression are detected in peripheral regions of BMP4-treated aggregates (arrowheads; n = 11/13 and 7/7 aggregates, respectively), but not in aggregates without BMP4 (n = 0/6 and 0/3 aggregates). Expression often occurs in vesicles that coexpress Ttr and Aqp1 (adjacent sections in G and G') and localize Aqp1 to the apical (luminal) surface (G', arrowheads) as seen in CPECs in vivo. H–I, ICC of sectioned M2 aggregates. Ttr::RFP (native fluorescence) and Aqp1 expression (arrowheads) are BMP4-dependent (n = 6/8 and 0/6 aggregates with and without BMP4, respectively), and colocalize in cell clusters toward the periphery of aggregates (I). J, K, Electron microscopy of M1-derived BMP4-treated aggregates. Vesicle-lining cells have extensive microvilli (white arrowheads), rare cilia, and juxtalumenal tight junctions (black arrowheads) characteristic of CPECs in vivo. Scale bars: C–I, 50 μm; J, 2 μm; K, 0.5 μm.
We then examined the BMP4-treated aggregates by immunocytochemistry (ICC) and ISH for evidence of CPEC differentiation. Ttr and Aqp1 expression was observed in M1 aggregates treated with 50 ng/ml BMP4 (Fig. 2D,F), but not in controls (Fig. 2C,E). Within the BMP4-treated aggregates, Ttr- or Aqp1-expressing cells were located peripherally below the aggregate surface and often formed “vesicles” (Fig. 2D–G'). In M2 aggregates, Ttr::RFP and Aqp1 expression colocalized in peripheral cell clusters (Fig. 2I). Within M1-derived vesicles, Aqp1, which localizes to apical CPEC microvilli (Huang et al., 2009; data not shown), was expressed toward vesicle lumens (Fig. 2G'). This suggested cell polarity with apical surfaces facing inward, which was confirmed by electron microscopy of vesicle-lining cells (Fig. 2J,K). Vesicle-lining cells also displayed multiple other characteristics of secretory and absorptive epithelial cells, including basally located nuclei, ZO1 immunoreactivity (data not shown), apically enriched mitochondria, extensive lumenal microvilli, rare cilia, and juxtalumenal tight junctions (Fig. 2J,K). These features, especially the microvilli, rare cilia, and tight junctions, are characteristic of CPECs, but not of other epithelia-forming neural cell types, such as ependyma or retinal pigmented epithelium (see below). These data indicate BMP4 sufficiency to induce cells with molecular, cellular, and ultrastructural features of CPECs, which differentiated in peripheral regions of aggregates composed almost entirely of neural progenitors.
ICC images yielded visual estimates of 5–10% induction efficiency. To estimate more accurately, we measured the percentages of RFP-expressing M2-derived cells treated with 0, 5, or 50 ng/ml BMP4 by flow cytometry (n = 2 independent runs for each condition; see Materials and Methods). Consistent with the observations from sections, significant cell death occurred in the aggregates based on forward/side scatter and live/dead cell staining, with cell death being highest in the 50 ng/ml BMP4 samples, as expected (Furuta et al., 1997; Mabie et al., 1999). We therefore calculated RFP expression as percentages of total or live cells. As a percentage of total cells/events, RFP-positive cells represented 3.74 ± 0.89% (5 ng/ml) or 2.71 ± 0.75% (50 ng/ml) compared with 0.72 ± 0.20% for the no BMP4 controls; these numbers rose to 20.8 ± 3.65% and 32.4 ± 9.70%, respectively, when live cell denominators were used. Thus, consistent with the ICC-based estimates, CPEC induction efficiency with BMP4 alone was relatively low, but was more moderate among viable cells.
Peak competency for CPEC gene induction correlates with early stage neuroepithelial cells
CPEC specification in mice occurs between embryonic day (E) 8.5 and E9.5 (Thomas and Dziadek, 1993), with definitive CPEC differentiation being evident (Currle et al., 2005) by the time cortical neurogenesis ensues at E11.5 (Caviness et al., 1995). This indicates that CPECs are specified from preneurogenic neuroepithelial cells (NECs) rather than later-stage neurogenic radial glia (RG), and raise the possibility that preneurogenic NECs are selectively competent for CPEC differentiation. To test for CPEC competency in vitro, we applied BMP4 to aggregates that had undergone neural induction for different amounts of time. Five-day neural aggregates (M1 and M2) strongly upregulated Ttr and Lmx1a expression, but little to no upregulation was seen in 7-, 9-, or 11-day aggregates (Fig. 3A). (Although neither Ttr nor Lmx1a is entirely specific to CPECs—e.g., Lmx1a is also expressed in the embryonic cortical hem adjacent to CPECs—the pairwise Ttr-Lmx1a combination is highly specific to CPECs within the body.) Baseline Ttr and Lmx1a levels in the absence of BMP4 did not vary significantly across the M1 aggregate time points; these increased by 2–16× in M2 aggregates, but the BMP4-mediated inductions in 5-day M2 aggregates were much greater (100–250× increases; data not shown). Hence, competency for CPEC gene induction was high for 5-day aggregates, but was markedly reduced in aggregates cultured for ≥7 DIV. This suggested a critical period for CPEC gene induction that was restricted to relatively early stages following neural induction.
Figure 3.

Temporally restricted CPEC competency correlates with NEC rather than RG fate. A, SFEBq aggregates treated with BMP4 for 7 DIV (M1) or 5 DIV (M2) (qRT-PCR normalized to no BMP4 controls). Five-day M1 and M2 aggregates show much stronger CPEC gene induction than 7-, 9-, or 11-day aggregates. B–E, RC2 ICC of sectioned M1 and M2 aggregates. Five-day aggregrates exhibit low RC2 expression levels characteristic of NECs, while strong RC2 expression typical of RG is present throughout 7-day aggregates. F–G, Sox1 and Nestin ICC of sectioned M2 aggregates. Five-day aggregates exhibit the strong Sox1 and weak Nestin patterns characteristic of NECs, whereas 7-day aggregates display the opposite RG-like pattern. H, Scoring of Sox1/Nestin staining patterns in M2 aggregates. Scale bars, 100 μm.
