

Abstract
Isoform-selective agonists and antagonists of the lysophosphatidic acid (LPA) G-protein-coupled receptors (GPCRs) have important potential applications in cell biology and therapy. LPA GPCRs regulate cancer cell proliferation, invasion, angiogenesis, and biochemical resistance to chemotherapy- and radiotherapy-induced apoptosis. LPA and its analogues are also feedback inhibitors of the enzyme lysophospholipase D (lysoPLD, also known as autotaxin), a central regulator of invasion and metastasis. For cancer therapy, the ideal therapeutic profile would be a metabolically stabilized pan-LPA receptor antagonist that also inhibits lysoPLD. Herein we describe the synthesis of a series of novel α-substituted methylene phosphonate analogues of LPA. Each of these analogues contains a hydrolysis-resistant phosphonate mimic of the labile monophosphate of natural LPA. The pharmacological properties of these phosphono-LPA analogues were characterized in terms of LPA receptor subtype-specific agonist and antagonist activity using Ca2+ mobilization assays in RH7777 and CHO cells expressing the individual LPA GPCRs. In particular, the methylene phosphonate LPA analogue is a selective LPA2 agonist, whereas the corresponding α-hydroxymethylene phosphonate is a selective LPA3 agonist. Most importantly, the α-bromomethylene and α-chloromethylene phosphonates show pan-LPA receptor subtype antagonist activity. The α-bromomethylene phosphonates are the first reported antagonists for the LPA4 GPCR. Each of the α-substituted methylene phosphonates inhibits lysoPLD, with the unsubstituted methylene phosphonate showing the most potent inhibition. Finally, unlike many LPA analogues, none of these compounds activate the intracellular LPA receptor PPARγ.
Keywords: autotaxin, lysophospholipase D, phosphonates, PPARγ, receptors
Introduction
Lysophosphatidic acid (LPA, 1-radyl-sn-glycerol-3-phosphate) elicits growth-factor-like effects in almost every cell type.[1-4] At the organ system level, LPA is implicated in complex physiological responses that include immunological competence, brain development, wound healing, coagulation, and regulation of blood pressure.[5, 6] The pleiotropic physiological functions of LPA suggest that LPA could contribute to a number of pathophysiological states including cancer, atherosclerosis, hypertension, ischemia reperfusion injury, diabetes, cardiovascular diseases, stroke, prevention of toxicity of chemotherapy and radiation therapy, immunomodulation, and others.[7]
LPA can be produced either extracellularly or intracellularly in response to various stimuli including growth factors, LPA itself, phorbol esters, and epidermal growth factor (EGF). In the course of blood coagulation, LPA is mainly generated sequentially by two enzymatic reactions. First, the action of phospholipase A1 and A2 (PLA) on phosphatidylcholine (PC) gives lysophosphatidylcholine (LPC).[8] Second, the lysophospholipase D (lysoPLD) activity of autotaxin (ATX) converts LPC to LPA.[9, 10] ATX is one of the 40 most upregulated genes in invasive cancers,[11] and has been implicated in cell motility and tumor invasion, metastasis, and neovascularization.[10] LPA signals through the activation of specific receptors which in turn leads to distinct cellular events depending on the receptor subtype expressed by the targeted cell. Cell-surface LPA receptors belong to the membrane G-protein-coupled receptor (GPCR) protein family. There are five different LPA GPCRs thus far characterized on the surface of mammalian cells: LPA1, LPA2, LPA3, LPA4, and LPA5.[7, 12-15] The first three were formerly called endothelial differentiation genes (EDG), EDG2, EDG4, and EDG7, whereas GPR23/P2Y9[14] and GPR92,[15, 16] tentatively designated LPA4 and LPA5, respectively, are members of the purinergic cluster in the GPCR superfamily. Cancer cells of different cellular origins express LPA GPCR subtypes of LPA1 in differing amounts; however, LPA1 is the most widely expressed in almost every cancer cell type, whereas LPA4 seems to be expressed at very low levels.[14] Ovarian and breast cancer cells express multiple isoforms of the LPA GPCRs, and LPA accumulates in tumor cell ascites[3] and in tumor cell effusates.[17] LPA also activates the nuclear transcription factor peroxisome proliferator-activated receptor γ (PPARγ).[18, 19] Through activation of these GPCRs and PPARγ, LPA regulates multiple physiological and pathological responses. Our understanding of these complex responses at present is limited by the lack of specific probes for the receptor types and subtypes.
The involvement of LPA receptors in many pathophysiologies has implicated them as attractive targets for therapeutic intervention. As with many other GPCRs, LPA receptors should be amenable to the development of highly specific and potent agonists or antagonists that have favorable pharmacokinetics, bioavailability, and metabolic characteristics. Currently available compounds represent a promising but limited start to the development of useful chemical tools, although none can be considered definitive in determining receptor selectivity or biological functions, especially for studies in vivo. Several groups have reported the characterization of the LPA agonists and antagonists.[20-29] Appropriately validated compounds are essential to advance in vivo studies, particularly in view of potential off-target effects. The development of more selective, more stable, more potent, and more druglike agonists and antagonists is eagerly awaited, and is the bottleneck in therapeutic exploration.
We have designed metabolically stabilized analogues of LPA, some of which function as agonists and antagonists that simultaneously resist degradation while exhibiting their effects only on one or more isoforms of the LPA GPCRs.[30] One major catabolic fate of LPA is the hydrolysis of its phosphate head group by ubiquitous phosphatases.[7] LPA and its structural analogues all have a polar head group, a linker, and a hydrophobic tail. As reported, of these three motifs, modifications to the polar head group may be the least well tolerated.[31] Only modifications of the phosphate head group that retain negative charge under physiological conditions have been found to retain receptor activation. Of these, only two classes of phosphomimetics have been identified as micromolar or submicromolar LPA agonists.[30] One class is the phosphonate derivatives, such as α-hydroxymethylene phosphonate and α-ketomethylene phosphonate with ethanolamine linker,[20] and α-fluoromethylene phosphonate.[26] The second class comprises the phosphorothioate analogues such as OMPT and alkyl OMPT.[25, 32] Herein, we describe the synthesis and pharmacology of a series of α-substituted methylene phosphonate analogues, in which the CH2 moiety is replaced by CHOH, CHCl, or CHBr (Figure 1). Using cell-based assays, we explore the structure–activity relationships (SARs) for transfected human LPA GPCRs expressed in RH7777 and CHO cells, the activation of PPARγ in CV-1 cells expressing a luciferase reporter construct, and the inhibition of recombinant ATX.
Figure 1.
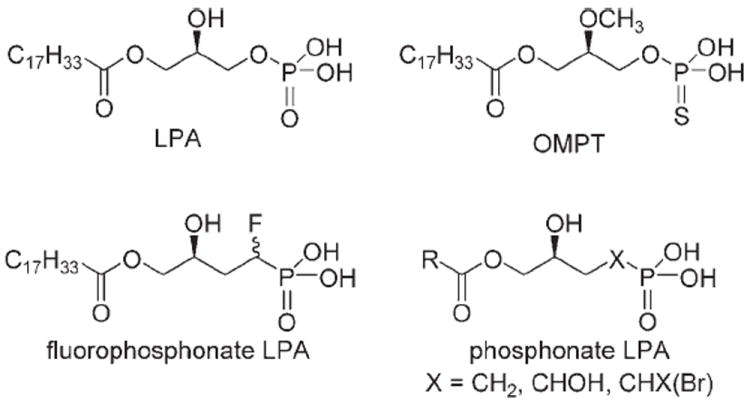
Structures of LPA and its analogues.
Results and Discussion
Chemical synthesis
Phosphonates in which the bridging oxygen atom of a phosphate ester is replaced by a methylene or substituted methylene group are attractive as metabolically stabilized analogues that can mimic a given ligand.[33, 34] Phosphonates are geometrically and electronically similar to the natural ligand, but cannot be hydrolyzed by normal phosphatase activity. To further refine the phosphomimetic properties, the bridging oxygen atom can be replaced by α-hydroxymethylene, α-fluoromethylene, α-bromomethylene, or α-chloromethylene groups. Such a substitution partially restores electronegativity that was lost as a result of the replacement of the oxygen atom.[26] Although the α-fluorophosphonates have been widely investigated over the last two decades,[26] few other α-halomethylene derivatives have been studied as potential enzyme inhibitors or as agonists or antagonists for receptor activation.
Scheme 1 illustrates the synthesis of the unsubstituted methylene phosphonate analogues 5a and 5b. Phosphonate 3[35] was prepared by the Arbuzov reaction of the known iodide[36] 2 with triethyl phosphite followed by removal of the acetonide with para-toluenesulfonic acid (p-TsOH) in methanol. Monoacylation of the intermediate diol 3 was carried out in the presence of 2,4,6-collidine at low temperature.[25] Treatment of the monoacylated diethyl phosphate 4 with trimethylsilyl bromide (TMSBr) and trifluoromethyl bistrimethylsilyl acetamide (TFBSA) in methylene chloride followed by hydrolysis with aqueous methanol afforded the desired product 5. The free acid was transformed into the more stable sodium salt by ion exchange.
Scheme 1.
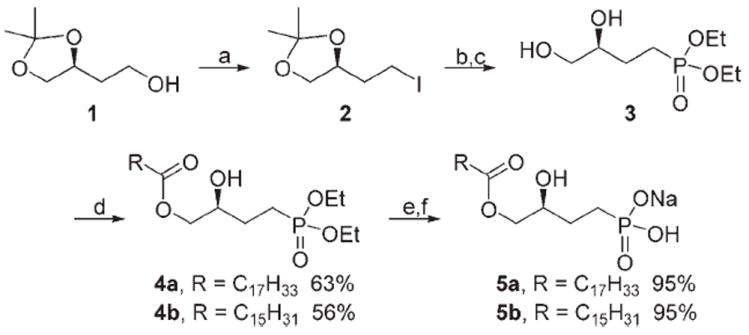
Synthesis of two methylene phosphonate analogues of LPA; reagents and conditions: a) PPh3, I2, imidazole, CH2Cl2; b) (EtO)3P, reflux; c) p-TsOH, CH3OH, 80%; d) RCOCl, 2,4,6-collidine, CH2Cl2, −78°C; e) TMSBr, TFBSA, CH2Cl2, CH3OH/H2O; f) Dowex ion-exchange resin.
The α-hydroxymethylene phosphonates are generally obtained by the Pudovik reaction, which involves the nucleophilic addition of a dialkyl phosphite to a carbonyl compound in the presence of base.[37, 38] As shown in Scheme 2, the α-hydroxymethylene phosphonate 7 was prepared by the addition of aldehyde 6 to dimethylphosphite in the presence of triethylamine. This addition reaction occurred non-stereospecifically, as two single sharp resonances of equal intensity at δ = 25.37 and 24.47 ppm were observed in the 31P NMR spectrum. The α-hydroxymethylene phosphonate diastereomers could not be separated chromatographically at this stage, and the mixture was used to obtain final compounds for assessing biological activity. Treatment of 7 with p-TsOH in methanol gave triol 8 in good yield. The monoacylation of triol 8 was achieved selectively by using the hindered base 2,4,6-collidine at −78 °C; deprotection of phosphonate esters 9 and ion exchange as above afforded the α-hydroxymethylene phosphonate analogues 10.
Scheme 2.
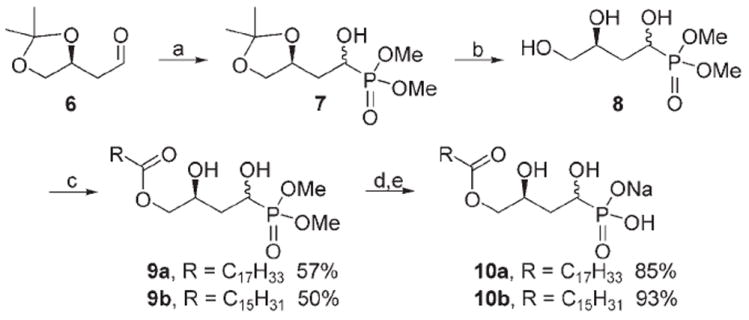
Synthesis of α-hydroxymethylene phosphonate analogues of LPA; reagents and conditions: a) TEA, HP(O)(OCH3)2; b) p-TsOH, CH3OH, 81%; c) RCOCl, 2,4,6-collidine, CH2Cl2, −78°C; d) TMSBr, TFBSA, CH2Cl2, CH3OH/H2O; e) Dowex ion-exchange resin.
