

Abstract
Epigenetic modifications are heritable chromatin alterations that contribute to the temporal and spatial interpretation of the genome. The epigenetic information is conveyed through a multitude of chemical modifications, including DNA methylation, reversible modifications of histones, and ATP-dependent nucleosomal remodeling. Deregulation of the epigenetic machinery contributes to the development of several pathologies, including cancer. Chromatin modifications are multiple and interdependent and they are dynamically modulated in the course of various biological processes. Combinations of chromatin modifications give rise to a complex code that is superimposed on the genetic code embedded into the DNA sequence to regulate cell function. This review addresses the role of epigenetic modifications in cancer, focusing primarily on histone methylation marks and the enzymes catalyzing their removal.
I. INTRODUCTION
The dramatic phenotypic changes associated with the establishment of fully differentiated cells during development, and the reprogramming required for the establishment of germ cells, occur in the absence of changes in DNA sequence and they are heritable. Such changes are referred to as epigenetic and they are responsible for major shifts in gene expression and consequently, for major phenotypic shifts during development (Surani et al., 2007). Epigenetic regulation of gene expression is the basic force driving stem cell biology and development. Epigenetic mechanisms also contribute to aging (Collado et al., 2007) and to the development of several pathologies, including cancer (Jones and Baylin, 2007), inflammation (Foster and Medzhitov, 2009), and degenerative diseases (Wang et al., 2008b). Epigenetic mechanisms contributing to the regulation of gene expression include the remodeling and repositioning of nucleosomes, the modification of histone amino acid residues, and the methylation of DNA (Bernstein et al., 2007). These processes are functionally linked and cross regulated.
Nuclear DNA is tightly packaged into chromatin fibers (Tremethick, 2007). The basic chromatin unit is the nucleosome, which consists of 147 bp of DNA wrapped 1.7 times in a left handed superhelix around the histone octamer, composed of two copies of each of the core histones, H2A, H2B, H3, and H4. Nucleosomes are separated by 10–60 bp of linker DNA, which is complexed with linker histones (Hansen, 2002; Kornberg and Thomas, 1974; Luger et al., 1997). Core histones contain a “histone fold” globular domain, which is responsible for histone–DNA and histone–histone interactions, and N-terminal and C-terminal tails. Histone tails contain sites that are targets of various posttranslational modifications, including phosphorylation of serine and threonine residues, acetylation of lysine side chains, methylation of lysine and arginine residues (Fig. 1), ubiquitination and sumoylation of lysine residues, and ADP ribosylation of glutamic acid residues. Posttranslational modifications of histone tails regulate the interaction of nucleosomes with other nucleosomes and with linker DNA and direct the folding of chromatin into higher order structures (Hansen, 2002; Luger et al., 1997). The same modifications regulate chromatin binding of various nonhistone chromatin-associated proteins. As a result, enzymes involved in the posttranslational modification of histone tails, in combination with adenosine-5′-triphosphate (ATP)-dependent chromatin remodeling enzymes, regulate transcription and other chromatin-dependent activities. The binding of these proteins is mediated by specific domains, such as chromodomains, bromodomains, plant homeodomains (PHD), WD, and Tudor domains that are present in these proteins and recognize modified histone residues (Berger, 2007; Strahl and Allis, 2000; Taverna et al., 2007). Modification of histone tails plays a dynamic functional role because all modifications are transient. The most recently discovered enzyme group responsible for the reversal of a histone modification is that of histone demethylases (Klose et al., 2006a; Shi and Whetstine, 2007).
Fig. 1.

The different methylation states of lysine and arginine residues. (A) In histones, lysine can be found in the mono-, di-, and trimethylated form. The methyl mark is established by the lysine methyltransferases (KMTs) and removed by the histone lysine demethylases (KDMs). (B) Arginine residues can be of three distinct forms, monomethylarginine, symmetric dimethyl-arginine, and asymmetric dimethylarginine. The methyl marks on the arginine side chain are established by the protein arginine methyltransferases (PRMTs) and removed by the histone arginine demethylases (RDMs). (See Page 2 in Color Section at the back of the book.)
Chromatin modifications regulate, and they are regulated by other chromatin modifications. In addition, they regulate, and they are regulated by other nuclear processes. As a result, they play dynamic roles in gene expression and in the regulation of other chromatin-dependent processes including DNA replication, recombination, and DNA repair. Regarding the interdependence between histone modifications, characteristic examples are the inhibition of histone H3 trimethylation at K4 by prior dimethylation of H3 at R2 and vice versa (Kirmizis et al., 2007; Vermeulen et al., 2007), as well as the promotion of histone H3 trimethylation at K4 and K79 by histone H2B ubiquitination at K123 (Lee et al., 2007). Histone modifications also interact functionally with the DNA methylation machinery. As a result, histone modifications and DNA methylation are also interdependent (D’Alessio and Szyf, 2006).
It was suggested in the preceding paragraph that chromatin modifications also regulate, and they are regulated by other nuclear processes. For example, trimethylation of histone H3 at K4 may contribute to RNA splicing. Thus, H3K4me3 is recognized by CHD1, and promotes the binding of CHD1, in association with factors involved in transcriptional elongation and RNA splicing (Sims et al., 2007). The contribution of this process to RNA splicing was confirmed by the knockdown of CHD1, which dramatically reduced splicing. Other experiments suggested that histone modifications may also regulate nucleocytoplasmic RNA transport. Specifically, the deubiquitination of histone H2B and the phosphorylation of histone H3 at Ser10 contribute to the recruitment of P-TEFb (Cyclin T1:Cdk9), which phosphorylates the carboxy terminal domain (CTD) of RNA PolII at Ser2. The latter recruits Iws1 and REF1/Aly, an RNA-binding protein that is involved in nucleocytoplasmic RNA transport (Bres et al., 2008). Experiments designed to address the regulation of chromatin modifications by other nuclear processes, suggested that small RNAs and proteins of the RNAi machinery may contribute to the recruitment of the histone H3K27 methyltransferase EZH2. One of these experiments showed that the human homolog of AGO1 recruits EZH2 to promoters targeted by siRNAs and silences them (Kim et al., 2006a).
Earlier studies had suggested that some chromatin modifications are associated with active and others with inactive chromatin. However, recent studies have challenged this view by showing that the repressive marks H3K9me3 and methylated CpG DNA are associated with active transcription, when localized in the body of a gene (Vakoc et al., 2005; Zilberman et al., 2007). Moreover, H3K36me, also in the body of a transcribed gene, recruits a deacetylase complex, which removes the nucleosome destabilizing acetyl groups (Carrozza et al., 2005; Joshi and Struhl, 2005; Keogh et al., 2005). It has been proposed that these marks are induced dynamically in the course of transcription in order to prevent unwanted initiation from cryptic start sites in the body of the gene. Therefore, transcription appears to involve a series of interlocking chromatin modification events that are associated with transcriptional initiation or transcriptional elongation and are separated, both spatially and temporally. These findings have suggested that the interpretation of the epigenetic code is multidimensional and includes the dimensions of space, time, and interpretation machinery, which may differ between cell types.
The cycling of stem cells and their differentiation depend on the balance between the opposing activities of the Polycomb group (PcG) and Trithorax group (TrxG) proteins. PcG and TrxG complexes were discovered in Drosophila as repressors (PcG) and activators (TrxG) of the Hox genes. More recent studies have shown that these complexes are conserved among species and that they play a critical role in stem cell identity and lineage commitment during differentiation. Deregulation of these complexes has been associated with various pathological conditions, including cancer (Pietersen and van Lohuizen, 2008; Schwartz and Pirrotta, 2007; Sparmann and van Lohuizen, 2006; Spivakov and Fisher, 2007). There are several PcG complexes of which two (Polycomb Repressive Complex 1 (PRC1) and Polycomb Repressive Complex 2 (PRC2)) were the first to be discovered and have received considerable attention. The recruitment of both to Polycomb responsive genes is mediated by Pleiohomeotic (PHO or YY1), a DNA-binding protein. However, the recruitment of PRC1 is reinforced by the binding of one of its components (Pc) to the H3K27me3 mark, which is introduced by PRC2 (Mohd-Sarip et al., 2002, 2005, 2006). TrxG complexes are more heterogeneous. One class of complexes contains SET (Suppression of variegation, Enhancer of zeste, Trithorax)-domain methyltransferases, while a second class contains ATP-dependent chromatin-remodeling complexes, such as SWI/SNF or NURF (nucleosome remodeling factor). The best studied repressive marks induced by the PcG complexes include the trimethylation of histone H3 at K27 and the ubiquitination of histone H2A at K119. Similarly, the best studied activation mark induced by TrxG complexes is the trimethylation of histone H3 at K4 (Pietersen and van Lohuizen, 2008). It is interesting that many of the genes that are repressed by PcG complexes in stem cells are characterized by bivalent chromatin domains that contain both repressive (H3K27me3) and activating (H3K4me3) marks (Mikkelsen et al., 2007; Pan et al., 2007; Zhao et al., 2007). Differentiation is associated with resolution of bivalence through the loss of one of these marks (Bernstein et al., 2006). These findings have been interpreted to suggest that the balance of PcG and TrxG complexes in stem cells gives rise to a state in which cells are poised to choose between two fates. The decision involves the loss of the one or the other mark (Pietersen and van Lohuizen, 2008).
The mechanism by which epigenetic marks are transmitted from one generation to the next varies depending on the mark. In the case of DNA methylation, the mechanism is clearly defined. During DNA replication, the parental DNA strand maintains its methylation status. As a result, one of the strands of a DNA sequence that was methylated in the parental cells will continue to be methylated in the double stranded DNA of the daughter cells. Hemimethylated DNA regions are recognized by the DNA methyltransferase DNMT1 (Hashimoto et al., 2008), which is recruited in these regions and methylates the unmethylated strand. However, the mechanism of inheritance of histone modifications has not been defined with the same clarity and various hypotheses have been proposed to explain it (Kouzarides, 2007; Trojer and Reinberg, 2006).
The preceding short discussion of chromatin modifications and their role in cell biology is not comprehensive and it is only meant as an introduction to the main topic of this review, which is the role of histone methylation marks and the enzymes catalyzing their removal in cancer. The regulation and interdependence of chromatin modifications and their dynamic role in transcription and other biological processes have been addressed in detail in a number of excellent recent reviews (Bhaumik et al., 2007; Fischle, 2008; Ito, 2007; Kouzarides, 2007).
II. HISTONE METHYLATION AND CHROMATIN STRUCTURE
A. Histone Methylation
Methylation of histone residues occurs at the side chains of arginines and lysines. The information encoded by histone methylation is interpreted via the specific binding of protein complexes to the modified histone tails. Association or dissociation of these proteins regulates both the establishment of the global chromatin environments of euchromatin and heterochromatin and DNA-based functions, including transcription, DNA repair, and DNA replication and condensation (Bhaumik et al., 2007; Kouzarides, 2007). Histone H3 is primarily methylated at four lysine residues within the N-terminal tail and one within the core (K4, K9, K27, and K36 in the tail and K79 in the core). In addition, histone H4 is methylated at K20, and histone H1 at K26. All these residues can be mono-, di-, or trimethylated, giving rise to endless combinations of methylation marks (Table I and Fig. 1). Histone methylation is not limited to lysine residues. Arginine side chains on the tails of histones H3 and H4 may also undergo methylation. Sites that can be methylated include R2, R8, R17, and R26 of histone H3, and R3 of histone H4 (Table II). Arginine side chains can be monomethylated or dimethylated (symmetrically or asymmetrically) (Fig. 1). Histone lysine methylation is catalyzed by histone lysine methyl transferases (KMTs), most of which contain a SET domain and catalyze the transfer of a methyl group from their cofactor S-adenosyl methionine to the targeted histone lysine side chain. Histone arginine methylation, on the other hand, is catalyzed by protein arginine methyltransferases (PRMTs) (Bedford and Richard, 2005; Kouzarides, 2007; Lee et al., 2005a; Wysocka et al., 2006a).
Table I.
Summary of Histone Lysine Methyltransferases and Demethylases
| Mark | Methylation | Demethylation | Catalytic specificity of the demethylase |
|---|---|---|---|
| H3K4 | MLL1/KMT2A | LSD1/KDM1 | me2/me1→me0 |
| MLL2/KMT2B | JARID1A/RBP2/KDM5A | me3/me2/me1→me0 | |
| MLL3/KMT2C | JARID1B/PLU-1/KDM5B | me3/me2/me1→me0 | |
| MLL4/KMT2D | JARID1C/SMCX/KDM5C | me3/me2→me1 | |
| MLL5/KMT2E | JARID1D/SMCY/KDM5D | me3/me2→me1 | |
| hSET1A/KMT2F | NDY1/JHDM1B/FBXL10/KDM2B | me3→me2 | |
| hSET1B/KMT2G | |||
| ASH1/KMT2H | |||
| SET7–9/KMT7 | |||
| H3K9 | SUV39H1/KMT1A | LSD1/KDM1 | me2/me1→me0 |
| SUV39H2/KMT1B | JHDM2A/JMJD1A/KDM3A | me2/me1→me0 | |
| G9a/KMT1C | JHDM2B/5qNCA/KDM3B | me2 | |
| EuHMTase/GLP/KMT1D | JHDM2C/TRIP8/KDM3C | ||
| ESET/SETDB1/KMT1E | JMJD2A/JHDM3/KDM4A | me3/me2→me1 | |
| CLL8/KMT1F | JMJD2B/KDM4B | me3/me2→me1 | |
| RIZ1/KMT8 | JMJD2C/GASC1/KDM4C | me3/me2→me1 | |
| JMJD2D/KDM4D | me3/me2/me1→me0 | ||
| H3K27 | KMT6/EZH2 | UTX/KDM6A | me3/me2→me1 |
| JMJD3/KDM6B | me3/me2→me1 | ||
| UTY | |||
| H3K36 | SET2/KMT3A | NDY2/JHDM1A/FBXL11/KDM2A | me2/me1→me0 |
| NSD1/KMT3B | NDY1/JHDM1B/FBXL10/KDM2B | me2/me1→me0 | |
| SMYD2/KMT3C | JMJD2A/JHDM3/KDM4A | me3/me2→me1 | |
| JMJD2B/KDM4B | me3 | ||
| JMJD2C/GASC1/KDM4C | |||
| H3K79 | DOT1L/KMT4 | ||
| H4K20 | PR-SET7–8/KMT5A | ||
| SUV4–20H1/KMT5B | |||
| SUV4–20H2/KMT5C |
Table II.
Summary of Histone Arginine Methyltransferases and Demethylases/Deiminases
| Mark | Methylation | Demethylation/deimination | Catalytic specificity of the demethylase or deiminase |
|---|---|---|---|
| H3R2 | CARM1 | PAD4/PADI4 | me1/me0→citrulline |
| PRMT6 | JMJD6 | me2→me1 | |
| H3R8 | PRMT5 | ||
| H3R17 | CARM1 | ||
| H3R26 | CARM1 | ||
| H4R3 | PRMT1 | PAD4/PADI4 | me1/me0→citrulline |
| PRMT5 | JMJD6 | me2(s)/me2(a)/me1→me0 |
The specific residue that is methylated, and the stoichiometry of methylation, dictates the overall structure of the chromatin and the encoded biological outcome. The multitude of histone modifications synergize or antagonize each other. For example, the lysine residues are methylated on the ε-nitrogen, which is also targeted by histone acetyl transferases. Thus, methylation and acetylation of a specific lysine residue are mutually exclusive. Moreover, modification of a specific arginine or lysine side chain may interfere with the modification or the recognition of a neighboring residue. For example, methylation of H3R2 prevents the trimethylation of H3K4 and vice versa (Guccione et al., 2007; Kirmizis et al., 2007). Similarly, the phosphorylation of a specific serine or threonine residue may regulate the recognition of a neighboring modification by chromatin-binding proteins. For example, H3S10 phosphorylation interferes with the binding of heterochromatin protein 1 (HP1) to methylated histone H3K9 (Fischle et al., 2005; Hirota et al., 2005). Another example of cross talk between histone modifications is illustrated by the isomerization of proline 38 of histone H3, which causes a conformational change in the histone tail that interferes with the binding of the H3K36 histone methyltransferase SET2 (Nelson et al., 2006). In the following paragraphs, we will summarize the histone methyltransferases and the biological role of the histone methylation marks they catalyze.
1. METHYLATION OF H3K4
Several H3K4-specific methyltransferases have been discovered in mammals. These include the MLL proteins MLL1–4, hSET1A and hSET1B, as well as ASH1 and SET7/9 (Table I). All these enzymes are components of large protein complexes that catalyze all methylation states of H3K4 (Shilatifard, 2006; Tenney and Shilatifard, 2005). The activities of the mammalian H3K4 methyltransferases are not redundant, since ablation of individual genes encoding these proteins causes embryonic lethality in the mouse (Glaser et al., 2006; Lubitz et al., 2007; Yagi et al., 1998; Yu et al., 1998). The developmental defects observed in homozygous mutant embryos are distinct for each gene, confirming the absence of redundancy. The requirement for multiple H3K4 methyltransferases underscores the complexity of gene regulation by histone methylation.