We then performed ICC to determine whether 5-day aggregates are enriched for NECs rather than RG. One useful antibody is RC2, which detects a post-translationally modified form of Nestin selectively expressed by RG, but not NECs (Hatakeyama et al., 2004). In 5-day aggregates, RC2 immunostaining was weak to undetectable (M1, n = 20/20 aggregates; M2, n = 21/21 aggregates) (Fig. 3B,D). On the other hand, RC2 expression was strong and detectable in most cells within 7-day aggregates (M1, n = 38/38 aggregates; M2, n = 24/24 aggregates) (Fig. 3C,E). These RC2 findings suggested that unlike the 5-day aggregates, 7-day aggregates were enriched for RG.
NECs and RG can also be distinguished by differences in Sox1 (high in NECs, low in RG) and Nestin levels (low in NECs, high in RG) (Mignone et al., 2004; Götz and Barde, 2005; Suter et al., 2009). In 5-day aggregates, Sox1 immunostaining was relatively strong in a high percentage of cells, while Nestin expression was weak and sparse (Fig. 3F). In contrast, Sox1 expression in 7-day aggregates was weak, whereas Nestin immunostaining was much stronger and evident in a larger fraction of cells (Fig. 3G). Blinded scoring yielded values of >80% of 5-day aggregates with high Sox1 and low Nestin, while >90% of 7-day aggregates expressed low Sox1 and high Nestin (Fig. 3H). This indicated that 5-day M2 aggregates are enriched for NECs, which differentiate into RG after two additional days in culture. Together with the qRT-PCR and RC2 data (Fig. 3A–E), these findings also suggest that CPEC competency is restricted to preneurogenic NECs.
Self-assembly and secretory functions of mouse dCPECs
To assess dCPEC function, we adopted an in vitro assay established for primary CP cells. In this assay, CPECs within dissociated CP cultures on Matrigel self-assemble into vesicles, which are composed of a single layer of CPECs facing inward. Due to CPEC secretion and tight junctions rather than proliferation, these vesicles expand, collapse upon treatment with the secretion inhibitors acetazolamide (carbonic anhydrase inhibitor) and ouabain (Na-K ATPase inhibitor), and regrow upon inhibitor withdrawal (Thomas and Dziadek, 1993; Swetloff et al., 2006). These properties were readily observed in primary CPEC-derived vesicles, which were also Aqp1-positive (Fig. 4A–F). Indistinguishable vesicles formed from dissociated SFEBq aggregates treated with BMP4 (M1: n = 6/6; M2: n = 3/3 independent cultures), but not from controls (M1: n = 0/6; M2: n = 0/3) (Fig. 4G,H,M,N). In the M2 cultures, most RFP-expressing cells were associated with the vesicles (Fig. 4N). The SFEBq-derived vesicles expressed Aqp1 (Fig. 4H, inset) and Ttr::RFP (Fig. 4N–R), grew in diameter, collapsed after exposure to the secretion inhibitors, and recovered after inhibitor removal (Fig. 4I–L,O–R). Thus, the dCPECs displayed self-assembly and secretory functions, as well as inhibitor responses, which were indistinguishable from those of primary CPECs. In addition, the BMP4 dependence for vesicle formation further confirmed the sufficiency of BMP4 for dCPEC induction in our system.
Figure 4.
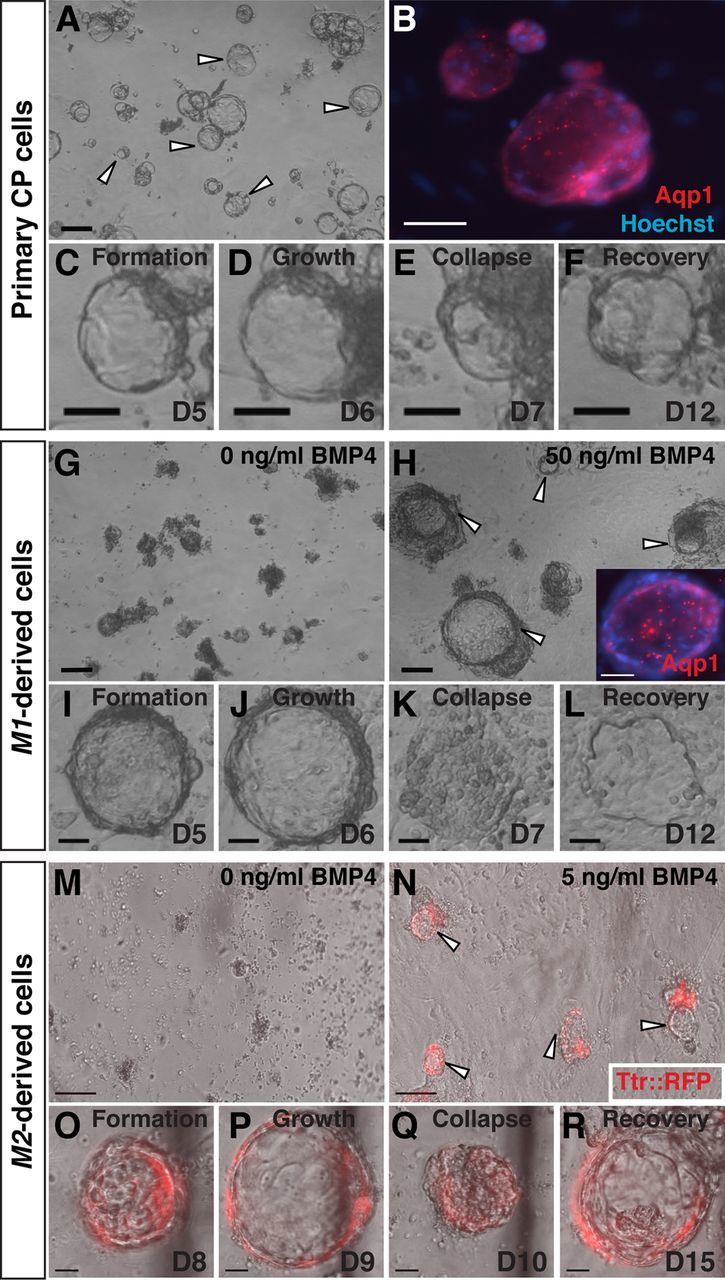
Vesicle self-assembly and secretion by primary and derived CPECs (ICC, phase contrast, and fluorescence microscopy; the same vesicle is shown in C–F, I–L, or O–R). A–F, Primary CPECs. CPECs from dissociated primary CP self-assemble into vesicles on Matrigel (A, arrowheads) that are Aqp1-positive (B). Due to their secretory activity, these vesicles enlarge (C, D), collapse upon treatment with the secretion inhibitors acetazolamide and ouabain (E), then regrow upon inhibitor withdrawal (F). G–L, M1-derived cells. M1 aggregates treated with 50 ng/ml BMP4 (n = 6/6 cultures), but not controls (n = 0/6 cultures), formed Aqp1-positive vesicles after dissociation and plating on Matrigel (G, H). Like primary CPEC vesicles, M1-derived vesicles expand (I, J), collapse upon secretion inhibitor treatment (K), and recover after inhibitor withdrawal (L). M–R, M2-derived cells. M2 aggregates treated with 5 ng/ml BMP4 (n = 3/3 cultures), but not controls (n = 0/3 cultures), form TTR::RFP-expressing vesicles (N, arrowheads) that expand, collapse upon secretion inhibitor treatment, and regrow after inhibitor withdrawal (O–R) in a fashion indistinguishable from that of primary CPECs. Scale bars: A–N, 100 μm; O–R, 50 μm.