The synthesis of α-halomethylene phosphonates by nucleophilic halogenation usually involves the reaction of α-hydroxymethylene phosphonates with different nucleophilic halogenating agents. As shown in Scheme 3, the α-bromomethylene phosphonates 13 were thus prepared by the reaction of the corresponding α-hydroxymethylene phosphonates with carbon tetrabromide and triphenylphosphine in toluene at reflux.[39] However, a similar attempt to prepare the α-chloromethylene phosphonates 12 with carbon tetrachloride and triphenylphosphine in toluene at reflux[40] gave only traces of the desired product. Several other commonly employed chlorination protocols such as SOCl2 [41] and SO2Cl2 [42] in the presence of a variety of amine bases also failed to give the desired chlorinated product. Further analysis determined that stored CCl4 often contained traces of HCl (1–10 ppm), which removed the acetonide at elevated temperatures and gave a triol that further decomposed. To remedy this, the chlorination reaction with CCl4/PPh3 was performed by using anhydrous pyridine as the solvent, and the diastereomeric α-chloromethylene phosphonates 12 were obtained in 72% yield. An analogous reaction sequence to that for the α-hydroxymethylene analogues was then employed for deprotection to diols 14 and 15, monoacylation to the α-halomethylene phosphonates 16 and 17, and finally phosphonate ester deprotection and ion exchange to give the final α-halomethylene phosphonate LPA analogues 18 and 19.
Scheme 3.
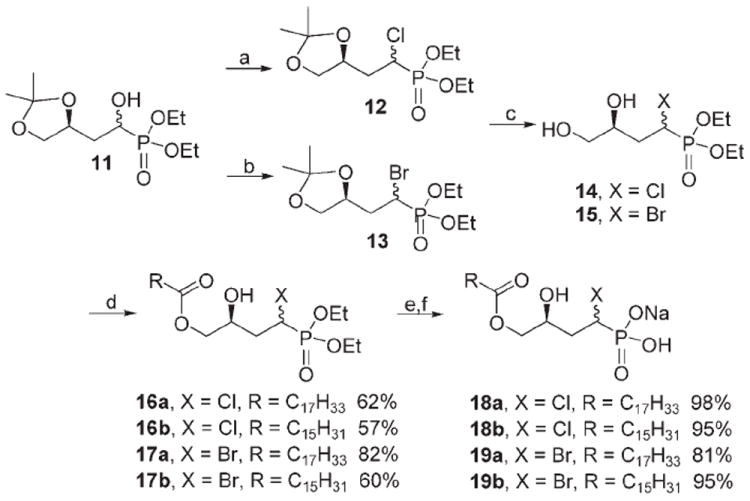
Synthesis of α-chloro- and α-bromomethylene phosphonate analogues of LPA; reagents and conditions: a) CCl4, PPh3, Py; b) CBr4, PPh3, toluene, 60%; c) p-TsOH, CH3OH, 81 %; d) RCOCl, 2,4,6-collidine, CH2Cl2, −78°C; e) TMSBr, CH2Cl2, CH3OH/H2O; f) Dowex ion-exchange resin.
Based on the biological results presented below, we prepared the separate diastereomers of analogue 10b. To separate the α-hydroxy epimers of 7, several derivatization reagents were tested (such as camphor chloride, methylated mandelic acid, tert-butyldiphenylsilyl chloride); the best results were obtained with tert-butyldimethylsilyl chloride (TBDMSCl; Scheme 4). The bulky TBDMS derivatives 20a and 20b were chromatographically separable; moreover, the TBDMS group was easy to introduce and remove under very mild conditions that minimized epimerization at the new stereocenter. Silylation with imidazole and 4-dimethylaminopyridine (DMAP) for 64 h produced the best results. Isomers 20a and 20b were separated by using repeated flash chromatography to afford > 99% de for each diastereomer as determined by 31P NMR. The TBDMS group was removed by NH4F in anhydrous methanol at 70°C to give intermediates 21a and 21b with > 96% de in high yields. Following the route above for 10b, compounds 24a and 24b were prepared in good yields and > 99% de.
Scheme 4.
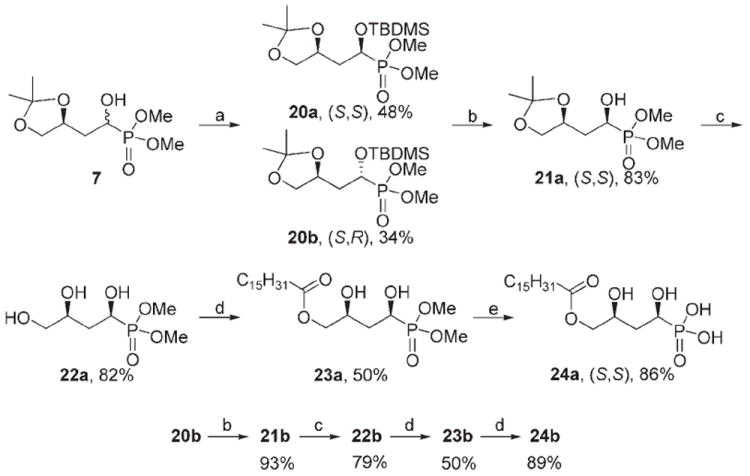
Synthesis of diastereomerically pure α-hydroxymethylene phosphonate analogues of LPA; reagents and conditions: a) TBDMSCl, imidazole, DMAP, DMF; b) NH4F, CH3OH, 70 °C; c) Dowex [H+], CH3OH; d) RCOCl, 2,4,6-collidine, CH2Cl2, −78°C; e) TMSBr, TFBSA, CH2Cl2, CH3OH/H2O.
We employed the asymmetric hydrophosphonylation[43] of aldehyde 6 with chiral complex Al(salalen) 27 to determine the configuration of the new stereogenic centers of 21a and 21b (Scheme 5). The Al(salalen) complex 27 induces S configuration at the newly formed α-hydroxy stereocenter. The chiral complex 27 was synthesized as described[43] by reductive amination of 3,5-di-tert-butylsalicylaldehyde with (1R,2R)-1,2-cyclohexanediamine (26), followed by N protection with the tert-butoxycarbonyl (Boc) group, N methylation of the amine, a second condensation with 3,5-di-tert-butylsalicylaldehyde, and treatment with Et2AlCl. Aldehyde 6 was then treated with dimethylphosphite in the presence of 10 mol% of the chiral complex 27 at −20°C for 48 h. The diastereomeric excess of the product was calculated to be 84% based on 31P NMR data, and the major product was the S,S diastereomer, as characterized by 1H, 13C, and 31P NMR spectroscopy. By comparing NMR data of the homogeneous diastereomers 21a and 21b with the data obtained for the product of asymmetric hydrophosphonylation, we were able to determine that 21a possesses the S,S configuration and that 21b has the S,R configuration.
Scheme 5.
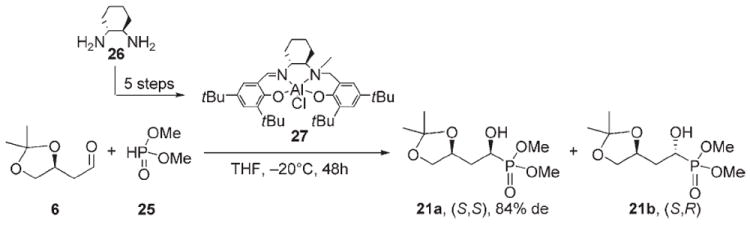
Diastereoselective hydrophosphonylation with chiral complex Al(salalen) 27.
Receptor activation assays
The ligand properties of the compounds were evaluated using Ca2+ mobilization assays for assessing the activation and inhibition of LPA1, LPA2, and LPA3 expressed in RH7777 cells, and LPA4 expressed in CHO cells. Table 1 illustrates calcium responses elicited through the activation of human LPA1, LPA2, LPA3, and LPA4 receptors. These cell lines have been used extensively for the characterization of LPA GPCR ligands because RH7777 cells are intrinsically unresponsive to LPA, and CHO cells show minimal endogenous responses to LPA unless transfected with LPA4.[14, 24, 44]
Table 1.
Effects of methylene phosphonate analogues at LPA GPCRs and in the PPARγ–PPRE luciferase reporter gene assay.[a]
| Compd | LPA1 | LPA2 | LPA3 | LPA4 | PPARγ | ||||
|---|---|---|---|---|---|---|---|---|---|
| EC50 [nm] (Emax) [nm][b] | IC50[nm] (Ki) [nm] | EC50[nm] (Emax) [nm] | IC 50[nm] (Ki) [nm] | EC50[nm] (Emax) [nm] | IC50[nm] (Ki) [nm] | EC50[nm] (Emax) [nm] | IC50[nm] (Ki) [nm] | ||
| LPA (18:1) | 100.2±13.2 | NE | 4.21±0.7 | NE | 150.2±15.5 | NE | 256.1±33.5 | NE | agonist |
| LPA (16:0) | 90.8±20.9 (88.7±3.6) | NE | 55.1±4.3 (100.3±1.9) | NE | 1527±194.2 (71.2±2.9) | NE | 843.4±130.1 (102.4±4.1) | NE | NE |
| 5a | >2520 (52.9±11.3) | NE[c] | >281 (94.7±4.3) | NE | >1710 (63.5±4.8) | NE | 3900±104 (110±1.3) | NE | NE |
| 5b | NE | PA[d] | NE | 2590±875 (1296±438) | NE | 2560±1160 (961±435) | 5400±526 (29.3±1.1) | NE | NE |
| 10a | >9250 (39.1±3.15) | NE | >2160 (71.9±6.0) | NE | 393±89 (116±9.5) | NE | >1150 (71.7±2.0) | NE | NE |
| 10b | NE | PA[e] | NE | PA[f] | NE | PA[g] | NE | NE | NE |
| 18a | 528±72 (63.2±9.5) | NE | NE | 1690±85 (845±42.5) | >2670 (63.5±3.7) | NE | 3480±561 (40.5±2.7) | NE | NE |
| 18b | NE | 2490±690 (919±255) | NE | 855±113 (428±56.5) | NE | 175±190 (98.3±105) | NE | PA[h] | NE |
| 19 a | NE | 1620±116 (694±49.7) | NE | 815±60 (174±12.8) | >2470 (44.2±3.5) | NE | NE | 4000±424 (2500±266) | NE |
| 19b | NE | 1500±559 (751±280) | NE | 1420±83 (304±17.7) | NE | 1160±259 (380±84.9) | NE | 266±124 (167±77.8) | NE |
| 24 a | NE | PA[i] | NE | PA[j] | NE | PA[k] | NE[l] | NT[m] | |
| 24 b | NE | PA[n] | NE | PA[o] | NE | PA[p] | NE[l] | NT | |
Data represent the average of four independent measurements (mean ±SD).
Emax=(maximal efficacy of compound/maximal efficacy of LPA 18:1)× 100.
NE=no effect was shown at the highest concentration (30 μm) tested.
PA=partial antagonist with 39.0±3.2% inhibition of 200 nm LPA response at the highest concentration (30 μm) tested.
PA=partial antagonist with 37.1±6.8% inhibition of 200 nm LPA response at the highest concentration (30 μm) tested.
PA=partial antagonist with 57.8±4.9% inhibition of 10 nm LPA response at the highest concentration (30 μm) tested.
PA= partial antagonist with 63.3±3.7% inhibition of 200 nm LPA response at the highest concentration (30 μm) tested.
PA=partial antagonist with 80.0±11.2% inhibition of 400 nm LPA response at the highest concentration (30 μm) tested.
PA=partial antagonist with 35.6±9.7% inhibition of 200 nm LPA response at the highest concentration (30 μm) tested.
PA=partial antagonist with 54.8±6.1% inhibition of 10 nm LPA response at the highest concentration (30 μm) tested.
PA=partial antagonist with 73.3±7.5% inhibition of 200 nm LPA response at the highest concentration (30 μm) tested.
NE alone, but increased LPA response when applied together with LPA.
NT=not tested.
PA=partial antagonist with 41.1±5.4% inhibition of 200 nm LPA response at the highest concentration (30 μm) tested.
PA=partial antagonist with 52.8±8.5% inhibition of 10 nm LPA response at the highest concentration (30 μm) tested.
PA=partial antagonist with 73.5±8.0% inhibition of 200 nm LPA response at the highest concentration (30 μm) tested.
The oleoyl chain-containing methylene phosphonate LPA analogue 5a, in which a methylene unit replaces the oxygen atom, is a selective, full agonist for LPA2 with an EC50 value of 281 nm. Interestingly, replacement of the oleoyl chain in 5a with the palmitoyl chain in 5b switched the activity of this partially selective agonist to that of modest antagonist. Analogue 5b had antagonist activity on all LPA receptor subtypes 1–3, with the relatively higher antagonist activity observed toward the LPA2 receptor (IC50 = 2.59 μm, Ki = 1 296 nm). The oleoyl α-hydroxymethylene phosphonate analogue 10a is a selective and potent LPA3 agonist (Table 1), with an EC50 value close to that of LPA (18:1) toward LPA3. In contrast, the palmitoyl analogue 10b exhibited a switch in activity to that of a weak partial antagonist of LPA1–3. It is clear that replacement of the oxygen atom of LPA with methylene or hydroxymethylene retains receptor recognition but with acylchain-dependent pharmacology. The biological activities of 24a and 24b, the chemically synthesized pure diastereomers of 10b, showed little difference from each other or that of the epimeric mixture 10b in terms of activity towards the LPA GPCRs.