Global histone modification maps provide evidence that actively transcribed genes exhibit a characteristic histone modification signature. This consists of H3K4 trimethylation in the promoter region plus H3K36 methylation within the body of the gene. This signature is further complemented by H3K9 and H3K14 acetylation (Guenther et al., 2007). Although many active genes exhibit this characteristic signature, there is a subset of such genes that bear the H3K4 trimethylation mark but lack significant H3K36 trimethylation. Many of these genes, however, fail to generate full-length transcripts because of the stalling of PolII during transcriptional elongation (Guenther et al., 2007; Zeitlinger et al., 2007). Binding of the polymerase at the transcription start site, combined with the failure of transcriptional elongation, is observed in a large number of developmental genes, which are either repressed or poised for activation at later stages of development.
Histone H3K4me3, in the promoter region of active genes, promotes transcriptional initiation by providing the docking site for the binding of TAF3, a component of TFIID. TAF3 binds histone H3K4me3 via its PHD finger domain and brings the entire TFIID complex to the promoter region. Selective loss of H3K4me3 indeed reduces TFIID binding and transcription from a subset of promoters in vivo (Vermeulen et al., 2007). The binding of TFIID to H3K4me3 is regulated via a complex series of functional interactions with other histone modifications. Specifically, asymmetric dimethylation of H3R2 selectively inhibits TFIID binding, whereas acetylation of H3K9 and H3K14 enhances it (Vermeulen et al., 2007). The H3K4 mark is, therefore, in the center of an elaborate mechanism that regulates RNA polymerase II-mediated transcription in higher eukaryotes.
Histone H3K4me3 also interacts functionally with other pathways, including the DNA damage response pathway and the regulation of gene expression by the ATP-dependent chromatin remodeling machinery. For example, members of the ING (INhibitor of Growth) family of tumor suppressor proteins bind specifically to H3K4me3 and H3K4me2 via their PHD domains. In response to DNA damage, recognition of H3K4me3 by the ING2 PHD domain stabilizes the repressive SIN3a–HDAC1 complex in the promoters of genes promoting proliferation, thus repressing their transcription (Shi et al., 2006). NURF, an ISWI-containing ATP-dependent chromatin-remodeling complex, associates directly with H3K4me3 through its PHD finger (Wysocka et al., 2006b). In the absence of H3K4 trimethylation, the BPTF (bromodomain and PHD finger transcription factor) subunit of NURF is released from the complex, compromising target-gene expression (Wysocka et al., 2006b). H3K4 trimethylation, therefore, may couple with NURF-mediated ATP-dependent chromatin remodeling to promote gene expression.
Histone H3K4me3 may also regulate processes that modify DNA, such as DNA recombination. Thus, RAG2, an essential component of the RAG1/2 V(D)J recombinase, contains a PHD finger that specifically recognizes histone H3K4me3. More important, mutations that abolish the ability of RAG2 to associate with H3K4me3 have a significant impact on V(D)J recombination in vivo (Matthews et al., 2007). Mutations of RAG2 at a conserved tryptophan residue that is essential for the association of RAG2 with H3K4me3 cause severe immunodeficiency in humans (Matthews et al., 2007).
2. METHYLATION OF H3K9
Histone H3 methylation at lysine 9 is implemented by the SET-domain-containing SUV39 family of histone methyltransferases and by the PRDI-BF1-RIZ1 (PR)-type SET-domain protein RIZ1/KMT8 (Kim et al., 2003). The mammalian SUV39 family is composed of SUV39H1, SUV39H2, G9a, EuHMTase/GLP/KMT1D, ESET/SetDB1/KMT1E, and CLL8/KMT1F (Table I) (Shilatifard, 2006). Deletion of individual H3K9 methyltransferases in mice causes embryonic lethality suggesting that, similar to H3K4 methyltransferases, H3K9 methyltransferases are not redundant (Dodge et al., 2004; Tachibana et al., 2002).
H3K9 methylation has long been linked to silencing of both heterochromatic and euchromatic regions (Kouzarides, 2007; Shilatifard, 2006). However, recent studies have reported H3K9 trimethylation in the transcribed region of active genes. This methylation mark was found to increase during activation of transcription and to be rapidly removed upon gene repression (Eissenberg and Shilatifard, 2006; Vakoc et al., 2005). Thus, the interpretation of the H3K9 mark appears to be context-specific. The methylation status of H3K9 is recognized by heterochromatin protein 1 (HP1), which associates with H3K9me3 and H3K9me2 via its chromodomain (Bannister et al., 2001; Lachner et al., 2001). There are three HP1 isoforms, α, β, and γ. HP1α localizes primarily in pericentric heterochromatin, HP1β is associated with promoters of silent euchromatic genes, and HP1γ is found within the coding regions of transcribed genes (Hediger and Gasser, 2006). The diversification of function of the different HP1 proteins may play a central role in the context-dependent interpretation of the H3K9 mark. The molecular mechanism that allows the different isoforms to distinguish between H3K9 marks in the different chromatin environments is not clear, but it has been proposed that it is the hinge region and the chromoshadow domain of each HP1 protein that may be responsible for the differential interaction (Smothers and Henikoff, 2001). The hinge domain of HP1 also contains several phosphorylation sites, suggesting that the specificity may be regulated, in part, by posttranslational modifications.
3. METHYLATION OF H3K27
The tri- and dimethylation of H3K27 is catalyzed by EZH1 and EZH2, two SET-domain-containing histone methyltransferases. Of these, EZH1 is expressed widely in nonproliferating cells, while EZH2 expression is tightly linked to cell proliferation (Margueron et al., 2008). EZH2 is a component of PRC2 (Cao and Zhang, 2004), which contains the Polycomb group (PcG) proteins EZH2, EED, SUZ12 and the nucleosome binding protein RbpAp48 (Schuettengruber et al., 2007). EZH1 also forms complexes that are similar to the PRC2 complex of EZH2. However, the knockdown of EZH2 is associated with the global downregulation of histone H3K27me2/me3, whereas the knockdown of EZH1 is not. Instead, EZH1 promotes H3K27 methylation and represses transcription of a subset of EZH2 target genes (Margueron et al., 2008). Interestingly, EZH1 is able to promote chromatin compaction in vitro in the absence of S-adenosyl methionine (and consequently histone methyltransferase activity). This is an intriguing observation that suggests that EZH1 may function as a transcriptional repressor by promoting the compaction of nucleosomal arrays (Margueron et al., 2008).
The H3K27 mark is specifically recognized by the chromodomain of the Polycomb protein (PC) (Cao and Zhang, 2004), a subunit of the PRC1. In addition to Polycomb, the core of PRC1 also contains Polyhomeotic (PH or HPH in human), BMI1 (the mammalian homolog of posterior sex combs, PSC), and RING (the mammalian homolog of sex combs extra, SCE). The RING proteins have E3 ubiquitin ligase activity that targets K119 of histone H2A (Cao et al., 2005; Li et al., 2006; Wang et al., 2004a). Ubiquitination of H2A appears to be a critical event in gene silencing.
In addition to promoting PRC1 binding, it was recently shown that EZH2 also recruits DNA methyltransferases to specific target genes, thus providing a direct link between histone and DNA methylation (Vire et al., 2006).
4. METHYLATION OF H3K36
Methylation of histone H3 at K36 is catalyzed by the SET2/KMT3 family of histone methyltransferases, which in mammals contains three members, SET2/KMT3A, NSD1/KMT3B, and SMYD2/KMT3C (Shilatifard, 2006). Global histone modification studies revealed that the H3K36 methylation mark is usually detected within the body of transcribed genes (Guenther et al., 2007). In agreement with this finding, several studies suggested that H3K36 methylation has a role in transcriptional elongation and in the suppression of transcriptional initiation within the body of a gene. SET2 methyltransferases are recruited, through their interaction with RNA polymerase II, at sites of transcription, where they catalyze H3K36 methylation (Gerber and Shilatifard, 2003; Hampsey and Reinberg, 2003). These methylation marks appear to provide a transcriptional memory that directs the deacetylation of open reading frames by specific histone deacetylases. Removal of acetyl marks from the transcribed region of a given gene is believed to result in the suppression of intragenic transcriptional initiation (Carrozza et al., 2005; Joshi and Struhl, 2005; Keogh et al., 2005).
5. METHYLATION OF H3K79
Histone H3K79 methylation is catalyzed by DOT1 (Disruptor of Telomeric Silencing 1), a histone methyltransferase originally identified in the yeast Saccharomyces cerevisiae, and DOT1L/KMT4 (DOT1-Like), its mammalian homolog. DOT1 and DOT1L are the only histone methyltransferases to date that lack the characteristic SET domain (Ng et al., 2002; van Leeuwen et al., 2002). Lysine 79 is located in the core of histone H3, on an accessible surface that appears not to be making contact with the DNA or with other histones (Luger et al., 1997). No activity capable of reversing H3K79 methylation has been reported to date.
DOT1 was discovered in a screen for genes whose overexpression disrupts telomeric gene silencing (Singer et al., 1998). More recent studies revealed that mammalian fibroblasts with an ablated DOTL1 gene lack H3K79 di- and trimethylation, suggesting that DOT1L is the only enzyme responsible for these methylation marks in mammalian cells (Steger et al., 2008). DOT1L preferentially occupies the proximal regions of active genes. More important, di- and trimethylation of histone H3 at K79 was linked to activation of transcription. Similarities in the pattern of H3K4 and H3K79 methylation in mammalian cells, suggested that MLL proteins, which mediate H3K4 methylation, and DOT1L, may be recruited in parallel or sequentially to common targets (Steger et al., 2008). The latter is consistent with the results of other studies, suggesting that DOT1L contributes to the pattern of gene expression induced by leukemia-derived MLL fusion proteins (Krivtsov et al., 2008). A molecular mechanism for the coordinate activities of DOT1L and MLL proteins was suggested by experiments in yeast. These experiments showed that histone H3K79 methylation by DOT1 and H3K4 methylation by COMPASS, the yeast H3K4 methyltransferase complex, depend on the Rad6-mediated monoubiquitination of histone H2B at K123, which provides a docking site for Cps35, a COMPASS subunit (Lee et al., 2007).
6. METHYLATION OF H4K20
H4K20 can be mono-, di-, or trimethylated. The monomethylated state is catalyzed by PR-SET7/KMT5A (Couture et al., 2005; Fang et al., 2002; Nishioka et al., 2002; Xiao et al., 2005), while the trimethylated state is catalyzed by SUV4–20H1 and SUV4–20H2. Histone H4K20 trimethylation is observed in pericentric heterochromatin (Schotta et al., 2004).
7. ARGININE METHYLATION
Methylation of histone tails is not limited to lysines. Several arginine residues are also modified by methylation. These include, R2, R8, R17, and R26 of histone H3, and R3 of histone H4. Arginine residues may undergo monomethylation, symmetric dimethylation, or asymmetric dimethylation (Fig. 1). The enzymes that catalyze the methylation of specific arginine residues are summarized in Table II. Thus, H3R2 is asymmetrically dimethylated by CARM1/PRMT4 (Chen et al., 1999; Schurter et al., 2001) and PRMT6 (Guccione et al., 2007). H3R8 is methylated by PRMT5 (Dacwag et al., 2007), while H3R17 and H3R26 are asymmetrically dimethylated by CARM1/PRMT4, which also methylates H3R2 (Chen et al., 1999; Schurter et al., 2001). Finally, H4R3 is monomethylated by PRMT1 (Stallcup et al., 2000; Strahl et al., 2001; Wang et al., 2001) and dimethylated, both symmetrically and asymmetrically by PRMT5 (Ancelin et al., 2006).
The methylation of specific arginine residues contributes to the regulation of cell fate. Ectopic expression of CARM1 in mouse blastomers increases the levels of arginine methylation and promotes the dramatic upregulation of the pluripotency genes NANOG and SOX2. This, in turn, promotes the cycling of pluripotent cells and the expansion of the inner cell mass of the blastocyst (Torres-Padilla et al., 2007).
The molecular mechanisms by which arginine methylation contributes to chromatin structure and transcriptional regulation are not yet clear. However, it has been shown that arginine methylation may regulate the modification or recognition of neighboring histone residues. Thus, it has been shown that methylation of H3R2 prevents the trimethylation of H3K4 and vice versa (Guccione et al., 2007; Kirmizis et al., 2007). Furthermore, it has been shown that asymmetric methylation of H3R2 inhibits the association of the TFIID subunit TAF3 with H3K4me3 (Vermeulen et al., 2007).
B. The Reversibility of Histone Methylation
The turnover of histone methyl groups in cultured mammalian cells was shown to be very slow, suggesting that histone methylation may be an irreversible or very slow process (Borun et al., 1972; Byvoet et al., 1972; Duerre and Lee, 1974). In the absence of an active mechanism, the only way to remove histone methylation would be to exchange the methylated histones with unmethylated ones, or to proteolytically remove the modified histone tails (Bannister et al., 2002). The stability of the methyl marks fitted perfectly with their proposed role as carriers of epigenetic information (Jenuwein and Allis, 2001; Zhang and Reinberg, 2001). However, as early as in 1995, it was observed that methyl groups may be removed from histone H3 during cell cycle progression (Annunziato et al., 1995), suggesting that methyl marks may not be as stable as initially thought. More recent studies showed that methyl arginines can be removed through the action of a peptidyl arginine deiminase (PAD4/PADI4) that converts monomethylated arginine to citrulline (Cuthbert et al., 2004; Wang et al., 2004b), although an activity that converts citrulline back into arginine has not been discovered to date. The static view of histone methylation was finally changed with the identification of the amine oxidase LSD1/KDM1 as an H3K4me2/me1-specific demethylase (Shi et al., 2004).
1. THE AMINE OXIDASE FAMILY OF HISTONE DEMETHYLASES
The first report for a lysine demethylase was published more than 40 years ago. In 1964, Paik and coworkers reported the detection of a demethylase activity directed against free mono- and dimethyllysine (Kim et al., 1964). Several years later, they partially purified the histone demethylase activity (Paik and Kim, 1973, 1974). However, the molecular identity of this putative lysine demethylase could not be determined. In an insightful report, Bannister and coworkers proposed that the chemical reaction catalyzed by FAD-dependent enzymes may remove methyl groups from modified histone side chains through the formation of an intermediate carbinolamine, which is unstable and degrades releasing formaldehyde and nonmethylated lysine or arginine (Bannister et al., 2002) (Fig. 2). Soon after, Shi and colleagues discovered that one of the components of the CtBP corepressor complex shared significant sequence homology with FAD-dependent amine oxidases (Shi et al., 2003). The same protein had been found earlier in a number of other corepressor complexes, including NRD (Tong et al., 1998) and CoR-EST (You et al., 2001). Using a combination of molecular and biochemical approaches, they confirmed that LSD1/KDM1 (previously known as p110b, BHC110, or NPAO) is a lysine-specific demethylase with specificity for methylated H3K4 and that it functions as a transcriptional corepressor (Shi et al., 2004).
Fig. 2.

The reaction mechanism of the amine oxidase family of histone demethylases. The demethylation of histone lysine residues by the amine oxidase group proceeds through the cleavage of the α-carbon bond of the substrate to generate an imine intermediate. This intermediate is subsequently hydrolyzed via a nonenzymatic process to produce a carbinolamine, which is unstable and degrades spontaneously to release formaldehyde and amine. This reaction reduces cofactor FAD to FADH2, which is then reoxidized by molecular oxygen to produce H2O2.
a. The Demethylation Reaction of the Amine Oxidase Family of Histone Demethylases
The demethylation of histone lysine residues by the amine oxidase histone demethylase family is characterized by the generation of an imine intermediate (Fig. 2). This intermediate is subsequently hydrolyzed via a nonenzymatic reaction to produce a carbinolamine, which is unstable and degrades spontaneously to release formaldehyde and demethylated lysine. This reaction reduces cofactor FAD to FADH2, which is then reoxidized by molecular oxygen to produce H2O2 (Fig. 2). The LSD1/KDM1 demethylation reaction requires a free lone pair of electrons on the nitrogen of the substrate to form the imine intermediate and for this it cannot reverse all three lysine methyl states. Thus, LSD1 is active against mono- or dimethylated peptides but cannot remove the methyl mark from trimethylated substrates.
b. Structure of LSD1/KDM1
LSD1/KDM1 contains three distinct structural domains: an N-terminal SWIRM domain, a central Tower domain, and a C-terminal AOL (amine oxidase-like) domain (Fig. 3) (Chen et al., 2006a; Stavropoulos et al., 2006; Yang et al., 2006). The SWIRM domain and the AOL domain form a core from which the Tower domain protrudes. The AOL domain contains two subdomains. The one is responsible for the binding of the FAD cofactor, while the other is dedicated to the binding of the peptide substrate. These two subdomains form a large cavity with the catalytic center located at the interface between the two substructures. The modular structure of the AOL domain is due to the insertion of the Tower domain, which forms a long helix-turn-helix that protrudes from the enzyme forming a surface for the binding of the corepressor CoREST (Chen et al., 2006a; Stavropoulos et al., 2006; Yang et al., 2006). CoREST promotes the demethylase activity of LSD1/KDM1 by allowing its accessibility to nucleosomal substrates and is required for the efficient demethylation of nucleosomal H3K4 (Lee et al., 2005b; Shi et al., 2005). Furthermore, CoREST also stabilizes and protects LSD1 from proteasomal degradation in vivo (Lee et al., 2005b; Shi et al., 2005). CoREST contains two successive SANT domains, SANT1 and SANT2, of which SANT2 alone is sufficient to confer LSD1/KDM1 with the ability to demethylate nucleosomal substrates. The crystal structure of the complex of LSD1 with the C-terminal region of CoREST revealed the details of this interaction, with the SANT2 domain wrapping around the protruding Tower domain of LSD1 (Fig. 3).
Fig. 3.