CP engraftment by primary and derived CPECs following intraventricular injection
We then sought an in vivo system for evaluating CPEC identity and function. We reasoned that CP engraftment by exogenous CPECs might be possible via intracerebroventricular (i.c.v.) injection. Such injections have been described using primary cells (Matsumoto et al., 2010), although it is not known whether integration into host CP occurred. For our initial studies, we used primary dissociated CP cells from CAG::H2B-GFP mice (on a mostly CD1 background), in which all nucleated cells are fluorescent, or from wild-type CD1 mice, which were exogenously labeled with Vybrant CFDA-SE dye. Two days after i.c.v. injection into CD1 hosts, many H2B-GFP or dye-labeled cells were associated with host CP (Fig. 5A–C). Most of the fluorescent cells were associated with ipsilateral CP, but some were also seen in the contralateral and third ventricles, consistent with the injected cells traveling through the ventricular system. Scattered supraependymal cells and rare intraparenchymal cells were also detected (data not shown). Interestingly, confocal microscopy of host CP revealed that H2B-GFP and CFDA-SE fluorescence was largely present on the nuclear side of the apical Aqp1 border (rather than the ventricular side), and this border was continuous between fluorescent and nonfluorescent cells (Fig. 5C). This indicated that within 2 days, injected primary CPECs had integrated into host CP epithelium with appropriate apicobasal polarity and with reasonable efficiency.
Figure 5.
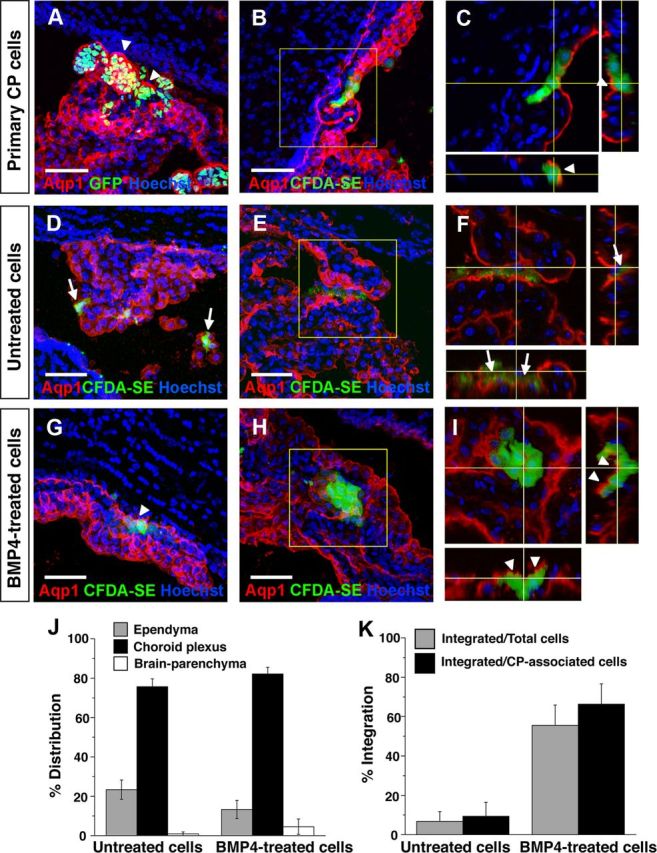
Engraftment of endogenous CP by primary and derived CPECs following intraventricular injection. A–C, Primary CP cell injections, single or collapsed z-stack images of ipsilateral telencephalic choroid plexus 2 days after injection. Injected H2B-GFP (A) or CFDA-SE-labeled CP cells (B) preferentially associate with host CP, which expresses Aqp1 (red). Confocal projections reveal labeled cells on the nuclear side of the continuous Aqp1-positive apical border (C, arrowheads), indicating integration of injected cells into host CP with appropriate apicobasal polarity. D–F, Mouse ESC-derived (M1) aggregates, no BMP4 treatment, 2 d after injection. Dye-labeled cells are associated with ipsilateral host CP, but remain on the ventricular side of the Aqp1-expressing apical border (F, arrows), indicating a failure to engraft host CP. G–I, Mouse ESC-derived (M1) aggregates, with BMP4 treatment and Ara-C enrichment, 1–2 d after injection. Like the primary cells, most of the dye-labeled cells associate with host CP and are appropriately polarized on the nuclear side of the endogenous Aqp1 border (G, I, arrowheads), indicating host CP engraftment. J, K, Quantification of distributions and integration of injected BMP4-treatedand-untreatedcells. Cells from both groups are found mostly in association with CP (J), but the BMP4-treated cells integrate into host CP with much greater efficiency (K), consistent with their dCPEC identity (see Results for numbers; error bars represent SEM.). Scale bars, 50 μm.
