

Abstract
SN1 DNA methylating agents are genotoxic agents that methylate numerous nucleophilic centers within DNA including the O6 position of guanine (O6meG). Methylation of this extracyclic oxygen forces mispairing with thymine during DNA replication. The mismatch repair (MMR) system recognizes these O6meG:T mispairs and is required to activate DNA damage response (DDR). Exonuclease I (EXO1) is a key component of MMR by resecting the damaged strand; however, whether EXO1 is required to activate MMR-dependent DDR remains unknown. Here we show that knockdown of the mouse ortholog (mExo1) in mouse embryonic fibroblasts (MEFs) results in decreased G2/M checkpoint response, limited effects on cell proliferation, and increased cell viability following exposure to the SN1 methylating agent N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), establishing a phenotype paralleling MMR deficiency. MNNG treatment induced formation of γ–H2AX foci with which EXO1 co-localized in MEFs, but mExo1-depleted MEFs displayed a significant diminishment of γ–H2AX foci formation. mExo1 depletion also reduced MSH2 association with DNA duplexes containing G:T mismatches in vitro, decreased MSH2 association with alkylated chromatin in vivo, and abrogated MNNG-induced MSH2/CHK1 interaction. To determine if nuclease activity is required to activate DDR we stably overexpressed a nuclease defective form of human EXO1 (hEXO1) in mExo1-depleted MEFs. These experiments indicated that expression of wildtype and catalytically null hEXO1 was able to restore normal response to MNNG. This study indicates that EXO1 is required to activate MMR-dependent DDR in response to SN1 methylating agents; however, this function of EXO1 is independent of its nucleolytic activity.
Keywords: DNA damage response, Mismatch Repair, Cell Cycle Checkpoints
1. INTRODUCTION
SN1 DNA methylating agents, such as the cancer therapeutics temozolomide and cyclophosphamide and the laboratory agents N-methyl-N′-nitrosourea (MNU) and N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), are potent genotoxic and mutagenic compounds that methylate numerous nucleophilic centers within DNA. While SN1 methylating agents most commonly methylate ring nitrogens, in particular the N7 position of guanine and the N3 position of adenine, methylation of the extracyclic O6 position of guanine (O6meG) is widely viewed as the most cytotoxic lesion induced by genotoxins of this class [1].
Methylguanine methyltransferase (MGMT) is the principal repair molecule responsible for resolving O6meG adducts. This repair is catalyzed by direct demethylation of the O6meG adduct and transfer of the methyl group to a cysteine within the primary structure of MGMT [2]. As this reaction is catalyzed in a 1:1 stoichiometry between O6meG and MGMT, and methyl-MGMT is quickly subjected to ubiquitin-mediated proteolysis [3], the cell possesses a finite capability to repair O6meG adducts until MGMT levels are restored. Thus, O6meG repair is temporally saturable and SN1 methylator exposure will force persistent O6meG adducts. Commonly, DNA replication results in the mispairing of O6meG with thymine and it is these O6meG:T mispairs that are widely viewed as the primary reason for the potently mutagenic nature of SN1 methylating agents [1, 4].
The DNA mismatch repair (MMR) system is a highly conserved DNA repair mechanism principally responsible for the repair of small loops or single base mismatches arising during replication [5]. Unsurprisingly, inactivation of MMR results in increased spontaneous mutation rates, genomic instability, and cancer predisposition [6, 7]. In addition to its role in mutation avoidance, MMR also plays a critical role in the activation of DNA damage response (DDR) in response to persistent O6meG lesions as well as some other types of DNA damage (such as cisplatin adducts). Specifically, MMR recognizes O6meG:T and, to a lesser extent, O6meG:C base pairs [8] and the recognition of these lesions by the MMR system is critical to the activation of responses such as the triggering of cell cycle checkpoints or apoptosis [9]. Among the evidence supporting this view is that MMR-deficient cells are resistant to the cytotoxic effects stemming from exposure to SN1 methylating agents [10-13], a phenotype that has been termed “alkylation tolerance” [12, 14].
Recent work from our group and others indicates that alkylation tolerant phenotype stems, in part, from defective activation of canonical DDR mechanisms; specifically, the ATM/ATR and CHK2/CHK1 signaling axes that activate the G2/M checkpoint in response to SN1 methylating agents [15-20]. While the requirement for an intact MMR system to activate DDR is well established, how the MMR system mechanistically functions in the activation of DDR remains unresolved and two non-mutually exclusive models have been proposed [21]. The “futile-repair” model hypothesizes that O6meG:T mismatches activate MMR following replication; however, since MMR specifically targets the nascent strand for repair, the O6meG adduct within the parental strand remains intact following strand resection. The persistence of the O6meG:T mispairing arising during repair-initiated DNA re-synthesis will again trigger another round of MMR. Such iterative repair cycles will generate repair intermediates leading to replication fork collapse during replication. These structures, as well as the associated accumulation of DNA strand breaks within the genome, will subsequently activate the ATM/ATR damage-signaling network. The second model, termed “direct-signaling”, proposes that the assembly of a MMR complex on alkylated DNA provides a molecular scaffold onto which response transducers such as ATM and ATR and signal effectors such as CHK1 and CHK2 associate. The assembly of this macromolecular complex at the site of damage is hypothesized to be critical for the timely activation of these signaling kinases. Substantial data obtained from numerous groups seemingly support either model [15, 19, 22-24], thus raising the possibility that elements of each model function in MMR-dependent activation of DDR following exposure to SN1 methylating agents.
EXO1 is a member of the RAD2 nuclease family, possesses 5’ to 3’ exonuclease and flap structure-specific endonuclease activity, and to date is the only exonuclease found to associate with MMR components in eukaryotes [25, 26]. EXO1 functions in resection of the mismatched nascent strand during both 5’ and 3’ nick-directed MMR [27, 28]. Despite this key role in eukaryotic MMR, exo1 depletion in yeast results in a weaker mutator phenotype when compared to depletion of msh2 or mlh1 [29-32]. Similarly, Exo1 knockout in mice results in only a modest cancer predisposition, and EXO1 deficiency is associated with a 30-fold elevation of hypoxanthine-guanine phosphoribosyltransferase (HGPRT) mutability, significantly less than the 150-fold increase observed in Msh2 deficient cells [33].
The weak mutator phenotype associated with EXO1 deficiency suggests that other exonucleases may function in a redundant fashion during eukaryotic MMR, just as is the case in E. coli where at least four exonucleases (ie, Exo1, ExoVII, RecJ and ExoX) function during MMR [34-37]. In support of this notion, analysis of EXO1-deficient cell lines has indicated that these cells display significant levels (up to 25% the level observed in EXO1-proficient cells) of residual mismatch repair activity [28, 33, 38, 39]. It was proposed that EXO1-independent but MutLα and DNA polymerase δ-dependent strand displacement synthesis may account for the EXO1-independent mode of mismatch repair in a purified system [39]. While an attractive mechanism, attempts to visualize strand displacement products produced upon incubation of heteroduplex substrates with EXO1-deficient cell extracts yielded negative results [39]. One possible explanation for these findings would be existence of one or more alternate nucleolytic activities that can substitute for EXO1; however, genetic screens designed to uncover such redundant exonucleases have proven unsuccessful [40].
In addition to its enzymatic role in strand resection, a number of studies support the view that EXO1 may also play a structural role in MMR-mediated mutation avoidance. Interaction of EXO1 with core MMR proteins is highly conserved, for example, EXO1 directly associates with MSH2, MSH3, MLH1, PMS2 [41-47] and co-localizes with PCNA [44, 48] in humans. Moreover, several lines of evidence suggest that interaction between EXO1 and core MMR components is necessary for optimal MMR activity. Specifically, two groups identified missense mutations in mlh1, msh2 or pms1 that, in isolation, exert a moderate effect on mutation rates in yeast; however, when these mutations where introduced into an exo1Δ strain, each produced a strong mutator phenotype [40, 49]. Further, when these missense alleles where examined in combination with an exonuclease-defective exo1 mutant (D173A), or exo1 mutated within the mlh1 binding motif (FF447AA), the resulting mutator phenotype was consistently less pronounced than that measured in exo1Δ cells [30, 49]. Moreover, double exo1 mutants (mlh1 binding and catalytic mutation) combined with these mlh1 or pms1 missense mutations phenocopied exo1Δ [45]. A similar study established that mutation of mlh1 at the site of exo1 interaction significantly reduced, but did not completely abolish, MMR-dependent mutation avoidance [42]. In humans, recent findings also suggest that EXO1-MMR interaction is likely of biological and clinical significance. For example, mutations in MLH1 linked to Hereditary Non-Polyposis Colorectal Cancer (HNPCC) have been characterized as affecting interaction with EXO1 [46, 47]. Another study identified MSH2 mutations in HNPCC patients that similarly alter interaction with EXO1 [50]. Conversely, three mutations in EXO1 that decreased its interaction with MSH2 have been characterized in colorectal tumors displaying no MSH2, MLH1, or MSH6 mutations [51]. Collectively, this body of evidence suggests that mutator phenotypes stemming from loss of EXO1 are not necessarily attributable to dysregulation of EXO1 catalytic activity; rather, these phenotypes may be linked to structural defects such as loss of EXO1 interaction with core MMR components.
Despite these advances, if and/or how EXO1 functions in activating MMR-dependent cytotoxic response to O6meG adducts remains largely unaddressed. Here we show that RNAi-mediated EXO1 knockdown in mouse fibroblasts directly results in an alkylation-tolerant phenotype, paralleling documented defects in MMR-deficient cells. Furthermore, findings presented support our conclusion that EXO1 is a critical mediator of cellular response to SN1 methylator-induced damage response, MSH2 interaction with mismatched DNA in vitro and alkylated chromatin in vivo, and MNNG-induced MSH2/CHK1 association. Moreover, these functions of EXO1 are independent of its exonucleolytic activity. In sum, this study leads us to conclude that the EXO1 protein is a critical component in MMR-dependent signaling in response to SN1 methylator-induced genotoxic stress.
2. MATERIALS AND METHODS
2.1 Cell culture
Mouse embryonic fibroblasts (MEFs) were a gift from Dr. J. Lu (Univ of Florida) and maintained in DMEM supplemented with 10% (v/v) fetal bovine serum and penicillin/streptomycin in a humidified 5% CO2 environment at 37°C. Growth media for C3-ΔExo1 and C6-ΔExo1 cell lines was supplemented with puromycin (2μg/ml). C3-HA-hEXO1, C3-HA-mut-hEXO1, C3-HA-VC, C6-HA-hEXO1 and C6-HA-VC were cultured with both puromycin (2 μg/ml) and G418 sulfate (300 μg/ml). Cells were pretreated with 10 μM O6-Benzylguanine (O6BG) for 6 hours prior to addition of MNNG to fully inhibit repair of O6meG adducts by MGMT. MNNG and O6BG were purchased from Sigma (St. Louis, MO). Stock solutions were prepared in DMSO and stored at -20°C prior to use.
2.2 RNA interference
Lentiviral shRNA clones were obtained from Open Biosystems (Lafayette, CO). Five independent mExo1 or mMsh6 shRNA clones were initially screened for Exo1 or Msh6 knockdown. mExo1 shRNA clones (TRCN0000071124 (C3) and TRCN0000071126 (C6)) one mMsh6 clone (TRCN0000071163) were characterized as producing the most diminished target gene expression.