The structure of LSD1 bound to the C-terminal part of CoREST and the targeted histone peptide. The structure of LSD1/KDM1 revealed a protein with three distinct structural domains: an N-terminal SWIRM domain, a central Tower domain, and a C-terminal AOL (amine oxidase-like) domain. The FAD cofactor and the peptide substrate bind to the AOL domain. The graphic was created with PyMOL (DeLano Scientific LLC) based on PDB: 2V1D (Forneris et al., 2007). (See Page 3 in Color Section at the back of the book.)
The N-terminal SWIRM domain was named after the Swi3p, Rsc8p, and Moira proteins in which it is also found. Although the function of this domain in LSD1/KDM1 has not been conclusively determined, some SWIRM domains in other proteins have been shown to bind DNA and promote the nucleosomal association of the corresponding protein complexes (Da et al., 2006; Qian et al., 2005).
2. JUMONJI-DOMAIN-CONTAINING DEMETHYLASES
As initially suggested by Bannister, the chemical mechanism of histone demethylation by the flavin-dependent amine oxidases cannot demethylate trimethyllysine due to the absence of a protonated nitrogen in the substrate to form the imine intermediate (Bannister et al., 2002). As an alternative activity to that of amine oxidases, Kubicek and Jenuwein proposed that the α-keto-glutarate-Fe(II)-dioxygenases may have an activity compatible with the demethylation of histones(Kubicek and Jenuwein, 2004). Some members of the α-ketoglutarate-Fe(II)-dioxygenase family demethylate DNA. For example, the bacterial enzyme AlkB repairs DNA by demethylating 1-methyladenine and 3-methylcytosine through the iron-dependent oxidization of the methyl group, which is resolved by the release of formaldehyde (Falnes et al., 2002; Trewick et al., 2002). By analogy, therefore, it was proposed that other members of this family of enzymes may use chemistry similar to AlkB to demethylate histone methyllysine residues (Kubicek and Jenuwein, 2004). Soon after the discovery of LSD1/KDM1, Trewick and coworkers proposed that the Jumonji (JmjC)-domain-containing Fe(II)-dioxygenases may possess such an activity (Trewick et al., 2005). This was based on the ability of FIH (Factor Inhibiting HIF), a member of this group, to hydroxylate protein residues, thus catalyzing the first step in such a putative mechanism.
Using a classical biochemical purification scheme based on monitoring the release of formaldehyde from appropriately labeled histone substrates, Zhang and colleagues identified the first JmjC-domain-containing histone demethylase (Tsukada et al., 2006). This protein, JHDM1A/FBXL11/KDM2A, was able to revert di- and monomethylated H3K36 to the non-methylated form. In agreement with the earlier proposals, the catalytic activity of this protein was JmjC-domain dependent. Soon after the discovery of KDM2A, the same group also reported the isolation of another JmjC-domain-containing demethylase, JHDM2A/JMJD1A/KDM3A, which promotes the demethylation of H3K9me2 (Yamane et al., 2006). Since then, several additional JmjC-domain-containing histone demethylases have been reported. JHDM3A/JMJD2A/KDM4A converts H3K9me3 and H3K36me3 to H3K9me2 and H3K36me2 (Whetstine et al., 2006), respectively; JMJD2B/KDM4B demethylates H3K9me3 at pericentric heterochromatin (Fodor et al., 2006); JMJD2C/KDM4C or GASC1, a protein encoded by a gene originally noticed because it was amplified and overexpressed in esoph-ageal squamous cell carcinoma (Yang et al., 2000, 2001), converts H3K9me3 to H3K9me2 and H3K9me1 (Cloos et al., 2006). Moreover, JARID1A/RBP2/KDM5A, an interactor of the retinoblastoma protein, catalyzes the demethylation of H3K4me3 (Klose et al., 2007) and JARID1B/PLU-1/KDM5B, a transcriptional repressor implicated in breast cancer, demethylates H3K4me3 (Yamane et al., 2007). A complete list of JmjC-domain-containing histone demethylases whose activity and specificity has been determined is presented in Tables I and II.
a. Catalytic Mechanism and Substrate Specificity of JmjC-Domain-Containing Histone Demethylases
JmjC-domain histone demethylases catalyze histone lysine demethylation through an oxidative reaction that requires Fe(II) and α-ketoglutarate as cofactors. The catalytic reaction begins with the coordination of molecular oxygen (O2) by Fe(II) and the conversion of α-ketoglutarate to succinate and CO2 with the concomitant hydroxylation of the methyl group of the peptide substrate. The resulting carbinolamine is unstable and degrades spontaneously to unmethylated peptide and formaldehyde (Fig. 4). For the interested reader, the radical mechanism and the role of the iron-oxo intermediate are elegantly detailed in recent reviews by Ozer and Bruick (2007) and Shi and Whetstine (2007). Unlike LSD1/KDM1, the JmjC-domain demethylases do not require the presence of a protonated nitrogen in the substrate, and therefore, demethylate not only mono- and dimethylated but also trimethylated lysine residues.
Fig. 4.

The reaction mechanism of the JmjC-domain-containing family of histone demethylases. The catalytic mechanism of JmjC-domain demethylases involves molecular oxygen (O2) in the conversion of α-ketoglutarate to succinate and CO2 with the concomitant hydroxylation of the methyl group of the peptide substrate. The resulting carbinolamine is unstable and breaks down to the unmethylated peptide with the release of formaldehyde.
With the specificity of several JmjC-domain-containing histone demethylases now known, several interesting observations can be made. (a) Several histone demethylases have multiple substrate specificities. For example, JHDM3/JMJD2A/KDM4A, JMJD2B/KDM4B, and JMJD2C/GASC1/KDM4C demethylate both K9 and K36 of histone H3 (Klose et al., 2006b; Whetstine et al., 2006), as well as K26 of histone H1.4 (Trojer et al., 2009). NDY1/KDM2B is also able to demethylate both the dimethylated form of H3K36 and the trimethylated form of H3K4 (Frescas et al., 2007; Tzatsos et al., 2009). Chromatin immunoprecipitation studies have shown that NDY1 not only catalyzes this reaction in vitro but also in vivo in the local environment of the Ink4a-Arf locus, which is a target of this demethylase (Tzatsos et al., 2009). (b) Two related enzymes may target the same methylated residue but give rise to a different product. Thus, both JHDM3/JMJD2A/KDM4A and JMJD2D/KDM4D, two enzymes that belong to the same demethylase family, demethylate H3K9me3. However, JMJD2A produces H3K9me2 while JMJD2D produces H3K9me (Table I). (c) A JmjC-domain-containing histone demethylase may target a mono- or dimethylated lysine residue but not the trimethylated residue despite the fact that it may be chemically compatible for this reaction. Thus, NDY2/KDM2A and NDY1/KDM2B demethylate H3K36me2 and H3K36me, but not H3K36me3 (Table I). In addition, JHDM2A/JMJD1A/KDM3A demethylates H3K9me2, but not H3K9me3. (d) The in vitro specificity of JmjC-domain-containing histone demethylases depends on the substrate used and may differ from their specificity in vivo. Recombinant JARID1A/RBP2/KDM5A or JARID1B/PLU-1/KBM5B failed to initiate the demethylation reaction when incubated with H3 peptide substrates harboring a mono-methylated lysine 4. However, the same enzymes efficiently initiated the reaction on di- or the trimethylated peptides and the reaction proceeded processively until lysine residues were fully demethylated (Klose et al., 2007; Yamane et al., 2007). Moreover, both these enzymes efficiently catalyzed the demethylation of histone H3K4me1 in vivo (Klose et al., 2007; Yamane et al., 2007). Finally, native and recombinant JARID1B/PLU-1/KBM5B catalyzed the demethylation of H3K4me1 in bulk histones in vitro (Iwase et al., 2007; Xiang et al., 2007). By contrast, JHDM3A converts H3K36me3 and H3K9me3 to the monomethylated forms when nucleosomal substrates are used but the reaction can reach the unmethylated state when peptide substrates are used (Klose et al., 2006b). Similarly, UTX/KDM6A and JMJD3/KDM6B remove the H3K27me1 mark from peptide substrates but not from histones or nucleosomes (Agger et al., 2007; Lan et al., 2007). Thus, in vitro experiments might not be conclusive in determining the specificity of the in vivo reaction.
b. Structure and Substrate Recognition of the JmjC-Domain Histone Demethylases
The crystal structure of a fragment that spans amino acids 1–350 of JHDM3A/JMJD2A/KDM4A was the first structure of a JmjC-domain-containing histone demethylase to be reported (Chen et al., 2006b). Consistent with being a member of the α-ketoglutarate-Fe(II)-dependent dioxygenases, the JmjC domain of JHDM3A/JMJD2A/KDM4A forms a typical jelly roll-like structure composed of eight conserved antiparallel β-strands (Fig. 5A). In the active site of JMJD2A/KDM4A an iron metal ion is chelated by His188, Glu190, and His 276. These residues are conserved among most JmjC-domain-containing demethylases and appear to be essential for activity. There is no evidence to date that any member of the group that lacks one of these residues possesses demethylase activity. Moreover, point mutations of the equivalent sites abolish activity in several enzymes of this class (Pfau et al., 2008; Tsukada et al., 2006; Tzatsos et al., 2009). In the complex, α-ketoglutarate forms hydrogen bonds with the side chains of Tyr132, Asn198, and Lys206 and associates with the Fe2+ via its C-1 carboxylate and C-2 ketone groups.
Fig. 5.

The structure of the N-terminal domain of JMJD2A/KDM4A. (A) Consistent with being a member of the α-ketoglutarate-Fe(II)-dependent dioxygenases, the JmjC domain (blue) forms a typical jelly roll-like structure composed of eight conserved antiparallel β-strands. The conserved JmjN domain (cyan) comprises three helices positioned between two β-strands and associates with the catalytic JmjC domain. The C-terminal domain (yellow) interacts with the JmjC domain through a zinc-finger. (B) The details of molecular recognition between demethylase and histone peptide. Surface representation of the JMJD2A catalytic core bound to a methylated H3K36 peptide (Chen et al., 2007b). The graphics were created with PyMOL based on PDB entries 2GP5 and 2PXJ. (See Page 4 in Color Section at the back of the book.)
The conserved JmjN domain was first identified as an N-terminal extension of the JmjC domain (Balciunas and Ronne, 2000), but has since been found to be absent from many JmjC-domain-containing proteins. In the structure of JMJD2A/KDM4A, the JmjN domain associates with the catalytic JmjC domain, forming an extensive interaction interface (Fig. 5A). Deletion of residues 1–55 (corresponding to the JmjN domain) resulted in an unstable, catalytically inactive, protein, suggesting that the JmjN domain contributes to the structural integrity of the enzyme (Chen et al., 2006b). The C-terminal domain (amino acid residues 294–350) interacts with the JmjC domain through a uniquely structured zinc-finger. This zinc-finger is formed by two cysteine residues at the tip of a loop in the C-terminal domain (Cys306 and Cys308) and His240 and Cys234 of the JmjC domain, which together coordinate the binding of a zinc ion (Chen et al., 2006b).
The structure of the catalytic core of the JHDM3/JMJD2A/KDM4A JmjC domain, cocrystallized with the methylated H3K36 peptide provided the details of molecular recognition between the demethylase and the histone peptide (Chen et al., 2007b). This structure revealed that 8 out of the 11 contacts between the peptide and the demethylase correspond to interactions between the main chain of the peptide and the main chain of the enzyme, and that the binding specificity depends primarily on the conformation of the peptide (Chen et al., 2007b). The active site is located in a deep pocket, to which the peptide must bend to fit. Proline at position 38 of the peptide is important for its correct bend (Fig. 5B). The importance of peptide bending was confirmed by experiments showing that glycine substitutions at positions 30 and 31 of a trimethylated H3K27 peptide, that is not a physiological substrate of JHDM3A/JMJD2A/KDM4A, allowed the peptide to be demethylated by this enzyme. Introduction of the two glycine residues is believed to mimic the flexibility of the efficiently demethylated H3K9 sequence. The same peptide was also demethylated by the JMJD2D/KDM4D, which normally targets trimethylated and dimethylated H3K9 (Chen et al., 2007b).
JHDM3/JMJD2A/KDM2A exhibits demethylase activity against both K36 and K9 methylated histone H3. The cocrystallization of the JMJD2A/KDM4A catalytic core with H3K9me3 and H3K36me3 peptides shed light on the selectivity of histone demethylases for particular lysyl-containing sequences (Ng et al., 2007). Although the position and conformation of the H3K9 and H3K36 trimethyllysine residue in the active pocket was very similar in the two structures, the peptide backbones of the two substrates were bound in distinct conformations. The H3K9 substrate bound in a broad “W-shaped” conformation while the H3K36 peptide adopted a tighter bend leading to a “U-shaped” conformation (Fig. 6).
Fig. 6.

The cocrystal structure of JMJD2A with trimethylated H3K9 (yellow) and H3K36 (cyan) histone peptides. The position and conformation of the H3K9 and H3K36 tri-methyllysine residue is very similar in the two structures. However, the two substrates are bound in distinct conformations. The H3K9 substrate binds in a broad “W”-shaped conformation while the H3K36 peptide adopts a tighter bend leading to a “U”-shaped binding conformation (Ng et al., 2007). The graphic was created with PyMOL based on PDB entries 2OS2 and 2OQ6. (See Page 5 in Color Section at the back of the book.)
The JmjC-domain structures discussed above also provided mechanistic insights into the cross talk between demethylation and other histone modifications. In the H3K9 peptide substrate, the side chain of Ser10 and the main chain amide of Gly12 form a hydrogen bond that stabilizes the bent conformation. The Ser10Ala mutant of the H3K9me3 peptide was a poor demethylation substrate, while phosphorylation of the peptide at Ser10 completely abolished demethylation (Ng et al., 2007).
The structures of JHDM3/JMJD2A/KDM4A with a set of mono-, di-, and trimethyl H3K9 and H3K36 histone peptides provided insights into the mechanism that dictates the specificity of these enzymes toward the three methylation states. In the complex with the monomethylated peptide, two water molecules take up the positions occupied by the two additional methyl groups of the trimethylated lysine. These water molecules help to direct the single methyl group away from the Fe(II) ion, thus preventing its catalytic removal. The dimethyllysine of the H3K9me2 and H3K36me2 peptides, on the other hand, adopts two possible conformations, one that is nonproductive and another that is catalytically favorable, with a water molecule taking the place of the missing methyl group in both conformations. The interchange between these two conformations may be responsible for the lower activity of the enzyme against H3K36me2 than against H3K36me3 (Ng et al., 2007).
In addition to the JmjC and JmjN domains, JMJD2A/KDM4A also contains two tandem Tudor domains. This double Tudor domain binds methylated histone H3K4 or H4K20 (Kim et al., 2006b). The two Tudor domains of JMJD2A/KDM4A combine into a bilobal, saddle-shaped structure (Huang et al., 2006b). The third and fourth β-strands of the first canonical Tudor domain exchange with third and fourth β-strands of the second, thus forming two connected lobes (Fig. 7). The two lobes generated by this elaborate structural arrangement are termed hybrid Tudor domain 1 and 2 (HTD-1 and HTD-2). The H3K4me3 peptide is bound in a cleft of HTD-2 formed by an arrangement of aromatic residues (Trp967 and Tyr973 of HTD-2 and Phe932 of HTD-1) and a negatively charged amino acid (Asp934) (Huang et al., 2006b). The tandem Tudor domain of JMJD2A/KDM4A binds both H3K4me3 and H4K20me3 with similar affinities (Lee et al., 2008). However, H3K4me3 and H4K20me3 peptides adopt opposite-binding orientations despite the fact that the methyllysine is caged by the same three aromatic residues (Fig. 7). The different peptide recognition surfaces revealed by the solution structures were confirmed by specific single point mutations in the JMJD2A/KDM4A Tudor domain that inhibited the recognition of H3K4me3 but not H4K20me3, and vice versa (Lee et al., 2008).
Fig. 7.

The structure of the tandem Tudor domains of JMJD2A bound to trimethylated histone H3K4 and H4K20. (A) The crystal structure of the double Tudor domain of JMJD2A bound to a trimethylated H3K4 peptide. The two Tudor domains combine into a bilobal, saddle-shaped structure (Huang et al., 2006b). Each lobe is formed by the exchange of the third and fourth β-strands with respect to the canonical Tudor domain. The two lobes (which each resemble the canonical Tudor domain structure) are termed hybrid Tudor domain 1 and 2 (HTD-1 and HTD-2). The H3K4me3 peptide is bound in a cleft of HTD-2. (B) Surface representation of the structure of the tandem Tudor domains complexed with an H4K20me3 peptide. The two peptides adopt radically different binding modes (Lee et al., 2008). Although both are bound to the same hybrid Tudor domain (HTD-2) with the methyllysine caged by the same three aromatic residues, they adopt opposite relative orientations. Graphic created with PyMOL based on PDB entries 2GFA and 2QQS. (See Page 6 in Color Section at the back of the book.)
III. HISTONE METHYLATION AND CANCER
Mounting evidence suggests that deregulated histone methylation plays an important role in oncogenesis. A list of methyltransferases involved in oncogenesis, with a brief description of their mechanisms of action, is presented in Table III. Here, we will outline briefly the oncogenic properties of some members of this group that have been studied best.
Table III.