The α-halomethylene phosphonate analogues showed a similar curious partial agonist–antagonist duality based on the acyl chain employed. Thus, α-chloromethylene phosphonate 18a with an oleoyl chain showed a mixed agonist–antagonist profile, with agonist effects toward LPA1, LPA3, and LPA4, showing quite high selectivity for activation of LPA1 as a partial agonist with an EC50 value of 528 nm. Surprisingly, 18a showed an IC50 value of 1.7 μm as an antagonist of the LPA2 receptor. In contrast to the mixed activities of the oleoyl α-chloromethylene phosphonate, the palmitoyl analogue 18b was a pan-antagonist with highest potency towards LPA2 (IC50 = 855 nm) and LPA3 (IC50 = 175 nm). An analogous mixed profile of agonist and antagonist effects was observed for the oleoyl α-bromomethylene analogue 19a, with partial LPA3 agonist activity, but had strong antagonist activities towards LPA1, LPA2, and LPA4 receptors. As observed for 18b, the palmitoyl α-bromomethylene phosphonate 19b was an LPA GPCR pan-antagonist with highest potency towards the non-EDG LPA receptor LPA4.
To date, there have been no reports of selective agonists or antagonists for LPA4 (p2y9/GPR23),[14] thereby limiting the search for the physiological role of this new receptor. In this series of substituted methylene phosphonate analogues, we noted that the methylene phosphonate analogues 5a and 5b, as well as the oleoyl α-hydroxymethylene phosphonate analogue 10a were weak LPA4 agonists. In general, analogues with oleoyl chains were more potent than those with palmitoyl chains, consistent with the preference of LPA4 for unsaturated acyl chains.[14] The most significant activity observed, however, was the discovery that both α-bromomethylene phosphonate analogues 19a and 19b were LPA4 antagonists, with the palmitoyl analogue 19b showing an IC50 value of 266 nm for LPA4. Analogue 19b is therefore the first reported antagonist of the LPA4 receptor and may be a useful pharmacological tool.
In addition to LPA GPCRs, LPA activates the nuclear transcription factor PPARγ.[18] Many agents have been reported to be agonists of PPARγ, including the thiazolidinedione family represented by rosiglitazone (Rosi), oxidized phospholipids, fatty acids, eicosanoids, and oxidized LDL. LPA and the alkyl ether analogue of LPA directly bind to the ligand binding domain of PPARγ.[45] The activation of PPARγ is direct and is enhanced when LPA entry into cells is facilitated by carrier amine sulfonamides capable of increasing the transmembrane movement of LPA.[18] Each of the unsubstituted and α-substituted methylene phosphonate LPA analogues 5, 10, 18, and 19, both oleoyl and palmitoyl analogues, were tested for PPARγ activation in CV-1 cells expressing an acyl-coenzyme A oxidase–luciferase (PPRE–Acox–Rluc) reporter gene construct as previously reported.[19] As shown in Table 1, none of these compounds activated the PPRE–Acox–Rluc reporter. These results are consistent with our previously reported results, in that LPA receptor and PPARγ have different structure–activity profiles.[46]
Inhibition of ATX
Recent evidence shows that LPA is produced extracellularly from LPC by the lysoPLD activity of ATX.[47] The local production of LPA by ATX/lysoPLD could support the invasion of tumor cells, promoting metastasis.[7] The mechanisms of enhanced tumor cell invasion by LPA include two important molecular mechanisms. First, LPA receptor-mediated activation of the Rho and Rac GTPase pathways are essential for the regulation of the actin cytoskeleton and cell motility.[48] Second, LPA has been shown to regulate the activity of matrix metalloproteinases, which are also intricately involved in metastasis as well as in the LPA-induced transphosphorylation of the epidermal growth factor (EGF) receptor.[49] Thus, LPA receptor antagonists and ATX inhibitors may have potential in cancer therapy by blocking the growth-supporting and anti-apoptotic effects of LPA and decreasing its titer. Indeed, a compound with pan-antagonist effects as well as ATX inhibitory effects would be a strong dual effect candidate for drug development.
The inhibition of ATX by the α-substituted methylene phosphonate analogues 5, 10, 18, and 19 was tested at a single dosage (10 μm) and compared with the ATX inhibitory effects of LPA (18:1) and LPA (16:0) at the same concentration (Table 2). ATX activity was measured by the hydrolysis of the fluorogenic lysoPC analogue FS-3, which has a KM value of 6.3 μm.[50] The results showed that all of the analogues, the unsubstituted as well as each of the α-substituted phosphonates, inhibited ATX at a concentration of 10 μm. Although dose–response data remain to be determined, in this preliminary screen, three compounds (5a, 19a, and 19b) were revealed as potent inhibitors of > 90% enzyme activity. The oleoyl unsubstituted phosphonate analogue 5a inhibited 99.8% of ATX activity. In contrast, the oleoyl α-hydroxy analogue 10a gave 74.7% inhibition. The mixed epimers of the palmitoyl α-hydroxy analogue 10b showed 54.1% inhibition; when the pure synthetic diastereomers were tested, the S,S isomer 24a showed 56.6% inhibition, and the S,R isomer 24b showed 43.4% inhibition. Only a marginal discrimination of diastereomers was found. Finally, both the oleoyl and palmitoyl α-bromo analogues 19a and 19b inhibited > 90% of ATX activity. Thus, in terms of therapeutic potential, the palmitoyl α-bromo analogue 19b is by far the most exciting new agent for studying LPA physiology in vitro and in vivo. As both a pan-LPA GPCR antagonist and a potent inhibitor of ATX, this single molecule can pack a “one-two” punch in both significantly lowering LPA production and by blocking activation of all cell-surface LPA receptors. Such a molecule has clear therapeutic potential to inhibiting the role of LPA in promoting tumor growth and metastasis.[51, 52]
Table 2.
Inhibition of autotaxin (ATX) phosphodiesterase activity by LPA analogues.
| Compd | c [μm] | Inhibition [%] | Compd | c[μm] | Inhibition [%] |
|---|---|---|---|---|---|
| LPA (18:1) | 10.0 | 88.4±1.3 | 18a | 10.0 | 92.5±2.1 |
| LPA (16:0) | 10.0 | 38.1±3.4 | 18b | 10.0 | 83.0±2.1 |
| 2ccPA (16:1) | 10.0 | 78.5±2.3 | 19a | 10.0 | 90.7±2.3 |
| 5a | 10.0 | 99.8±2.3 | 19b | 10.0 | 93.7±2.0 |
| 5b | 10.0 | 60.6±4.6 | 24a | 10.0 | 56.6±6.0 |
| 10a | 10.0 | 74.7±4.3 | 24b | 10.0 | 43.4±6.0 |
| 10b | 10.0 | 54.1±5.7 |
Conclusions
We have synthesized a series of α-substituted methylene phosphonate analogues of LPA. Furthermore, we have demonstrated that these analogues are recognized by LPA GPCRs and show a variety of agonist and antagonist effects. The initial SAR study with these novel analogues led to the discovery of agents that show selective agonist activity for LPA2 (methylene phosphonate analogue 5a), selective LPA3 agonist activity (ahydroxymethylene phosphonate 10a), and the LPA4 antagonist activity (α-bromomethylene phosphonate 19b). Methylene phosphonate 5a was the most potent ATX inhibitor and was as effective as natural LPA. However, the pan-antagonism of LPA GPCRs by 19b combined with its ATX inhibitory effect suggest potential utility in anticancer and anti-metastasis models for cancer therapy. The α-substituted methylene phosphonate analogues provided a number of interesting examples of agonist (oleoyl) to antagonist (palmitoyl) switching, indicating the importance of acyl chain length and unsaturation in defining the LPA GPCR activity for a ligand. The LPA receptor subtype-specific agonists and antagonists identified in this study can inform and encourage the development of more potent and more selective compounds. In particular, separate diastereomers of the α-halomethylene phosphonates are required for further study. The results of these synthetic challenges, the biological activities of the individual diastereomers, and computational modeling of ligand activation[53] by individual diastereomers will be reported in due course.
Experimental Section
General
Chemicals were purchased and used without prior purification. Solvents were reagent grade and distilled before use. CH2Cl2 was distilled from CaH2 and THF was distilled from sodium wire. Pre-coated silica gel aluminum sheets were used for TLC. Flash chromatography (FC) was performed with Whatman 230–400 mesh ASTM silica gel. NMR spectra were recorded on a Varian INOVA 400 at 400 MHz (1H), 101 MHz (13C), 162 MHz (31P), and 376 MHz (19F) at 25 °C. Chemical shifts are reported in ppm with TMS as internal standard (δ = 0.00); 31P, 85% H3PO4 (δ = 0.00); 19F, CFCl3 (δ = 0.00).
Diethyl [3(S)-4-dihydroxybutyl]phosphonate (3)
A solution of iodide 2[36] (600 mg, 2.34 mmol) in triethyl phosphite (6 mL) was heated at 130°C under nitrogen for 2 h. Excess triethylphosphite was evaporated and thoroughly dried under vacuum overnight. The crude mixture was then dissolved in 3 mL CH3OH containing p-TsOH monohydrate (7.7 mg, 0.042 mmol). The mixture was stirred at room temperature for 6 h. Next, NaHCO3 (12 mg) was added, and the solvent was evaporated. The residue was purified by silica gel FC (CH2Cl2/CH3OH, 10:1, v/v) and gave 3 as colorless oil (425 mg, 80%): Rf = 0.19 (CH2Cl2/CH3OH, 10:1, v/v); 1H NMR (CDCl3): δ = 4.15 (br, 2 H), 3.97 (m, 4 H), 3.60 (m, 1 H), 3.49 (m, 1H), 3.35 (m, 1 H), 1.90 (m, 1H), 1.51–1.71 (m, 3 H), 1.22 (t, J = 7.2 Hz, 6H); 13C NMR (CDCl3): δ = 71.6 (d, J = 14.7 Hz), 66.2 (s), 61.7 (d, J = 6.2 Hz), 25.7 (d, J = 4.6 Hz), 22.2 (s), 20.7 (s), 16.2 (d, J = 6.2 Hz); 31P NMR (CDCl3): δ = 34.30 (s); MS (ESI) m/z 227.15 [M++1]; HRMS (MALDI) for C8H20O5P [M++1]: found 227.1043, calcd 227.1048.
General procedure for preparing phosphonate monoesters 4, 9, 16, 17, and 23
The procedure is described for 9a. The diol 8 (100 mg, 0.467 mmol) was dissolved in anhydrous CH2Cl2 (4.5 mL), followed by the addition of oleoyl chloride (132 mg, 0.45 mmol) and 2,4,6-collidine (108 mg, 0.90 mmol) under N2. After the reaction mixture was stirred at −78°C for 2.5 h, the solvent was evaporated and EtOAc was added. After filtration, the solution was washed successively with 1n HCl, 10% NaHCO3, and brine. The organic layer was dried over Na2SO4, concentrated, and purified by FC (acetone/hexanes, 1:2, v/v) to give 122 mg of 9a as a colorless oil (57 %).
General procedure for preparing phosphonate analogues 5, 10, 18, 19, and 24
The general protocol is described for preparing 5a. The thoroughly dried precursor 4a (31 mg, 0.063 mmol) was dissolved in dry CH2Cl2 (1 mL) at room temperature, followed by addition of TFBSA (0.1 mL) and TMSBr (44 μL, 0.25 mmol) with dry syringes. The mixture was stirred overnight. When TLC indicated that all of the reactant had been consumed, the solvents were removed in vacuo. The residue was dissolved in CH3OH/H2O (95:5, 1.0 mL) and stirred for 1 h, and then concentrated in vacuo to give the final product 5a (26 mg, 0.060 mmol, 95%).