Lentivirus were produced by transfecting HEK 293T with the packaging plasmids psPAX2 and pMD2.G (Addgene, Cambridge, MA) using Lipofectamine 2000 (Invitrogen, Grand Island, NY) per manufacturer’s instructions. 48 hr post-transfection, virus-containing media was collected, centrifuged at 3000 rpm for 5 min and passed through a syringe filter. Polybrene was added to a final concentration of 5μg/ml and medium added to cultures of MEFs. 48 hr post-infection, puromycin was added and selection maintained for > 2 weeks, at which time gene depletion were determined by RT-PCR and immunoblotting.
2.3 Cell viability and proliferation assays
Relative cell viability following MNNG treatment was determined using an AlamarBlue assay as outlined by the manufacturer (AbDSerotec, Raleigh, NC). Briefly, cells were plated into 24-well plates and allowed to adhere overnight. The next day, cells were pretreated with O6BG, MNNG was later added to the media and at indicated times cells were rinsed with 1X PBS, new media containing 1/10 volume of AlamarBlue reagent was added to the wells and cells were incubated at 37°C for an additional 2 hr. After this, 100 μl of media from each well was pipetted (in triplicate) into a 96 well plate and fluorescence (545 nm excitation, 590 nm emission wavelength) measured using a BMG Labtech fluorometer. Cell viability was calculated relative to an untreated culture of cells incubated in parallel.
Cell proliferation rate was measured by plating 5×103 cells into 24 well plates and the following day cells were either treated with MNNG or were untreated. Cells were grown for indicated time, harvested by trypsinization, and live cells counted using Trypan Blue. Alternatively, phase contrast images of attached cells were acquired using a Zeiss Axioplan 2 microscope equipped with an AxioCam digital camera.
2.4 RT-PCR and Q-PCR
Total RNA was extracted from cells using TRI Reagent (Molecular Research Center, Cincinnati, OH) following the manufacturer’s protocol. 2μg of total RNA was used to synthesize cDNA using an AffinityScript Multi Temperature cDNA synthesis kit (Aglient, Santa Clara, CA) and random hexamer primers. Subsequently, PCR amplification was conducted using GoTaq polymerase (Promega, Madison WI) and a PTC-200 Thermal Cycler (MJ Research, Waltham, MA). PCR products were resolved by agarose electrophoresis and visualized with ethidium bromide. Q-PCR was conducted on cDNA using Power SYBR Green PCR master Mix (Applied Biosystems Co., Carlsbad, CA) and an ABI StepOne Real-time PCR thermocycler. Data analyzed with supplied ABI software and relative mRNA concentrations calculated using the ΔΔCt method and GAPDH as the internal standard. RT-PCR and Q-PCR were conducted using the following primers: mExo1 (forward: 5’-CTGAAGGAAAAGCCAAGGAC-3’; reverse: 5’-CCACAGCCATTTTCCTTGAT-3’); hEXO1 (forward: 5’-GAGAAAGACGACAAGCCAATC-3’; reverse: 5’-CTAATCCAATCCCACGCAGT-3’); mGAPDH (forward: 5’-AACGACCCCTTCATTGAC-3’; reverse: 5’-TCCACGACATACTCAGCAC -3’); hGAPDH (forward: 5’-ACCACAGTCCATGCCATCAC-3’; reverse: 5’-TCCACCACCCT GTTGCTGTA-3’).
2.5 Flow cytometry
Aliquots of 1 × 106 cells were washed twice with ice-cold PBS and cells subsequently fixed in 75% ethanol at −20°C for at least 24 hr. Following this, cells were washed twice with PBS, and incubated with 1 μg/ml RNase A and 10 μg/ml propidium iodide for 40 min at 37°C. Cells were subsequently analyzed using a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ). Cell cycle distribution was determined using ModFit LT software (Verity Software House, Topsham, ME).
2.6 Immunoblotting
Cell lysates were prepared by adding an appropriate volume of PBS-TDS lysis buffer (1x PBS, 1% Triton-X 100, 0.1% SDS, 20% Glycerol) supplemented with Halt protease inhibitor cocktail (Thermo Scientific, Rockford, IL) to pelleted cells followed by incubation on ice for 10 min. Lysates were cleared by centrifugation, protein concentrations were determined by the BCA method, and lysates diluted in SDS sample buffer (80 mM Tris-HCl, pH 6.8 / 2% sodium dodecyl sulfate / 10% glycerol / 5% β-mercaptoethanol / bromphenol blue) prior to SDS-PAGE. Immunoblotting was conducted using anti-EXO1 from Sigma Aldrich (cat# SAB4503568) or Thermo Scientific (cat# MS-1534), anti-MSH6 (cat# A300-022A), anti-MSH2 (cat# A300-452A), and anti-MLH1 (cat# A300-015A) (Bethyl, Montgomery, TX), anti-pCDC2-Tyr15 (cat# 9111), and anti-pCHK1-Ser345 (cat# 2341) (Cell Signaling, Danvers, MA), anti-CDC2 (cat# sc-54), anti-CHK1 (cat# sc-8408 or sc-7898) and anti-HA (cat# sc-7392) (Santa Cruz Biotech, Santa Cruz, CA). Mouse monoclonal anti-β-tubulin was prepared from culture supernatants of E7 hybridoma obtained from University of Iowa Developmental Studies Hybridoma Bank. Secondary horseradish peroxidase (HRP)-conjugated antibodies were from KPL (Gaithersburg, MD) and signals developed using West-Pico chemiluminescence substrate (Thermo Scientific). ImageJ (ver 1.46) software was used to quantify immunoblot signals on exposed films. To assure the significance of observed changes in immunoblot signal intensities, films from at least 3 independent experiments were measured. Unless reported otherwise, all reported changes in immunoblot signal intensity were found to be statistically significant (p<0.05, Student’s t-test).
2.7 Immunoprecipitation
Lysates used in immunoprecipitation (IP) procedures were prepared using PBS-TDS lysis buffer as outlined above. Lysates were pre-cleared by centrifugation, anti-CHK1 (mouse) antibody was added to 500 μg of lysate and then incubated at 4°C O/N. The following day, A/G-sepharose beads (Thermo Scientific) were added and incubated for 2 hr at room temperature with end-over-end rocking. Beads were collected by centrifugation, washed 3 times in RIPA lysis buffer (Tris 50 mM, pH 7.4/NaCl 150 mM, SDS 0.1 %/Na Deoxycholate 0.5 %/ Triton X 100 1%/ HALT protease inhibitor cocktail), SDS sample buffer added, and after boiling, the supernatant analyzed by immunoblotting.
2.8 Immunofluorescence microscopy
Cells were grown on sterilized glass coverslips and fixed by sequential immersion in 4% paraformaldehyde (10 min, room temperature) and methanol (5 min, -20°C). Cells were subsequently permeabilized with 1% Triton X-100 in PBS, blocked in PBS with 3% BSA, and incubated with anti-γ-H2AX rabbit monoclonal antibody (Bethyl, Montgomery, TX) (2 hr, room temperature) followed by Alexa Fluor 488-labeled anti-rabbit antibody (Life Technologies, Grand Island, NY) and counterstaining with DAPI (Vector Labs, Burlingame, CA). Where indicated, cells were also incubated with mouse anti-EXO1 (cat# MS-1534; Thermo Scientific), and Alexa Fluor 594-labeled anti-mouse secondary antibody (Life Technologies). Cells were viewed using a Zeiss Axioplan 2 microscope and images acquired using OpenLab software (Perkin Elmer). For quantification of γ–H2AX foci, >600 total cells were counted at indicated time points and the percentage of cells displaying ≥ 5 foci/cell were recorded in three independent experiments.
2.9 DNA duplex-protein precipitation assay
PBS washed cells were incubated in hypotonic lysis buffer (10 mM HEPES, pH 7.4 / 0.1 mM EDTA / 10 mM KCl / 2% NP40) supplemented with HALT protease inhibitor cocktail for 5 min on ice. Lysates were centrifuged at 13,000 × g, supernatant discarded and pelleted nuclei resuspended in high-salt nuclear extraction buffer (20 mM HEPES, pH 7.4 / 25% glycerol / 420 mM NaCl / 1 mM EDTA) supplemented with protease inhibitor cocktail. Extracted nuclei were centrifuged at 13,000 × g, pellets discarded, and lysates stored at -80°C prior to use.
DNA duplexes were formed by mixing 2 μg of 5’ biotinylated forward oligo (5’-ATATTTTATAGATAGATAGATGGTATgCAAGTCAGAAAAA-3) with 2 μg either control reverse oligo (5’-TTTTTCTGACTTGcATACCATCTATCTATCTATAAAATAT-3’) or reverse mismatched oligo (5’- TTTTTCTGACTTGtATACCATCTATCTATCTATAAA ATAT-3’), briefly heating to 95°C, and slowly cooling to room temperature. (NB: Site of mismatched base pair is denoted in lower case and all oligos were synthesized with phosphorothioated bases to eliminate exonuclease digestion).
Biotinylated DNA duplexes were incubated with streptavidin beads (GE Healthcare, Piscataway, NJ) in binding buffer (10mM Tris-HCL, pH 7.6 / 2M NaCl / 1mM EDTA / 0.1% Tween 20) for 30 minutes at room temperature with end-over-end rocking. Beads were subsequently washed 3 times, resuspended in reaction buffer (200mM Tris-HCL, pH 7.6 / 10 mM reduced glutathione / 100mM MgCl2 / 500μg acetylated bovine serum albumin / 100mM KCl / 20mM HEPES / HALT protease inhibitor) and added to 100 μg of nuclear extract. The reaction was incubated for 15 min at 37°C with end-over-end rocking. Following this, beads were collected by centrifugation, washed 3X in reaction buffer, resuspended in SDS sample buffer, subjected to SDS-PAGE, and probed with anti-Msh2 antibody. As an internal control for equal oligo input, a small aliquot of a precipitated sample was loaded on the agarose gel and stained by ethidium bromide.
2.10 Biochemical analysis of chromatin-associated proteins
Small-scale chromatin isolation was performed as previously described [52]. Briefly, cells were resuspended in buffer A (10 mM HEPES, pH 7.9/10 mM KCl/1.5 mM MgCl2/0.34 M sucrose/10% glycerol/1 mM DTT/0.1% Triton X-100/HALT protease and phosphatase inhibitors) and incubated at 4°C for 5 min followed by low speed centrifugation to collect nuclei. Nuclei were then washed with buffer A followed by lysis in buffer B (3 mM EDTA/0.2 mM EGTA/1 mM DTT/ HALT protease and phosphatase inhibitor) at 4°C for 15 minutes. Chromatin was harvested by centrifugation, washed once with buffer B, and resuspended in SDS sample buffer prior to SDS-PGE and immunoblot analysis.
2.11 Recombinant hEXO1 cloning and expression
A full-length human EXO1 cDNA was obtained from Open Biosystems. Following validation by automated sequencing the cDNA was subcloned as a BamHI/ApaI fragment into the expression vector pcDNA3.1-6HA. This vector, a generous gift from Dr. E. Golemis (Fox Chase Cancer Center), is a derivative of pcDNA3.1 that places six tandem hemagglutinin (HA) epitope tag sequences in an amino terminal fusion with the inserted cDNA.