Histone Methyltransferases Implicated in Cancer
| Methyltransferase | Role | References |
|---|---|---|
| MLL1/KMT2A | Frequently altered by chromosomal translocations in acute leukemias, particularly in infant and therapy-related leukemias | Daser and Rabbitts (2004), Harper and Aplan (2008), Rowley (1993) |
| MLL2/KMT2B | Amplified in glioblastoma and pancreatic carcinoma | Huntsman et al. (1999) |
| MLL3/KMT2C | Frequently mutated in glioblastomas, melanomas, and pancreatic carcinomas | Balakrishnan et al. (2007) |
| RIZ1/KMT8 | Genetically or epigenetically inactivated in a variety of human cancers, including B cell lymphomas, hepatocellular, gastric, ovarian, prostate, and thyroid carcinomas. Mice lacking RIZ1 develop diffuse large B cell lymphomas and a broad spectrum of other tumors | Akahira et al. (2007), Carling et al. (2004), Chen et al. (2007a), Deng and Huang (2004), Du et al. (2005), Hasegawa et al. (2007), Kim et al. (2003), Lakshmikuttyamma et al. (2008), Lal et al. (2006), Piao et al. (2008), Steele-Perkins et al. (2001), Yoon et al. (2007) |
| EZH2/KMT6 | Overexpressed in a variety of human tumors, including aggressive prostate and breast cancers, transitional cell carcinomas of the bladder, and cutaneous melanomas. EZH2 overexpression promotes cell proliferation in culture, induces colony formation of immortalized cells, and is oncogenic in a mouse xenograft model | Bachmann et al. (2005, 2006), Bracken et al. (2003), Bryant et al. (2007), Croonquist and Van Ness (2005), Kleer et al. (2003), Raman et al. (2005), Saramaki et al. (2006), Sellers and Loda (2002), Simon and Lange (2008), Varambally et al. (2002, 2008) |
| NSD1/KMT3B | Mutated in human acute myeloid leukemia (AML), multiple myeloma, and lung cancers and overgrowth syndromes. A frequent translocation in AML fuses NSD1 to nucleoporin-98. The fusion protein induces AML in vivo and sustains self renewal of myeloid stem cells in vitro | La Starza et al. (2004), Rosati et al. (2002), Tatton-Brown et al. (2005), Wang et al. (2007) |
| G9a/KMT1C | Depletion of G9a induces centrosome disruption and chromosomal instability in cancer cells, and promotes the oncogenic transformation of immortalized primary human cells | Kondo et al. (2008), Mulligan et al. (2008) |
| DOT1L/KMT4 | Fusion of DOT1L to MLL results in leukemic transformation that depends on the DOT1L methyltransferase activity | Okada et al. (2005) |
| ASH1/KMT2H | Essential for neuroendocrine differentiation in the lung. Overexpressed in a diverse array of lung cancers with neuroendocrine features, including small cell lung cancer. Constitutive expression in the mouse promotes airway dysplasia and lung neuroendocrine tumors | Borges et al. (1997), Linnoila et al. (2000) |
| SMYD2/KMT3C | Methylates p53 and represses its activity | Huang et al. (2006a) |
| SET7–9/KMT7 | Methylates p53 and estrogen receptor alpha and regulates their activity in vivo. A breast cancer-associated mutation in the estrogen receptor abolishes its methylation by SET7–9 | Kurash et al. (2008), Subramanian et al. (2008) |
| CARM1/PRMT4 | Promotes prostate cancer cell viability and estrogen-stimulated breast cancer growth, and it is upregulated in grade-3 breast tumors | El Messaoudi et al. (2006), Frietze et al. (2008), Majumder et al. (2006) |
| PRMT1 | Methylates H4R3 and cooperates with MLL to enhance MLL-mediated transformation of hematopoietic cells | Cheung et al. (2007) |
| PRMT5 | Methylates H3R8 and H4R3 and interacts with the SWI/SNF complex to repress the transcription of RB family members. It is overexpressed in several human leukemias and lymphomas | Pal et al. (2007), Wang et al. (2008a) |
The H3K4 methyltransferase MLL1 is frequently altered by chromosomal translocation in acute myeloid and lymphoid leukemias. Some of these translocations give rise to MLL1 proteins without a SET domain, and therefore without methyltransferase activity (Daser and Rabbitts, 2004; Rowley, 1993). The tumor suppressor gene encoding the histone H3K9 methyltransferase RIZ1 (Retinoblastoma interacting zinc-finger protein 1) is a common target of frameshift and missense mutations that inactivate its PR methyltransferase domain in a variety of human cancers (Kim et al., 2003), and it is epigenetically silenced by promoter methylation in B cell lymphomas, hepatocellular, gastric, ovarian, prostate, and thyroid carcinomas (Akahira et al., 2007; Chen et al., 2007a; Hasegawa et al., 2007; Lal et al., 2006; Piao et al., 2008). Mice with an inactivating mutation in RIZ1 develop diffuse large B cell lymphomas and a broad spectrum of other tumors (Steele-Perkins et al., 2001). The histone H3K27 methyltransferase EZH2 is frequently overexpressed via amplification (Bracken et al., 2003), downregulation of microRNA 101 which targets EZH2 (Varambally et al., 2008), or other factors (Varambally et al., 2002), in a variety of human tumors. EZH2 overexpression enhances the proliferation of primary cells (Bracken et al., 2003), and promotes anchorage-independent growth and invasiveness of immortalized cells in culture (Kleer et al., 2003). Finally, EZH2 is oncogenic in an NIH 3T3-based mouse xenograft model. Tumor formation in this model depends on the integrity of the histone methyltransferase domain (Croonquist and Van Ness, 2005).
In addition to histone methyltransferases, several histone demethylases have also been implicated in cancer. The oncogenic potential of most of them has been revealed by genomic alterations, such as proviral insertion in retrovirus-induced tumors, amplification, or deletion. The oncogenic potential of other histone demethylase genes has been revealed by upregulation of their expression in various types of tumors, or by their functional interaction with known oncogenes or tumor suppressor genes.
A. Histone Demethylase Genes Genetically Altered During Oncogenesis
1. HISTONE DEMETHYLASES ENCODED BY GENES THAT ARE TARGETS OF PROVIRUS INTEGRATION IN RETROVIRUS-INDUCED TUMORS
Retroviral-insertional mutagenesis is a powerful genetic tool for the identification of novel oncogenes (Tsichlis and Lazo, 1991). Integration of a provirus into the genome causes alterations that result in the induction and progression of retrovirus-induced tumors. Given that the sites of provirus integration are virtually random, provirus integration at common sites (CIS) in multiple tumors suggests that the mutation caused by integration at these sites confers a selective advantage to the affected cells, and that the associated loci have oncogenic function.
Four JmjC-domain-containing proteins were identified in a genome-wide screen from this laboratory for targets of provirus integration in MoMuLV-induced rat T cell lymphomas (Pfau et al., 2008). Most of these integrations occurred at 5′ of a gene which, because of its characteristic phenotype (see below), was named Not-dead-yet-1 (NDY1; also known as FBXL10, JHDM1B, or KDM2B). In the same screen, single provirus insertions were detected immediately upstream of the NDY1 homolog NDY2/KDM2A (also known as FBXL11 or JHDM1A), and upstream of the JmjC-domain-containing proteins PHF2 and PHF8 (Pfau et al., 2008). NDY1/KDM2B was identified as a CIS in three additional studies (Suzuki et al., 2002, 2006; Uren et al., 2008), while PHF2 was also identified as a CIS in a large-scale retrovirus-mediated insertional mutagenesis screen in p19ARF and p53-deficient mice (Uren et al., 2008). Finally, multiple provirus insertions in the NDY2/FBXL11 gene were detected in screens for common integration sites of the HIV provirus in human cells infected in culture (Bushman et al., 2005).
Two more JmjC-domain-containing proteins, JMJD5 and JMJD1A/KDM3A, were identified in a separate retrovirus-mediated insertional mutagenesis screen in Blm-deficient mice (Suzuki et al., 2006). The argument for using Blm-deficient mice was that the conventional screens in wild-type mice preferentially target oncogenes, whereas the Blm-deficient mouse screen may preferentially target tumor suppressor genes.
a. NDY1/KDM2B and NDY2/KDM2A
i. Molecular Characterization of NDY1 and NDY2
NDY1/KDM2B belongs to the family of KDM2 histone demethylases, which has two members, NDY1/KDM2B and NDY2/KDM2A. Both of them have been shown to be the targets of provirus integration in retrovirus-induced tumors in rodents (NDY1 and NDY2) (Pfau et al., 2008; Suzuki et al., 2002, 2006; Uren et al., 2008), or in HIV-infected human lymphocytes in culture (NDY2) (Bushman et al., 2005). Both members of this family are nuclear proteins that contain an N-terminal JmjC domain, a CXXC zinc-finger domain, a PHD zinc-finger, an F-box, and a leucine-rich repeat (LRR). In addition, they both contain a region rich in proline residues (PRR), which is located N-terminally to the F-box (Fig. 8). The CXXC motif binds unmethylated CpG DNA and contributes to the association of these proteins with their target genes (Koyama-Nasu et al., 2007; Yamagishi et al., 2008). The PHD domain is a protein–protein interaction domain. By analogy with the PHD domains of other histone demethylases, it may mediate the binding of NDY1 and NDY2 to methylated histone tails (Iwase et al., 2007; Shi et al., 2006; Taverna et al., 2007; Vermeulen et al., 2007; Wysocka et al., 2006b), or to other regulators of chromatin structure, such as histone deacetylases (Barrett et al., 2007). The F-box domain identifies NDY1 and NDY2 as components of ubiquitin ligase complexes. The F-box is frequently coupled with C-terminal LRRs, and both domains function coordinately in the assembly of the ubiquitination machinery and the recognition of the target protein (Kipreos and Pagano, 2000). The potential function of the proline-rich motif will be discussed briefly in subsequent sections.
Fig. 8.

Domain structure of the JHDM1/KDM2 family.
Differential transcriptional initiation and alternative splicing give rise to several NDY1 isoforms (Pfau et al., 2008). One of these isoforms, termed the short isoform (Pfau et al., 2008), is particularly interesting because it is relatively abundant and lacks the JmjC-demethylase domain. The experiments we have reported to date have been based primarily on the mouse NDY1v1 isoform, which is referred to as the long isoform and contains all the above-mentioned functional domains (Pfau et al., 2008). The relative roles of these isoforms in cell biology and animal physiology remain to be determined.
Both NDY1/KDM2B and NDY2/KDM2A demethylate histone H3K36me2 and H3K36me1, but not H3K36me3 (He et al., 2008; Tsukada et al., 2006; Tzatsos et al., 2009). Although the substrate specificity of NDY2 appears to be limited to histone H3K36me2, the precise substrate specificity of NDY1 is being debated. Thus, whereas one group reported that NDY1 targets histone H3K4me3 (Frescas et al., 2007), others provided evidence that both the mammalian NDY1 and its Drosophila homolog dKDM2 target specifically histone H3K36me2 (He et al., 2008; Lagarou et al., 2008). Finally, our group showed that although NDY1 targets primarily histone H3K36me2, it also has a weak demethylase activity against histone H3K4me3 (Tzatsos et al., 2009). While this controversy remains to be resolved, we favor the dual specificity of this enzyme. A strong argument in support of our preference has been provided by genetic experiments in Drosophila. These experiments have shown that heterozygous loss of function mutations of dKDM2 reduce the frequency of homeotic transformation of heterozygous loss of function mutations of Trithorax group genes encoding ASH1 (an H3K36 methyltransferase) or TRX1 (an H3K4 methyltransferase) (Lagarou et al., 2008). The genetic interaction between dKDM2 and ASH1 suggests that downregulation of the H3K36me2 demethylase activity of dKDM2 ameliorates the effects of the H3K36me2 downregulation caused by loss of function mutations of ASH1. A similar explanation could be given to the genetic interaction between dKDM2 and TRX1. Instead, it was attributed to the fact that the binding of TRX1 to chromatin depends on ASH1 (Lagarou et al., 2008; Rozovskaia et al., 1999). However, the ASH1 dependence of TRX1 chromatin binding is difficult to explain the genetic interaction between dKDM2 and TRX1. The ASH1 dependence of TRX1 binding would suggest that TRX1 binds chromatin by recognizing ASH1 or methylated H3K36, of which none will be lost if dKDM2 undergoes a loss of function mutation.
The overexpression and the knockdown of NDY1 have diametrically opposite phenotypes in different cell types. Thus, in normal cells, such as mouse embryonic fibroblasts (MEFs), NDY1 promotes cell proliferation and functions as a physiological inhibitor of senescence (Pfau et al., 2008). In HeLa cells, on the other hand, overexpression of NDY1 inhibits, and knockdown promotes, cellular proliferation (Frescas et al., 2007; Koyama-Nasu et al., 2007). Interestingly, some of the cell proliferation inhibitory effects of NDY1 appear to be JmjC-domain independent (Koyama-Nasu et al., 2007). Differences in phenotype correlate with differences in the subcellular distribution of the protein in the two cell types. In both types of cells the protein exhibits primarily a nuclear distribution. However, in MEFs it is excluded from the nucleoli (Pfau et al., 2008), whereas in HeLa cells it is primarily nucleolar (Frescas et al., 2007). The subcellular distribution of NDY1 may explain the phenotypic differences of its overexpression in different cell types. In addition, it suggests that the protein may normally shuttle between the nucleoplasm and the nucleoli and that in some tumor cells the protein may be sequestered in the nucleolus because of defects in nucleolar retention or transport.
ii. Inhibition of Senescence; NDY1-Mediated Immortalization
NDY1 is progressively downregulated in passaged MEFs, as they undergo senescence (Tzatsos et al., 2009). Moreover, cells engineered to express exogenous NDY1 proliferate faster and they undergo immortalization in the absence of replicative senescence. Finally, knockdown of NDY1 and expression of JmjC-domain mutants of NDY1 promote senescence (Pfau et al., 2008). Based on these findings, we have concluded that in normal cells, such as MEFs, NDY1 promotes cell proliferation and functions as a physiological inhibitor of senescence (Pfau et al., 2008).
Expression of single domain mutants of NDY1 or NDY2 in MEFs revealed that the immortalization phenotype depends on the JmjC domain and the CXXC motif, but is independent of the PHD domain, the F-box, or the C-terminal LRR domain. Point mutants of the JmjC domain that are histone demethylase-deficient fail to immortalize MEFs, further supporting that it is the JmjC-domain-encoded histone demethylase activity of the protein that is responsible for the immortalization phenotype. Another domain that was deleted in the course of the preceding experiments was the PRR (Fig. 8). Surprisingly, deletion of the proline-rich region reproducibly enhanced the immortalization phenotype. We interpreted this finding to suggest that the PRR domain may be a docking site for NDY1-binding proteins that inhibit immortalization, or that it may have a function in regulating protein stability (Pfau et al., 2008).
Although both the JmjC-domain and the CXXC-motif mutants fail to promote immortalization, they differ in that the JmjC-domain mutants promote senescence while the CXXC-motif mutant does not. This suggests that only the JmjC-domain mutants interfere with the function of the endogenous protein. Based on these findings, we have concluded that the JmjC domain provides the immortalizing function of the protein and that the DNA-binding CXXC motif is involved in the association of the protein with its nucleosomal targets. The CXXC-motif mutant does not induce immortalization because it does not bind DNA, but it does not have a dominant-negative phenotype because due to its inability to bind DNA it does not interfere with the function of the endogenous protein.
The cellular senescence machinery and the role of NDY1
Cellular senescence is due to progressive telomere shortening, to the activation of the Ink4a-Arf-Ink4b locus, and to the activation of the DNA damage response. Telomere shortening and the activation of the Ink4a-Arf-Ink4b locus may be developmentally programmed in dividing cells. Alternatively, they may be induced by factors that activate the DNA damage response, including activated oncogenes, telomere shortening, oxidative stress, and the aberrant firing of DNA replication origins (Fig. 9). To address the mechanism by which NDY1 inhibits senescence, we systematically addressed its effects on the components of the senescence machinery illustrated in Fig. 9.
Fig. 9.
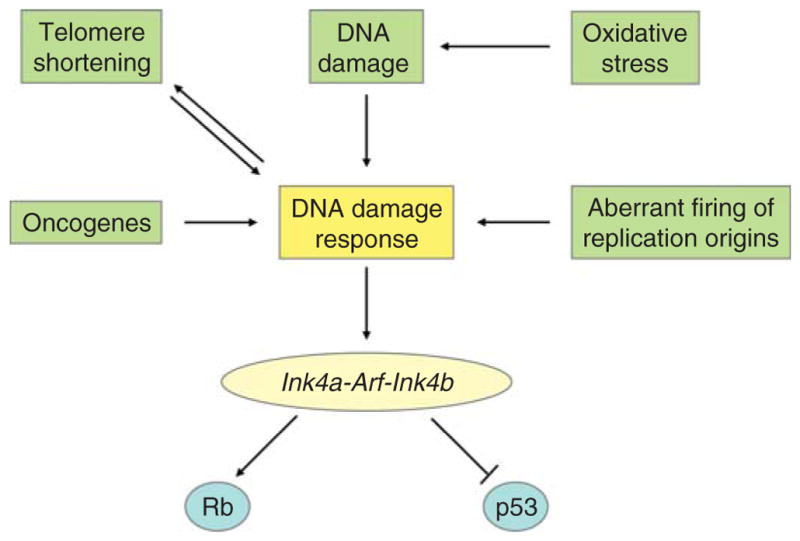
Different mechanisms promote cellular senescence through the Ink4a-Arf-Ink4b locus.