Diethyl-[3(S)-hydroxy-4-(oleoyloxy)butyl]phosphonate (4 a) was obtained as a colorless oil in 63% yield from precursor 3 after purification by FC. 1H NMR (CDCl3): δ = 5.29 (m, 2 H), 4.05 (m, 5H), 3.95 (m, 1 H), 3.83 (m, 1 H), 2.29 (t, J = 7.8 Hz, 2H), 1.96 (m, 5 H), 1.75 (br, 3H), 1.55 (m, 2 H), 1.05–1.20 (m, 26H), 0.83 ppm (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3): δ = 173.9 (s), 129.8 (d, J = 28.5 Hz), 69.3 (d, J = 13.8 Hz), 67.7 (s), 61.7 (m), 34.1 (s), 31.8 (s), 29.7 (s), 29.6 (s), 29.4 (s), 29.2 (s), 29,1 (m), 27.1 (m), 26.4 (d, J = 4.6 Hz), 24.8 (s), 22.6 (s), 22.4 (s), 21.0 (s), 16.4 (d, J = 6.2 Hz), 14.0 ppm (s); 31P NMR (CDCl3): δ = 33.66 ppm (s); MS (CI) m/z 491.3 [M++1]; HRMS (CI) for C26H52O6P [M++1]: found 491.3495, calcd 491.3503.
Diethyl-[3(S)-hydroxy-4-(palmitoyloxy)butyl]phosphonate (4 b) was obtained in 56% yield from precursor 3. 1H NMR (CDCl3): δ = 4.02 (m, 6H), 3.82 (s, 1H), 2.27 (t, J = 7.8 Hz, 2H), 1.75 (br, 4 H), 1.55 (m, 2 H), 1.05–1.20 (m, 30H), 0.81 ppm (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3): δ = 173.8 (s), 69.2 (d, J = 13.8 Hz), 67.7 (s), 61.7 (m), 34.1 (s), 31.8 (s), 29.6 (s), 29.5 (m), 29.4 (s), 29.3 (s), 29.2 (s), 29.1 (s), 26.4 (d, J = 4.8 Hz), 24.8 (s), 22.6 (s), 22.4 (s), 20.9 (s), 16.3 (d, J = 5.2 Hz), 14.0 ppm (s); 31P NMR (CDCl3): δ = 33.65 ppm (s); MS (CI) m/z 465.3 [M++1]; HRMS (CI) for C24H50O6P [M++1]: found 465.3328, calcd 465.3347.
[3(S)-Hydroxy-4-(oleoyloxy)butyl]phosphonate (5 a) was obtained as colorless oil in 95% yield from precursor 4a. 1H NMR (CDCl3/CD3OD): δ = 5.25 (m, 2 H), 3.95 (m, 2 H), 3.75 (m, 1 H), 2.24 (t, J = 7.8 Hz, 2 H), 1.91 (m, 4 H), 1.75 (br, 4 H), 1.50 (m, 2 H), 1.05–1.20 (m, 20H), 0.78 ppm (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3/CD3OD): δ = 174.2 (s), 129.7 (d, J = 27.7 Hz), 69.0 (d, J = 14.6 Hz), 67.5 (s), 33.9 (s), 31.7 (s), 29.6 (s), 29.3 (s), 29.1 (s), 29.0 (s), 29.0 (s), 27.0 (d, J = 3.1 Hz), 26.4 (d, J = 3.8 Hz), 24.7 (s), 22.5 (s), 13.9 ppm (s); 31P NMR (CDCl3/CD3OD): δ = 32.61 ppm (s); MS (CI) m/z 417.3 [M+−OH]; HRMS (CI) for C22H42O5P [M+ −OH]: found 417.2767, calcd 417.2770.
[3(S)-Hydroxy-4-(palmitoyloxy)butyl]phosphonate (5 b) was obtained in 95% yield from precursor 4b. 1H NMR (CDCl3/CD3OD): δ = 4.95 (m, 1 H), 3.44 (m, 1 H), 3.35 (m, 1 H), 2.26 (dt, J = 7.8 Hz, 2.4 Hz, 2 H), 1.93 (m, 2H), 1.61 (m, 2 H), 1.55 (m, 2H), 1.05–1.20 (m, 24H), 0.79 ppm (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3/CD3OD): δ = 173.5 (s), 72.0 (d, J = 17.7 Hz), 34.2 (s), 33.2 (s), 31.8 (s), 29.5 (s), 29.5 (s), 29.5 (s), 29.3 (s), 29.2 (s), 29.1 (s), 29.0 (s), 25.9 (s), 24.8 (s), 22.5 (s), 13.9 ppm (s); 31P NMR (CDCl3/CD3OD): δ = 31.20 ppm (s); MS (CI) m/z 391.3 [M+ −OH]; HRMS (CI) for C20H40O5P (M+-OH): found 391.2591, calcd 391.2613.
Dimethyl-[1,3(S)-4-trihydroxybutyl]phosphonate (8)
Compound 7 (150 mg, 0.590 mmol) was dissolved in 1.5 mL CH3OH containing p-TsOH monohydrate (5.4 mg, 0.05 equiv, 0.03 mmol). After the mixture was stirred at room temperature overnight, NaHCO3 (8.1 mg) was added, and the solvent was evaporated. The residue was purified by silica gel FC (EtOAc/CH3OH, 3:1, v/v) to give 8 as a colorless oil (102 mg, 81 %): Rf = 0.20 (EtOAc/CH3OH, 4:1, v/v); 1H NMR (CDCl3): δ = 4.20 (br, 2 H), 3.90 (m, 1H), 3.80 (m, 6H), 3.50 (m, 2H), 1.80 ppm (br, 2 H); 13C NMR (CDCl3): δ = 127.8 (s), 125.0 (s), 69.0 (s), 68.8 (s), 66.7 (s), 66.5 (s), 65.6 (s), 64.6 (d, J = 3.1 Hz), 63.2 (s), 63.0 (s), 61.5 (s), 59.5 (s), 52.3 (m), 51.8 (s), 51.7 (s), 34.0 (m), 19.3 (s), 12.5 ppm (s); 31P NMR (CDCl3): δ = 28.51 (s), 29.52 ppm (s); MS (ESI) m/z 215.16 [M++1]; HRMS (MALDI) for C6H16O6P [M++1]: found 215.0679, calcd 215.0685.
Dimethyl-[1,3(S)-dihydroxy-4-(oleoyloxy)butyl]phosphonate (9 a) was obtained as colorless oil in 57% yield from precursor 8 after purification by FC (Rf = 0.16, EtOAc). 1H NMR (CDCl3): δ = 5.17 (m, 2 H), 4.04 (m, 2H), 3.90 (m, 2 H), 3.65 (m, 6H), 2.18 (t, J = 7.8 Hz, 2 H), 1.81 (m, 4.4 H), 1.70 (br, 1.6 H), 1.47 (m, 2H), 1.05–1.20 (m, 20H), 0.71 ppm (t, J = 6.4 Hz, 3H); 13C NMR (CDCl3): δ = 174.1 (s), 129.5 (d, J = 27.7 Hz), 68.0 (m), 67.3 (s), 66.3 (s), 64.6 (m), 63.8 (s), 62.1 (s), 53.1(m), 48.6 (s), 34.1 (s), 33.8 (s), 31.6 (s), 29.4 (s), 29.4 (s), 29.2 (s), 29.0 (m), 28.9 (s), 28.8 (s), 26.8 (d, J = 3.1 Hz), 24.5 (s), 22.3 (s), 13.7 ppm (s); 31P NMR (CDCl3): δ = 32.94 (s), 31.40 ppm (s); MS (ESI) m/z 479.47 [M++1]; HRMS (MALDI) for C24H47NaO7P [M++Na]: found 501.2964, calcd 501.2957.
Dimethyl-[1,3(S)-dihydroxy-4-(palmitoyloxy)butyl]phosphonate (9 b) was obtained in 50% yield from precursor 8. 1H NMR (CDCl3): δ = 4.15–4.30 (m, 2 H), 4.00–4.15 (m, 2 H), 3.77 (m, 6 H), 2.29 (t, J = 7.8 Hz, 2H), 1.90 (m, 1.1 H), 1.80 (m, 0.9 H), 1.57 (m, 2H), 1.16–1.32 (m, 24H), 0.83 ppm (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3): δ = 173.9 (d, J = 2.3 Hz), 69.6 (d, J = 16 Hz), 67.1 (s), 67.6 (m), 66.0 (s), 65.7 (s), 65.5 (s), 64.8 (s), 63.1 (s), 53.4 (m), 34.1 (m), 31.9 (s), 29.6 (s), 29.6 (m), 29.4 (s), 29.3 (s), 29.2 (s), 29.1 (s), 24.8 (s), 22.6 (s), 14.0 ppm (s); 31P NMR (CDCl3): δ = 28.43 (s), 26.67 ppm (s); MS (ESI) m/z 453.42 [M++1]; HRMS (MALDI) for C22H45NaO7P [M+Na+]: found 475.2825, calcd 475.2801.
[1,3(S)-Dihydroxy-4-(oleoyloxy)butyl]phosphonate (10 a): was obtained in 85% yield from precursor 9a. 1H NMR (CDCl3/CD3OD): δ = 5.26 (m, 2H), 4.00 (m, 4 H), 2.26 (t, J = 8.0 Hz, 2 H), 1.93 (m, 4.8 H), 1.82 (m, 1.2 H), 1.52 (m, 2 H), 1.16–1.32 (m, 20H), 0.79 ppm (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3/CD3OD): δ = 174.5 (s), 67.8 (s), 67.0 (s), 50.6 (s), 37.0 (s), 34.1 (s), 32.9 (s), 31.9 (s), 29.7 (s), 29.3 (m), 29.2 (s), 24.8 (s), 22.7 (s), 14.1 ppm (s); 31P NMR (CDCl3): δ = 29.45 (s), 28.07 ppm (s); MS (ESI) m/z 451.35 [M++1]; HRMS (MALDI) for C22H43NaO7P [M+Na+]: found 473.2639, calcd 473.2644.
[1,3(S)-Dihydroxy-4-(palmitoyloxy)butyl]phosphonate (10 b) was obtained in 93% yield from precursor 9b. 1H NMR (CDCl3/CD3OD): δ = 4.00 (m, 4 H), 2.23 (t, J = 7.8 Hz, 2H), 1.86 (m, 0.6 H), 1.73 (m, 1.4 H), 1.50 (m, 2H), 1.16–1.32 (m, 24H), 0.76 ppm (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3/CD3OD): δ = 174.3 (s), 68.3 (m), 67.5 (s), 67.0 (s), 65.3 (m), 64.6 (s), 63.0 (s), 53.4 (s), 34.0 (m), 31.7 (s), 29.5 (m), 29.3 (s), 29.2 (s), 29.1 (s), 29.0 (s), 24.7 (s), 22.5 (s), 13.8 ppm (s); 31P NMR (CDCl3/CD3OD): δ = 29.66 (s), 28.27 ppm (s); MS (ESI) m/z 425.42 [M++1]; HRMS (MALDI) for C20H41NaO7P [M+Na+]: found 447.2474, calcd 447.2488.
1-Diethylphosphonyl-1-chloro-3,4-O-isopropylidene-1(R,S)-3(S),4-butanetriol (12)
Pyridine (1 mL) was added in one portion to a mixture of diethyl-1-hydroxyalkyphosphonate 11 (1.35 g, 4.77 mmol) and triphenylphosphine (1.53 g, 5.73 mmol) in CCl4 (12 mL). After the mixture was held at reflux for 15 h, the solvent was removed in vacuo, and the crude was extracted with EtOAc/hexanes (7:3, v/v). The organic phase was filtered through a 2.54-cm bed of celite 521, and the solvent was evaporated under reduced pressure. Silica gel FC (EtOAc/hexanes, 2:3, v/v) gave 12 as a colorless oil (960 mg, 71%): Rf = 0.55 (EtOAc); 1H NMR (CDCl3): δ = 4.30 (m, 1 H), 4.15 (m, 4H), 4.05 (m, 1.5 H), 3.85 (m, 0.5 H), 3.55 (m, 1 H), 2.25 (m, 1.50 H), 1.85 (m, 0.50 H), 1.25–1.37 ppm (m, 12H); 13C NMR (CDCl3): δ = 109.2 (d, J = 14.1 Hz), 73.8 (s), 72.7 (d, J = 14.1 Hz), 68.7 (d, J = 18.2 Hz), 63.6 (dd, J = 42.4 Hz, 7.1 Hz), 37.5 (dd, J = 36.4 Hz, 20.8 Hz), 27.1 (s), 26.9 (s), 25.5 (s), 16.3 ppm (s); 31P NMR (CDCl3): δ = 21.24 (s), 20.64 ppm (s); MS (CI) m/z 300.9 [M++1]. HRMS (CI) for C11H23ClO5P [M++1]: found 301.0985, calcd 301.0971.