We then tested whether dCPECs could similarly adhere and integrate into endogenous CP epithelium. To enrich for dCPECs, we dissociated BMP4-treated M1 aggregates into adherent monolayers, then applied Ara-C to enrich for postmitotic cells (Matsumoto et al., 2010; Menheniott et al., 2010). As expected, Ara-C caused massive cell death, but a significant number of BMP4-treated cells survived; 60–90% of these were positive for Aqp1, compared with <20% in controls without Ara-C treatment (n = 3). Almost no cells survived Ara-C treatment when BMP4 was omitted, suggesting that BMP4 was needed (and Ara-C alone was insufficient) to generate significant numbers of postmitotic cells. We then injected 5000 dye-labeled M1-derived cells (mixed CD1/B6 background) with or without BMP4 and Ara-C treatments into CD1 hosts. Between the two treatment groups, dye-labeled cells preferentially associated with CP to similar degrees (Fig. 5J), but the extent of epithelial integration differed dramatically. While the vast majority of labeled control cells lacked Aqp1 expression and were seen on the anuclear (ventricular) side of the endogenous Aqp1 border (90.6 ± 7.0%, n = 126/139 CP-associated cells from two brains) (Fig. 5D–F,K), the majority of BMP4-treated, Ara-C-enriched cells had integrated into host CP epithelium (66.3 ± 9.4%, n = 339/511 CP-associated cells from four brains) (Fig. 5G–I,K) in patterns indistinguishable from the injected primary CPECs. These findings supported the identity of mouse ESC-derived dCPECs, which, like primary CPECs, can engraft endogenous CP epithelium from the ventricular space with appropriate cell polarity.
CPEC induction from human ESC-derived neuroepithelial cells
We then attempted to derive CPECs from human ESCs using two different approaches. First, we applied the SFEBq aggregation method to H9 human ESCs (Fig. 6A). Unlike mouse ESCs, H9 cells usually formed multiple rather than single aggregates in each U-bottom well, and expression levels of neural progenitor genes did not vary significantly between 3000 and 12,000 cells/well by qRT-PCR (data not shown). Using 9000 cells/well, temporal qRT-PCR analysis revealed significant induction of neural progenitor genes by 6 DIV, which continued to increase through 24 DIV, the last time point tested (Fig. 6B). In contrast, OCT4 levels decreased progressively over the same time period (data not shown). Like mouse SFEBq aggregates, H9 aggregates usually cavitated centrally and expressed neural progenitor antigens peripherally, where virtually all cells were NESTIN-positive (Fig. 6C), as previously described (Eiraku et al., 2008). Hence, neural induction from human ESCs was also efficient using the SFEBq method.
Figure 6.

BMP4 sufficiency to induce CPEC markers from human ESC-derived neural progenitors. A, Schematic of the differentiation method. The human ESCs form multiple and less uniform SFEBq aggregates than mouse ESCs. B, Temporal analysis of neural induction in human SFEBq aggregates (9000 human ESCs/well, qRT-PCR normalized to human ESCs). Dramatic increases in forebrain (LHX2, FOXG1) and generic neural progenitor gene expression (SOX1, NESTIN) occur by 6 DIV and are stably maintained thereafter. C, ICC of 9000 cells/well human SFEBq aggregates at 21 DIV for PAX6 and NESTIN demonstrate efficient neural induction. D, Temporal-dependent and BMP4 concentration-dependent induction of CPEC genes in human SFEBq aggregates (9000 cells/well, qRT-PCR normalized to no BMP4 controls). Eighteen-day aggregates show marked BMP4-mediated upregulation of TTR and LMX1A, while little to no induction is seen in 9-, 12-, or 15-day aggregates. TTR and LMX1A baselines also increased with aggregation time (∼64× for Ttr; 4× for Lmx1a), but these baseline changes were much less than the BMP4-mediated changes in 18-day aggregates (>1000× for Ttr; >100× for Lmx1a). E, CPEC and neural progenitor markers, 18-day aggregates (qRT-PCR normalized to no BMP4 controls). BMP4 induces CPEC markers TTR, LMX1A, and OTX2 at the expense of neural progenitor markers FOXG1 and NESTIN. Scale bars, 50 μm.
We then treated H9 aggregates neuralized for different amounts of time with BMP4 (5 or 15 ng/ml for 14 DIV, replaced every third day). Upregulation of the CPEC markers TTR and LMX1A was much stronger in 18-day aggregates than in 9-, 12-, or 15-day aggregates (Fig. 6D). In the 18-day aggregates, TTR and LMX1A upregulation occurred in a BMP4 concentration-dependent fashion at the expense of neural progenitor markers NESTIN and FOXG1 (Fig. 6E). As with the mouse cells, this suggested BMP4 sufficiency to induce CPEC gene expression from ESC-derived neural progenitors.