First, we addressed the effects of NDY1 on senescence induced by the Ha-Ras oncogene, which activates the Ink4a-Arf-Ink4b locus in the absence of DNA damage. Data from experiments conducted by both our and another group, showed that NDY1 inhibits the activation of this locus and the induction of senescence, and suggested that it may function in this pathway downstream of the DNA damage response (He et al., 2008; Tzatsos et al., 2009).
In other experiments, we addressed whether NDY1 regulates redox homeostasis and the cellular response to oxidative stress. NDY1 indeed protects cells from H2O2-induced apoptosis and G2/M arrest, and inhibits cellular signaling and DNA damage induced by reactive oxygen species (ROS). Knockdown of NDY1 has the opposite effects. Further studies showed that NDY1 promotes the expression of genes encoding the antioxidant enzymes aminoadipic semialdehyde synthase (Aass), NAD(P)H quinone oxidoreductase-1 (Nqo1), peroxiredoxin-4 (Prdx-4), and serine peptidase inhibitor b1b (Serpin b1b), and inhibits the expression of IL-19. Simultaneous knockdown of Aass, Nqo1, Prdx4, and Serpinb1b, in NDY1-expressing cells to levels equivalent to those in control cells fully repressed the NDY1 redox phenotype. NDY1 binds at specific sites on the promoters of at least two of these genes (Nqo1 and Prdx4), suggesting that their regulation by NDY1 may be direct (Polytarchou et al., 2008). The inhibition of ROS-induced DNA damage by NDY1 is in agreement with other studies that had also suggested that NDY1 may protect cells from DNA damage (Pothof et al., 2003; Suzuki et al., 2006).
To determine whether NDY1 regulates telomere shortening, we examined its ability to immortalize IMR90 human fibroblasts. MEFs and human fibroblasts differ with regard to the relative importance of telomere erosion in the induction of senescence, with telomere erosion being the primary cause of senescence in human fibroblasts, but not in MEFs (Blackburn, 2000; Bodnar et al., 1998). Our experiments showed that the expression of NDY1 in human fibroblasts prevents early senescence but fails to immortalize them, suggesting that while it prevents cell cycle arrest induced by telomere shortening, it does not affect telomere shortening per se (Pfau et al., 2008).
The Ink4a-Arf-Ink4b locus is activated by senescence inducing processes (Fig. 9). NDY1 may inhibit the activation of this locus in passaged MEFs, either by targeting pathways that regulate the DNA damage response, as suggested in the preceding paragraphs, or directly. In either case, repression of either p15INK4B or p16INK4A, or both, should result in the upregulation of pRB phosphorylation. Repression of p19ARF, on the other hand, should result in the downregulation of p53 and its targets. Experiments addressing these questions in passaged MEFs revealed that pRB is indeed hyperphosphorylated at Ser807/811. However, p53, and its target p21CIP1, are upregulated rather than downregulated. These findings suggest that NDY1 may target primarily the p15INK4B and p16INK4A encoding genes rather than the overlapping p19ARF-encoding gene. Further studies showed that the expression of p21CIP1 in NDY1-expressing cells is not counterselected during passage. This finding suggests that NDY1 expression renders MEFs resistant to the antiproliferative effects of p53 and p21CIP1. The molecular mechanism of the resistance, however, has not been determined.
NDY1 represses the Ink4a-Arf-Ink4b locus: Coupling of histone H3K36me2 and H3K4me3 demethylation to histone H3K27 trimethylation and histone H2A K119 ubiquitination
Activation of the Ink4a-Arf-Ink4b locus, which occurs in MEFs during passaging or in response to a variety of signals (Fig. 9), promotes senescence. Consistent with this observation, p19ARF−/− MEFs are immortal (Kamijo et al., 1997). However, p16Ink4a−/− and p15Ink4b−/− MEFs are not (Krimpenfort et al., 2007; Latres et al., 2000; Sharpless et al., 2001). The upregulation of the genes encoded by the Ink4a-Arf-Ink4b locus and the parallel downregulation of NDY1 in passaged MEFs undergoing senescence suggested that NDY1 may be a direct repressor of this locus and that the downregulation of NDY1 may be directly responsible for its activation. Data to date, support this hypothesis. However, the NDY1-mediated repression is stronger toward p16Ink4a and p15Ink4b than toward p19ARF (He et al., 2008; Tzatsos et al., 2009).
To exert its transcriptional silencing function, NDY1 accesses chromatin as a component of chromatin modifying complexes. Evidence to date suggests that NDY1 is associated with a novel Polycomb complex that shares components with, but is distinct from PRC1. In extracts derived from Drosophila embryos, this complex (dRAF; dRING-associated factors) was shown to contain dRING (a homolog of the mammalian RING proteins), PSC (a homolog of the mammalian BMI proteins), dKDM2 (a homolog of the mammalian NDY1 and NDY2) and other, not confirmed proteins (Lagarou et al., 2008). Complexes containing the mammalian homologs of the core components of dRAF were also isolated from mammalian cells. One group purified a complex bound to BCOR (BCL6 corepressor) from HeLaS3 and HEK293 cells stably transduced with an epitope-tagged BCOR construct. The complex contained RING1A and RING1B/RNF2, NSPC1, a homolog of BMI1, NDY1, and several other proteins, including RYBP/YAF2, and SKP1 (Gearhart et al., 2006). RING1B and NDY1 complexes isolated by another group from MEL erythroleukemia cells, also contained NSPC1 and its homolog BMI1, RING1A and RING1B, NDY1, BCOR, RYBP/YAF2, SKP1, and several additional proteins (Sanchez et al., 2007). These data combined, suggest that distinct complexes containing NDY1 and several PRC1 proteins, can be detected both in Drosophila and in mammalian cells. Our studies have shown that NDY1 also interacts with the histone methyltransferase EZH2, a component of PRC2 (Tzatsos et al., 2009). However, we do not know whether this interaction places NDY1 into the PRC2 complex, or into complexes, related to, but distinct from PRC2. Complexes containing PRC2 components including EZH2 and the Polycomb-like protein PCL (PHF1 in mammals) have been detected both in Drosophila and in mammalian cells (Kuzmichev et al., 2005; Nekrasov et al., 2007; Sarma et al., 2008).
NDY1 inhibits the upregulation of p16INK4a in passaged MEFs, through a complex mechanism (Fig. 10) that couples histone H3K36me2 and H3K4me3 demethylation to histone H3K27 trimethylation (Tzatsos et al., 2009) and perhaps to histone H2AK119 monoubiquitination (Lagarou et al., 2008). MEFs transduced with NDY1, upregulate instead of down-regulating EZH2 during passaging via a JmjC-domain-dependent process. Knockdown of NDY1 in primary MEFs and deletion of exogenous NDY1 in NDY1-immortalized MEFs, on the other hand, downregulate EZH2. Changes in the level of expression of EZH2 alter the level of histone H3K27 trimethylation, both globally and locally within the Ink4a-Arf-Ink4b locus. NDY1 also binds the Ink4a-Arf-Ink4b locus and promotes histone H3K36me2 and H3K4me3 demethylation. Given that it also interacts with EZH2, its binding to the locus may be facilitated by the observed EZH2 upregulation (Tzatsos et al., 2009). Finally, histone H3K27me3 may promote the binding of PRC1, or a dRAF-like complex, to the Ink4a-Arf-Ink4b locus (Tzatsos et al., 2009), which by analogy with observations in Drosophila (Lagarou et al., 2008), may promote the monoubiquitination of histone H2A at K119 (Fig. 10). The interdependence of these events is suggested by the similar distribution of trimethylated histone H3K27 and NDY1 and BMI1 binding within the locus. Another observation supporting the interdependence is the gradual increase of the levels of H3K27me3 and of the NDY1 and BMI1 binding to the locus with each passage, which suggests a feed forward mechanism of histone modification and complex binding. Although questions remain, it appears that NDY1 coordinates several histone modifications and contributes to the fine tuning of transcriptional regulation (Tzatsos et al., 2009).
Fig. 10.

Schematic representation of the coordinated action of NDY1/KDM2B that leads to the repression of the Ink4a-Arf-Ink4b locus. NDY1/KDM2B represses the Ink4a-Arf locus by an elaborate mechanism. On the one hand, NDY1/KDM2B expression counteracts the senescence-associated downregulation of EZH2, resulting in elevated global H3K27me3 levels. Increased trimethylation of H3K27 is also observed specifically in the Ink4a-Arf locus and results in the recruitment of the Polycomb group protein BMI1 to the locus (Tzatsos et al., 2009). However, BMI1 recruitment alone cannot account for the immortalization phenotype, since knockdown of BMI1 in NDY1/KDM2B-immortalized cells caused only a partial reversion of the NDY1-mediated repression of the Ink4a-Arf locus. NDY1/KDM2B is at the center of an additional event that is essential for the immortalization phenotype. It is specifically recruited to the Ink4a-Arf-Ink4b locus, where it removes local H3K36me2 and H3K4me3 marks. This recruitment of NDY1/KDM2B is likely mediated by its association with PRC-type complexes. In Drosophila, the homolog of NDY2/KDM2A, dKDM2, is part of the Polycomb group complex dRAF. Except for dKDM2, dRAF also includes the histone H2A ubiquitin ligase dRING, and the Drosophila BMI1 homolog PSC. In mammalian cells, NDY1/KDM2B interacts with the ubiquitin ligase RING1B/RNF2, the human counterpart of dRING, and the mammalian Polycomb group proteins RING1A/RNF1, RYBP, and NSPC1/PCGF1 (Gearhart et al., 2006; Sanchez et al., 2007). Thus, the NDY1-induced upregulation of EZH2 and the trimethylation of H3K27 promote the binding of BMI1 to the Ink4a-Arf-Ink4b locus, which in turn recruits NDY1 as part of a PRC1-like complex that possibly also contains BMI1. The Drosophila homolog of NDY2/KDM2A, dKDM2, is involved in a coordinated mechanism of gene silencing, in which the removal of histone H3K36 dimethylation is coupled to histone H2A monoubiquitylation (Lagarou et al., 2008). It is possible that the repressive function of NDY1/KDM2B also involves the ubiquitination of H2A. (See Page 6 in Color Section at the back of the book.)
One of the histone modifications that appear to be regulated by NDY1 is the monoubiquitination of histone H2A. By extrapolation from the Drosophila data, NDY1 may play an obligatory role in this process in mammalian cells. The importance of this modification, however, for the regulation of the Ink4a-Arf-Ink4b locus and for MEF immortalization is uncertain. Assuming that the F-box of NDY1 is required for this activity, monoubiquitination of histone H2A at K119 should be dispensable for the regulation of Ink4a-Arf-Ink4b and for the immortalization phenotype, because the F-box is dispensable for both (Pfau et al., 2008).
iii. NDY1 Inhibits Cell Growth and Proliferation in HeLa Cells by JmjC-Dependent and -Independent Mechanisms
It was suggested in the preceding paragraphs that the subcellular distribution of NDY1 may differ between cell types and that the phenotype of NDY1 overexpression and knockdown may depend on its subcellular distribution in a given cell type. Studies in HeLa cells have indeed shown that NDY1 is localized in the nucleoli and that it binds the transcribed regions of ribosomal DNA to repress the transcription of ribosomal RNA genes via a JmjC-dependent process. Given that cell growth and proliferation depend on ribosomal biogenesis, repression of ribosomal RNA genes inhibits both cell growth and proliferation (Frescas et al., 2007).
In other studies, also in HeLa cells, it was shown that NDY1 binds c-Jun and that through this binding it is recruited to the c-jun promoter. The binding of the c-Jun/NDY1 complex inhibits c-jun transcription and cellular proliferation, as demonstrated by knockdown of NDY1 via RNA interference. The same was observed upon UV irradiation of HeLa cells, which also downregulates NDY1 expression. These data are unique in that they show that the NDY1-mediated repression of c-jun is JmjC-domain independent (Koyama-Nasu et al., 2007).
iv. NDY1 may Function Either as an Oncogene or as a Tumor Suppressor Gene
The functional activities of NDY1, which were presented in the preceding paragraphs, suggest that it may function either as an oncogene or as a tumor suppressor gene, depending on the cellular context. Favoring its prooncogenic role is its ability to inhibit senescence, which is a tumor protective process (Pfau et al., 2008; Polytarchou et al., 2008; Tzatsos et al., 2009). Favoring its tumor suppressing role are its ability to protect the genome against mutations (Polytarchou et al., 2008; Pothof et al., 2003; Suzuki et al., 2006) and its ability to inhibit the growth and proliferation of tumor cell lines in culture (Frescas et al., 2007; Koyama-Nasu et al., 2007). In agreement with its prooncogenic role, NDY1 is genetically modified by provirus integration and is overexpressed in retrovirus-induced lymphomas in rodents (Pfau et al., 2008; Polytarchou et al., 2008; Tzatsos et al., 2009). In addition it is overexpressed in human B and T acute lymphoblastic leukemias (T and B ALL), acute myeloid leukemias (AML), as well as in breast cancer and seminomas (Tzatsos et al., 2009). In agreement with its antioncogenic role, it is expressed at very low levels in aggressive glioblastomas (Frescas et al., 2007).
b. PHF2 and PHF8
PHF2 is ubiquitously expressed in adult tissues. However, during embryonic development in the mouse, it is expressed primarily in the neural tube and dorsal root ganglia (Hasenpusch-Theil et al., 1999). In human cancer, the PHF2 gene may be silenced either by deletions or by DNA methylation. Gene silencing correlates with poor patient survival in late-onset breast cancer (Sinha et al., 2008). The PHF2 protein contains a JmjC and a PHD finger domain (Fig. 11). However, sequence comparisons reveal that the JmjC domain of PHF2 lacks one of the residues essential for catalytic activity. Thus, it is likely that the PHF2 protein does not have histone demethylase activity. Evidence from other JmjC-domain-containing proteins that lack crucial catalytic residues suggests that an active catalytic domain is not a requirement for these proteins to impact chromatin structure and to regulate gene expression. For example, the yeast protein Epe1 profoundly restricts the spreading of heterochromatin in the MAT locus of the fission yeast (Ayoub et al., 2003). Also, the prototypical JmjC-domain protein Jumonji (JARID2), which also carries a JmjC domain that lacks demethylase activity, represses cyclin D1 gene expression by recruiting the histone methyltransferases G9a and GLP to the cyclin D1 promoter and promoting H3K9 methylation (Shirato et al., 2009).
Fig. 11.

Domain structure of PHF2 and PHF8.
PHF8 is expressed strongly in mouse embryonic and adult brain. Truncations or point mutations in the gene encoding PHF8 have been found in patients with X-linked mental retardation associated with cleft lip/palate (Abidi et al., 2007; Koivisto et al., 2007; Laumonnier et al., 2005; Qiao et al., 2008). The PHF8 protein contains a PHD finger and a JmjC domain (Fig. 11). Although the JmjC domain of PHF8 contains all the residues required for histone demethylase activity, its exact specificity has not yet been determined. Notably, most of the point mutations or deletions in patients with X-linked mental retardation and cleft lip/palate affect the JmjC domain.
c. JMJD5
JMJD5 belongs to the JmjC-domain-only family of histone demethylases. Although sequence analysis shows that the JmjC domain of JMJD5 contains all the residues required for histone demethylase activity, the exact substrate specificity of JMJD5 is still unknown. JMJD5 was identified by retrovirus-mediated insertional mutagenesis in Blm-deficient mice (Suzuki et al., 2006). Functionally, it appears to contribute to genomic stability in cultured cells and its depletion results in a mutator phenotype (Pothof et al., 2003; Suzuki et al., 2006).
d. JMJD1A/JHDM2A/KDM3A
JMJD1A/JHDM2A/KDM3A was first isolated as a gene that is highly expressed in male germ cells (Hoog et al., 1991). The encoded protein contains a JmjC domain and a zinc-finger motif (Fig. 12), and demethylates both H3K9me2 and H3K9me1, but not H3K9me3 (Yamane et al., 2006). JMJD1A/KDM3A regulates the expression of the genes encoding transition nuclear protein 1 (Tnp1) and protamine 1 (Prm1), two proteins that play an essential role in the packaging and condensation of sperm chromatin. As a result, mice deficient in JMJD1A/KDM3A exhibit postmeiotic chromatin condensation defects and are infertile (Okada et al., 2007). Other studies have shown that JMJD1A/KDM3A is induced by OCT4 and contributes to the maintenance of pluripotency in stem cells by epigenetically regulating the expression of Tcl1, Tcfcp211, and Zfp57, and perhaps other genes associated with pluripotency and self renewal in stem cells (Loh et al., 2007). Furthermore, JMJD1A/KDM3A interacts with the androgen receptor in a hormone-dependent manner, and promotes the transcription of androgen receptor target genes by removing local H3K9me2 marks (Yamane et al., 2006). Finally, JMJD1A/KDM3A is induced by hypoxia, both in vitro and in vivo, via the direct binding of HIF-1 to a specific response element in the promoter of the JMJD1A/KDM3A gene (Beyer et al., 2008; Pollard et al., 2008; Wellmann et al., 2008).
Fig. 12.

Domain structure of the JMJD1/JHDM2/KDM3 family.