1-Diethylphosphonyl-1-bromo-3,4-O-isopropylidene-1(R,S)-3(S),4-butanetriol (13)
Carbon tetrabromide (555 mg, 1.671 mmol) was added in one portion to a stirred solution of diethyl-1-hydroxyalkyphosphonate 11 (480 mg, 1.453 mmol) and triphenylphosphine (476 mg, 1.817 mmol) in dry toluene (2 mL) at 0 °C. Stirring was continued for 15 min at this temperature, and then the mixture was held at reflux for 9 h. The solvent was removed in vacuo, and the crude was extracted with EtOAc/hexanes (7:3, v/v). The organic phase was filtered through a 2.54-cm bed of celite 521, and the solvent was evaporated under reduced pressure. Silica gel FC (EtOAc/hexanes, 2:3, v/v) gave 13 as a colorless oil (351 mg, 60%): Rf = 0.28 (EtOAc/hexanes, 2:1, v/v); 1H NMR (CDCl3): δ = 4.33 (m, 1 H), 4.15 (m, 4 H), 4.05 (m, 2H), 3.55 (m, 1 H), 2.25 (m, 1.30 H), 1.92 (m, 0.70 H), 1.25–1.37 ppm (m, 12H); 13C NMR (CDCl3): δ = 109.2 (d, J = 14.1 Hz), 73.8 (s), 72.7 (d, J = 14.1 Hz), 68.7 (d, J = 18.2 Hz), 63.6 (dd, J = 42.4 Hz, 7.1 Hz), 37.5 (dd, J = 36.4 Hz, 20.8 Hz), 27.1 (s), 26.9 (s), 25.5 (s), 16.3 ppm (s); 31P NMR (CDCl3): δ = 21.02 (s), 20.49 ppm (s); MS (ESI) m/z 345.11, 347.10 [M++1]. HRMS (MALDI) for C11H23BrO5P [M++1]: found 345.0461, 347.0380, calcd 345.0388, 347.0368.
Diethyl-[1-chloro-3(S)-4-dihydroxybutyl]phosphonate (14)
Compound 12 (900 mg, 3.0 mmol) was dissolved in 20 mL CH3OH containing p-TsOH monohydrate (57 mg, 0.1 equiv, 0.3 mmol). The mixture was stirred at room temperature overnight. Next, NaHCO3 (25 mg) was added, and the solvent was evaporated. The residue was purified by silica gel FC (EtOAc/CH3OH, 10:1, v/v) to give 14 as a colorless oil (750 mg, 96 %): Rf = 0.23 (EtOAc/CH3OH, 6:1, v/v); 1H NMR (CDCl3): δ = 4.12–4.26 (m, 4 H), 3.90 (m, 3H), 3.60 (m, 1 H), 3.45 (m, 1 H), 2.10 (m, 1.50 H), 1.75 (m, 0.50 H), 1.25–1.37 ppm (m, 6H); 13C NMR (CDCl3): δ = 68.7 (d, J = 8.4 Hz), 67.7 (d, J = 13.0 Hz), 66.5 (s), 65.5 (s), 64.3 (dd, J = 47.0 Hz, 7.7 Hz), 63.5 (s), 60.3 (s), 39.2 (s), 48.6 (dd, J = 163.0 Hz, 27.0 Hz), 35.9 (d, J = 97.8 Hz), 16.3 ppm (m); 31P NMR (CDCl3): δ = 22.35 (s), 21.89 ppm (s); MS (CI) m/z 261.0 [M++1]; HRMS (CI) for C8H19ClO5P [M++1]: found 261.0656, calcd 261.0659.
Diethyl-[1-bromo-3(S)-4-dihydroxybutyl]phosphonate (15)
Compound 13 (80 mg, 0.233 mmol) was dissolved in 1.5 mL CH3OH containing p-TsOH monohydrate (2.2 mg, 0.05 equiv, 0.012 mmol). The mixture was stirred at room temperature overnight. Next, NaHCO3 (3.6 mg) was added, and the solvent was evaporated. The residue was purified by silica gel FC (EtOAc/CH3OH, 10:1, v/v) to give 15 as a colorless oil (67 mg, 95 %): Rf = 0.42 (EtOAc/CH3OH, 6:1, v/v); 1H NMR (CDCl3): δ = 4.12–4.26 (m, 4 H), 4.00 (m, 0.5 H), 3.93 (m, 0.5 H), 3.64 (m, 1 H), 3.49 (m, 2H), 2.18 (m, 1.30 H), 1.85 (m, 0.70 H), 1.25–1.37 ppm (m, 6H); 13C NMR (CDCl3): δ = 69.2 (d, J = 40.0 Hz), 66.5 (s), 64.0 (s), 63.7 (s), 39.2 (s), 36.5 (dd, J = 106.6 Hz, 64.6 Hz), 16.4 ppm (s); 31P NMR (CDCl3): δ = 22.07 ppm (s); HRMS (MALDI) for C8H19BrO5P [M++1]: found 305.0148, 307.0129, calcd 305.0154, 307.0133.
Diethyl-[1-chloro-3(S)-hydroxy-4-(oleoyloxy)butyl]phosphonate (16 a) was obtained as a colorless oil in 62% yield from precursor 14 after purification by FC. Rf = 0.17 (EtOAc/hexanes, 1:1, v/v); 1H NMR (CDCl3): δ = 5.30 (m, 2H), 4.00–4.30 (m, 9H), 2.31 (t, J = 7.2 Hz, 2H), 2.25 (br, 1.5 H), 1.93 (m, 4 H), 1.63 (m, 2.5 H), 1.16–1.32 (m, 26H), 0.84 ppm (t, J = 6.4 Hz, 3H); 13C NMR (CDCl3): δ = 173.9 (d, J = 7.7 Hz), 129.8 (d, J = 27.8 Hz), 68.0 (s), 67.2 (s), 66.2 (d, J = 6.9 Hz), 65.8 (s), 65.7 (s), 64.7 (d, J = 7.0 Hz), 63.6 (m), 37.2 (s), 35.9 (s), 34.1 (d, J = 3.0 Hz), 33.9 (s), 29.7 (s), 29.6 (s), 29.5 (s), 29.3 (s), 29.1 (m), 27.1 (m), 25.6 (s), 24.9 (d, J = 4.5 Hz), 22.6 (s), 16.4 (m), 14.1 ppm (s); 31P NMR (CDCl3): δ = 21.98 (s), 21.78 ppm (s); MS (CI) m/z 525.3 [M++1]; HRMS (CI) for C26H51ClO6P [M++1]: found 525.3105, calcd 525.3112.
Diethyl-[1-chloro-3(S)-hydroxy-4-(palmitoyloxy)butyl]phosphonate (16 b) was obtained in 57% yield from precursor 14. 1H NMR (CDCl3): δ = 4.15–4.25 (m, 5H), 4.00–4.15 (m, 3H), 2.32 (t, J = 7.8 Hz, 2 H), 2.20 (br, 1.4 H), 1.90 (m, 0.6 H), 1.63 (m, 2H), 1.16–1.32 (m, 30H), 0.85 ppm (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3): δ = 173.9 (s), 68.1 (s), 67.2 (s), 67.0 (m), 64.5 (s), 63.9 (s), 63.4 (s), 37.2 (s), 36.0 (s), 34.1 (s), 31.9 (s), 29.7 (s), 29.6 (m), 29.4 (s), 29.3 (s), 29.2 (s), 29.1 (s), 24.9 (s), 22.7 (s), 16.4 (d, J = 6.2 Hz), 14.1 ppm (s); 31P NMR (CDCl3): δ = 22.02 (s), 21.75 ppm (s); HRMS (CI) for C24H49ClO6P [M++1]: found 499.2902, calcd 499.2955.
Diethyl-[1-bromo-3(S)-hydroxy-4-(oleoyloxy)butyl]phosphonate (17 a) was obtained as a colorless oil in 82% yield from precursor 15 after purification by FC. 1H NMR (CDCl3): δ = 5.31 (m, 2 H), 4.00–4.28 (m, 7.5 H), 3.45 (m, 0.5 H), 2.31 (t, J = 8.0 Hz, 2H), 2.20 (br, 1.4 H), 1.93 (m, 4.6 H), 1.63 (m, 2H), 1.16–1.32 (m, 26 H), 0.84 ppm (t, J = 6.4 Hz, 3H); 13C NMR (CDCl3): δ = 173.9 (d, J = 7.8 Hz), 129.9 (d, J = 30.8 Hz), 68.0 (s), 67.1 (m), 63.9 (d, J = 7.0 Hz), 63.6 (m), 39.0 (s), 36.1 (s), 34.1 (s), 33.9 (s), 31.9 (s), 29.7 (m), 29.5 (s), 29.3 (s), 29.1 (m), 27.1 (m), 25.6 (s), 24.9 (s), 22.6 (s), 16.4 (m), 14.1 ppm (s); 31P NMR (CDCl3) 21.93 (s), 21.55 ppm (s); HRMS (MALDI) for C26H50BrO6P [M+Na+]: found 591.2414, 593.2416, calcd 591.2426, 593.2406.
Diethyl-[1-bromo-3(S)-hydroxy-4-(palmitoyloxy)butyl]phosphonate (17 b) was obtained in 60% yield from precursor 15. 1H NMR (CDCl3): δ = 4.15–4.25 (m, 4H), 4.00–4.15 (m, 3.5 H), 3.45 (m, 0.5 H), 2.31 (t, J = 8.0 Hz, 2 H), 2.20 (br, 1H), 1.90 (m, 1H), 1.63 (m, 2H), 1.16–1.32 (m, 30H), 0.84 ppm (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3): δ = 173.9 (d, J = 8.5 Hz), 67.9 (s), 67.0 (m), 63.5 (d, J = 7.0 Hz), 63.7 (m), 39.0 (s), 37.6 (m), 36.1 (s), 34.1 (s), 33.9 (s), 31.9 (s), 29.6 (m), 29.4 (s), 29.3 (s), 29.2 (s), 29.1 (s), 25.6 (s), 24.9 (s), 22.6 (s), 16.4 (d, J = 5.4 Hz), 14.1 ppm (s); 31P NMR (CDCl3): δ = 21.90 (s), 21.56 ppm (s); HRMS (MALDI) for C24H48BrO6P [M++1]: found 543.2445, 545.2428, calcd 543.2450, 545.2430.
[1-Chloro-3(S)-hydroxy-4-(oleoyloxy)butyl]phosphonate (18 a) was obtained in 98% yield from precursor 16a. 1H NMR (CDCl3/CD3OD): δ = 5.24 (m, 2 H), 3.90–4.05(m, 4H), 2.25 (t, J = 7.8 Hz, 2H), 1.91 (m, 4 H), 1.52 (m, 4H), 1.12–1.28 (m, 20 H), 0.78 ppm (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3/CD3OD): δ = 174.2 (s), 129.8 (d, J = 27.0 Hz), 67.8 (s), 67.0 (s), 65.5 (s), 65.4 (s), 36.7 (s), 36.0 (s), 33.9 (s), 33.4 (s), 31.7 (s), 29.1–30.2 (m), 27.0 (s), 24.0 (d, J = 3.1 Hz), 25.4 (s), 24.7 (s), 22.5 (s), 13.9 ppm (s); 31P NMR δ = (CDCl3/CD3OD): 20.68 (s), 20.32 ppm (s); MS (CI) m/z 469.3 [M++1]; HRMS (CI) for C22H43ClO6P [M++1]: found 469.2456, calcd 469.2486.
[1-Chloro-3(S)-hydroxy-4-(palmitoyloxy)butyl]phosphonate (18 b) was obtained in 95% yield from precursor 16b. 1H NMR (CDCl3/CD3OD): δ = 4.05–4.25 (m, 3H), 2.27 (t, J = 7.8 Hz, 2 H), 2.10 (br, 1.2 H), 1.85 (m, 0.8 H), 1.54 (m, 2H), 1.16–1.32 (m, 24 H), 0.80 ppm (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3/CD3OD): δ = 174.6 (s), 68.2 (s), 67.4 (s), 66.0 (s), 37.1 (s), 36.4 (s), 34.3 (s), 32.1 (s), 29.9 (s), 29.7 (m), 25.0 (s), 22.9 (s), 22.7 (s), 14.3 ppm (s); 31P NMR (CDCl3/CD3OD): δ = 20.75 ppm (s); HRMS (ESI) for C20H41ClO6P [M++1]: found 443.2326, calcd 443.2329.
[1-Bromo-3(S)-hydroxy-4-(oleoyloxy)butyl]phosphonate (19 a) was obtained in 81% yield from precursor 17a. 1H NMR (CDCl3/CD3OD): δ = 5.31 (m, 2 H), 4.05–4.21(m, 3.5 H), 3.36 (m, 0.5 H), 2.34 (t, J = 7.8 Hz, 2 H), 2.08 (br, 0.8 H), 1.97 (m, 5.2 H), 1.59 (m, 2H), 1.16–1.32 (m, 20H), 0.85 ppm (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3/CD3OD): δ = 174.4 (s), 129.8 (d, J = 23.9 Hz), 67.8 (s), 50.9 (s), 34.1 (s), 32.8 (s), 31.9 (s), 29.1–30.2 (m), 27.2 (s), 24.8 (s), 22.7 (s), 14.1 ppm (s); 31P NMR (CDCl3/CD3OD): δ = 21.57 (s), 21.25 ppm (s); HRMS (MALDI) for C22H42BrNaO6P [M+Na+]: found 535.1803, 537.1813, calcd 535.1800, 537.1779.