As a second dCPEC generation approach, we treated H1 human ESCs that had been differentiated into NRs (Zhang et al., 2001) with predominant forebrain character (Elkabetz et al., 2008). BMP4 (0.5–15 ng/ml) was then added for another 20 DIV (Fig. 7A), which resulted in a concentration-dependent upregulation of TTR and LMX1A gene expression by qRT-PCR (Fig. 7B). Compared with the mouse and human SFEBq cultures (Figs. 2B, 6E), baseline TTR and LMX1A levels were higher in the NR cultures, which probably reflected spontaneous CPEC differentiation due to BMP production by cells in the more heterogeneous NR cultures (data not shown) (Wilson and Stice, 2006). Strikingly, beginning ∼7 DIV after BMP4 addition (0.5–15 ng/ml), the NRs transformed directly into expanding vesicular structures (Fig. 7C,D) reminiscent of the mouse dCPEC vesicles (Figs. 2, 4). Eventually, the H1-derived vesicles transformed into epithelial monolayer sheets (data not shown). H9 cultures showed similar morphologic changes, although NR-to-vesicle transformations were less prominent. Interestingly, H1-derived vesicular regions had a papillary-like histology reminiscent of CP epithelium in vivo (Fig. 7E), and both H1-derived and H9-derived vesicular/monolayer regions exhibited prominent TTR and ZO1 coexpression (Fig. 7F–H; data not shown). Ultrastructurally, abundant cells in the vesicular/monolayer regions exhibited features characteristic of CPECs, including extensive lumenal microvilli, rare cilia, and juxtalumenal tight junctions (Fig. 7I,J). Together, findings from the two human ESC culture systems suggest CPEC differentiation from human ESC-derived neural progenitors and NRs, which is facilitated by exogenous BMP4.
Figure 7.

CPEC differentiation from human ESC-derived NRs. A, Schematic of the NR differentiation method (Zhang et al., 2001). After 16 DIV, NR-containing cultures are treated for 20 DIV with BMP4. B, BMP4 effects on CPEC markers in H9-derived NR cultures (qRT-PCR normalized to no BMP4 controls). Similar induction curves are seen for CPEC genes LMX1A and TTR. C, D, Phase contrast images of H1-derived NR cultures (same field in C and D). In the presence of BMP4, NRs (arrowheads) transform into phase-bright vesicular structures, which eventually form flat epithelial sheets. E, H&E-stained cross section of an H1-derived BMP4-treated colony. The peripheral NR-containing region consists of cells with apical cytoplasm that form a papillary-like tissue reminiscent of endogenous CP. F, ISH of an H9-derived BMP4-treated colony. Strong Ttr expression is evident in epithelial cells toward the colony periphery. G, H, ICC of an H1-derived BMP4-treated colony. Extensive colocalization of TTR and ZO1 occurs at the colony periphery (G and H from two different fields). I, J, Electron microscopy of an H1-derived BMP4-treated colony. Abundant epithelial cells demonstrate extensive lumenal microvilli (arrows), rare cilia, and juxtalumenal tight junctions (arrowheads). Scale bars: C, D, 100 μm; E–H, 50 μm; I, 5 μm; J, 0.5 μm.
Discussion
In this report, we presented molecular, cellular, and ultrastructural evidence for CPEC differentiation from mouse and human ESC-derived NECs. For the mouse dCPECs, CPEC functions indistinguishable from those of primary cells were also demonstrated. These functions included those previously described for CPECs in vitro (vesicle self-assembly and secretion) and the newly discovered capability of CPECs to engraft host CP epithelium following i.c.v. injection. Consistent with normal mouse CPEC development, peak competency for mouse dCPEC differentiation correlated with preneurogenic NECs, and this report provides the first evidence for BMP4 sufficiency to specify CPEC fate outside of a rescue setting (Cheng et al., 2006). In addition to these basic insights, dCPEC generation in culture should enable new translational applications, including dCPEC drug screens, transplants, and engineering, although higher efficiency dCPEC derivation and purification methods will be needed. The CP engraftment capabilities of primary and induced CPECs also provide for a novel CPEC replacement approach in neurological disorders characterized by CPEC dysfunction, such as AD.
Evidence for mouse and human dCPEC differentiation
The multiple lines of evidence for dCPEC identity exclude non-CPEC fates as possibilities. The neural origin of the induced cells is clear: not only did they arise in peripheral regions of SFEBq aggregates that were virtually 100% Sox1 or Nestin positive, but the human dCPECs also differentiated directly from NRs. Among the markers used, Ttr was particularly informative, since the induced cells expressed Ttr strongly and, within the CNS, such strong Ttr expression is exclusive to CPECs (Herbert et al., 1986; Sousa et al., 2007). Among CNS cell types, ependymal and retinal pigmented epithelial (RPE) cells have similarities to CPECs, but the dCPECs bear little resemblance to these cell types. For example, ependymal cells do not express Ttr, have abundant cilia rather than microvilli, and do not form tight junctions; RPE cells do not express Lmx1a, are pigmented, contain melanosomes, and do not form vesicles on Matrigel (Thomas and Dziadek, 1993). Importantly, the in vitro and in vivo functional capabilities of the dCPECs, which mimic those of primary CPECs, provide strong support for their CPEC identity.
BMP4 sufficiency for CPEC differentiation
The qRT-PCR, ICC, ISH, and vesicle formation data (Figs. 2–4, 6) provide evidence for BMP4 sufficiency to specify CPEC fate. This complements previous findings of BMP4 sufficiency to partially rescue CPECs in roof plate-ablated explants (Cheng et al., 2006) and to upregulate CPEC genes in a concentration-dependent fashion in cortical progenitor cells (Hu et al., 2008). While both instructive and permissive factors are required to specify cell fates, only instructive molecules demonstrate sufficiency. Our findings therefore support BMP4 acting as an instructive molecule for CPEC fate in the dorsal telencephalon. Together with the known BMP signaling gradient within the dorsal telencephalon (Cheng et al., 2006; Hu et al., 2008), these findings further support the concept of the dorsal telencephalon being a classic BMP morphogen gradient system (Grove and Monuki, 2012).
While BMP4 is sufficient for dCPEC induction, BMP4 alone led to only low to moderate induction efficiency. Given the plethora of developmental signals present in and around the CPEC anlagen, including multiple BMPs, WNTs, IGFs, FGFs, SHH, and others, the low-to-moderate efficiency is perhaps unsurprising. Regardless, for clinical applications, higher efficiency dCPEC derivation and enrichment methods will be needed. In addition to optimizing the current method (e.g., conversion to monolayer systems and finer-grained BMP4 concentration testing), it will be important to test BMP4 together with other signals, such as WNT3A (Eiraku et al., 2008), and to develop FACS-based or other purification methods; these and other process optimizations are currently in progress.