Site-directed mutagenesis was conducted on full-length human EXO1 cDNA subcloned into pBluescript using Quikchange II XL Site-Directed Mutagenesis kit (Agilent, Santa Clara CA) and the following mutagenic primers: (E150D: 5’-CTCGTGGCTCCCTATGAcGCTGATGC-3’; D171A/D173A: 5’-CATAATTACAGA GGcCTCGGcTCTCCTAGCTTTT-3’). Mutations were subsequently validated by automated sequencing and mutant cDNA (designated mut-hEXO1) subcloned into pcDNA3.1-6HA.
C3-ΔExo1 and C6-ΔExo1 cells were transfected with pcDNA3.1-6HA-hEXO1, pcDNA3.1-6HA-mut-hEXO1 or empty (pcDNA3.1-6HA) plasmids using Lipofectamine 2000 (Invitrogen) following the manufacturer’s instructions. 48 hr post-transfection, 300 μg/ml G418 sulfate was added and after at least 2 weeks of selection transgene expression within the polyclonal population was confirmed by RT-PCR and immunoblotting.
3. RESULTS
3.1 Exo1 depletion results in alkylation tolerance
To study a potential function for EXO1 in MMR-dependent response to SN1 methylating agents, we stably depleted endogenous mEXO1 expression in mouse embryonic fibroblasts (MEFs) by RNA interference. This was accomplished by expressing short hairpin RNA (shRNA) molecules specific for mExo1 by lentiviral transduction. Cell lines transduced with two different mExo1-specific shRNAs (C3-ΔExo1 and C6-ΔExo1), show a sharp reduction in endogenous mExo1 expression when compared to cells transduced with control shRNA virus as demonstrated by RT-PCR (Fig 1A). Quantitative RT-PCR (Q-PCR) measured a ~5-fold decrease in mExo1 mRNA levels in both C3-ΔExo1 and C6-ΔExo1 lines compared to controls (Fig 1B). Further, immunoblotting confirmed decreased mEXO1 protein expression in both C3-ΔExo1 and C6-ΔExo1 cell lines (Fig 1C). As MSH6 plays a fundamental role in mismatch recognition, and loss of this protein confers an alkylation tolerant phenotype [53], we stably depleted mMsh6 in MEFs using shRNA to function as a control (Fig 1D).
Figure 1. Exo1 depletion in MEFs results in increased cellular viability and proliferation in response to MNNG exposure.
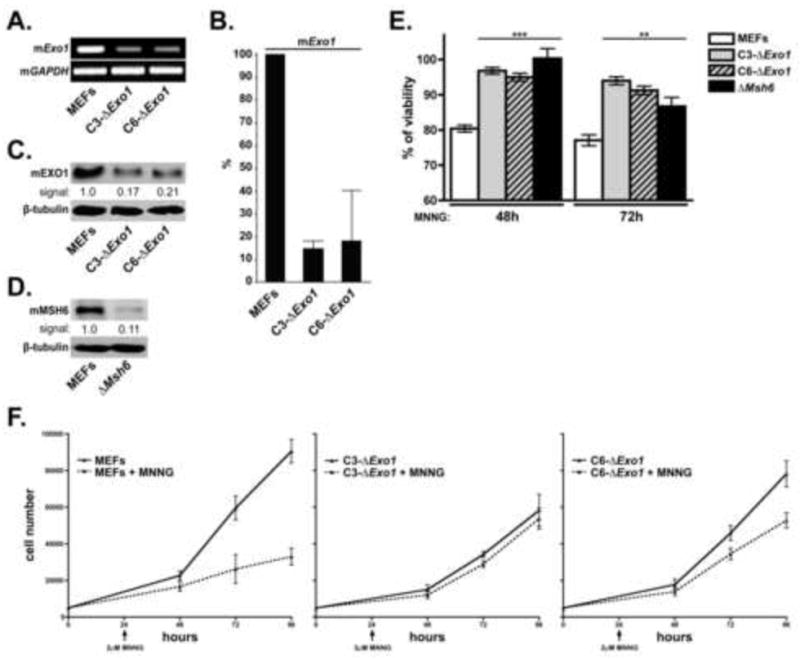
A. mExo1 expression was knocked down in MEFs using lentivirus-encoded shRNA. Cells transduced with control virus (MEFs) or two independent Exo1-specific shRNA sequences (designated C3-ΔExo1 and C6-ΔExo1) were analyzed by RT-PCR using mExo1 (top) and Gapdh (bottom) specific primer sets. B. Cells outlined in (A) were analyzed for relative Exo1 mRNA levels by Q-PCR using GAPDH as the internal standard. C. Cells outlined in (A) were analyzed by immunoblot analysis for mEXO1 (top) or tubulin (bottom) (loading control). D. MEFs with shRNA-mediated knockdown of Msh6 (ΔMsh6) were analyzed by immunoblotting for MSH6 (top) or tubulin (bottom). E. Control MEFs, C3-ΔExo1, C6-ΔExo1, and ΔMsh6 cells were treated with 2μM MNNG. 48 and 72 hrs after drug addition cell viability was assessed by Alamar Blue assay. Graphed is the % viability compared to untreated cells cultured in parallel (n=5, error bars = 1.0 SD). F. Cultures of control MEFs, C3-ΔExo1 and C6-ΔExo1 cells were seeded into tissue culture plates and allowed to attach overnight. 24 hrs after seeding, cultures were either treated with 2μM MNNG (dotted line) or were untreated (solid line). Cells were harvested at indicated time points and viable cells counted. n= 3-5 independent measurements, error bars = 1.0 SD, (** indicates p<0.01; *** indicates p<0.001, Student’s t-test).
Following the characterization of these cell lines, we sought to determine the effects of mExo1 depletion on cell viability in response to the SN1 methylating agent MNNG. Alamar Blue viability assays conducted at 48 and 72 hrs after treatment with 2 μM MNNG indicated that both C3-ΔExo1 and C6-ΔExo1, as well as ΔMsh6 cell lines, displayed statistically significant increases in cell viability compared to control cells at both time points (Fig 1E). Moreover, we measured no significant difference in viability between the two ΔExo1 lines and the ΔMsh6 line following treatment with MNNG.
We next examined rates of cell proliferation in the C3-ΔExo1 and C6-ΔExo1 lines following MNNG exposure. 2 μM MNNG was added to the cultures 24 hrs after plating and was found to dramatically reduce the rate of cellular proliferation in cultures of MEFs transduced with control virus (Fig 1F). While we measured slower rates of proliferation in cultures of untreated C3-ΔExo1 cells compared to either control MEFs or C6-ΔExo1 cells, the rates of proliferation in MNNG-treated and untreated C3-ΔExo1 cells were indistinguishable, and while a significant difference in cell number was measured in treated/untreated C6-ΔExo1 cells 72 and 96 hrs after plating (ie, 48 and 72 hrs after MNNG treatment), this effect was clearly less pronounced than that observed in control MEFs. Taken together, these findings indicate that Exo1 depletion in MEFs results in a sharply reduced cytotoxic/cytostatic response to MNNG.
To examine the effects of Exo1 depletion on G2/M checkpoint activation we treated control MEFs, C3-ΔExo1, C6-ΔExo1, and ΔMsh6 cells with 2μM MNNG and 48 hr after addition of the drug cells were analyzed by flow cytometry. This revealed a strong G2/M checkpoint response in MNNG-treated control MEFs (Fig 2A). However, diminished accumulation of cells in G2/M phase was observed in MNNG treated C3-ΔExo1, C6-ΔExo1, and ΔMsh6 cells. When the results of multiple experiments were compiled, we measured statistically significant decreases in G2/M arrest in both Exo1-depleted cell lines as well as in the Msh6-depleted line compared to controls (Fig 2B).
Figure 2. Depletion of Exo1 results in blunted MNNG-induced checkpoint activation.
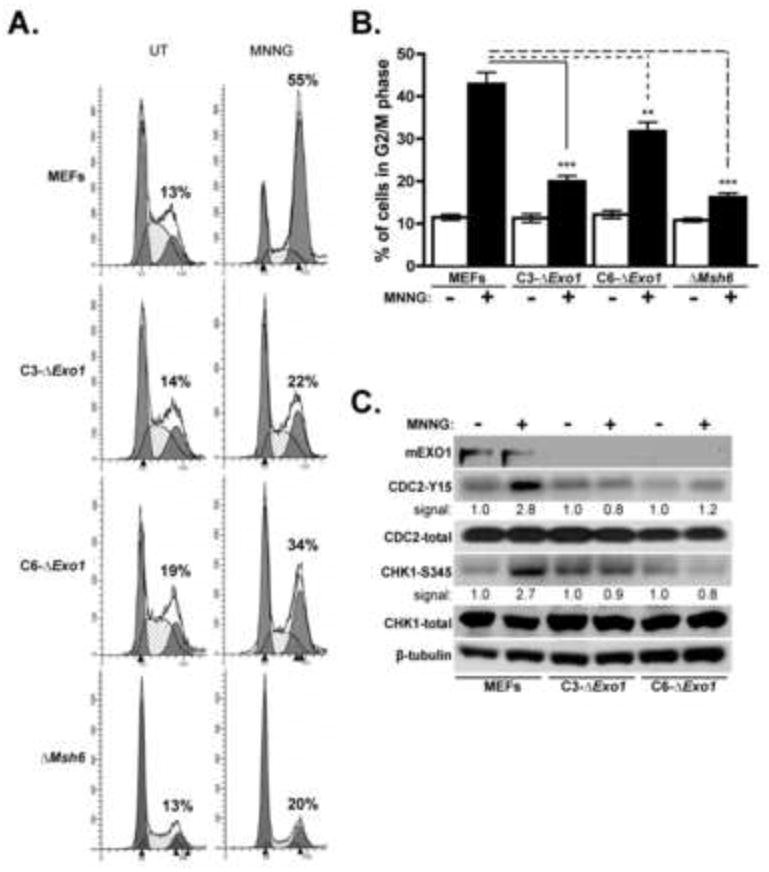
A. Asynchronous cultures of control MEFs, C3-ΔExo1, C6-ΔExo1, and ΔMsh6 cells were untreated (UT) or treated with 2 μM MNNG for 48 hrs. Following this, cells were harvested and subjected to cell cycle analysis by flow cytometry. Indicated is the percentage of cells in the G2/M phase of the cell cycle. B. The mean percentage of G2/M cells from multiple experiments (n ≥ 10) outlined in (A) is graphed, error bars = 1.0 SD. C. 2 μM MNNG treated and untreated control MEFs, C3-ΔExo1 and C6-ΔExo1 cells were subjected to immunoblot analysis with antibodies specific for phosphorylated and total CHK1 and CDC2, and tubulin as indicated. Immunoblot signal intensity for phospho-CDC2 and CHK1 are provided. (** indicates p<0.01; *** indicates p<0.001, Student’s t-test).
Our group and others have previously reported that MNNG-induced G2/M cell cycle arrest is activated through the ATR/CHK1 checkpoint pathway [15, 16, 18]. Analysis of CHK1 phosphorylation (Ser 345) by immunoblotting with a phospho-specific antibody indicates a ~ 2-fold elevation in baseline CHK1 phosphorylation in C3-ΔExo1 cells relative to control MEFs and C6-ΔExo1 cells (Fig 2C). In response to MNNG treatment we observed a clear increase (~ 3-fold) in CHK1 phosphorylation in control MEFs but this response was undetectable in both C3-ΔExo1 and C6-ΔExo1 cells (Fig 2C).