2. HISTONE DEMETHYLASE GENES AMPLIFIED IN CANCER
a. GASC1/JMJD2C/JHDM3/KDM4C
GASC1/KDM4C was initially identified as a gene amplified in cell lines from esophageal squamous cell carcinomas (Yang et al., 2000). It belongs to the JMJD2/JHDM3/KDM4 family of histone demethylases, which consists of four members: JMJD2A/JHDM3/KDM4A, JMJD2B/JHDM3/KDM4B, GASC1/JMJD2C/JHDM3/KDM4C, and JMJD2D/JHDM3/KDM4D. The domain structure of these proteins is characterized by N-terminal JmjN and JmjC domains, two PHD fingers and two tandem Tudor domains (Fig. 13). The latter are known to recognize methylated histone peptides, and the Tudor domain of the GASC1/KDM4C-related histone demethylase JMJD2A/KDM4A recognizes histone H3K4me3 and histone H4K20me3 (Huang et al., 2006b; Lee et al., 2008). Although the structure of the JmjC domain of GASC1 has not been determined, elucidation of the structure of this domain in the related histone demethylase JMJD2A provided valuable insights into the binding of the JmjC domain with methylated histone peptides and into the molecular mechanism of catalysis (see Section II.E.2). Other studies showed that GASC1/KDM4C forms multimers with itself and JMJD2A/KDM4A, but not with JMJD2A/KDM4D (Shin and Janknecht, 2007). However, the physiological role of these multimers has not been determined.
Fig. 13.

Domain structure of the JMJD2/JHDM3/KDM4 family.
Several observations provide clues on the mechanism(s) by which GASC1/KDM4C may contribute to oncogenesis. First, recent evidence suggests that GASC1 may be involved in a cascade of events that contributes to the maintenance of pluripotency and self renewal in stem cells. Thus, OCT4-regulated GASC1/KDM4C promotes the expression of NANOG, by demethylating histone H3K9me3 and by interfering with the binding of HP1 and KAP1 to the NANOG promoter (Loh et al., 2007). GASC1/KDM4C, therefore, may contribute to oncogenesis by promoting the establishment of a stem cell renewal program in adult somatic cells. Other studies suggest that the oncogenic potential of GASC1/KDM4C may depend on its ability to promote genomic instability and transcriptional deregulation. Specifically, ectopic expression of GASC1, JMJD2A/KDM4A, or JMJD2B/KDM4B, both in primary and immortalized cells, abrogates the recruitment of all three isoforms of HP1 to heterochromatin (Cloos et al., 2006; Fodor et al., 2006; Klose et al., 2006b), suggesting a physiologically important role of this family of histone demethylases in heterochromatin assembly. Heterochromatin assembly represses transcription and contributes to the maintenance of genomic integrity (Kouzarides, 2007), suggesting that GASC1-amplification may promote genomic instability and transcriptional deregulation, both of which may contribute to oncogenesis.
i. GASC1/KDM4C Catalytic Activity and Specificity
The histone demethylase GASC1/KDM4C has a broad substrate specificity, targeting both H3K9me3/me2 and H3K36me3/me2, as well as H1.4me3 (Cloos et al., 2006; Trojer et al., 2009). The H3K9 demethylase activity of GASC1 explains its role in HP1 binding and suggests that it may function both as a transcriptional corepressor and a transcriptional coactivator. The latter is due to the context-dependent interpretation of the H3K9 methylation marks and the functional diversity of the HP1 proteins. Thus, if the H3K9me3/me2 mark is associated with the coding region of a transcribed gene, its removal may result in transcriptional repression. If on the other hand, the H3K9me3/me2 mark is associated with the promoter of a silenced euchromatic gene, its removal may result in transcriptional activation. The dual specificity of GASC1/KDM4C for both H3K36me3/me2 and H3K9me3/me2 may promote transcriptional repression by directing the coordinate removal of the H3K36me3/me2 and H3K9me3/me2 methylation marks, both of which promote transcriptional elongation when associated with the transcribed region of active genes.
Recently, it was shown that GASC1/KDM4C, and other members of the KDM4 histone demethylase family, also promote the demethylation of K26 trimethylated linker histone variant H1.4. GASC1/KDM4C converts the H1.4K26me3 mark to H1.4K26me2, while its relative JMJD2D/KDM4D converts both the H1.4K26me3 and H1.4K26me2 marks to H1.4K26me (Trojer et al., 2009). Histone H1 consists of seven somatic isotypes, all of which are involved in transcriptional regulation. The methylation of H1.4 by the histone methyltransferase G9a/KMT1C generates HP1 and L3MBTL1 recognition marks and contributes to transcriptional repression (Trojer et al., 2009).
The broad specificity of GASC1/KDM4C raises questions about substrate recognition by histone demethylases in general. Structural studies suggest that the recognition of the histone substrate depends on peptide conformation rather than on amino acid sequence (Chen et al., 2007b; Couture et al., 2007; Ng et al., 2007). Thus, a histone demethylase may have a broad target specificity because it could recognize multiple structurally compatible peptides. In addition to GASC1/KDM4C, other histone demethylases, such as LSD1/KDM1 (Metzger et al., 2005; Perillo et al., 2008; Shi et al., 2004) and NDY1/KDM2B (Frescas et al., 2007; Tsukada et al., 2006; Tzatsos et al., 2009), have also been reported to have multiple histone targets. In the case of LSD1/KDM1 the shift in specificity is cofactor dependent (Metzger et al., 2005; Perillo et al., 2008; Shi et al., 2004).
ii. Cross Talk of GASC1/KDM4C with Other Histone-Modifying Enzymes
GASC1/KDM4C cooperates with LSD1 and protein-kinase-C-related kinase 1 (PRK1) to demethylate histone H3 methylated at K9, in the promoters of androgen receptor-targeted genes. LSD1/KDM1 demethylates histone H3K9me2 and H3K9me, but not H3K9me3, which can be demethy-lated by GASC1. Recent studies have shown that GASC1/KDM4C interacts with LSD1 and with ligand-bound androgen receptor, and that all three proteins colocalize in the promoter region of androgen-regulated genes. The coordinated activities of LSD1/KDM1 and GASC1/KDM4C in these complexes result in the removal of all three methylation marks from K9 of histone H3. The functional significance of the interaction between LSD1 and GASC1/KDM4C is supported by experiments showing that the depletion of either protein interferes with the efficient transcription of androgen receptor-dependent genes (Wissmann et al., 2007).
PRK1 phosphorylates histone H3 at Thr11, following its recruitment to the promoter of androgen-responsive genes by ligand-bound androgen receptors (Metzger et al., 2008). Histone H3 phosphorylation at this site promoted histone demethylation by GASC1/KDM4C, which is also recruited to these promoters by binding to androgen receptor. Inhibition of PRK1 on the other hand, abrogated H3T11 phosphorylation and down-regulated androgen-induced demethylation of histone H3. This suggests that histone H3 phosphorylated at this site is a better substrate for GASC1/KDM4C than nonphosphorylated histone H3. This hypothesis is further supported by in vitro findings showing that nucleosomes phosphorylated at Thr11 of histone H3 by PRK1 are better substrates for GASC1/KDM4C than unmodified nucleosomes (Metzger et al., 2008).
The preceding data raise the question whether the GASC1/KDM4C histone demethylase, or other members of this histone demethylase family, cooperate with other histone modifying enzymes. For example, one could envisage that GASC1 may interact with additional histone H3 demethylases, expanding their specificities.
3. HISTONE DEMETHYLASE GENES GENETICALLY INACTIVATED IN CANCER
a. JHDM2B/5qNCA/KDM3B
Interstitial deletion of chromosome 5q (Hu et al., 2001) gives rise to a distinct myelodysplastic syndrome, the 5q-syndrome, which is characterized by thrombocytosis, macrocytic anemia, and hypolobulated megakaryocytes. Like other myelodysplastic syndromes, the 5q-syndrome usually progresses into AML. Recent studies revealed that the gene whose deletion may be responsible for this syndrome is the gene encoding the ribosomal protein RPS14 (Gondek et al., 2008). However, del(5q) deletes different sets of genes in different patients, in addition to the gene encoding RPS14. Some of these genes may contribute to the 5q-phenotype, including its progression to leukemia. One of these genes encodes the histone H3 demethylase JHDM2B/KDM3B which belongs to the KDM3 demethylase family (Fig. 12). Members of this family demethylate histone H3 methylated at K9 (Klose et al., 2006a; Yamane et al., 2006). However, whereas JHDM2A/JMJD1A/KDM3A, another member of the family, is known to demethylate H3K9me2 and H3K9me1, but not H3K9me3 (Yamane et al., 2006), the specificity of JHDM2B/KDM3B has not been defined further. The biological importance of the deletion of JHDM2B/KDM3B has been confirmed by experiments showing that ectopic expression of JHDM2B/KDM3B in cell lines derived from leukemias arising in 5q-patients, inhibits their clonogenic growth (Hu et al., 2001).
B. Histone Demethylase Genes Overexpressed in Cancer
1. LSD1/KDM1
LSD1/KDM1 has been shown to function as a nuclear cofactor, which interacts with the androgen receptor and stimulates androgen receptor-dependent transcription both in the normal prostate and in prostate tumors (Metzger et al., 2005). More importantly, high levels of nuclear LSD1 in organ-confined prostate adenocarcinomas correlate with relapse after radical prostatectomy (Kahl et al., 2006). Locally confined prostate tumors may have low or high risk for relapse after surgical intervention. LSD1 therefore may serve as a marker for the prediction of the clinical outcome of these tumors. The preceding data also suggest that LSD1 may be a good molecular target for prostate cancer. Small-molecule inhibitors for this enzyme (Huang et al., 2007b; Lee et al., 2006; Schmidt and McCafferty, 2007) may allow us to address this question in the near future.
a. Catalytic Activity and Substrate Specificity of LSD1/KDM1
Both the activity and substrate specificity of LSD1/KDM1 are controlled by protein–protein interactions and they are context dependent. The histone demethylase activity of LSD1 depends on the corepressor CoREST, which renders nucleosomal substrates accessible to the enzyme and stabilizes and protects LSD1 from proteasomal degradation in vivo (Lee et al., 2005b; Shi et al., 2005). The PHD-domain protein BHC80, on the other hand, inhibits CoREST/LSD1-mediated demethylation in vitro and may, therefore, be a negative regulator of the demethylation reaction in vivo (Metzger et al., 2005).
The interaction of LSD1 with the androgen receptor alters the substrate specificity of LSD1. Thus, it was shown that in the presence of the androgen receptor, LSD1 demethylates histone H3 methylated at K9, but has no effect on the methylation status of histone H3 methylated at K4. This results in the derepression of androgen receptor target genes (Metzger et al., 2005). The substrate specificity of LSD1 is similarly altered by the estrogen receptor. As a result, LSD1 also contributes to the induction of estrogen-responsive genes (Perillo et al., 2008). Although the molecular details that dictate this shift in specificity remain to be determined, these findings suggest a potentially general regulatory strategy where LSD1/KDM1 and other demethylases can expand their substrate repertoire by participating in distinct complexes.
Histone demethylation by LSD1/KDM1 is also regulated by other histone modifications (Forneris et al., 2005). Thus, the acetylation, but not the methylation, of histone H3 at K9 downregulates LSD1-promoted demethylation of H3K4me2 by reducing the binding of LSD1 to the peptide substrate. Similarly, phosphorylation of histone H3 at serine 10 abolishes LSD1-mediated H3K4 demethylation (Forneris et al., 2005).
b. DNA Modifications Regulated by LSD1/KDM1
LSD1 contributes to the regulation of DNA methylation by at least two distinct mechanisms. The first mechanism is indirect. Here, LSD1 demethylates histone H3K4me3 and facilitates the recruitment of the DNA methyltransferase regulator DNMT3L, which recognizes histone H3 tails unmethylated at K4. This results in the recruitment or activation of the DNA methyltransferase DNMT3A2, which promotes de novo DNA methylation (Ooi et al., 2007). The second mechanism is more direct. Here, LSD1 demethylates the DNA methyltransferase DNMT1, which is methylated by the histone methyltransferase Set7/9 (KMT7). Demethylation stabilizes DNMT1 and stimulates DNA methylation (Wang et al., 2009). The role of LSD1 in DNA methylation was confirmed by experiments showing that the deletion of LSD1 in embryonic stem cells results in reduced stability and downregulation of DNMT1 and in the progressive loss of DNA methylation (Wang et al., 2009).
LSD1 also promotes DNA oxidation (Perillo et al., 2008). Recent studies have shown that demethylation of H3K9me2 by LSD1 in the promoter region of estrogen receptor-targeted genes, produces hydrogen peroxide that oxidizes the surrounding DNA, mainly at guanine bases. This results in the recruitment of 8-oxoguanine–DNA glycosylase 1 and topoisomerase IIβ to the region. The topoisomerase relaxes the DNA and allows chromatin to bend, thus facilitating the contact of the estrogen receptor with RNA polymerase II (Perillo et al., 2008). This ingenious use of what appeared to be a “reaction by-product” underlines the economy and complexity of biological systems and raises questions about the potential role of the highly reactive formaldehyde that is produced by the JmjC-domain-containing histone demethylases.
c. Regulation of Oncogenes and Tumor Suppressor Genes by LSD1/KDM1
The role of LSD1 in tumorigenesis is underscored by its specific interaction with known oncogenes and tumor suppressors. The tumor suppressor protein p53 is maintained at a low level in unstimulated cells, but becomes rapidly stabilized and activated in response to DNA damage and other types of cellular stress. Depletion of LSD1/KDM1 resulted in a delay in the DNA damage-induced stabilization of p53 (Scoumanne and Chen, 2007). The stability and activity of p53 are regulated through multiple posttranslational modifications, including lysine methylation (Scoumanne and Chen, 2008). The connection between LSD1/KDM1 depletion and p53 stabilization became clear with the discovery that LSD1 also demethylates p53. LSD1 interacts with p53 and removes both monomethylation and dimethylation at K370, resulting in its stabilization (Huang et al., 2007a). In an additional aspect of the p53-LSD1 interplay, LSD1 cooperates with p53 in the repression of the tumor-marker alpha-fetoprotein (AFP). p53 targets LSD1 to specific p53-response elements to mediate transcriptional repression. In mice lacking p53, the binding of LSD1 to the AFP locus is inhibited, resulting in increased levels of H3K4 dimethylation in the AFP promoter (Tsai et al., 2008).
The human telomerase reverse transcriptase protein (hTERT) plays a central role in cellular immortalization and transformation. The gene encoding for hTERT is silent in most normal human cells but it is activated in the vast majority of human cancers. LSD1 was found to play a direct role in the repression of the hTERT gene in both normal and cancer cells (Zhu et al., 2008). LSD1 removed local H3K4 methylation and cooperated with histone deacetylases to stably repress the hTERT gene, while inhibition of LSD1 resulted in the significant upregulation of hTERT.
2. PLU-1/KDM5B
The gene encoding the histone demethylase PLU-1/KDM5B was initially identified as a gene whose expression is restricted in normal adult tissues but is consistently expressed in human breast and testicular cancer (Barrett et al., 2002; Lu et al., 1999). More recent studies have also linked PLU-1/KDM5B to prostate cancer (Xiang et al., 2007). In agreement with the evidence that it is involved in mammary oncogenesis, knockdown of PLU-1/KDM5B in MCF-7 breast cancer cells resulted in growth inhibition and in the derepression of several negative regulators of cell growth, including 14–3–3σ, BRCA1, CAV1, and HOXA5. ChIP experiments confirmed that PLU-1/KDM5B associates with evolutionarily conserved target sites in the promoter regions of these genes, with the exception of 14–3–3σ. Knockdown of PLU-1/KDM5B reduced the PLU-1/KDM5B occupancy of the target sites (Yamane et al., 2007). Further support to the function of PLU-1/KDM5B as an oncogene comes from experiments revealing its direct interaction with the protooncogene MYC, which is a strong activator of cell growth and cell cycle progression (Secombe and Eisenman, 2007). The human PLU-1/KDM5B homolog RBP2/KDM5A, and the Drosophila JARID1/KDM5 homolog Little imaginal discs (Lid), also interact with MYC, suggesting that this interaction is evolutionarily conserved. In Drosophila, Lid functions as a transcriptional coactivator of MYC and it is required for the transcription of the MYC-induced growth regulatory gene Nop60B. Interestingly, MYC inhibits the histone demethylase activity of Lid, suggesting that Lid does not function as a histone demethylase when it cooperates with MYC in activating gene expression (Secombe and Eisenman, 2007).
PLU-1/KDM5B is a member of the JARID1/KDM5 family of histone demethylases that in humans consists of four members: JARID1A/RBP2/KDM5A, JARID1B/PLU-1/KDM5B, JARID1C/SMCX/KDM5C, and JAR-ID1D/SMCY/KDM5D (Table I and Fig. 14). The proteins that belong to this family, apart from the catalytic JmjC domain, also contain a JmjN domain, a BRIGHT or ARID (AT-rich interactive) domain, a C5HC2 zinc-finger, and two or three PHD-fingers (Fig. 14). ARID domains bind DNA (Kortschak et al., 2000; Wilsker et al., 2002), and PHD fingers are protein–protein interaction domains, some of which bind methylated histone tails (Iwase et al., 2007; Shi et al., 2006; Taverna et al., 2007; Vermeulen et al., 2007; Wysocka et al., 2006b). The function of the C5HC2 zinc-fingers is still unknown; however, a fragment of Lid that contains the C5HC2 zinc-finger binds strongly to dMYC (Secombe and Eisenman, 2007).
Fig. 14.