The NEC as the CPEC-competent progenitor cell
Peak competency for CPEC fate in response to BMP4 correlated to NECs rather than RG (Fig. 3), which mirrors in vivo development. In the developing mouse forebrain, CPEC specification occurs during the E8.5–E9.5 period (Thomas and Dziadek, 1993), which corresponds to preneurogenic NECs rather than neurogenic RG (Götz and Huttner, 2005). The relatively rapid loss of dCPEC competency in culture further suggests that neurogenic RG are refractory to CPEC specification. Previous studies of cortical RG in dissociated cultures (Hu et al., 2008) and explants (Furuta et al., 1997; Monuki et al., 2001) would support the concept of RG being refractory to CPEC specification and, accordingly, of preneurogenic NECs being selectively competent for this fate.
Indeed, based on these in vitro and in vivo findings, one could argue that NECs represent the only true “neural” stem cell (NSC) in the CNS. The term NSC is generally applied to cells that can self-renew and differentiate in culture into three neural cell types: neurons, astrocytes, and oligodendrocytes. According to this definition, NECs, radial glia, and adult progenitors of the hippocampus and subventricular zone are NSCs (Reynolds and Weiss, 1992; Gage, 2000). Our studies add a fourth and very distinctive cell type, the CPEC, to the list of neural derivatives in culture. Since CPECs are derived selectively from NECs not only in vivo, but also in vitro, NECs would then represent the only NSC capable of giving rise to all neural derivatives in culture in the absence of reprogramming.
The clinical potential for dCPECs at the blood–CSF–brain interface
Importantly, in addition to the basic insights, dCPEC derivation in culture enables brand-new translational approaches with significant conceptual promise. As mentioned earlier, CPEC dysfunction is associated with several neurodegenerative conditions (Weller, 1998; Serot et al., 2003; Sousa et al., 2007), and proof-of-concept for dCPEC transplants has been established from primary CP transplant studies in animal models of neurodegeneration (Emerich et al., 2005a; Skinner et al., 2006). As long-lived cellular pumps that also absorb toxins, dCPECs should also be an ideal cell type for tissue engineering and protein delivery, and their derivation from human ESCs provides a scalable source to generate large numbers of dCPECs for such applications.
The ability of primary and induced CPECs to integrate into endogenous CP epithelium after i.c.v. injection, which occurs with appropriate polarity and with surprisingly high efficiency within the first 2 days (Fig. 5), also provides proof-of-concept for CP epithelial engraftments and replacements as novel translational approaches. These approaches are noteworthy because they could enable therapies aimed at the unique interface between the blood, CSF, and brain that CPECs occupy. While CPECs generate the blood–CSF barrier, there is no blood–CPEC barrier. In fact, CPECs are supplied by fenestrated capillaries like those in the liver and kidney, which allows for relatively free exchange between peripheral blood and CPECs. (This free exchange accounts for the strong contrast enhancement of CP, a characteristic well known to neuroradiologists.) In addition, while there is a blood–brain barrier (BBB), there is no CSF–brain barrier (i.e., molecules in the CSF can readily penetrate the brain across ependymal, pial, and certain perivascular surfaces). Indeed, the lack of a CSF–brain barrier was critical to the identification of the BBB near the beginning of the twentieth century (the same dye denied entry into the brain from peripheral circulation could readily enter the brain when injected into the CSF; Goldmann, 1913). Engineered dCPECs within host CP could therefore enable peripheral blood-based therapies to bypass the BBB, which might otherwise be impossible.
Footnotes
This work was supported by a California Institute for Regenerative Medicine (CIRM) Training Grant TG2-01152 (M.W.), CIRM New Faculty Award RN2-00915-1, National Institutes of Health Grant RO1 NS064587, a UCI Institute for Clinical and Translational Science Pilot Project Award, and a UCI Alzheimer's Disease Research Center Pilot Project Award (E.S.M.). We thank Drs. Lisa Flanagan, Robin Wesselschmidt (Primogenix), Grant MacGregor, and Tom Fielder (UCI Transgenic Mouse Facility) for deriving and expanding mouse ESC lines; Farah Akhtar and the UCI Pathology Services Core for electron microscopy; the UCI Optical Biology Core for access to confocal microscopes; Vanessa Scarfone for assistance with flow cytometry, and the UCI Sue and Bill Gross Stem Cell Research Center for access to equipment and assistance with human ESC cultures. We also thank Basam Barkho and other members of the Monuki laboratory for support and comments on the manuscript.
M.W and E.S.M. are coinventors on a patent application filed by UCI for the process of deriving choroid plexus epithelial cells from human pluripotent stem cells. The authors declare no other competing financial interests.