We also examined the levels of baseline and MNNG-induced accumulation of the inhibited (Y15 phosphorylated) form of CDC2 kinase by immunoblotting (Fig 2C). We observed a suppressed basal level of phosphorylated CDC2 in C6-ΔExo1 cells relative to C3-ΔExo1 cells and MEFs. In response to MNNG, however, a ~3-fold increase in phosphorylated CDC2 was measured in control MEFs whereas no accumulation was observed in C3-ΔExo1, and a statistically insignificant increase in phosphorylated CDC2 was measured in C6-ΔExo1 cells.
To assure that the observed phenotype in C3-ΔExo1 and C6-ΔExo1 cell lines was directly attributable to reduced expression of EXO1 we expressed recombinant human EXO1 (hEXO1) in each line. Consistent with previous in vitro studies [39], expression of hEXO1 complemented the measured defects in MNNG-induced cytotoxicity, G2/M arrest, and CDC2 phosphorylation (Supplemental Figure 1), indicating that the phenotypes observed in C3-ΔExo1 and C6-ΔExo1 are directly due to Exo1 depletion. Collectively, these experiments indicate that mExo1 depletion in MEFs results in diminished cytotoxicity, reduced growth suppression, and abrogated checkpoint activation following exposure to MNNG and lead us to conclude that reduced mExo1 expression results in an alkylation-tolerant phenotype similar to defects observed in MMR-deficient cells.
3.2 Exo1 depletion results in delayed formation of γ-H2AX foci in response to MNNG
ATM phosphorylates the histone variant H2AX on residue Ser139 at the site of DNA double-strand breaks [54]. Phosphorylated H2AX, termed γ-H2AX, forms foci that function as sites of assembly of DNA repair and response complexes following genotoxic insult [55]. Although MNNG does not directly induce double-strand breaks, γ-H2AX foci are generated in response to MNNG through a MMR-dependent mechanism [17, 18]. To examine mEXO1 function in γ–H2AX foci formation we treated control and Exo1-depleted MEFs with MNNG, then, at 12, 24, and 48 hr after drug addition conducted indirect immunofluorescence microscopy with anti-γ–H2AX (Fig 3A). At the 12 hr time point we observed ~40% of control MEFs scored positive for multiple (>5/cell) γ–H2AX foci, compared to ~5% in untreated cells (Fig 3B). At 24 and 48 hr after drug treatment we observed that this γ–H2AX-positive population in control MEFs displayed further increases over the 12 hr time point. In contrast, C3-ΔExo1, C6-ΔExo1, and ΔMsh6 cells displayed a significantly diminished number of γ–H2AX foci-positive cells following MNNG (Fig 3B). Specifically, Msh6-depleted MEFs showed a very modest increase in γ–H2AX foci formation over the time period interrogated, and C3-ΔExo1 cells showed no measureable increase in γ–H2AX foci 12 hr post-treatment. Although at both the 24 and 48 hr time point C3-ΔExo1 cells displayed increased numbers of γ–H2AX foci, these measurements were statistically lower than those made on MNNG-treated MEFs. During the time period studied, C6-DExo1 cells showed a steady increase in γH2AX foci although the number of γ-H2AX positive cells was significantly lower than in control cells. These findings indicate that EXO1 knockdown does not result in as dramatic a phenotype as knockdown of Msh6, perhaps suggesting that EXO1 does not play as critical a role in MNNG-induced γ-H2AX foci formation as Msh6. Nevertheless, these findings clearly indicate that EXO1 is a molecular component in the MMR-dependent formation of γ–H2AX foci during response to MNNG exposure.
Figure 3. Exo1 depletion results in reduced formation of γ-H2AX foci in response to MNNG treatment.

A. Control MEFs, C3-ΔExo1, C6-ΔExo1, and ΔMsh6 cells were either untreated (UT) or treated with 2 μM MNNG. At the indicated time point, cells were fixed, stained with anti-γ–H2AX (green) and counterstained with DAPI (blue) to visualize nuclei. Scale bars = 100 μm. B. Cells positive (foci ≥ 5) for γ–H2AX foci were counted at indicated time points (≥ 600 cells per time point per cell line). Graphed is the mean percentage of foci-positive cells in the population counted in three independent experiments. Error bars = 1.0 SD. C. Control MEFs were either untreated or treated with 2 μM MNNG for 24 hrs and subsequently fixed and stained with anti-γ–H2AX (green) anti-EXO1 (red) or counterstained with DAPI (blue). Images were also merged to highlight the co-localization of EXO1 and γ–H2AX.
We next sought to determine if EXO1 is a component of γ–H2AX foci formed during response to MNNG. To answer this question we treated MEFs with 2 μM MNNG and 24 hrs after drug administration cells were fixed and stained with anti-EXO1, anti-γ–H2AX, and counterstained with DAPI. As expected, no γ–H2AX foci were present in untreated cells, and no specific EXO1 localization was observed within these cells (Fig 3C). However, in MNNG treated cells, abundant foci were observed in cells stained with anti-EXO1 and these foci co-localized with γ–H2AX. From these experiments we conclude that EXO1 co-localizes with γ–H2AX in response to MNNG treatment.
3.3 EXO1 is required for association of MMR components with base-pair mismatches in vitro and alkylated chromatin in vivo
As recruitment and assembly of a MMR complex at the site of mismatched bases is a critical first step in both repair and response, we examined the effects of mExo1 depletion on the association of MSH2 with synthetic DNA duplexes containing either a single G:T mismatch or possessing perfect complementarity. In brief, 5’ biotinylated DNA duplexes were incubated with nuclear extracts from either control or Exo1-depleted MEFs. Protein-DNA complexes were precipitated with streptavidin beads, subjected to SDS-PAGE, and probed with anti-MSH2, MSH6, MLH1 or EXO1 (Fig 4A). We observed modest background precipitation of each protein with streptavidin beads in the absence of duplex DNA, but this binding was clearly reduced compared to precipitation in the presence of duplex DNA. As MMR complexes have previously been shown to associate nonspecifically with the arms or ends of linear DNA fragments [56, 57], we posit that the binding of these MMR components to G:C duplex is attributable to DNA end association. Nevertheless, in control MEFs we measured a clear increase (~ 2-fold) in MSH2, MSH6, MLH1 and EXO1 association with duplex DNA containing a G:T mismatch compared to duplex DNA not containing a mismatch (ie, G:C duplex) (Fig 4A).
Figure 4. Exo1 promotes association of MSH2 with G:T mismatches in vitro and alkylated chromatin in vivo.
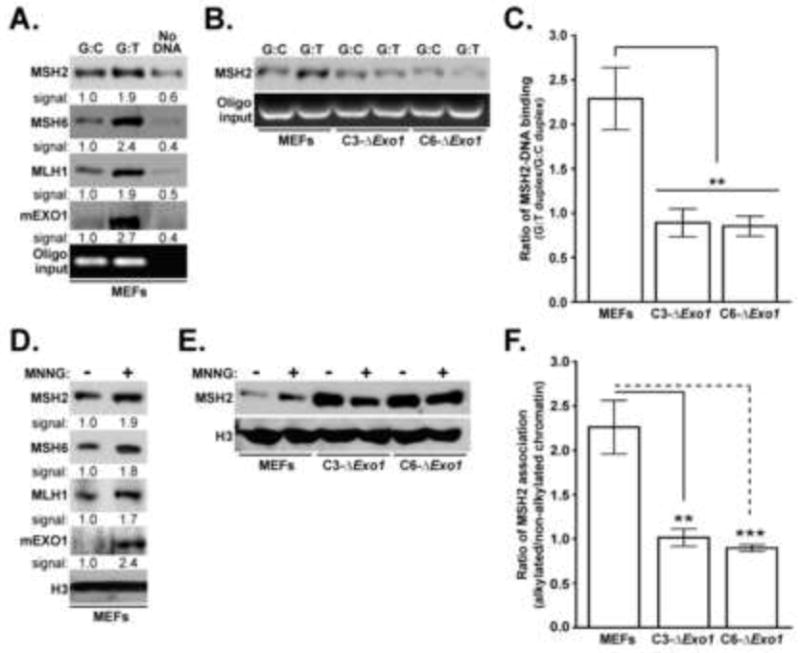
A. Nuclear extracts prepared from control MEFs were incubated with 5’ biotinylated 40bp DNA duplex containing a single G:T mismatch, control duplex (G:C at this site), or no added DNA. Precipitation with streptavidin beads was conducted, this material subjected to SDS-PAGE, and subsequently probed with anti-MSH2, MSH6, MLH1 or EXO1 as indicated. As a control for equal duplex input, a small aliquot of precipitated sample was loaded on an agarose gel and stained with ethidium bromide (bottom). B. Nuclear extracts from control MEFs, C3-ΔExo1, or C6-ΔExo1 cells were incubated with indicated duplex, precipitated with streptavidin beads, and precipitates analyzed for MSH2 (top) or DNA content (bottom). C. The ratio of MSH2 bound to G:T vs. G:C duplex was obtained by quantification of immunoblot signals. Graph reflects mean ratios calculated from 5 independent experiments, error bars = 1.0 SD. D. Chromatin fractions were isolated from MEFs either untreated or treated with 2 μM MNNG for 48 hrs. Material was subsequently subjected to immunoblot analysis with anti-MSH2 MSH6, MLH1 or EXO1 as indicated. As a loading control, fractions were immunoblotted with anti-histone H3 (bottom). E. Chromatin fractions were isolated from MEFs, C3-ΔExo1, or C6-ΔExo1 cells either untreated or treated with 2 μM MNNG for 48 hrs. Material was subsequently subjected to immunoblot analysis with anti-MSH2 (top) or anti-histone H3 (bottom). F. The ratio of MSH2 co-fractionating with alkylated vs. non-alkylated chromatin was obtained by quantification of immunoblot signals. Graph reflects mean ratios calculated from 5 independent experiments, error bars = 1.0 SD. (** indicates p<0.01; *** indicates p<0.001, Student’s t-test).
Nuclear extracts from Exo1 knockdown cells were used to measure MSH2 association with mismatched DNA duplexes (Fig 4B). In contrast to results obtained using nuclear extracts from control MEFs (Fig 4A), nuclear extracts from either C3-ΔExo1 or C6-ΔExo1 cells failed to show G:T duplex binding above the observed background association measured using G:C duplex. Compilation of data obtained from 5 independent experiments indicated that nuclear extracts from MEFs displayed an approximate 2-fold increase in MSH2 precipitation with G:T duplex compared to G:C duplex (Fig 4C). However, no increase in G:T duplex binding was measured in C3-ΔExo1 or C6-ΔExo1 extracts, and these values were statistically significant from those measured in control MEFs.