Domain structure of the JARID1/KDM5 family.
a. Enzymatic Activity, Substrate Specificity, and Targeting of PLU-1/KDM5B
PLU-1/KDM5B, and other members of this histone demethylase family, demethylate histone H3 peptides that are tri- or dimethylated at K4. The same enzyme targets peptides monomethylated at this site and demethylates them in vivo, but not in vitro, suggesting that its in vivo activity may depend on factors that are absent from the in vitro reaction (Yamane et al., 2007). Other demethylases that target histone H3 methylated at K4 include LSD1/KDM1 and NDY1/KDM2B. LSD1/KDM1, however, cannot remove the H3K4me3 mark because it requires a protonated nitrogen in the substrate. Thus, the JARID1/KDM5 family members, and NDY1/KDM2B, may complement LSD1/KDM1 for the removal of all three H3K4 methylation marks. The potential complementation among these demethylases may play an essential role in the regulation of gene expression.
The genomic targeting of PLU-1/KDM5B, and other members of the JARID1/KDM5 family of histone demethylases, is mediated by sequence-specific DNA binding, which is ARID domain dependent, and by binding to posttranslationally modified histones, which is PHD domain dependent. The consensus binding site for the ARID domain of PLU-1/KDM5B has the sequence GCACA/C. This was demonstrated via PCR-assisted selection of PLU-1 binding oligonucleotides from a random oligonucleotide library. The GCACA/C sequence was present in the promoter region of PLU-1/KDM5B target genes. PLU-1/KDM5B binding to this site in the promoter of the genes encoding metallothionein 1H, 1F, and 1X (MT1H, MT1F, and MT1X) was confirmed by chromatin immunoprecipitation (ChIP). The same ChIP experiments demonstrated a correlation between PLU-1/KDM5B binding and downregulation of the histone H3K4me3 mark in the vicinity of the PLU-1/KDM5B-binding site (Scibetta et al., 2007).
The two C-terminal PHD domains of PLU-1/KDM5B do not play an essential role in chromatin recognition, since their removal did not affect PLU-1 demethylase activity in vivo (Yamane et al., 2007). Instead, these two PHD domains are essential for the association of PLU-1/KDM5B with class I and class IIa histone deacetylases (Barrett et al., 2007). Given that HDAC4 and PLU-1/KDM5B are coordinately expressed in breast cancer and in mammary epithelia, differentiating in the course of pregnancy involution and lactation, the PHD finger-dependent interaction between PLU-1/KDM5B and HDAC4 is likely to be physiologically relevant.
Clues on the functional role of the N-terminal PHD finger of PLU-1/KDM5B can be obtained from studies on the mode of chromatin recognition by the related histone demethylase SMCX/KDM5C. SMCX/KDM5C contains two PHD domains. PHD1 is analogous to the N-terminal PHD domain of PLU-1, while PHD2 corresponds to one of the PLU-1 C-terminal PHD domains (Fig. 14). Recombinant PHD1, but not PHD2, binds preferentially to histone H3K9me3 (Iwase et al., 2007), suggesting that the corresponding PHD domain of PLU-1/KDM5B may also recognize H3K9me3. The binding of KDM5 PHD fingers to K9-methylated histone H3 may direct KDM5 proteins to their genomic targets, where the cross talk between histone H3K4 demethylation and H3K9 trimethylation may coordinate the establishment of a repressive chromatin state. The phenotype of SMCX/KDM5C mutations in humans provides strong support to the hypothesis that the binding of its PHD1 finger to H3K9me3 in target genes serves a biologically important function. Mutations of this gene cause severe X-linked mental retardation, short stature, and hyperreflexia (Abidi et al., 2008; Jensen et al., 2005; Santos et al., 2006; Tzschach et al., 2006). Several of these mutations result in SMCX/KDM5C enzymes with either compromised demethylase activity or low-histone binding affinity (Iwase et al., 2007; Tahiliani et al., 2007). Importantly, one such PHD1 finger mutation (A388P) reduced both the binding of PHD1 to H3K9me3 and the demethylation activity of this protein (Iwase et al., 2007).
3. MINA53/NO52
MINA53 (MYC-induced nuclear antigen of 53 kDa), a direct target of MYC (Tsuneoka et al., 2002), is a member of the JmjC-domain-only protein family. Although it is a likely histone demethylase, its substrate specificity is currently unknown. MINA53/NO52 appears to have a physiological role in ribosomal biogenesis, as it was found to localize in the granular component of nucleoli and in free preribosomal particles but not on cytoplasmic ribosomes (Eilbracht et al., 2005). Its induction by MYC and its apparent contribution to ribosomal biogenesis, suggest that MINA53 may function as an oncogene. Clinical studies have indeed shown that MINA53 is overexpressed in several types of human cancer including, colorectal cancer (Teye et al., 2004), esoph-ageal squamous cell carcinoma (Tsuneoka et al., 2004), primary gingival squamous cell carcinoma (Kuratomi et al., 2006), neuroblastoma (Fukahori et al., 2007), renal cell carcinoma (Ishizaki et al., 2007), lymphoma (Teye et al., 2007), and gastric carcinoma (Zhang et al., 2008). More important, knockdown of MINA53 in human promyelocytic leukemia cells severely suppresses cell proliferation (Tsuneoka et al., 2002).
4. MAPJD/NO66
Myc-associated protein with JmjC domain (MAPJD) is a MINA53-related protein that is upregulated in the majority of cases of nonsmall cell lung cancer (NSCLC) (Suzuki et al., 2007). Like MINA53, MAPJD/NO66 is localized in the granular part of nucleoli, and may contribute to ribosomal biogenesis. In addition, however, it is localized in nucleoplasmic entities associated with late replicating chromatin (Eilbracht et al., 2004). Another cancer relevant property of MAPJD/NO66, also suggested by its name, is its interaction with c-MYC. Through this interaction, it may contribute to the regulation of MYC-target genes (Suzuki et al., 2007). The preceding data suggest that MAPJD/NO66 functions as an oncogene. This hypothesis is supported by findings showing that overexpression of this protein in NIH3T3 cells promotes cellular proliferation, while its knockdown in high expressor NSCLC lines, suppresses proliferation (Suzuki et al., 2007).
C. Histone Demethylases that Functionally Interact with Known Oncogenes or Tumor Suppressors
1. RBP2/KDM5A
RBP2/KDM5A belongs to the KDM5 or JARID1 family of histone demethylases, together with PLU-1/KDM5B, SMCX/KDM5C, and SMCY/KDM5D (Fig. 14). Its domain structure, enzymatic activity, and substrate specificity (Klose et al., 2007) are similar to those of other members of this family, including PLU-1, which was discussed in the preceding paragraphs. The RBP2 genomic targeting, similar to that of PLU-1, is mediated by ARID domain-dependent sequence-specific DNA binding and, possibly, PHD domain-dependent binding to posttranslationally modified histones. The consensus DNA-binding motif of RBP2 was shown to have the sequence CCGCCC (Tu et al., 2008).
The structure of the ARID domain of RBP2/KDM5A was elucidated by NMR spectroscopy and the molecular determinants of DNA recognition were confirmed by site-directed mutagenesis (Tu et al., 2008). The N-terminal PHD finger may recognize methylated H3K9 (as in SMCX/KDM5C), while the two C-terminal PHD fingers may bind histone deace-tylases (as in PLU-1/KDM5B). In agreement with this hypothesis, RBP2 binds HDAC1 in the context of the multisubunit SIN3 corepressor complex (Hayakawa et al., 2007). In mammalian cells, the SIN3 complex consists of at least eight subunits, including HDAC1, E2F4, and members of the SWI/SNF-remodeling group (Hassig et al., 1997). The activity of SIN3 and RBP2 on a subset of E2F4 target genes leads to their permanent repression by coordinated histone deacetylation and demethylation, and by repositioning of nucleosomes (van Oevelen et al., 2008).
RBP2/KDM5A was originally isolated in a screen for proteins that interact with the retinoblastoma protein (pRB) (Defeo-Jones et al., 1991). Recent studies have shown that the relationship between pRB and RBP2 is complex, but informative for the function of both. Experimental modulation of the expression of these proteins in osteoblast and myeloid differentiation models showed that their effects on cellular proliferation and differentiation are diametrically opposite, with RBP2 promoting proliferation and inhibiting differentiation. This conclusion is supported by findings showing that the knockdown of RBP2 phenocopies the effects of pRB expression, on cellular proliferation, differentiation, and gene expression (Benevolenskaya et al., 2005; Lopez-Bigas et al., 2008). The cause of the opposing effects of RBP2 and pRB is that, by binding to RBP2, pRB inhibits the RBP2-mediated transcriptional regulation of differentiation and cell cycle regulatory genes. Alternatively, RBP2 inhibits the expression of differentiation-specific genes, which are induced by pRB via E2F-independent transcription. An example of the latter mechanism is the activation of differentiation genes by pRB/RUNX2 complexes in differentiating osteoblasts, which is inhibited by RBP2 (Gutierrez et al., 2005). Although the interaction between pRB and RBP2 may play a central role in RBP2 function, recent studies suggest that RBP2 also functions as a pRB-independent transcriptional regulator, whose activity depends on interaction with other cofactors. One such cofactor may be PAX-6 (Lopez-Bigas et al., 2008; Tan et al., 2003). Another cofactor may be MYC. RBP2/KDM5A, like PLU-1/KDM5B, interacts directly with MYC and appears to be required for the transcription of MYC-induced genes. This function of RBP2/KDM5A appears to be independent of its histone demethylase activity (Secombe and Eisenman, 2007).
Knocking out RBP2/KDM5A in mice resulted in behavioral abnormalities and neutrophilia. Analysis of the hematopoietic stem cell and myeloid progenitor compartments in these mice, revealed a decrease in the rate of apoptosis and stimulation of cell cycle progression (Klose et al., 2007). Genome wide location analysis and gene expression profiling in U937 cells, before and after induction of differentiation with TPA, showed that RBP2 targets genes whose promoters contain histone H3K4me3. These genes belong to two functionally distinct classes. The first class is targeted by RBP2 in both differentiated and undifferentiated cells and is rich in genes that encode mitochondrial proteins, suggesting that RBP2 contributes to mitochondrial biogenesis. The second class, which is rich in cell cycle genes, is targeted by RBP2 upon differentiation (Lopez-Bigas et al., 2008). Genome-wide location analysis in mouse embryonic stem cells, revealed an association of RBP2/KDM5A with a large number of Polycomb group (PcG) target genes (Pasini et al., 2008). The recruitment of RBP2 to these genes is due to its interaction with the PRC2 which is responsible for the trimethylation of histone H3 at K27. The interaction of RBP2 with the PRC2 complex is required for the repression of developmental genes by PRC2 during mouse embryonic stem cell differentiation (Pasini et al., 2008).
The ability of RBP2 to inhibit apoptosis, stimulate cell cycle progression, and inhibit differentiation, suggests that, when overexpressed, RBP2 may function as an oncogene. Its antagonistic interaction with pRB, one of the best characterized tumor suppressors, strongly supports this hypothesis. Importantly, the association of RBP2/KDM5A with the protooncogene MYC, which regulates cell growth and cell cycle progression, and their coordinated function in gene activation (Secombe and Eisenman, 2007), further supports the oncogenic role of RBP2.
In addition to RBP2/KDM5A and PLU-1/KDM5B, two other histone demethylases, MINA53 and MAPJD, have also been implicated in MYC function. Thus, several histone demethylases regulate cell proliferation by functioning as transcriptional cofactors for MYC.
IV. HISTONE DEMETHYLASES AS THERAPEUTIC TARGETS
The central role of epigenetic abnormalities in tumor evolution has prompted efforts for the development of therapeutic approaches that aim to reverse epigenetic changes. The cytosine analogs 5-azacytosine (azacytidine) and 2-deoxy-5-azacytidine (decitabine) are currently the most advanced drugs for epigenetic cancer therapies. These DNA methyltransferase inhibitors have shown remarkable efficiency in the derepression of epigenetically silenced tumor suppressor genes (Constantinides et al., 1977; Santi et al., 1983). Following the success of DNA methyltransferase inhibitors, the antitumor potential of another class of proteins involved in epigenetic regulation, that of histone deacetylases, has received significant attention. A series of histone deacetylase inhibitors has been developed and several first-generation compounds are currently being tested in phase I/II clinical trials (Bolden et al., 2006; Minucci and Pelicci, 2006).
With inhibitors for other main components of the epigenetic machinery already showing clinical promise, histone demethylases are emerging as new possible drug targets. Several lines of evidence suggest that histone demethylases may indeed be good candidates for the development of epigenetic cancer therapies. PLU-1/KDM5B is implicated in breast cancer and its depletion from MCF-7 breast cancer cells resulted in growth inhibition and promoted the derepression of several genes (Yamane et al., 2007). Knockdown of GASC1, which is amplified in squamous cell carcinoma, inhibited the proliferation of two esophageal carcinoma cell lines (Cloos et al., 2006). Moreover, depletion of MINA53 in human promyelocytic leukemia cells severely suppressed their proliferation (Tsuneoka et al., 2002), while knockdown of MAPJD by RNAi effectively suppressed the growth of nonsmall cell lung cancer cells (Suzuki et al., 2007). Finally, the knockdown of NDY1 and the ablation of exogenous NDY1 in MEFs, downregulated the expression of EZH2, induced the expression of p16INK4A and promoted senescence (Pfau et al., 2008; Tsukada et al., 2006; Tzatsos et al., 2009).
LSD1/KDM1 is a member of the FAD-dependent amine oxidases, a group of enzymes that includes both polyamine and monoamine oxidases. Several known inhibitors for monoamine and polyamine oxidases exist, such as the clinically used antidepressant trans-2-phenylcyclopropylamine (tranylcypromine) and several polyamine analogs. When these compounds were tested against LSD1, they were found to efficiently inhibit its histone demethylase activity (Huang et al., 2007b; Lee et al., 2006; Schmidt and McCafferty, 2007). Treatment of P19 embryonic carcinoma cells with tranylcypromine resulted in a global increase in H3K4 methylation, as well as in the transcriptional derepression of two LSD1 target genes, EGR1 and the pluripotent stem cell marker OCT4 (Lee et al., 2006). Furthermore, biguanide and bisguanidine polyamine analogs inhibit LSD1 in human colon carcinoma cells and cause the reexpression of several aberrantly silenced genes implicated in the development of colon cancer, including members of the GATA and SFRP (secreted frizzle-related proteins) families (Huang et al., 2007b). Thus, histone demethylase inhibitors, either alone or in combination with histone deacetylase or DNA methyltransferase inhibitors, may prove to be effective epigenetic therapeutic agents.
V. CONCLUDING REMARKS
Cancer is a genetic disease. Emerging evidence, however, is challenging this concept by showing that nongenetic heritable changes affecting chromatin structure and gene expression may play an equally important role in the biology of the cancer cell. Based on this evidence, tumor development and evolution depends on the combination of genetic and epigenetic changes. The two of course are not independent. Thus, epigenetic silencing of genes involved in the response to DNA damage may give rise to genetic instability and the accumulation of genetic changes in cancer cells. Genetic changes affecting genes involved in epigenetic regulation, on the other hand, may alter the epigenome of the cancer cell.
In this review, we have focused on one type of epigenetic regulators, namely the enzymes that regulate the demethylation of methylated histones. However, it is clear that epigenetic mechanisms are interdependent and integrated into the regulatory machinery of the cell. Epigenetic regulators can be modified by the complex signaling pathways that become deregulated in the cancer cell. Future studies on the cancer epigenome and its regulation will redefine our understanding of the cancer cell and may reveal new targets for therapeutic intervention.
Acknowledgments
Work on histone demethylases in the PNT laboratory is supported by National Institutes of Health Grant R01CA109747.