[1-Bromo-3(S)-hydroxy-4-(palmitoyloxy)butyl]phosphonate (19 b) was obtained in 95% yield from precursor 17b. 1H NMR (CDCl3/CD3OD): δ = 4.05–4.25 (m, 3.5 H), 3.70 (br, 0.5 H), 2.33 (t, J = 7.8 Hz, 2 H), 2.20 (br, 1.2 H), 2.00 (m, 0.8 H), 1.59 (m, 2H), 1.16–1.32 (m, 24H), 0.85 ppm (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3/CD3OD): δ = 174.5 (s), 67.8 (s), 67.0 (s), 50.6 (s), 37.0 (s), 34.1 (s), 32.9 (s), 31.9 (s), 29.7 (s), 29.3 (m), 29.2 (s), 24.8 (s), 22.7 (s), 14.1 ppm (s); 31P NMR (CDCl3/CD3OD): δ = 22.09 ppm (s), 21.84 (s); HRMS (MALDI) for C20H40BrNaO6P [M+Na+]: found 509.1641, 511.1515, calcd 509.1644, 511.1623.
(S)-Dimethyl-1-O-tert-butyldimethylsilyl-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)ethylphosphonate (20 a)
Compound 7 (1 g, 3.92 mmol) was dissolved in anhydrous DMF (8 mL), and imidazole (0.53 g, 7.84 mmol) and DMAP (0.29 g, 2.35 mmol) were added followed by a solution of TBDMSCl (0.76 g, 5.1 mmol) in DMF (2 mL) at room temperature. The reaction was conducted for 64 h, then stopped by removing of the solvent in vacuo. The mixture of the diastereomers was separated by FC on SiO2 using a gradient of the solvents hexanes/EtOAc: 6:4, 5:5, and 4:6, v/v. The total yield of silylation was 80% (1.16 g). The pure isomer 20a was isolated in 48% yield (0.69 g) in the form of a colorless oil. Rf = 0.27 (EtOAc/hexanes, 4:6, v/v); (c = 1.68, CHCl3); 1H NMR (CD3OD): δ = 4.32–4.26 (m, 1H), 4.09–4.02 (m, 2 H), 3.74 (d, J = 10 Hz, 6 H), 3.52–3.48 (m, 1 H), 2.03–1.92 (m, 2 H), 1.36 (s, 3H), 1.29 (s, 3H), 0.86 (s, 9 H), 0.09 ppm (d, J = 10.8 Hz, 6H); 13C NMR (CD3OD): δ = 108.6, 72.7, 72.6, 69.6, 67.3, 65.6, 53.1 (m), 38.8 (m), 26.9, 25,6, 25.5, 18.0, −5.0 ppm (m); 31P NMR (CD3OD): δ = 26.48 ppm (s); MS (MALDI) m/z 369.17 [M+H+]; HRMS (MALDI) for C15H34O6PSi [M+H+]: found 369.1864, calcd 369.1862.
(R)-Dimethyl-1-O-tert-butyldimethylsilyl-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)ethylphosphonate (20 b)
Isomer 20b was isolated in 34% yield (0.49 g) from precursor 7 in the form of a white solid. Rf = 0.31 (EtOAc/hexanes, 4:6, v/v); (c = 1.07, CHCl3); 1H NMR (CD3OD): δ = 4.21–4.15 (m, 2H), 4.03–3.99 (m, 1H), 3.73 (t, J = 10 Hz, 6H), 3.48 (t, J = 7.6 Hz, 1H), 1.86–1.79 (m, 2 H), 1.33 (s, 3 H), 1.27 (s, 3 H), 0.86 (s, 9 H),) 0.12 (s, 3H), 0.08 ppm (s, 3H); 13C NMR (CD3OD): δ = 108.9 (s), 71.1 (s), 70.9 (s), 69.4 (s), 66.7 (s), 65.0 (s), 53.1 (d, J = 6.9 Hz), 53.7 (d, J = 6.9 Hz), 36.6 (s), 27.0 (s), 25.7 (s), 25.7 (s), 18.2 (s), −4.8 (s), −5.2 ppm (s); 31P NMR (CD3OD): δ = 27.1 ppm (s); MS (MALDI) m/z 369.18 [M+H+]; HRMS (MALDI) for C15H34O6PSi [M+H+]: found 369.1864, calcd 369.1862.
(S)-Dimethyl-1-hydroxy-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-ethylphosphonate (21 a)
Compound 20a (202 mg, 548 mmol) was dissolved in anhydrous CH3OH (2 mL), and solid NH4F (121.4 mg, 3.28 mmol) was added. The reaction was heated at 70°C and stirred overnight (16 h) at that temperature. After it was cooled, the solvent was removed in vacuo, and the crude product 21a was purified on SiO2 (EtOAc/CH3OH, 9:1, v/v) to 83% yield (115.4 mg, 0.45 mmol) as a colorless oil (96% de based on 31P NMR). Rf = 0.39 (EtOAc/CH3OH, 9:1); (c = 0.53, CHCl3); 1H NMR (CDCl3): δ = 4.40 (dd, J = 4.4 Hz, 12.4 Hz, 1H), 4.30–424 (m, 1H), 4.06–4.00 (m, 2 H), 3.74 (s, 3 H), 3.72 (s, 3H), 3.58–3.54 (m, 1 H), 1.99–1.91 (m, 2H), 1.33 (s, 3 H), 1.27 ppm (s, 3H); 13C NMR (CDCl3): δ = 109.1 (s), 74.5 (s), 74.3 (s), 69.0 (s), 66.7 (s), 65.0 (s), 53.3 (s) (m), 34.9 (s), 26.7 (s), 25.5 ppm (s); 31P NMR (CDCl3): δ = 26.9 ppm (s); HRMS (MALDI) for C9H19NaO6P [M+Na+]: found 277.0819, calcd 277.0817.
(R)-Dimethyl-1-hydroxy-2-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-ethylphosphonate (21 b)
Following the procedure described above, compound 21b was obtained in 93% yield (97.7% de based on 31P NMR) from precursor 20b in form of white solid. Rf = 0.39 (EtOAc/CH3OH, 9:1); (c = 0.53, CHCl3); 1H NMR (CDCl3): δ = 4.68 (br, 1H), 4.35–4.29 (m 1 H), 4.12–4.07 (m, 1 H), 4.05–4.01 (m, 1H), 3.75–3.72 (m, 6H), 3.56–3.53 (m, 1 H), 1.92–1.78 (m, 2 H), 1.33 (s, 3 H), 1.27 ppm (s, 3H); 13C NMR (CDCl3): δ = 108.7 (s), 72.6 (s), 72.5 (s), 69.4 (s), 65.4 (s), 63.7 (s), 53.2 (m), 35.3 (s), 26.9 (s), 25.5 ppm (s); 31P NMR (CDCl3): δ = 27.8 ppm (s); HRMS (MALDI) for C9H19NaO6P [M+Na+]: found 277.0815, calcd 277.0817.
Dimethyl-[(1S,3S)-4-trihydroxybutyl]phosphonate (22 a)
Ion-exchange resin (Dowex [H+] 50WX8–200, 150 mg) was added to the solution of 21a (115.4 mg, 0.45 mmol) in CH3OH (5 mL), and the reaction was stirred for 3 h at room temperature. Dowex was filtered off, the organic solvent was removed, and the pure product 22a was dried under high vacuum prior to the next step. 22a was obtained in 82% yield (80 mg) in the form of a colorless oil (94.4% de based on 31P NMR). Rf = 0.49 (CH2Cl2/CH3OH, 8:2, v/v); (c = 1.8, CH3OH);1H NMR (CD3OD): δ = 4.13–4.08 (m, 1 H), 3.86–3.80 (m, 1H), 3.77–3.72 (m, 6 H), 3.49–3.41 (m, 2H), 1.96–1.88 (m, 1 H), 1.80–1.69 ppm (m, 1H); 13C NMR (CD3OD): δ = 70.9 (s), 70.8 (s), 66.6 (s), 64.9 (s), 54.3 (d, J = 7.6 Hz), 53.7 (d, J = 7.6 Hz), 36.2 ppm (s); 31P NMR (CD3OD): δ = 28.57 ppm (s); HRMS (MALDI) for C6H16O6P [M+H+]: found 215.0690, calcd 215.0684.
Dimethyl-[(1R,3S)-4-trihydroxybutyl]phosphonate (22 b) was obtained in 79% yield from precursor 21b as a colorless oil (94.9% de based on 31P NMR) following the procedure provided for compound 22a: Rf = 0.49 (CH2Cl2/CH3OH, 8:2, v/v); (c = 1.24, CH3OH); 1H NMR (CD3OD): δ = 4.17–4.12 (m, 1H), 3.84–3.17 (overlapping signals, m, 7 H), 3.46–3.38 (m, 2H), 1.78–1.69 (m, 1 H), 1.68–1.55 ppm (m, 1H); 13C NMR (CD3OD): δ = 68.7 (s), 68.5 (s), 67.6 (s), 65.2 (s), 63.3 (s), 54.2 (s), 53.7 (s), 36.0 ppm (s); 31P NMR (CD3OD): δ = 29.57 ppm (s); HRMS (MALDI) for C6H16O6P [M+H+]: found 215.0691, calcd 215.0684.
Dimethyl-[(1S,3S)-dihydroxy-4-(palmitoyloxy)butyl]phosphonate (23 a) was obtained in 50% yield from precursor 22a as a white sticky oil. Rf = 0.26 (hexanes/acetone, 5:5, v/v); (c = 0.95, CHCl3, purity: 95% based on 31P NMR); 1H NMR (CDCl3): δ = 4.24–4.20 (m, 1H), 4.15–4.10 (m, 1H), 4.08–4.02 (m, 2 H), 3.81–3.77 (m, 6 H), 2.31 (t, J = 7.6 Hz, 2 H), 1.94–1.87 (m, 2 H), 1.61–1.55 (m, 2 H), 1.34–1.16 (m, 24H), 0.84 ppm (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3): δ = 174.0 (s), 67.0 (s), 69.8 (s), 67.9 (s), 67.8 (s), 66.3 (s), 53.5 (m), 34.1 (m), 31.9 (s), 29.6 (s), 29.6 (s), 29.6 (s), 29.4 (s), 29.3 (s), 29.2 (s), 29.1 (s), 24.8 (s), 22.7 (s), 14.1 ppm (s); 31P NMR (CDCl3): δ = 26.57 ppm (s); MS (MALDI) m/z 475.30 [M+Na+]; HRMS (MALDI) for C22H45NaO7P [M+Na+]: found 475.2817, calcd 475.2801;
Dimethyl-[(1S,3S)-dihydroxy-4-(palmitoyloxy)butyl]phosphonate (23 b) was obtained in 50% yield from precursor 22b as a white sticky oil. Rf = 0.26 (hexanes/acetone, 5:5, v/v); (c = 1.03, CHCl3, purity: 94% based on 31P NMR); 1H NMR (CDCl3): δ = 4.30–4.18 (m, 2 H), 4.10 (dd, J = 3.6 Hz, 11.6 Hz, 1H), 4.02 (dd, J = 6.8 Hz, 11.6 Hz, 1H), 3.80–3.77 (m, 6H), 2.31 (t, J = 7.6 Hz, 2 H), 1.84– 1.76 (m, 2 H), 1.62–1.55 (m, 2H), 1.32–1.16 (m, 24H), 0.84 ppm (t, J = 0.84 Hz, 3H); 13C NMR (CDCl3): δ = 174.0 (s), 68.1 (s), 66.0 (s), 65.8 (s), 65.0 (s), 63.4 (s), 53.4 (m), 34.3 (s), 34.1 (s), 31.9 (s), 29.7 (s), 29.6 (s), 29.6 (s), 29.4 (s), 29.3 (s), 29.3 (s), 29.1 (s), 24.9 (s), 22.6 (s), 14.1 ppm (s); 31P NMR (CDCl3): δ = 28.41 ppm (s); MS (MALDI) m/z 475.28 [M+Na+]; HRMS (MALDI) for C22H45NaO7P [M+Na+]: found 475.2816, calcd 475.2801;
[(1S,3S)-Dihydroxy-4-(palmitoyloxy)butyl]phosphonate (24 a) was obtained in 86% yield from precursor 23a in the form of white flakes. 1H NMR (CD3OD): δ = 4.02–3.92 (m, 3H), 3.86–3.80 (m, 1H), 2.21 (t, J = 7.6 Hz, 2H), 1.91–1.84 (m, 1 H), 1.79–1.68 (m, 1H), 1.50– 1.44 (m, 1 H), 1.24–1.08 (m, 24H), 0.76 ppm (t, J = 6.8 Hz, 3H); 13C NMR (CD3OD): δ = 175.5 (s), 68.7 (s), 68.6 (s), 67.3 (s), 36.3 (s), 34.9 (s), 33.1 (s), 30.8 (s), 30.7 (s), 30.7 (s), 30.6 (s), 30.5 (s), 30.4 (s), 30.3 (s), 26.0 (s), 23.7 (s), 14.4 ppm (s); 31P NMR (CD3OD): δ = 24.24 ppm (s); MS (MALDI) m/z 447.26 [M+Na+]; HRMS (MALDI) for C20H41NaO7P [M+Na+]: found 447.2504, calcd 447.2488;
[(1R,3S)-Dihydroxy-4-(palmitoyloxy)butyl]phosphonate (24 b) was obtained in 89% yield from precursor 23b in the form of white sand. 1H NMR (CD3OD): δ = 3.90–3.80 (m, 4H), 2.15 (t, J = 7.6 Hz, 2 H), 1.63–1.55 (m, 2H), 1.43–1.38 (m, 2 H), 1.17–1.02 (m, 24H), 0.69 ppm (t, J = 6.8 Hz, 3H); 13C NMR (CD3OD): δ = 175.5 (s), 69.6 (s), 66.4 (s), 66.3 (s), 36.2 (m), 34.9 (s), 33.1 (s), 30.8 (s), 30.8 (s), 30.7 (s), 30.6 (s), 30.5 (s), 30.4 (s), 30.2 (s), 26.9 (s), 23.7 (s), 14.5 ppm (s); 31P NMR (CD3OD): δ = 25.28 ppm (s); MS (MALDI) m/z 447.25 [M+Na+]; HRMS (MALDI) for C20H41NaO7P [M+Na+]: found 447.2488, calcd 447.2488.