We next analyzed the association of MMR components with chromatin isolated from MEFs treated or untreated with 2 μM MNNG. 48 hrs after MNNG treatment, cells were harvested, chromatin isolated and analyzed for MSH2, MSH6, MLH1 or EXO1 content by immunoblotting (Fig 4D, lanes 1,2). This analysis indicated a ~2-fold increase in the association of these MMR components with chromatin harvested from MNNG-treated MEFs when compared to untreated MEFs. Chromatin was also isolated from MNNG-treated C3-ΔExo1, C6-ΔExo1, and control MEFs and co-fractionating MSH2 analyzed by immunoblotting (Fig 4E). We observed that, unlike control MEFs, MNNG treatment had no measureable effect on the association of MSH2 with chromatin in the EXO1-depleated cells (Fig 4F).
Of note, we consistently observed an increase in basal levels of MSH2 and MSH6 (data not shown) association with chromatin in untreated C3-ΔExo1 or C6-ΔExo1 cells when compared to untreated MEF controls. While we do not fully understand the mechanistic underpinnings of this observation, it is possible that as EXO1-deficient cells display reduced, but not absent, repair activity [28, 33, 38, 39], the kinetics of MMR in the absence of EXO1 could be altered. In turn, this could change the retention of MMR components within chromatin. Nevertheless, our findings indicate that the association of core MMR components with alkylated chromatin is dysregulated upon depletion of EXO1.
3.4 EXO1 is required to promote association of MSH2 with CHK1
We have previously shown that MNNG treatment dramatically upregulates the association of the cell cycle checkpoint activator CHK1 with the MMR component MSH2 [15]. To test whether EXO1 influences this association, we prepared extracts from untreated and MNNG-treated control, C3-ΔExo1, or C6-ΔExo1 MEFs and immunoprecipitated CHK1. These immunocomplexes were subsequently analyzed by immunoblotting with anti-MSH2 and we observed a notable increase in the co-precipitation of MSH2 with CHK1 in extracts prepared from MEFs following MNNG treatment (Fig 5A). In contrast, we observed no detectable co-precipitation of MSH2 with CHK1 in either of our mExo1-depleted lines. Control experiments indicated that knockdown of mExo1 did not effect MSH2 expression (Fig 5B), supporting the conclusion that Exo1-depletion abrogates MNNG-induced CHK1/MSH2 interaction. We observed co-precipitation of EXO1 with CHK1 in MNNG-treated MEFs (Fig 5A) establishing EXO1 as a component of this CHK1/MSH2 damage-induced complex and, collectively, indicating that EXO1 is required to facilitate association of MSH2 with CHK1 in response to SN1 methylators exposure.
Figure 5. EXO1 is required for the co-association of MSH2 with CHK1 in response to MNNG.
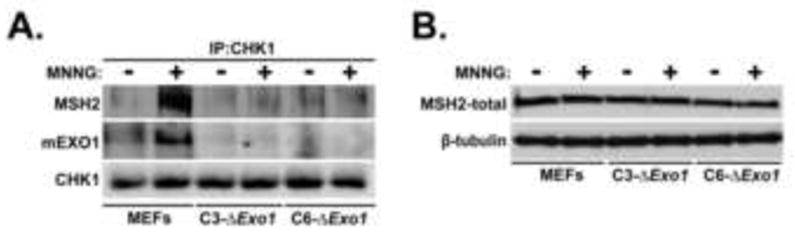
A. Control MEFs, C3-ΔExo1, or C6-ΔExo1 cells were either untreated or treated with 2 μM MNNG for 48 hrs, whole cell lysates prepared, and immunoprecipitated with anti-CHK1 antibody. Resultant immunocomplexes were immunoblotted with anti-MSH2 (top), EXO1 (middle), or CHK1 (bottom). B. Total cell lystaes were formed from Control MEFs, C3-ΔExo1, or C6-ΔExo1 cells either untreated or treated with MNNG (2 μM, 48 hrs). Lysates were immunoblotted with anti-MSH2 (top) or tubulin (bottom).
3.5 MMR-dependent signaling occurs independently of EXO1 catalytic activity
Data gathered clearly indicated that EXO1 is required to activate cell cycle checkpoint signaling and other molecular events in response to MNNG; however, it was undetermined if this signaling was attributable to EXO1 nucleolytic activity. To test this, we engineered human EXO1 (hEXO1) cDNA with three distinct missense mutations (D171A, D173A and E150D) within the highly conserved catalytic domain of hEXO1. Numerous biochemical studies clearly demonstrate that each of these mutations will independently result in multi-fold reduction, or complete elimination, of EXO1 exonuclease and flap-endonuclease activity, while having no effect on EXO1 substrate binding [31, 49, 58-60]. We stably expressed N-terminally HA-tagged nuclease-defective hEXO1 (C3-HA-mut-hEXO1), wild type human hEXO1 (C3-HA-hEXO1), or empty vector (C3-HA-VC) in the C3-ΔExo1 line. Both RT-PCR (Fig 6A), and immunoblotting (Fig 6B) confirmed expression of wild type or mutant transgene expression in both polyclonal lines. As an additional control, we PCR amplified the catalytic domain of the hEXO1 transgene and confirmed transgene identity by DNA sequencing in both lines (Supplemental Fig 2A). Further, Q-PCR, using mouse-specific Exo1 primers, assured that endogenous mExo1 expression was equivalently suppressed in each line (Supplemental Fig 2B).
Figure 6. Catalytically null hEXO1 complements the alkylation tolerant phenotype in Exo1 depleted MEFs.
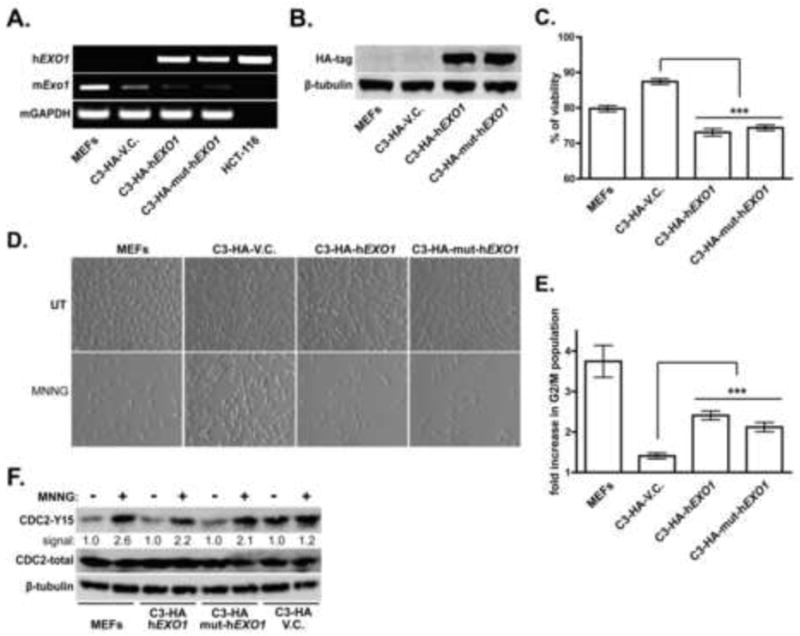
A. C3-ΔExo1 cells were stably transfected with plasmids encoding HA-tagged, recombinant human EXO1 (C3-HA-hEXO1), exonuclease mutant hEXO1 (C3-HA-mut-hEXO1) or control vector (C3-HA-VC). Following drug selection of polyclonal lines, RT-PCR was conducted on RNA harvested from indicated cell lines using human-specific (top) or mouse-specific (middle) EXO1 primers, or mouse-specific GAPDH primers (bottom). The human colorectal tumor line HCT116 was included as a positive control for h EXO1 expression. B. Lysates of control MEFs, C3-HA-hEXO1, C3-HA-mut-hEXO1, and C3-HA-VC cells were analyzed by immunoblotting with anti-HA (top) or anti-tubulin (bottom) antibodies. C. Cell viability after exposure of indicated cell lines to 2 μM MNNG for 48 hrs was assessed by Alamar Blue assay. Graphed is the % viability compared to untreated cells cultured in parallel, mean ratios calculated from 5 independent experiments, error bars = 1.0 SD. D. Control MEFs, C3-HA-hEXO1, C3-HA-mut-hEXO1 or C3-HA-VC cells were either untreated or treated with 2 μM MNNG. Representative photomicrographs were taken using phase-contrast optics 48 hrs after MNNG treatment. E. Cell cycle analysis of asynchronously growing control MEFs, C3-HA-hEXO1, C3-HA-mut-hEXO1 or C3-HA-VC cells untreated or treated with 2 μM MNNG was conducted. Fold-increase in the G2/M population compared to untreated cells cultured in parallel was calculated. Mean fold changes were calculated from 5 independent experiments, error bar = 1.0 SD. F. Control MEFs, C3-HA-hEXO1, C3-HA-mut-hEXO1 and C3-HA-VC cells were either untreated or treated with 2 μM MNNG. 48 hrs after treatment cells were harvested and lysates immunoblotted with anti-phospho (Y15) CDC2 (top), total CDC2 (middle) or tubulin (bottom). (*** indicates p<0.001, Student’s t-test).
Consistent with previous findings (see Supplemental Fig 1) expression of wild type human hEXO1 complemented the increased viability observed in MNNG-treated, mExo1-depleted cells (Fig 6C). Of note, we also observed that the catalytic mutant hEXO1 similarly complemented this defect in the C3-ΔExo1 line. While expression of both, wild type and mutant hEXO showed a statistically significant decrease in viability in response to MNNG when compared to control cells (C3-HA-VC), there was no measurable difference in viability between the C3-HA-mut-hEXO1 and C3-HA-hEXO1 lines. Similarly, we observed reduced cellular proliferation in MNNG-treated cultures of MEFs, C3-HA-mut-hEXO1 and C3-HA-hEXO1, but no apparent decrease in the C3-HA-VC control line (Fig 6D). Flow cytometry conducted on cells treated with 2 μM MNNG revealed an approximate two-fold increase in G2/M phase cells in both C3-HA-hEXO1 and C3-HA-mut-hEXO1 lines when compared to MNNG-treated C3-HA-VC cells (Fig 6E). While we observed increased baseline levels of phosphorylated CDC2 in the control C3-HA V.C. line (Fig 6F), both C3-HA-hEXO1 and C3-HA-mut-hEXO1 lines displayed sharp increases in CDC2 phosphorylation in response to MNNG compared to the abrogated response detected in C3-HA-VC cells. These findings clearly indicate that catalytically inactive hEXO1 is fully capable of complementing the alkylation tolerant phenotype resulting from depletion of mExo1.
We next examined other cellular phenotypes associated with reduced mEXO1 expression in MEFs. CHK1 was immunoprecipitated from extracts of MNNG treated and untreated MEFs, C3-HA-hEXO1, C3-HA-mut-hEXO1, or control C3-HA-VC cells. Immunoblotting with anti-MSH2 clearly indicates that co-precipitation of MSH2 and CHK1 was restored in mExo1-depleted MEFs expressing either wild type or mutant hEXO1 (Fig 7A). Consistent with results obtained on the parental C3-ΔExo1 line (see Fig 5), no co-precipitation of MSH2 and CHK1 was observed in C3-HA-VC cells treated with MNNG.
Figure 7. EXO1 catalytic activity is not required for association of MSH2 with DNA mismatches, MNNG-induced CHK1-MSH2 association, or γ-H2AX foci formation.