References
- Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T. Transthyretin protects Alzheimer's mice from the behavioral and biochemical effects of Abeta toxicity. Proc Natl Acad Sci U S A. 2008;105:2681–2686. doi: 10.1073/pnas.0712197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviness VS, Jr, Takahashi T, Nowakowski RS. Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. Trends Neurosci. 1995;18:379–383. doi: 10.1016/0166-2236(95)93933-o. [DOI] [PubMed] [Google Scholar]
- Chauhan AN, Lewis PD. A quantitative study of cell proliferation in ependyma and choroid plexus in the postnatal rat brain. Neuropath Appl Neurobiol. 1979;5:303–309. doi: 10.1111/j.1365-2990.1979.tb00629.x. [DOI] [PubMed] [Google Scholar]
- Cheng X, Hsu CM, Currle DS, Hu JS, Barkovich AJ, Monuki ES. Central roles of the roof plate in telencephalic development and holoprosencephaly. J Neurosci. 2006;26:7640–7649. doi: 10.1523/JNEUROSCI.0714-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodobski A, Szmydynger-Chodobska J. Choroid plexus: target for polypeptides and site of their synthesis. Microsc Res Tech. 2001;52:65–82. doi: 10.1002/1097-0029(20010101)52:1<65::AID-JEMT9>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Choi SH, Leight SN, Lee VM, Li T, Wong PC, Johnson JA, Saraiva MJ, Sisodia SS. Accelerated Abeta deposition in APPswe/PS1Delta E9 mice with hemizygous deletions of TTR (Transthyretin) J Neurosci. 2007;27:7006–7010. doi: 10.1523/JNEUROSCI.1919-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa R, Ferreira-da-Silva F, Saraiva MJ, Cardoso I. Transthyretin protects against A-beta peptide toxicity by proteolytic cleavage of the peptide: a mechanism sensitive to the kunitz protease inhibitor. PLoS One. 2008;3:e2899. doi: 10.1371/journal.pone.0002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currle DS, Cheng X, Hsu CM, Monuki ES. Direct and indirect roles of CNS dorsal midline cells in choroid plexus epithelia formation. Development. 2005;132:3549–3559. doi: 10.1242/dev.01915. [DOI] [PubMed] [Google Scholar]
- Eiraku M, Watanabe K, Matsuo-Takasaki M, Kawada M, Yonemura S, Matsumura M, Wataya T, Nishiyama A, Muguruma K, Sasai Y. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell. 2008;3:519–532. doi: 10.1016/j.stem.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Elkabetz Y, Panagiotakos G, Al Shamy G, Socci ND, Tabar V, Studer L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008;22:152–165. doi: 10.1101/gad.1616208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerich DF, Skinner SJ, Borlongan CV, Thanos CG. A role of the choroid plexus in transplantation therapy. Cell Transplant. 2005a;14:715–725. doi: 10.3727/000000005783982576. [DOI] [PubMed] [Google Scholar]
- Emerich DF, Skinner SJ, Borlongan CV, Vasconcellos AV, Thanos CG. The choroid plexus in the rise, fall and repair of the brain. Bioessays. 2005b;27:262–274. doi: 10.1002/bies.20193. [DOI] [PubMed] [Google Scholar]
- Fernandes M, Gutin G, Alcorn H, McConnell SK, Hébert JM. Mutations in the BMP pathway in mice support the existence of two molecular classes of holoprosencephaly. Development. 2007;134:3789–3794. doi: 10.1242/dev.004325. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Piston DW, Hogan BL. Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development. 1997;124:2203–2212. doi: 10.1242/dev.124.11.2203. [DOI] [PubMed] [Google Scholar]
- Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- Goldmann EE. Vitalfarbung am Zentral-nervensystem. Abh Preuss Akad Wissensch, Physkol Mathem Klass. 1913;1:1–60. [Google Scholar]
- Götz M, Barde YA. Radial glial cells defined and major intermediates between embryonic stem cells and CNS neurons. Neuron. 2005;46:369–372. doi: 10.1016/j.neuron.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Götz M, Huttner W. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–788. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- Grove EA, Monuki ES. Morphogens, patterning centers, and their mechanisms of action. In: Rakic P, Rubenstein J, editors. Comprehensive Developmental Neuroscience—Basic and Clinical Mechanisms. Elsevier, Oxford: United Kingdom; in press. [Google Scholar]
- Hadjantonakis AK, Papaioannou VE. Dynamic in vivo imaging and cell tracking using a histone fluorescent protein fusion in mice. BMC Biotechnol. 2004;4:33. doi: 10.1186/1472-6750-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama J, Bessho Y, Katoh K, Ookawara S, Fujioka M, Guillemot F, Kageyama R. Hes genes regulate size, shape and histogenesis of the nervous system by control of the timing of neural stem cell differentiation. Development. 2004;131:5539–5550. doi: 10.1242/dev.01436. [DOI] [PubMed] [Google Scholar]
- Herbert J, Wilcox JN, Pham KT, Fremeau RT, Jr, Zeviani M, Dwork A, Soprano DR, Makover A, Goodman DS, Zimmerman EA. Transthyretin: a choroid plexus-specific transport protein in human brain: the 1986 S. Weir Mitchell Award. Neurology. 1986;36:900–911. doi: 10.1212/wnl.36.7.900. [DOI] [PubMed] [Google Scholar]
- Hu JS, Doan LT, Currle DS, Paff M, Rheem JY, Schreyer R, Robert B, Monuki ES. Border formation in a Bmp gradient reduced to single dissociated cells. Proc Natl Acad Sci U S A. 2008;105:3398–3403. doi: 10.1073/pnas.0709100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Ketova T, Fleming JT, Wang H, Dey SK, Litingtung Y, Chiang C. Sonic hedgehog signaling regulates a novel epithelial progenitor domain of the hindbrain choroid plexus. Development. 2009;136:2535–2543. doi: 10.1242/dev.033795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson CE, Duncan JA, Stopa EG, Baird A. Enhanced prospects for drug delivery and brain targeting by the choroid plexus-CSF route. Pharm Res. 2005;22:1011–1037. doi: 10.1007/s11095-005-6039-0. [DOI] [PubMed] [Google Scholar]
- Kaplan MS. Proliferation of epithelial cells in the adult primate choroid plexus. Anat Rec. 1980;197:495–502. doi: 10.1002/ar.1091970411. [DOI] [PubMed] [Google Scholar]