Procedure for diastereoselective hydrophosphonylation of aldehyde 6 with chiral complex Al(salalen) 27
Complex 27 (12.5 mg, 0.02 mmol) and dimethyl phosphite (10.1 μL, 0.21 mmol) were dissolved in anhydrous THF (1 mL) under Ar atmosphere, and the solution was stirred for 10 min at room temperature. After cooling to −20°C, aldehyde 6 (27 mg, 0.2 mmol) was added, and the solution was stirred for 48 h at −20°C. The reaction was quenched with water and extracted with ethyl acetate. Combined organic phases were dried over Na2SO4, filtered, and concentrated in vacuo. Purification on SiO2 using EtOAc and EtOAc/MeOH 5:1 afforded a colorless oily product. 1H and 31P NMR spectra confirmed the structure of the product. Chemical shifts and integration of the two diastereomers in 31P NMR spectrum: major signal (S,S diastereomer 21a): δ = 26.6 ppm (integration: 92.22 %), minor signal (S,R diastereomer 21b): δ = 27.5 ppm (integration: 7.78 %). Diastereomeric excess for obtained compound was calculated to be 84%.
Receptor activation using Ca2+ mobilization assay
The assay for mobilization of intracellular Ca2+ was performed as described.[29] Briefly, rat hepatoma RH7777 cells stably expressing human LPA1, LPA2, or LPA3, and CHO cells stably expressing LPA4, were loaded with Fura-2 AM (Molecular Probes). Using a FLEXStation (Molecular Devices, Sunnyvale, CA), changes in intracellular Ca2+ concentration were monitored. Ligand-induced changes in fluorescence were monitored for 80–120 s. Ca2+ transients were quantified automatically by calculating the difference between maximum and baseline ratio values for each sample. To determine antagonist properties, different concentrations of each analogue were mixed with either palmitoyl (16:0) or oleoyl (18:1) LPA (200 nm for LPA1 and LPA3, 10 nm for LPA2, and 400 nm for LPA4) (Avanti Polar Lipids, Inc., Shearwater, AL, USA), and responses were recorded. Each test was performed in quadruplicate. EC50, IC50, and Ki values were calculated as described.[24]
PPARγ activation assay
PPARγ activation was performed using CV-1 cells transfected with an acyl-coenzyme A oxidase–luciferase (PPRE–Acox–Rluc) reporter gene construct as previously reported.[45] Briefly, CV-1 cells were plated in 96-well plates at a density of 1×104 cells per well in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. The next day, the cells were transiently transfected with 125 ng of pGL3 PPRE–Acox–Rluc, 62.5 ng of pcDNA3.1 PPARγ, and 12.5 ng of pSV β-galactosidase (Promega) using LipofectAMINE 2000 (Invitrogen). Twenty-four hours after transfection, cells were treated with OptiMEM (Invitrogen) containing 10 μm rosiglitazone or 10 μm of the test analogue dissolved in DMSO for 20 h. Luciferase and β-galactosidase activities were measured with Steady-Glo Luciferase Assay System (Promega) and the Galacto-Light Plus system (Applied Biosystems), respectively. Samples were run in quadruplicate, and the mean values ± SE were calculated. Data are representative of at least two independent transfections.
Autotaxin assay
This assay uses FS-3[50] (Echelon Biosciences, Inc., Salt Lake City, UT, USA) as substrate and recombinant ATX-HA (generously provided by Dr. Tim Clair (NCI)). For analysis, 50 μL of ATXHA (0.25 μg) in assay buffer (Tris 50 mm, NaCl 140 mm, KCl 5 mm, CaCl2 1 mm, MgCl2 1 mm, pH 8.0) was mixed with 25 μL of FS-3 (1 μm final concentration in assay buffer) and 25 μL of test compound dissolved in assay buffer containing 1:1.5 bovine serum albumin in a 96-well plate. FS-3 fluorescence was monitored using a FLEXStation fluorescence plate reader at time zero and after 2 h of incubation at 37°C at excitation and emission wavelengths of 485 nm and 538 nm, respectively. Data was normalized to the corresponding vehicle control, and the mean ±SD of triplicate wells were expressed as percent ATX inhibition. IC50 and Ki values were calculated as described.[24]
Acknowledgments
We thank the Human Frontier Science Program (RG0073-2000B), the NIH (HL070231 and NS29632), and Echelon Biosciences, an Aeterna–Zentaris Company, for financial support to G.D.P. We also thank the NIH for CA921160 and HL61469 to G.T. We are grateful to Dr. T. Shimizu (University of Tokyo) for providing the CHO cell line expressing LPA4, Dr. T. Clair (NIH) for HA-tagged ATX, and Dr. D. Baker for his advice with the ATX assays. We also thank Dr. C. Ferguson (Echelon Biosciences, Inc.) for a generous gift of FS-3.
References
- 1.Moolenaar WH, Kranenburg O, Postma FR, Zondag GCM. Lysophosphatidic acid: G-protein signalling and cellular responses. Curr Opin Cell Biol. 1997;9:168–173. doi: 10.1016/s0955-0674(97)80059-2. [DOI] [PubMed] [Google Scholar]
- 2.Fishman DA, Liu YY, Ellerbroek SM, Stack MS. Lysophosphatidic acid promotes matrix metalloproteinase (MMP) activation and MMP-dependent invasion in ovarian cancer cells. Cancer Res. 2001;61:3194–3199. [PubMed] [Google Scholar]
- 3.Fang X, Schummer M, Mao M, Yu S, Tabassam FH, Swaby R, Hasegawa Y, Tanyi JL, LaPushin R, Eder A, Jaffe R, Erickson J, Mills GB. Lysophosphatidic acid is a bioactive mediator in ovarian cancer. Biochim Biophys Acta Mol Cell Biol Lipids. 2002;1582:257–264. doi: 10.1016/s1388-1981(02)00179-8. [DOI] [PubMed] [Google Scholar]
- 4.Xu Y, Xiao Y-J, Zhu K, Baudhuin LM, Lu J, Hong G, Kim K-S, Cristina KL, Song L, Williams FS, Elson P, Markman M, Belinson J. Unfolding the pathophysiological role of bioactive lysophospholipids. Curr Drug Targets Immune Endocr Metab Disord. 2003;3:23–32. [PubMed] [Google Scholar]
- 5.Fang X, Yu S, LaPushin R, Lu Y, Furui T, Penn LZ, Stokoe D, Erickson JR, Bast RC, Jr, Mills GB. Lysophosphatidic acid prevents apoptosis in fibroblasts via G(i)-protein-mediated activation of mitogen-activated protein kinase. Biochem J. 2000;352:135–143. [PMC free article] [PubMed] [Google Scholar]
- 6.Mills GB, Eder A, Fang X, Hasegawa Y, Mao M, Lu Y, Tanyi J, Tabassam FH, Wiener J, Lapushin R, Yu S, Parrott JA, Compton T, Tribley W, Fishman D, Stack MS, Gaudette D, Jaffe R, Furui T, Aoki J, Erickson JR. Critical role of lysophospholipids in the pathophysiology, diagnosis, and management of ovarian cancer. Cancer Treat Res. 2002;107:259–283. doi: 10.1007/978-1-4757-3587-1_12. [DOI] [PubMed] [Google Scholar]
- 7.Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3:582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 8.Sano T, Baker D, Virag T, Wada A, Yatomi Y, Kobayashi T, Igarashi Y, Tigyi G. Multiple mechanisms linked to platelet activation result in lysophosphatidic acid and sphingosine 1-phosphate generation in blood. J Biol Chem. 2002;277:21197–21206. doi: 10.1074/jbc.M201289200. [DOI] [PubMed] [Google Scholar]
- 9.van Meeteren LA, Ruurs P, Christodoulou E, Goding JW, Takakusa H, Kikuchi K, Perrakis A, Nagano T, Moolenaar WH. Inhibition of autotaxin by lysophosphatidic acid and sphingosine 1-phosphate. J Biol Chem. 2005;280:21155–21161. doi: 10.1074/jbc.M413183200. [DOI] [PubMed] [Google Scholar]
- 10.Hama K, Aoki J, Fukaya M, Kishi Y, Sakai T, Suzuki R, Ohta H, Yamori T, Watanabe M, Chun J, Arai H. Lysophosphatidic acid and autotaxin stimulate cell motility of neoplastic and non-neoplastic cells through LPA1. J Biol Chem. 2004;279:17634–17639. doi: 10.1074/jbc.M313927200. [DOI] [PubMed] [Google Scholar]
- 11.Euer N, Schwirzke M, Evtimova V, Burscher H, Jarsch M, Tarin D, Weidle UH. Identification of genes associated with metastasis of mammary carcinoma in metastatic versus non-metastatic cell lines. Anticancer Res. 2002;22:733–740. [PubMed] [Google Scholar]
- 12.Luquain C, Sciorra VA, Morris AJ. Lysophosphatidic acid signaling: how a small lipid does big things. Trends Biochem Sci. 2003;28:377–383. doi: 10.1016/S0968-0004(03)00139-7. [DOI] [PubMed] [Google Scholar]
- 13.Umezu-Goto M, Tanyi J, Lahad J, Liu S, Yu S, Lapushin R, Hasegawa Y, Lu Y, Trost R, Bevers T, Jonasch E, Aldape K, Liu J, James RD, Ferguson CG, Xu Y, Prestwich GD, Mills GB. Lysophosphatidic acid production and action: validated targets in cancer? J Cell Biochem. 2004;92:1115–1140. doi: 10.1002/jcb.20113. [DOI] [PubMed] [Google Scholar]
- 14.Noguchi K, Ishii S, Shimizu T. Identification of p2y9/GPR23 as a novel G-protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J Biol Chem. 2003;278:25600–25606. doi: 10.1074/jbc.M302648200. [DOI] [PubMed] [Google Scholar]
- 15.Kotarsky K, Boketoft A, Bristulf J, Nilsson NE, Norberg A, Hansson S, Sillard R, Owman C, Leeb-Lundberg FLM, Olde B. Lysophosphatidic acid binds to and activates GPR92, a G-protein-coupled receptor highly expressed in gastrointestinal lymphocytes. J Pharmacol Exp Ther. 2006;318:619–628. doi: 10.1124/jpet.105.098848. [DOI] [PubMed] [Google Scholar]
- 16.Lee C-W, Rivera R, Gardell S, Dubin A, Chun J. GPR92 as a new G12/13 and Gq coupled lysophosphatidic acid receptor that increases cAMP: LPA5. J Biol Chem. 2006;281:23589–23597. doi: 10.1074/jbc.M603670200. [DOI] [PubMed] [Google Scholar]
- 17.Baker DL, Morrison P, Miller B, Riely CA, Tolley B, Westermann AM, Bonfrer JMG, Bais E, Moolenaar WH, Tigyi G. Plasma lysophosphatidic acid concentration and ovarian cancer. JAMA J Am Med Assoc. 2002;287:3081–3082. doi: 10.1001/jama.287.23.3081. [DOI] [PubMed] [Google Scholar]