A. Lysates from control MEFs, C3-HA-hEXO1, C3-HA-mut-hEXO1 or C3-HA-VC cells either untreated or MNNG-treated (2 μM, 48 hrs) were subjected to immunoprecipitation using anti-CHK1 antibody. Immunocomplexes were subsequently immunoblotted with anti-MSH2 (top), stripped and re-probed with anti-CHK1 (bottom). B. Nuclear extracts from control MEFs, C3-HA-hEXO1, C3-HA-mut-hEXO1 or C3-HA-VC cells were incubated with either G:T or control (G:C) DNA duplexes. Following precipitation, material was immunoblotted with anti-MSH2 (top) or subjected to agarose electrophoresis/ethidium bromide staining (bottom). C. The ratio of MSH2 bound to G:T vs. G:C duplex was obtained by quantification of immunoblot signals. Graph reflects mean ratios calculated from 5 independent experiments, error bars = +/- 1.0 SD. D. Control MEFs, C3-HA-hEXO1, C3-HA-mut-hEXO1, or C3-HA-VC cells were either untreated (UT) or treated with MNNG (2 μM, 24 hrs) and subsequently fixed, stained with anti-γ-H2AX (green), and counterstained with DAPI (blue). E. Cells positive (foci ≥ 5) for γ–H2AX foci in untreated or MNNG-treated (2 μM, 24 hrs) cell lines were counted (≥ 600 per time point and cell line). Graphed is the mean percentage of foci-positive cells in the population counted in three independent experiments. Error bars = +/- 1.0 SD. (* indicates p<0.05; ** indicates p<0.01, Student’s t-test).
To test whether catalytic activity of hEXO1 is required for MSH2 association with base-pair mismatches in vitro, we conducted DNA duplex precipitation experiments using nuclear extracts derived from MEFs, C3-HA-hEXO1, C3-HA-mut-hEXO1 or C3-HA-VC cells (Fig 7B). Similar to previous results, we measured increased binding of MSH2 with the mismatch-containing G:T duplex, relative to G:C duplex, in nuclear extracts from MEFs and no measurable increase in G:T duplex binding in nuclear extracts from Exo1-depleted C3-HA-VC cells (Fig 7C). Nuclear extracts from both C3-HA-hEXO1 and C3-HA-mut-hEXO1 cells demonstrated statistically significant increases in the ratio of G:T / G:C duplex binding.
Finally, we quantified the formation of γ–H2AX foci in response to MNNG in this panel of engineered MEFs (Fig 7D). Data gathered indicated, consistent with prior findings, that cells with suppressed mEXO1 expression (ie, C3-HA-VC) exhibited significantly reduced foci formation 24 hr after MNNG when compared to control MEFs (Fig 7E). In contrast, both C3-HA-hEXO1 and C3-HA-mut-hEXO1 cells displayed increased γ–H2AX foci 24 hr after MNNG to levels quantitatively consistent with foci present in MNNG-treated MEFs at this time point (Fig 7E). Taken together, these observations clearly indicate that cellular phenotypes associated with reduced mEXO1 expression are complemented by expression of catalytically null hEXO1. This finding leads us to conclude that defects stemming from Exo1 depletion are not due to reduced EXO1 nuclease capacity, but rather are attributable to reduced expression of the EXO1 protein itself.
4. DISCUSSION
Cells deficient in core components of the MMR system (eg, MSH2, MSH6, MLH1, PMS2) fail to activate cellular responses to SN1 methylator-induced damage including the activation of cell cycle checkpoints or apoptosis [61]. This feature of MMR-deficiency, termed alkylation tolerance, stems from defects in activation of canonical ATM/ATR > CHK1/CHK2 signaling in response to persistent O6meG adducts within the genome [15, 16, 18, 20, 61, 62]. In this study we show that diminished EXO1 expression phenocopies MMR-deficiency. Specifically, we documented that knockdown of EXO1 expression resulted in blunted sensitivity, dysregulated cell growth and cell cycle checkpoint activation, and reduced CHK1 and CDC2 phosphorylation in response to MNNG. As these defects are also linked to MMR-deficiency, we conclude that EXO1 is an important component in MMR-dependent activation of DDR in response to SN1 methylating agents. Klapacz et al [63] similarly observed that EXO1-deficiency reduced SN1 methylator sensitivity in Mgmt-deficient mice. Of note, EXO1 has been implicated in the activation of DDR in response to an array of other forms of genotoxic insult [64-66], suggesting that EXO1 may play a broad role in the activation of cellular response to genotoxic stress.
Several studies have documented that phosphorylation of H2AX and the subsequent formation of foci within the nucleus mark sites of DNA DSB [67, 68]. While MNNG does not directly induce DNA DSBs [69], γ–H2AX foci are formed in response to SN1 methylating agent exposure [18, 70-72]. Moreover, Stojic et al [18] observed that γ–H2AX foci are formed in response to MNNG through a biphasic response. The rapid (4 hrs after drug) accumulation of γ–H2AX foci occurred in a MMR-independent manner and is likely a response to the repair of abundant N3meA and N7meG adducts through the base excision repair (BER) mechanism. At later times, these investigators, as well as others, observed that MMR-deficient cells did not contain γ–H2AX foci while these structures were present in MMR-proficient cells [18, 70]. In addition, numerous other proteins that co-localize with γ–H2AX in response to other forms of genotoxic stress form foci in response to SN1 methylating agents including 53BP1, ATM, ATR, RAD9, RAD51 and MRE11 [18, 73-75]. While the exact nature of the DNA structures formed in response to MNNG that trigger H2AX phosphorylation and the association of other damage-response molecules to the site of damage remains unknown, H2AX-deficiency abrogates G2/M cell-cycle arrest, increases mitotic irregularities and chromosomal instability after MNNG exposure [76], effects that are commonly seen in MMR-deficient cells [77, 78]. These findings support the idea that H2AX is essential in activating proper response to SN1 methylating agent exposure. In sum, it is straightforward to propose that γ–H2AX foci represent sites of MMR-dependent processing of persistent O6meG lesions and likely signify an important morphological feature in MMR-dependent DDR activation following SN1 methylator exposure.
EXO1 functions in resection of the mismatched DNA strand during MMR [27, 28], and we have observed that knockout of EXO1 results in attenuation of γ–H2AX foci formation; thus, it is reasonable to speculate that EXO1-mediated DNA strand resection may generate repair intermediates that trigger the formation of γ–H2AX foci. However, as expression of catalytically defective EXO1 restored normal foci formation, it is unlikely that such repair intermediates are directly formed by EXO1-associated nuclease activity. Rather, we propose that EXO1 recruits redundant exonuclease(s) to the sites of MMR-dependent lesion processing. Consistent with this view, recent findings show that in response to DNA DSBs, catalytically inactive (D173A) EXO1 co-localizes with γ–H2AX foci [48], clearly indicating that this localization property of EXO1 is independent of its nucleolytic activity. Alternatively, EXO1 may be required for the recruitment of ATM/ATR kinases to the site of MMR-dependent lesion processing and, subsequently, catalyze H2AX phosphorylation. This possibility is supported by the observation that in response to DSBs, EXO1-deficient cells show attenuated activation/recruitment of ATR to sites of DNA damage [64].
Repair of G:T or O6-meG:T base pairs is initiated by the binding of the MutSα complex (heterodimer of MSH2 and MSH6) to the mismatch [79]. Numerous studies conducted using yeast and mammalian cell extracts indicate that MutSα binds to DNA duplexes containing an O6meG:T or G:T mismatch and, to a lesser extent, O6meG:C base pairs [19, 57, 80, 81]. Additionally, available evidence demonstrates increased nuclear localization, and chromatin association of MMR proteins in cells treated with SN1 methylators [82-84]. In view of these findings, we examined the effects of mExo1 depletion on the association of MSH2 with mismatched DNA in vitro and alkylated chromatin in vivo. As expected, we found a marked increase in association of MSH2 with G:T containing DNA duplexes and increased association of MSH2 with alkylated chromatin in control MEFs. In contrast, MSH2 association with either mismatched DNA or alkylated chromatin was dysregulated following depletion of mExo1. As previously described, numerous studies support the idea that EXO1 functions in a structural capacity during MMR-mediated mutation avoidance [30, 40, 42, 44, 45, 48, 49]. Consistent with this, results presented in this study suggest that EXO1 is required for either the formation and/or stabilization of MutSα on damaged DNA and as we observed complementation of this defect with ectopic expression of mutant hEXO1, the nucleolytic activity of EXO1 appears to be dispensable for MSH2 association with mismatched DNA duplexes in vitro.
We have previously shown that both CHK1 and CHK2 proteins associate with MSH2 in response to genotoxic insult [15, 85]. Here, we show that MNNG-induced association of CHK1 with MSH2 was eliminated in mExo1-depleted lines. Moreover, this association was fully restored in cells stably expressing either wild type or catalytically-null hEXO1. These findings clearly indicate that EXO1, but not its nucleolytic activity, is required to promote association of MSH2 with CHK1 in response to SN1 methylator exposure. When interpreted along with the findings that EXO1 is required for association of MSH2 with G:T mismatched DNA duplexes in vitro, alkylated chromatin in vivo, and formation of MMR-dependent γ-H2AX foci, it is tempting to speculate that EXO1 facilitates DDR activation by promoting and/or stabilizing MMR complex assembly at the sites of persistent O6meG adducts. Moreover, while EXO1 catalytic activity is important in DNA repair, our findings indicate that this property is dispensable for EXO1-dependent DDR activation.
At present, two non-mutually exclusive models have been proposed to explain how the MMR system mechanistically activates DDR in response to persistent O6meG lesions [21]. The “futile-repair” model proposes that structures generated in the process of iterative repair of O6meG:T base pairs activate DDR. Conversely, the “direct-signaling” model proposes that the association of DDR signaling molecules with the MMR complex assembled at the sites of DNA lesions is a necessary step in the activation of DDR. We have observed that independent of its nuclease activity, EXO1 is required to induce formation of γ-H2AX foci, activate cell cycle checkpoints and promote the association of MSH2 with CHK1 following MNNG exposure. These findings, as well as studies that document that repair-defective mutants of MSH2 and MSH6 efficiently activate DDR [24, 86], lend support for the direct signaling model. While we cannot rule out that EXO1 is required for iterative cycles of MMR repair / excision necessary to activate DDR through the futile repair mechanism, EXO1 nucleolytic activity is clearly not required to activate response to MNNG. Thus, on balance, our study may lend additional evidence supporting the validity of the direct-signaling model.
Supplementary Material
Highlights.
EXO1 is required for activating cytotoxic/cytostatic response to MNNG.
EXO1 co-localizes with γ-H2AX foci, and EXO1 depletion results in blunted γ-H2AX foci formation in response to MNNG.
EXO1 is required for association of MMR with mismatched DNA and alkylated chromatin.
EXO1 co-precipitates with MSH2 and CHK1, and is required to promote their association during response to MNNG.
MMR-dependent signaling occurs independently of EXO1 catalytic activity.