- Kwon GS, Hadjantonakis AK. Transthyretin mouse transgenes direct RFP expression or Cre-mediated recombination throughout the visceral endoderm. Genesis. 2009;47:447–455. doi: 10.1002/dvg.20522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Zappaterra MW, Chen X, Yang YJ, Hill AD, Lun M, Maynard T, Gonzalez D, Kim S, Ye P, D'Ercole AJ, Wong ET, LaMantia AS, Walsh CA. The cerebrospinal fluid provides a proliferative niche for neural progenitor cells. Neuron. 2011;69:893–905. doi: 10.1016/j.neuron.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Tong C, Mehrian-Shai R, Jia L, Wu N, Yan Y, Maxson RE, Schulze EN, Song H, Hsieh CL, Pera MF, Ying QL. Germline competent embryonic stem cells derived from rat blastocysts. Cell. 2008;135:1299–1310. doi: 10.1016/j.cell.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabie PC, Mehler MF, Kessler JA. Multiple roles of bone morphogenetic protein signaling in the regulation of cortical cell number and phenotype. J Neurosci. 1999;19:7077–7088. doi: 10.1523/JNEUROSCI.19-16-07077.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangale VS, Hirokawa KE, Satyaki PR, Gokulchandran N, Chikbire S, Subramanian L, Shetty AS, Martynoga B, Paul J, Mai MV, Li Y, Flanagan LA, Tole S, Monuki ES. Lhx2 selector activity specifies cortical identity and suppresses hippocampal organizer fate. Science. 2008;319:304–309. doi: 10.1126/science.1151695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto N, Taguchi A, Kitayama H, Watanabe Y, Ohta M, Yoshihara T, Itokazu Y, Dezawa M, Suzuki Y, Sugimoto H, Noda M, Ide C. Transplantation of cultured choroid plexus epithelial cells via cerebrospinal fluid shows prominent neuroprotective effects against acute ischemic brain injury in the rat. Neurosci Lett. 2010;469:283–288. doi: 10.1016/j.neulet.2009.09.060. [DOI] [PubMed] [Google Scholar]
- Maurizi CP. Choroid plexus portals and a deficiency of melatonin can explain the neuropathology of Alzheimer's disease. Med Hypotheses. 2010;74:1059–1066. doi: 10.1016/j.mehy.2009.12.026. [DOI] [PubMed] [Google Scholar]
- Menheniott TR, Charalambous M, Ward A. Derivation of primary choroid plexus epithelial cells from the mouse. Methods Mol Biol. 2010;633:207–220. doi: 10.1007/978-1-59745-019-5_15. [DOI] [PubMed] [Google Scholar]
- Mignone JL, Kukekov V, Chiang AS, Steindler D, Enikolopov G. Neural stem and progenitor cells in nestin-GFP transgenic mice. J Comp Neurol. 2004;469:311–324. doi: 10.1002/cne.10964. [DOI] [PubMed] [Google Scholar]
- Monuki ES, Porter FD, Walsh CA. Patterning of the dorsal telencephalon and cerebral cortex by a roof plate-Lhx2 pathway. Neuron. 2001;32:591–604. doi: 10.1016/s0896-6273(01)00504-9. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Schwarzman AL, Gregori L, Vitek MP, Lyubski S, Strittmatter WJ, Enghilde JJ, Bhasin R, Silverman J, Weisgraber KH, Coyle PK. Transthyretin sequesters amyloid beta protein and prevents amyloid formation. Proc Natl Acad Sci U S A. 1994;91:8368–8372. doi: 10.1073/pnas.91.18.8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serot JM, Béné M, Foliguet B, Faure GC. Monocyte-derived IL-10-secreting dendritic cells in choroid plexus epithelium. J Neuroimmunol. 2000;105:115–119. doi: 10.1016/s0165-5728(99)00240-4. [DOI] [PubMed] [Google Scholar]
- Serot JM, Béné M, Faure GC. Choroid plexus, aging of the brain, and Alzheimer's disease. Front Biosci. 2003;8:s515–521. doi: 10.2741/1085. [DOI] [PubMed] [Google Scholar]
- Silverberg GD, Mayo M, Saul T, Rubenstein E, McGuire D. Alzheimer's disease, normal-pressure hydrocephalus, and senescent changes in CSF circulatory physiology: a hypothesis. Lancet Neurol. 2003;2:506–511. doi: 10.1016/s1474-4422(03)00487-3. [DOI] [PubMed] [Google Scholar]
- Skinner SJ, Geaney MS, Rush R, Rogers ML, Emerich DF, Thanos CG, Vasconcellos AV, Tan PL, Elliott RB. Choroid plexus transplants in the treatment of brain diseases. Xenotransplantation. 2006;13:284–288. doi: 10.1111/j.1399-3089.2006.00310.x. [DOI] [PubMed] [Google Scholar]
- Sousa JC, Cardoso I, Marques F, Saraiva MJ, Palha JA. Transthyretin and Alzheimer's disease: where in the brain? Neurobiol Aging. 2007;28:713–718. doi: 10.1016/j.neurobiolaging.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Suter DM, Tirefort D, Julien S, Krause KH. A Sox1 to Pax6 switch drives neuroectoderm to radial glia progression during differentiation of mouse embryonic stem cells. Stem Cells. 2009;27:49–58. doi: 10.1634/stemcells.2008-0319. [DOI] [PubMed] [Google Scholar]
- Swetloff A, Greenwood S, Wade AM, Ferretti P. Growth of choroid plexus epithelium vesicles in vitro depends on secretory activity. J Cell Physiol. 2006;208:549–555. doi: 10.1002/jcp.20687. [DOI] [PubMed] [Google Scholar]
- Thomas T, Dziadek M. Capacity to form choroid plexus-like cells in vitro is restricted to specific regions of the mouse neural ectoderm. Development. 1993;117:253–262. doi: 10.1242/dev.117.1.253. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Kamiya D, Nishiyama A, Katayama T, Nozaki S, Kawasaki H, Watanabe Y, Mizuseki K, Sasai Y. Directed differentiation of telencephalic precursors from embryonic stem cells. Nat Neurosci. 2005;8:288–296. doi: 10.1038/nn1402. [DOI] [PubMed] [Google Scholar]
- Weller RO. Pathology of cerebrospinal fluid and interstitial fluid of the CNS: significance for Alzheimer disease, prion disorders and multiple sclerosis. J Neuropathol Exp Neurol. 1998;57:885–894. doi: 10.1097/00005072-199810000-00001. [DOI] [PubMed] [Google Scholar]
- Wilson PG, Stice SS. Development and differentiation of neural rosettes derived from human embryonic stem cells. Stem Cell Rev. 2006;2:67–77. doi: 10.1007/s12015-006-0011-1. [DOI] [PubMed] [Google Scholar]
- Zhang SC, Wernig M, Duncan ID, Brüstle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol. 2001;19:1129–1133. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]