- 18.McIntyre TM, Pontsler AV, Silva AR, St Hilaire A, Xu Y, Hinshaw JC, Zimmerman GA, Hama K, Aoki J, Arai H, Prestwich GD. Identification of an intracellular receptor for lysophosphatidic acid (LPA): LPA is a transcellular PPARγ agonist. Proc Natl Acad Sci USA. 2003;100:131–136. doi: 10.1073/pnas.0135855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang C, Baker DL, Yasuda S, Makarova N, Balazs L, Johnson LR, Marathe GK, McIntyre TM, Xu Y, Prestwich GD, Byun HS, Bittman R, Tigyi G. Lysophosphatidic acid induces neointima formation through PPARγ activation. J Exp Med. 2004;199:763–774. doi: 10.1084/jem.20031619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santos WL, Heasley BH, Jarosz R, Carter KM, Lynch KR, Macdonald TL. Synthesis and biological evaluation of phosphonic and thiophosphoric acid derivatives of lysophosphatidic acid. Bioorg Med Chem Lett. 2004;14:3473–3476. doi: 10.1016/j.bmcl.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 21.Heise CE, Santos WL, Schreihofer AM, Heasley BH, Mukhin YV, Macdonald TL, Lynch KR. Activity of 2-substituted lysophosphatidic acid (LPA) analogues at LPA receptors: discovery of a LPA1/LPA3 receptor antagonist. Mol Pharmacol. 2001;60:1173–1180. doi: 10.1124/mol.60.6.1173. [DOI] [PubMed] [Google Scholar]
- 22.Ohta H, Sato K, Murata N, Damirin A, Malchinkhuu E, Kon J, Kimura T, Tobo M, Yamazaki Y, Watanabe T, Yagi M, Sato M, Suzuki R, Murooka H, Sakai T, Nishitoba T, Im DS, Nochi H, Tamoto K, Tomura H, Okajima F. Ki16425, a subtype-selective antagonist for EDG-family lysophosphatidic acid receptors. Mol Pharmacol. 2003;64:994–1005. doi: 10.1124/mol.64.4.994. [DOI] [PubMed] [Google Scholar]
- 23.Heasley RH, Jarosz R, Lynch KR, Macdonald TL. Initial structure–activity relationships of lysophosphatidic acid receptor antagonists: discovery of a high-affinity LPA1/LPA3 receptor antagonist. Bioorg Med Chem Lett. 2004;14:2735–2740. doi: 10.1016/j.bmcl.2004.03.076. [DOI] [PubMed] [Google Scholar]
- 24.Virag T, Elrod DB, Liliom K, Sardar VM, Parrill AL, Yokoyama K, Durgam G, Deng W, Miller DD, Tigyi G. Fatty alcohol phosphates are subtype-selective agonists and antagonists of lysophosphatidic acid receptors. Mol Pharmacol. 2003;63:1032–1042. doi: 10.1124/mol.63.5.1032. [DOI] [PubMed] [Google Scholar]
- 25.Qian L, Xu Y, Hasegawa Y, Aoki J, Mills GB, Prestwich GD. Enantioselective responses to OMPT, a phosphorothioate analog of lysophosphatidic acid with LPA3 receptor-selective agonist activity. J Med Chem. 2003;46:5575–5578. doi: 10.1021/jm034207p. [DOI] [PubMed] [Google Scholar]
- 26.Xu Y, Qian L, Prestwich GD. Synthesis of α-fluorinated phosphonates from α-fluorovinylphosphonates: a new route to analogues of lysophosphatidic acid. Org Lett. 2003;5:2267–2270. doi: 10.1021/ol034597+. [DOI] [PubMed] [Google Scholar]
- 27.Xu Y, Aoki J, Shimizu K, Umezu-Goto M, Hama K, Takanezawa Y, Yu S, Mills GB, Arai H, Qian L, Prestwich GD. Structure–activity relationships of fluorinated lysophosphatidic acid analogues. J Med Chem. 2005;48:3319–3327. doi: 10.1021/jm049186t. [DOI] [PubMed] [Google Scholar]
- 28.Tamaruya Y, Suzuki M, Kamura G, Kanai M, Hama K, Shimizu K, Aoki J, Arai H, Shibasaki M. Identifying specific conformations by using a carbohydrate scaffold: discovery of subtype-selective LPA-receptor agonists and an antagonist. Angew Chem. 2004;116:2894–2897. doi: 10.1002/anie.200454065. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:2834–2837. doi: 10.1002/anie.200454065. [DOI] [PubMed] [Google Scholar]
- 29.Durgam GG, Virag T, Walker MD, Tsukahara R, Yasuda S, Liliom K, van Meeteren LA, Moolenaar WH, Wilke N, Siess W, Tigyi G, Miller DD. Synthesis, structure–activity relationships, and biological evaluation of fatty alcohol phosphates as lysophosphatidic acid receptor ligands, activators of PPARγ and inhibitors of autotaxin. J Med Chem. 2005;48:4919–4930. doi: 10.1021/jm049609r. [DOI] [PubMed] [Google Scholar]
- 30.Prestwich GD, Xu Y, Qian L, Gajewiak J, Jiang G. New metabolically stabilized analogues of lysophosphatidic acid: agonists, antagonists, and enzyme inhibitors. Biochem Soc Trans. 2005;33:1357–1361. doi: 10.1042/BST0331357. [DOI] [PubMed] [Google Scholar]
- 31.Tigyi G, Parrill AL. Molecular mechanisms of lysophosphatidic acid action. Prog Lipid Res. 2003;42:498–526. doi: 10.1016/s0163-7827(03)00035-3. [DOI] [PubMed] [Google Scholar]
- 32.Hasegawa Y, Erickson JR, Goddard GJ, Yu S, Liu S, Cheng KW, Eder A, Bandoh K, Aoki J, Jarosz R, Schrier AD, Lynch KR, Mills GB, Fang X. Identification of a phosphothionate analogue of lysophosphatidic acid (LPA) as a selective agonist of the LPA3 receptor. J Biol Chem. 2003;278:11962–11969. doi: 10.1074/jbc.M209168200. [DOI] [PubMed] [Google Scholar]
- 33.Blackburn GM. Phosphonates as analogues of biological phosphates. Chem Ind. 1981;5:134–138. [Google Scholar]
- 34.Engel R. Phosphonates as analogues of natural phosphates. Chem Rev. 1977;77:349–367. [Google Scholar]
- 35.Thomas BN, Corcoran RC, Cotant CL, Lindemann CM, Kirsch JE, Persichini PJ. Phosphonate lipid tubules. 1. J Am Chem Soc. 1998;120:12178–12186. doi: 10.1021/ja012137a. [DOI] [PubMed] [Google Scholar]
- 36.Allingham MT, Howard-Jones A, Murphy PJ, Thomasa DA, Caulkett PWR. Synthesis and applications of C2-symmetric guanidine bases. Tetrahedron Lett. 2003;44:8677–8680. [Google Scholar]
- 37.Hanaya T, Miyoshi A, Noguchi A, Kawamoto H, Armour MA, Hogg AM, Yamamoto H. A convenient synthesis of (2R)-1-amino-1-deoxy-1-phosphinylglycerols. Bull Chem Soc Jpn. 1990;63:3590–3594. [Google Scholar]
- 38.Pudovik AN, Konovalova IV. Addition reactions of esters of phosphorus(III) acids with unsaturated systems. Synthesis. 1979:81–96. [Google Scholar]
- 39.Gajda T. Preparation of diethyl 1-bromoalkylphosphonates. Phosphorus Sulfur Silicon Relat Elem. 1990;53:327–331. [Google Scholar]
- 40.Gajda T. A convenient synthesis of diethyl 1-chloroalkylphosphonates. Synthesis. 1990:717–718. [Google Scholar]
- 41.Cabioch JL, Pellerin B, Denis JM. Synthesis of primary α-chlorophosphines by a chemoselective reduction of α-chlorophosphonates. Phosphorus Sulfur Silicon Relat Elem. 1989;44:27–32. [Google Scholar]
- 42.Yamashita M, Morizane T, Fujita K, Nakatani K, Inokawa S. Reaction of dimethyl 1-hydroxyalkylphosphonates with some chlorides. Bull Chem Soc Jpn. 1987;60:812–814. [Google Scholar]
- 43.Saito B, Katsuki T. Synthesis of an optically active C1-symmetric Al(salalen) complex and its application to the catalytic hydrophosphonylation of aldehydes. Angew Chem. 2005;117:4676–4678. doi: 10.1002/anie.200501008. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:4600–4602. doi: 10.1002/anie.200501008. [DOI] [PubMed] [Google Scholar]
- 44.Fischer DJ, Nusser N, Virag T, Yokoyama K, Wang DA, Baker DL, Bautista D, Parrill AL, Tigyi G. Short-chain phosphatidates are subtype-selective antagonists of lysophosphatidic acid receptors. Mol Pharmacol. 2001;60:776–784. [PubMed] [Google Scholar]
- 45.Tsukahara T, Tsukahara R, Yasuda S, Makarova N, Valentine WJ, Allison P, Yuan H, Baker DL, Li Z, Bittman R, Parrill AL, Tigyi G. Different residues mediate recognition of 1-O-oleyllysohosphatidic acid and rosiglitazone in the ligand binding domain of peroxisome proliferator-activated receptor gamma. J Biol Chem. 2005;281:3398–3407. doi: 10.1074/jbc.M510843200. [DOI] [PubMed] [Google Scholar]
- 46.Xu Y, Qian L, Pontsler AV, McIntyre TM, Prestwich GD. Synthesis of difluoromethyl substituted lysophosphatidic acid analogues. Tetrahedron. 2004;60:43–49. [Google Scholar]
- 47.Aoki J, Taira A, Takanezawa Y, Kishi Y, Hama K, Kishimoto T, Mizuno K, Saku K, Taguchi R, Arai H. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J Biol Chem. 2002;277:48737–48744. doi: 10.1074/jbc.M206812200. [DOI] [PubMed] [Google Scholar]
- 48.Fleming IN, Elliott CM, Collard JG, Exton JH. Lysophosphatidic acid induces threonine phosphorylation of Tiam1 in Swiss 3T3 fibroblasts via activation of protein kinase C. J Biol Chem. 1997;272:33105–33110. doi: 10.1074/jbc.272.52.33105. [DOI] [PubMed] [Google Scholar]
- 49.Gschwind A, Prenzel N, Ullrich A. Lysophosphatidic acid-induced squamous cell carcinoma cell proliferation and motility involves epidermal growth factor receptor signal transactivation. Cancer Res. 2002;62:6329–6336. [PubMed] [Google Scholar]
- 50.Ferguson CG, Bigman CS, Richardson RD, van Meeteren LA, Moolenaar WH, Prestwich GD. Fluorogenic phospholipid substrate to detect lysophospholipase D/autotaxin activity. Org Lett. 2006;8:2023–2026. doi: 10.1021/ol060414i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tanyi J, Croetzer D, Wolf J, Yu S, Hasegawa Y, Lahad J, Cheng KW, Umezu-Goto M, Prestwich GD, Morris AJ, Newman RA, Felix EA, Lapis R, Mills GB. Functional lipidomics: Lysophosphatidic acid as a target for molecular diagnosis and therapy of ovarian cancer. In: Feng L, Prestwich GD, editors. Functional Lipidomics. CRC Press/Taylor & Francis; New York: 2006. pp. 101–123. [Google Scholar]
- 52.Baker D, Fujiwara Y, Pigg KR, Tsukahara R, Kobayashi S, Murofushi H, Uchiyama A, Murakami-Murofushi K, Koh E, Bandle RW, Byun HS, Bittman R, Fan D, Murph M, Mills GB, Tigyi G. Carba analogues of cyclic phosphatidic acid are selective inhibitors of autotaxin and cancer cell invasion and metastasis. J Biol Chem. 2006;281:22786–22793. doi: 10.1074/jbc.M512486200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sardar VM, Bautista DL, Fischer DJ, Yokoyama K, Nusser N, Virag T, Wang D, Baker DL, Tigyi G, Parrill AL. Molecular basis for lysophosphatidic acid receptor antagonist selectivity. Biochim Biophys Acta Mol Cell Biol Lipids. 2002;1582:309–317. doi: 10.1016/s1388-1981(02)00185-3. [DOI] [PubMed] [Google Scholar]