Acknowledgments
The authors thank Dr. Lisa Dyer for technical assistance and Dr. Satya Narayan for critical review of the manuscript prior to submission. EI was supported by an NIH T32 training grant (5T32-CA9126). This work was funded by grants from the NIH (R01-CA102289) and the Florida Department of Health to KDB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bignami M, O’Driscoll M, Aquilina G, Karran P. Unmasking a killer: DNA O(6)-methylguanine and the cytotoxicity of methylating agents. Mutat Res. 2000;462:71–82. doi: 10.1016/s1383-5742(00)00016-8. [DOI] [PubMed] [Google Scholar]
- 2.Gerson SL. MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer. 2004;4:296–307. doi: 10.1038/nrc1319. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl T, Demple B, Robins P. Suicide inactivation of the E. coli O6-methylguanine-DNA methyltransferase. EMBO J. 1982;1:1359–1363. doi: 10.1002/j.1460-2075.1982.tb01323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eadie JS, Conrad M, Toorchen D, Topal MD. Mechanism of mutagenesis by O6-methylguanine. Nature. 1984;308:201–203. doi: 10.1038/308201a0. [DOI] [PubMed] [Google Scholar]
- 5.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 6.Kolodner RD, Marsischky GT. Eukaryotic DNA mismatch repair. Curr Opin Genet Dev. 1999;9:89–96. doi: 10.1016/s0959-437x(99)80013-6. [DOI] [PubMed] [Google Scholar]
- 7.Lynch HT, Smyrk T, Lynch JF. Overview of natural history, pathology, molecular genetics and management of HNPCC (Lynch Syndrome) Int J Cancer. 1996;69:38–43. doi: 10.1002/(SICI)1097-0215(19960220)69:1<38::AID-IJC9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 8.Duckett DR, Drummond JT, Murchie AI, Reardon JT, Sancar A, Lilley DM, Modrich P. Human MutSalpha recognizes damaged DNA base pairs containing O6-methylguanine, O4-methylthymine, or the cisplatin-d(GpG) adduct. Proc Natl Acad Sci U S A. 1996;93:6443–6447. doi: 10.1073/pnas.93.13.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stojic L, Brun R, Jiricny J. Mismatch repair and DNA damage signalling. DNA Repair (Amst) 2004;3:1091–1101. doi: 10.1016/j.dnarep.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 10.Aquilina G, Zijno A, Moscufo N, Dogliotti E, Bignami M. Tolerance to methylnitrosourea-induced DNA damage is associated with 6-thioguanine resistance in CHO cells. Carcinogenesis. 1989;10:1219–1223. doi: 10.1093/carcin/10.7.1219. [DOI] [PubMed] [Google Scholar]
- 11.Goldmacher VS, Cuzick RA, Jr, Thilly WG. Isolation and partial characterization of human cell mutants differing in sensitivity to killing and mutation by methylnitrosourea and N-methyl-N’-nitro-N-nitrosoguanidine. J Biol Chem. 1986;261:12462–12471. [PubMed] [Google Scholar]
- 12.Kat A, Thilly WG, Fang WH, Longley MJ, Li GM, Modrich P. An alkylation-tolerant, mutator human cell line is deficient in strand-specific mismatch repair. Proc Natl Acad Sci U S A. 1993;90:6424–6428. doi: 10.1073/pnas.90.14.6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, Boland CR. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N’-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 1994;54:4308–4312. [PubMed] [Google Scholar]
- 14.Aquilina G, Biondo R, Dogliotti E, Bignami M. Genetic consequences of tolerance to methylation DNA damage in mammalian cells. Carcinogenesis. 1993;14:2097–2103. doi: 10.1093/carcin/14.10.2097. [DOI] [PubMed] [Google Scholar]
- 15.Adamson AW, Beardsley DI, Kim WJ, Gao Y, Baskaran R, Brown KD. Methylator-induced, mismatch repair-dependent G2 arrest is activated through Chk1 and Chk2. Mol Biol Cell. 2005;16:1513–1526. doi: 10.1091/mbc.E04-02-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beardsley DI, Kim WJ, Brown KD. N-methyl-N’-nitro-N-nitrosoguanidine activates cell-cycle arrest through distinct mechanisms activated in a dose-dependent manner. Mol Pharmacol. 2005;68:1049–1060. doi: 10.1124/mol.105.013888. [DOI] [PubMed] [Google Scholar]
- 17.Stojic L, Cejka P, Jiricny J. High doses of SN1 type methylating agents activate DNA damage signaling cascades that are largely independent of mismatch repair. Cell Cycle. 2005;4:473–477. doi: 10.4161/cc.4.3.1528. [DOI] [PubMed] [Google Scholar]
- 18.Stojic L, Mojas N, Cejka P, Di Pietro M, Ferrari S, Marra G, Jiricny J. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 2004;18:1331–1344. doi: 10.1101/gad.294404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol Cell. 2006;22:501–510. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamane K, Taylor K, Kinsella TJ. Mismatch repair-mediated G2/M arrest by 6-thioguanine involves the ATR-Chk1 pathway. Biochem Biophys Res Commun. 2004;318:297–302. doi: 10.1016/j.bbrc.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 21.Wang JY, Edelmann W. Mismatch repair proteins as sensors of alkylation DNA damage. Cancer Cell. 2006;9:417–418. doi: 10.1016/j.ccr.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 22.York SJ, Modrich P. Mismatch repair-dependent iterative excision at irreparable O6-methylguanine lesions in human nuclear extracts. J Biol Chem. 2006;281:22674–22683. doi: 10.1074/jbc.M603667200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Qin J. MSH2 and ATR form a signaling module and regulate two branches of the damage response to DNA methylation. Proc Natl Acad Sci U S A. 2003;100:15387–15392. doi: 10.1073/pnas.2536810100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin DP, Wang Y, Scherer SJ, Clark AB, Yang K, Avdievich E, Jin B, Werling U, Parris T, Kurihara N, Umar A, Kucherlapati R, Lipkin M, Kunkel TA, Edelmann W. An Msh2 point mutation uncouples DNA mismatch repair and apoptosis. Cancer Res. 2004;64:517–522. doi: 10.1158/0008-5472.can-03-2957. [DOI] [PubMed] [Google Scholar]
- 25.Lieber MR. The FEN-1 family of structure-specific nucleases in eukaryotic DNA replication, recombination and repair. Bioessays. 1997;19:233–240. doi: 10.1002/bies.950190309. [DOI] [PubMed] [Google Scholar]
- 26.Lee BI, Wilson DM., 3rd The RAD2 domain of human exonuclease 1 exhibits 5’ to 3’ exonuclease and flap structure-specific endonuclease activities. J Biol Chem. 1999;274:37763–37769. doi: 10.1074/jbc.274.53.37763. [DOI] [PubMed] [Google Scholar]
- 27.Genschel J, Modrich P. Mechanism of 5’-directed excision in human mismatch repair. Mol Cell. 2003;12:1077–1086. doi: 10.1016/s1097-2765(03)00428-3. [DOI] [PubMed] [Google Scholar]
- 28.Genschel J, Bazemore LR, Modrich P. Human exonuclease I is required for 5’ and 3’ mismatch repair. J Biol Chem. 2002;277:13302–13311. doi: 10.1074/jbc.M111854200. [DOI] [PubMed] [Google Scholar]
- 29.Tishkoff DX, Boerger AL, Bertrand P, Filosi N, Gaida GM, Kane MF, Kolodner RD. Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proc Natl Acad Sci U S A. 1997;94:7487–7492. doi: 10.1073/pnas.94.14.7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tran PT, Fey JP, Erdeniz N, Gellon L, Boiteux S, Liskay RM. A mutation in EXO1 defines separable roles in DNA mismatch repair and post-replication repair. DNA Repair (Amst) 2007;6:1572–1583. doi: 10.1016/j.dnarep.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sokolsky T, Alani E. EXO1 and MSH6 are high-copy suppressors of conditional mutations in the MSH2 mismatch repair gene of Saccharomyces cerevisiae. Genetics. 2000;155:589–599. doi: 10.1093/genetics/155.2.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rudolph C, Fleck O, Kohli J. Schizosaccharomyces pombe exo1 is involved in the same mismatch repair pathway as msh2 and pms1. Curr Genet. 1998;34:343–350. doi: 10.1007/s002940050405. [DOI] [PubMed] [Google Scholar]
- 33.Wei K, Clark AB, Wong E, Kane MF, Mazur DJ, Parris T, Kolas NK, Russell R, Hou H, Jr, Kneitz B, Yang G, Kunkel TA, Kolodner RD, Cohen PE, Edelmann W. Inactivation of Exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev. 2003;17:603–614. doi: 10.1101/gad.1060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harris RS, Ross KJ, Lombardo MJ, Rosenberg SM. Mismatch repair in Escherichia coli cells lacking single-strand exonucleases ExoI, ExoVII, and RecJ. J Bacteriol. 1998;180:989–993. doi: 10.1128/jb.180.4.989-993.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Viswanathan M, Lovett ST. Single-strand DNA-specific exonucleases in Escherichia coli. Roles in repair and mutation avoidance. Genetics. 1998;149:7–16. doi: 10.1093/genetics/149.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Viswanathan M, Lovett ST. Exonuclease X of Escherichia coli. A novel 3’-5’ DNase and Dnaq superfamily member involved in DNA repair. J Biol Chem. 1999;274:30094–30100. doi: 10.1074/jbc.274.42.30094. [DOI] [PubMed] [Google Scholar]
- 37.Burdett V, Baitinger C, Viswanathan M, Lovett ST, Modrich P. In vivo requirement for RecJ, ExoVII, ExoI, and ExoX in methyl-directed mismatch repair. Proc Natl Acad Sci U S A. 2001;98:6765–6770. doi: 10.1073/pnas.121183298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Constantin N, Dzantiev L, Kadyrov FA, Modrich P. Human mismatch repair: reconstitution of a nick-directed bidirectional reaction. J Biol Chem. 2005;280:39752–39761. doi: 10.1074/jbc.M509701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kadyrov FA, Genschel J, Fang Y, Penland E, Edelmann W, Modrich P. A possible mechanism for exonuclease 1-independent eukaryotic mismatch repair. Proc Natl Acad Sci U S A. 2009;106:8495–8500. doi: 10.1073/pnas.0903654106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amin NS, Nguyen MN, Oh S, Kolodner RD. exo1-Dependent mutator mutations: model system for studying functional interactions in mismatch repair. Mol Cell Biol. 2001;21:5142–5155. doi: 10.1128/MCB.21.15.5142-5155.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schmutte C, Marinescu RC, Sadoff MM, Guerrette S, Overhauser J, Fishel R. Human exonuclease I interacts with the mismatch repair protein hMSH2. Cancer Res. 1998;58:4537–4542. [PubMed] [Google Scholar]
- 42.Dherin C, Gueneau E, Francin M, Nunez M, Miron S, Liberti SE, Rasmussen LJ, Zinn-Justin S, Gilquin B, Charbonnier JB, Boiteux S. Characterization of a highly conserved binding site of Mlh1 required for exonuclease I-dependent mismatch repair. Mol Cell Biol. 2009;29:907–918. doi: 10.1128/MCB.00945-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tishkoff DX, Amin NS, Viars CS, Arden KC, Kolodner RD. Identification of a human gene encoding a homologue of Saccharomyces cerevisiae EXO1, an exonuclease implicated in mismatch repair and recombination. Cancer Res. 1998;58:5027–5031. [PubMed] [Google Scholar]
- 44.Nielsen FC, Jager AC, Lutzen A, Bundgaard JR, Rasmussen LJ. Characterization of human exonuclease 1 in complex with mismatch repair proteins, subcellular localization and association with PCNA. Oncogene. 2004;23:1457–1468. doi: 10.1038/sj.onc.1207265. [DOI] [PubMed] [Google Scholar]
- 45.Tran PT, Erdeniz N, Symington LS, Liskay RM. EXO1-A multi-tasking eukaryotic nuclease. DNA Repair (Amst) 2004;3:1549–1559. doi: 10.1016/j.dnarep.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 46.Jager AC, Rasmussen M, Bisgaard HC, Singh KK, Nielsen FC, Rasmussen LJ. HNPCC mutations in the human DNA mismatch repair gene hMLH1 influence assembly of hMutLalpha and hMLH1-hEXO1 complexes. Oncogene. 2001;20:3590–3595. doi: 10.1038/sj.onc.1204467. [DOI] [PubMed] [Google Scholar]
- 47.Schmutte C, Sadoff MM, Shim KS, Acharya S, Fishel R. The interaction of DNA mismatch repair proteins with human exonuclease I. J Biol Chem. 2001;276:33011–33018. doi: 10.1074/jbc.M102670200. [DOI] [PubMed] [Google Scholar]
- 48.Liberti SE, Andersen SD, Wang J, May A, Miron S, Perderiset M, Keijzers G, Nielsen FC, Charbonnier JB, Bohr VA, Rasmussen LJ. Bi-directional routing of DNA mismatch repair protein human exonuclease 1 to replication foci and DNA double strand breaks. DNA Repair (Amst) 2011;10:73–86. doi: 10.1016/j.dnarep.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tran PT, Erdeniz N, Dudley S, Liskay RM. Characterization of nuclease-dependent functions of Exo1p in Saccharomyces cerevisiae. DNA Repair (Amst) 2002;1:895–912. doi: 10.1016/s1568-7864(02)00114-3. [DOI] [PubMed] [Google Scholar]
- 50.Lutzen A, de Wind N, Georgijevic D, Nielsen FC, Rasmussen LJ. Functional analysis of HNPCC-related missense mutations in MSH2. Mutat Res. 2008;645:44–55. doi: 10.1016/j.mrfmmm.2008.08.015. [DOI] [PubMed] [Google Scholar]
- 51.Sun X, Zheng L, Shen B. Functional alterations of human exonuclease 1 mutants identified in atypical hereditary nonpolyposis colorectal cancer syndrome. Cancer Res. 2002;62:6026–6030. [PubMed] [Google Scholar]
- 52.Pabla N, Ma Z, McIlhatton MA, Fishel R, Dong Z. hMSH2 recruits ATR to DNA damage sites for activation during DNA damage-induced apoptosis. J Biol Chem. 2011;286:10411–10418. doi: 10.1074/jbc.M110.210989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo G, Wang W, Bradley A. Mismatch repair genes identified using genetic screens in Blm-deficient embryonic stem cells. Nature. 2004;429:891–895. doi: 10.1038/nature02653. [DOI] [PubMed] [Google Scholar]
- 54.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 55.Dickey JS, Redon CE, Nakamura AJ, Baird BJ, Sedelnikova OA, Bonner WM. H2AX: functional roles and potential applications. Chromosoma. 2009;118:683–692. doi: 10.1007/s00412-009-0234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gradia S, Acharya S, Fishel R. The human mismatch recognition complex hMSH2-hMSH6 functions as a novel molecular switch. Cell. 1997;91:995–1005. doi: 10.1016/s0092-8674(00)80490-0. [DOI] [PubMed] [Google Scholar]
- 57.Marsischky GT, Kolodner RD. Biochemical characterization of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 complex and mispaired bases in DNA. J Biol Chem. 1999;274:26668–26682. doi: 10.1074/jbc.274.38.26668. [DOI] [PubMed] [Google Scholar]
- 58.Segurado M, Diffley JF. Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev. 2008;22:1816–1827. doi: 10.1101/gad.477208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci U S A. 2008;105:16906–16911. doi: 10.1073/pnas.0809380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dzantiev L, Constantin N, Genschel J, Iyer RR, Burgers PM, Modrich P. A defined human system that supports bidirectional mismatch-provoked excision. Mol Cell. 2004;15:31–41. doi: 10.1016/j.molcel.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 61.Stojic L, Cejka P, Jiricny J. High Doses of S(N)1 Type Methylating Agents Activate DNA Damage Signaling Cascades that are Largely Independent of Mismatch Repair. Cell Cycle. 2005;4 doi: 10.4161/cc.4.3.1528. [DOI] [PubMed] [Google Scholar]
- 62.Yoshioka K-I, Yoshioka Y, Hseih P. ATR kinase activation mediated by MutSa and MutLa in response to cytotoxic O6-methylguanine adducts. Molec Cell. 2006;22:501–510. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klapacz J, Meira LB, Luchetti DG, Calvo JA, Bronson RT, Edelmann W, Samson LD. O6-methylguanine-induced cell death involves exonuclease 1 as well as DNA mismatch recognition in vivo. Proc Natl Acad Sci U S A. 2009;106:576–581. doi: 10.1073/pnas.0811991106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tomimatsu N, Mukherjee B, Burma S. Distinct roles of ATR and DNA-PKcs in triggering DNA damage responses in ATM-deficient cells. EMBO Rep. 2009;10:629–635. doi: 10.1038/embor.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Giannattasio M, Follonier C, Tourriere H, Puddu F, Lazzaro F, Pasero P, Lopes M, Plevani P, Muzi-Falconi M. Exo1 competes with repair synthesis, converts NER intermediates to long ssDNA gaps, and promotes checkpoint activation. Mol Cell. 2010;40:50–62. doi: 10.1016/j.molcel.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 66.Tomimatsu N, Mukherjee B, Deland K, Kurimasa A, Bolderson E, Khanna KK, Burma S. Exo1 plays a major role in DNA end resection in humans and influences double-strand break repair and damage signaling decisions. DNA Repair (Amst) 2012;11:441–448. doi: 10.1016/j.dnarep.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kalamegham R, Warmels-Rodenhiser S, MacDonald H, Ebisuzaki K. O6-methylguanine-DNA methyltransferase-defective human cell mutant: O6-methylguanine, DNA strand breaks and cytotoxicity. Carcinogenesis. 1988;9:1749–1753. doi: 10.1093/carcin/9.10.1749. [DOI] [PubMed] [Google Scholar]
- 70.Rajesh P, Rajesh C, Wyatt MD, Pittman DL. RAD51D protects against MLH1-dependent cytotoxic responses to O(6)-methylguanine. DNA Repair (Amst) 2010;9:458–467. doi: 10.1016/j.dnarep.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Banath JP, Klokov D, MacPhail SH, Banuelos CA, Olive PL. Residual gammaH2AX foci as an indication of lethal DNA lesions. BMC Cancer. 2010;10:4. doi: 10.1186/1471-2407-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Staszewski O, Nikolova T, Kaina B. Kinetics of gamma-H2AX focus formation upon treatment of cells with UV light and alkylating agents. Environ Mol Mutagen. 2008;49:734–740. doi: 10.1002/em.20430. [DOI] [PubMed] [Google Scholar]
- 73.Mirzoeva OK, Kawaguchi T, Pieper RO. The Mre11/Rad50/Nbs1 complex interacts with the mismatch repair system and contributes to temozolomide-induced G2 arrest and cytotoxicity. Molecular cancer therapeutics. 2006;5:2757–2766. doi: 10.1158/1535-7163.MCT-06-0183. [DOI] [PubMed] [Google Scholar]
- 74.Bai H, Madabushi A, Guan X, Lu AL. Interaction between human mismatch repair recognition proteins and checkpoint sensor Rad9-Rad1-Hus1. DNA Repair (Amst) 2010;9:478–487. doi: 10.1016/j.dnarep.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Short SC, Giampieri S, Worku M, Alcaide-German M, Sioftanos G, Bourne S, Lio KI, Shaked-Rabi M, Martindale C. Rad51 inhibition is an effective means of targeting DNA repair in glioma models and CD133+ tumor-derived cells. Neurooncology. 2011;13:487–499. doi: 10.1093/neuonc/nor010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meador JA, Zhao M, Su Y, Narayan G, Geard CR, Balajee AS. Histone H2AX is a critical factor for cellular protection against DNA alkylating agents. Oncogene. 2008;27:5662–5671. doi: 10.1038/onc.2008.187. [DOI] [PubMed] [Google Scholar]
- 77.Campbell MR, Wang Y, Andrew SE, Liu Y. Msh2 deficiency leads to chromosomal abnormalities, centrosome amplification, and telomere capping defect. Oncogene. 2006;25:2531–2536. doi: 10.1038/sj.onc.1209277. [DOI] [PubMed] [Google Scholar]
- 78.Edelmann W, Cohen PE, Kane M, Lau K, Morrow B, Bennett S, Umar A, Kunkel T, Cattoretti G, Chaganti R, Pollard JW, Kolodner RD, Kucherlapati R. Meiotic pachytene arrest in MLH1-deficient mice. Cell. 1996;85:1125–1134. doi: 10.1016/s0092-8674(00)81312-4. [DOI] [PubMed] [Google Scholar]
- 79.Palombo F, Gallinari P, Iaccarino I, Lettieri T, Hughes M, D’Arrigo A, Truong O, Hsuan JJ, Jiricny J. GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science. 1995;268:1912–1914. doi: 10.1126/science.7604265. [DOI] [PubMed] [Google Scholar]
- 80.Christmann M, Tomicic MT, Kaina B. Phosphorylation of mismatch repair proteins MSH2 and MSH6 affecting MutSalpha mismatch-binding activity. Nucleic Acids Res. 2002;30:1959–1966. doi: 10.1093/nar/30.9.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cejka P, Mojas N, Gillet L, Schar P, Jiricny J. Homologous recombination rescues mismatch-repair-dependent cytotoxicity of S(N)1-type methylating agents in S. cerevisiae. Curr Biol. 2005;15:1395–1400. doi: 10.1016/j.cub.2005.07.032. [DOI] [PubMed] [Google Scholar]
- 82.Christmann M, Kaina B. Nuclear translocation of mismatch repair proteins MSH2 and MSH6 as a response of cells to alkylating agents. J Biol Chem. 2000;275:36256–36262. doi: 10.1074/jbc.M005377200. [DOI] [PubMed] [Google Scholar]
- 83.Schroering AG, Williams KJ. Rapid induction of chromatin-associated DNA mismatch repair proteins after MNNG treatment. DNA Repair (Amst) 2008;7:951–969. doi: 10.1016/j.dnarep.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hidaka M, Takagi Y, Takano TY, Sekiguchi M. PCNA-MutSalpha-mediated binding of MutLalpha to replicative DNA with mismatched bases to induce apoptosis in human cells. Nucleic Acids Res. 2005;33:5703–5712. doi: 10.1093/nar/gki878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brown KD, Rathi A, Kamath R, Beardsley DI, Zhan Q, Mannino JL, Baskaran R. The mismatch repair system is required for S-phase checkpoint activation. Nat Genet. 2003;33:80–84. doi: 10.1038/ng1052. [DOI] [PubMed] [Google Scholar]
- 86.Yang G, Scherer SJ, Shell SS, Yang K, Kim M, Lipkin M, Kucherlapati R, Kolodner RD, Edelmann W. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell. 2004;6:139–150. doi: 10.1016/j.ccr.2004.06.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.