References
- Abidi FE, Miano MG, Murray JC, Schwartz CE. A novel mutation in the PHF8 gene is associated with X-linked mental retardation with cleft lip/cleft palate. Clin Genet. 2007;72:19–22. doi: 10.1111/j.1399-0004.2007.00817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abidi FE, Holloway L, Moore CA, Weaver DD, Simensen RJ, Stevenson RE, Rogers RC, Schwartz CE. Mutations in JARID1C are associated with X-linked mental retardation, short stature and hyperreflexia. J Med Genet. 2008;45:787–793. doi: 10.1136/jmg.2008.058990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449:731–734. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- Akahira J, Suzuki F, Suzuki T, Miura I, Kamogawa N, Miki Y, Ito K, Yaegashi N, Sasano H. Decreased expression of RIZ1 and its clinicopathological significance in epithelial ovarian carcinoma: Correlation with epigenetic inactivation by aberrant DNA methylation. Pathol Int. 2007;57:725–733. doi: 10.1111/j.1440-1827.2007.02169.x. [DOI] [PubMed] [Google Scholar]
- Ancelin K, Lange UC, Hajkova P, Schneider R, Bannister AJ, Kouzarides T, Surani MA. Blimp1 associates with Prmt5 and directs histone arginine methylation in mouse germ cells. Nat Cell Biol. 2006;8:623–630. doi: 10.1038/ncb1413. [DOI] [PubMed] [Google Scholar]
- Annunziato AT, Eason MB, Perry CA. Relationship between methylation and acetylation of arginine-rich histones in cycling and arrested HeLa cells. Biochemistry. 1995;34:2916–2924. doi: 10.1021/bi00009a023. [DOI] [PubMed] [Google Scholar]
- Ayoub N, Noma K, Isaac S, Kahan T, Grewal SI, Cohen A. A novel jmjC domain protein modulates heterochromatization in fission yeast. Mol Cell Biol. 2003;23:4356–4370. doi: 10.1128/MCB.23.12.4356-4370.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann N, Hoegel J, Haeusler J, Kuefer R, Herkommer K, Paiss T, Vogel W, Maier C. Mutation screen and association study of EZH2 as a susceptibility gene for aggressive prostate cancer. Prostate. 2005;65:252–259. doi: 10.1002/pros.20296. [DOI] [PubMed] [Google Scholar]
- Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, Salvesen HB, Otte AP, Akslen LA. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24:268–273. doi: 10.1200/JCO.2005.01.5180. [DOI] [PubMed] [Google Scholar]
- Balakrishnan A, Bleeker FE, Lamba S, Rodolfo M, Daniotti M, Scarpa A, van Tilborg AA, Leenstra S, Zanon C, Bardelli A. Novel somatic and germline mutations in cancer candidate genes in glioblastoma, melanoma, and pancreatic carcinoma. Cancer Res. 2007;67:3545–3550. doi: 10.1158/0008-5472.CAN-07-0065. [DOI] [PubMed] [Google Scholar]
- Balciunas D, Ronne H. Evidence of domain swapping within the jumonji family of transcription factors. Trends Biochem Sci. 2000;25:274–276. doi: 10.1016/s0968-0004(00)01593-0. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Schneider R, Kouzarides T. Histone methylation: Dynamic or static? Cell. 2002;109:801–806. doi: 10.1016/s0092-8674(02)00798-5. [DOI] [PubMed] [Google Scholar]
- Barrett A, Madsen B, Copier J, Lu PJ, Cooper L, Scibetta AG, Burchell J, Taylor-Papadimitriou J. PLU-1 nuclear protein, which is upregulated in breast cancer, shows restricted expression in normal human adult tissues: A new cancer/testis antigen? Int J Cancer. 2002;101:581–588. doi: 10.1002/ijc.10644. [DOI] [PubMed] [Google Scholar]
- Barrett A, Santangelo S, Tan K, Catchpole S, Roberts K, Spencer-Dene B, Hall D, Scibetta A, Burchell J, Verdin E, Freemont P, Taylor-Papadimitriou J. Breast cancer associated transcriptional repressor PLU-1/JARID1B interacts directly with histone deacetylases. Int J Cancer. 2007;121:265–275. doi: 10.1002/ijc.22673. [DOI] [PubMed] [Google Scholar]
- Bedford MT, Richard S. Arginine methylation an emerging regulator of protein function. Mol Cell. 2005;18:263–272. doi: 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Benevolenskaya EV, Murray HL, Branton P, Young RA, Kaelin WG., Jr Binding of pRB to the PHD protein RBP2 promotes cellular differentiation. Mol Cell. 2005;18:623–635. doi: 10.1016/j.molcel.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Beyer S, Kristensen MM, Jensen KS, Johansen JV, Staller P. The histone demethylases JMJD1A and JMJD2B are transcriptional targets of hypoxia-inducible factor HIF. J Biol Chem. 2008;283:36542–36552. doi: 10.1074/jbc.M804578200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol. 2007;14:1008–1016. doi: 10.1038/nsmb1337. [DOI] [PubMed] [Google Scholar]
- Blackburn EH. Telomere states and cell fates. Nature. 2000;408:53–56. doi: 10.1038/35040500. [DOI] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Borges M, Linnoila RI, van de Velde HJ, Chen H, Nelkin BD, Mabry M, Baylin SB, Ball DW. An achaete-scute homologue essential for neuroendocrine differentiation in the lung. Nature. 1997;386:852–855. doi: 10.1038/386852a0. [DOI] [PubMed] [Google Scholar]
- Borun TW, Pearson D, Paik WK. Studies of histone methylation during the HeLa S-3 cell cycle. J Biol Chem. 1972;247:4288–4298. [PubMed] [Google Scholar]
- Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bres V, Yoh SM, Jones KA. The multi-tasking P-TEFb complex. Curr Opin Cell Biol. 2008;20:334–340. doi: 10.1016/j.ceb.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant RJ, Cross NA, Eaton CL, Hamdy FC, Cunliffe VT. EZH2 promotes proliferation and invasiveness of prostate cancer cells. Prostate. 2007;67:547–556. doi: 10.1002/pros.20550. [DOI] [PubMed] [Google Scholar]
- Bushman F, Lewinski M, Ciuffi A, Barr S, Leipzig J, Hannenhalli S, Hoffmann C. Genome-wide analysis of retroviral DNA integration. Nat Rev Microbiol. 2005;3:848–858. doi: 10.1038/nrmicro1263. [DOI] [PubMed] [Google Scholar]
- Byvoet P, Shepherd GR, Hardin JM, Noland BJ. The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Arch Biochem Biophys. 1972;148:558–567. doi: 10.1016/0003-9861(72)90174-9. [DOI] [PubMed] [Google Scholar]
- Cao R, Zhang Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev. 2004;14:155–164. doi: 10.1016/j.gde.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Cao R, Tsukada Y, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell. 2005;20:845–854. doi: 10.1016/j.molcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Carling T, Kim KC, Yang XH, Gu J, Zhang XK, Huang S. A histone methyltransferase is required for maximal response to female sex hormones. Mol Cell Biol. 2004;24:7032–7042. doi: 10.1128/MCB.24.16.7032-7042.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, Workman JL. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter BT, Aswad DW, Stallcup MR. Regulation of transcription by a protein methyltransferase. Science. 1999;284:2174–2177. doi: 10.1126/science.284.5423.2174. [DOI] [PubMed] [Google Scholar]
- Chen Y, Yang Y, Wang F, Wan K, Yamane K, Zhang Y, Lei M. Crystal structure of human histone lysine-specific demethylase 1 (LSD1) Proc Natl Acad Sci USA. 2006a;103:13956–13961. doi: 10.1073/pnas.0606381103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zang J, Whetstine J, Hong X, Davrazou F, Kutateladze TG, Simpson M, Mao Q, Pan CH, Dai S, Hagman J, Hansen K, et al. Structural insights into histone demethylation by JMJD2 family members. Cell. 2006b;125:691–702. doi: 10.1016/j.cell.2006.04.024. [DOI] [PubMed] [Google Scholar]
- Chen LB, Xu JY, Yang Z, Wang GB. Silencing SMYD3 in hepatoma demethylates RIZI promoter induces apoptosis and inhibits cell proliferation and migration. World J Gastroenterol. 2007a;13:5718–5724. doi: 10.3748/wjg.v13.i43.5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zang J, Kappler J, Hong X, Crawford F, Wang Q, Lan F, Jiang C, Whetstine J, Dai S, Hansen K, Shi Y, et al. Structural basis of the recognition of a methylated histone tail by JMJD2A. Proc Natl Acad Sci USA. 2007b;104:10818–10823. doi: 10.1073/pnas.0704525104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung N, Chan LC, Thompson A, Cleary ML, So CW. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol. 2007;9:1208–1215. doi: 10.1038/ncb1642. [DOI] [PubMed] [Google Scholar]
- Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Constantinides PG, Jones PA, Gevers W. Functional striated muscle cells from non-myoblast precursors following 5-azacytidine treatment. Nature. 1977;267:364–366. doi: 10.1038/267364a0. [DOI] [PubMed] [Google Scholar]
- Couture JF, Collazo E, Brunzelle JS, Trievel RC. Structural and functional analysis of SET8, a histone H4 Lys-20 methyltransferase. Genes Dev. 2005;19:1455–1465. doi: 10.1101/gad.1318405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couture JF, Collazo E, Ortiz-Tello PA, Brunzelle JS, Trievel RC. Specificity and mechanism of JMJD2A, a trimethyllysine-specific histone demethylase. Nat Struct Mol Biol. 2007;14:689–695. doi: 10.1038/nsmb1273. [DOI] [PubMed] [Google Scholar]
- Croonquist PA, Van Ness B. The polycomb group protein enhancer of zeste homolog 2 (EZH 2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype. Oncogene. 2005;24:6269–6280. doi: 10.1038/sj.onc.1208771. [DOI] [PubMed] [Google Scholar]
- Cuthbert GL, Daujat S, Snowden AW, Erdjument-Bromage H, Hagiwara T, Yamada M, Schneider R, Gregory PD, Tempst P, Bannister AJ, Kouzarides T. Histone deimination antagonizes arginine methylation. Cell. 2004;118:545–553. doi: 10.1016/j.cell.2004.08.020. [DOI] [PubMed] [Google Scholar]
- D’Alessio AC, Szyf M. Epigenetic tete-a-tete: The bilateral relationship between chromatin modifications and DNA methylation. Biochem Cell Biol. 2006;84:463–476. doi: 10.1139/o06-090. [DOI] [PubMed] [Google Scholar]
- Da G, Lenkart J, Zhao K, Shiekhattar R, Cairns BR, Marmorstein R. Structure and function of the SWIRM domain, a conserved protein module found in chromatin regulatory complexes. Proc Natl Acad Sci USA. 2006;103:2057–2062. doi: 10.1073/pnas.0510949103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacwag CS, Ohkawa Y, Pal S, Sif S, Imbalzano AN. The protein arginine methyltransferase Prmt5 is required for myogenesis because it facilitates ATP-dependent chromatin remodeling. Mol Cell Biol. 2007;27:384–394. doi: 10.1128/MCB.01528-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daser A, Rabbitts TH. Extending the repertoire of the mixed-lineage leukemia gene MLL in leukemogenesis. Genes Dev. 2004;18:965–974. doi: 10.1101/gad.1195504. [DOI] [PubMed] [Google Scholar]
- Defeo-Jones D, Huang PS, Jones RE, Haskell KM, Vuocolo GA, Hanobik MG, Huber HE, Oliff A. Cloning of cDNAs for cellular proteins that bind to the retinoblastoma gene product. Nature. 1991;352:251–254. doi: 10.1038/352251a0. [DOI] [PubMed] [Google Scholar]
- Deng Q, Huang S. PRDM5 is silenced in human cancers and has growth suppressive activities. Oncogene. 2004;23:4903–4910. doi: 10.1038/sj.onc.1207615. [DOI] [PubMed] [Google Scholar]
- Dodge JE, Kang YK, Beppu H, Lei H, Li E. Histone H3-K9 methyltransferase ESET is essential for early development. Mol Cell Biol. 2004;24:2478–2486. doi: 10.1128/MCB.24.6.2478-2486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Jenkins NA, Copeland NG. Insertional mutagenesis identifies genes that promote the immortalization of primary bone marrow progenitor cells. Blood. 2005;106:3932–3939. doi: 10.1182/blood-2005-03-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerre JA, Lee CT. In vivo methylation and turnover of rat brain histones. J Neurochem. 1974;23:541–547. doi: 10.1111/j.1471-4159.1974.tb06057.x. [DOI] [PubMed] [Google Scholar]
- Eilbracht J, Reichenzeller M, Hergt M, Schnolzer M, Heid H, Stohr M, Franke WW, Schmidt-Zachmann MS. NO66, a highly conserved dual location protein in the nucleolus and in a special type of synchronously replicating chromatin. Mol Biol Cell. 2004;15:1816–1832. doi: 10.1091/mbc.E03-08-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilbracht J, Kneissel S, Hofmann A, Schmidt-Zachmann MS. Protein NO52—A constitutive nucleolar component sharing high sequence homologies to protein NO66. Eur J Cell Biol. 2005;84:279–294. doi: 10.1016/j.ejcb.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Eissenberg JC, Shilatifard A. Leaving a mark: The many footprints of the elongating RNA polymerase II. Curr Opin Genet Dev. 2006;16:184–190. doi: 10.1016/j.gde.2006.02.004. [DOI] [PubMed] [Google Scholar]
- El Messaoudi S, Fabbrizio E, Rodriguez C, Chuchana P, Fauquier L, Cheng D, Theillet C, Vandel L, Bedford MT, Sardet C. Coactivator-associated arginine methyltransferase 1 (CARM1) is a positive regulator of the Cyclin E1 gene. Proc Natl Acad Sci USA. 2006;103:13351–13356. doi: 10.1073/pnas.0605692103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falnes PO, Johansen RF, Seeberg E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature. 2002;419:178–182. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]
- Fang J, Feng Q, Ketel CS, Wang H, Cao R, Xia L, Erdjument-Bromage H, Tempst P, Simon JA, Zhang Y. Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr Biol. 2002;12:1086–1099. doi: 10.1016/s0960-9822(02)00924-7. [DOI] [PubMed] [Google Scholar]
- Fischle W. Talk is cheap—Cross-talk in establishment, maintenance, and readout of chromatin modifications. Genes Dev. 2008;22:3375–3382. doi: 10.1101/gad.1759708. [DOI] [PubMed] [Google Scholar]
- Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005;438:1116–1122. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- Fodor BD, Kubicek S, Yonezawa M, O’Sullivan RJ, Sengupta R, Perez-Burgos L, Opravil S, Mechtler K, Schotta G, Jenuwein T. Jmjd2b antagonizes H3K9 trimethylation at pericentric heterochromatin in mammalian cells. Genes Dev. 2006;20:1557–1562. doi: 10.1101/gad.388206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forneris F, Binda C, Vanoni MA, Battaglioli E, Mattevi A. Human histone demethylase LSD1 reads the histone code. J Biol Chem. 2005;280:41360–41365. doi: 10.1074/jbc.M509549200. [DOI] [PubMed] [Google Scholar]
- Forneris F, Binda C, Adamo A, Battaglioli E, Mattevi A. Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J Biol Chem. 2007;282:20070–20074. doi: 10.1074/jbc.C700100200. [DOI] [PubMed] [Google Scholar]
- Foster SL, Medzhitov R. Gene-specific control of the TLR-induced inflammatory response. Clin Immunol. 2009;130:7–15. doi: 10.1016/j.clim.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frescas D, Guardavaccaro D, Bassermann F, Koyama-Nasu R, Pagano M. JHDM1B/FBXL10 is a nucleolar protein that represses transcription of ribosomal RNA genes. Nature. 2007;450:309–313. doi: 10.1038/nature06255. [DOI] [PubMed] [Google Scholar]
- Frietze S, Lupien M, Silver PA, Brown M. CARM1 regulates estrogen-stimulated breast cancer growth through up-regulation of E2F1. Cancer Res. 2008;68:301–306. doi: 10.1158/0008-5472.CAN-07-1983. [DOI] [PubMed] [Google Scholar]
- Fukahori S, Yano H, Tsuneoka M, Tanaka Y, Yagi M, Kuwano M, Tajiri T, Taguchi T, Tsuneyoshi M, Kojiro M. Immunohistochemical expressions of Cap43 and Mina53 proteins in neuroblastoma. J Pediatr Surg. 2007;42:1831–1840. doi: 10.1016/j.jpedsurg.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Gearhart MD, Corcoran CM, Wamstad JA, Bardwell VJ. Polycomb group and SCF ubiquitin ligases are found in a novel BCOR complex that is recruited to BCL6 targets. Mol Cell Biol. 2006;26:6880–6889. doi: 10.1128/MCB.00630-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber M, Shilatifard A. Transcriptional elongation by RNA polymerase II and histone methylation. J Biol Chem. 2003;278:26303–26306. doi: 10.1074/jbc.R300014200. [DOI] [PubMed] [Google Scholar]
- Glaser S, Schaft J, Lubitz S, Vintersten K, van der Hoeven F, Tufteland KR, Aasland R, Anastassiadis K, Ang SL, Stewart AF. Multiple epigenetic maintenance factors implicated by the loss of Mll2 in mouse development. Development. 2006;133:1423–1432. doi: 10.1242/dev.02302. [DOI] [PubMed] [Google Scholar]
- Gondek LP, Ishwaran H, Dunbar AJ, O’Keefe CL, McDevitt MA, Batista D, Sekeres MA, Mufti GJ, Maciejewski J. Array-based karyotyping and genotyping demonstrates a non random selection of allelic variants of genes in clones with 5q31 deletion mutants. ASH Annu Meet Abstr. 2008;112:2057. [Google Scholar]
- Guccione E, Bassi C, Casadio F, Martinato F, Cesaroni M, Schuchlautz H, Luscher B, Amati B. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature. 2007;449:933–937. doi: 10.1038/nature06166. [DOI] [PubMed] [Google Scholar]
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez GM, Kong E, Hinds PW. Master or slave: The complex relationship of RBP2 and pRb. Cancer Cell. 2005;7:501–502. doi: 10.1016/j.ccr.2005.05.021. [DOI] [PubMed] [Google Scholar]
- Hampsey M, Reinberg D. Tails of intrigue: Phosphorylation of RNA polymerase II mediates histone methylation. Cell. 2003;113:429–432. doi: 10.1016/s0092-8674(03)00360-x. [DOI] [PubMed] [Google Scholar]
- Hansen JC. Conformational dynamics of the chromatin fiber in solution: Determinants, mechanisms, and functions. Annu Rev Biophys Biomol Struct. 2002;31:361–392. doi: 10.1146/annurev.biophys.31.101101.140858. [DOI] [PubMed] [Google Scholar]
- Harper DP, Aplan PD. Chromosomal rearrangements leading to MLL gene fusions: Clinical and biological aspects. Cancer Res. 2008;68:10024–10027. doi: 10.1158/0008-5472.CAN-08-2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa Y, Matsubara A, Teishima J, Seki M, Mita K, Usui T, Oue N, Yasui W. DNA methylation of the RIZ1 gene is associated with nuclear accumulation of p53 in prostate cancer. Cancer Sci. 2007;98:32–36. doi: 10.1111/j.1349-7006.2006.00338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenpusch-Theil K, Chadwick BP, Theil T, Heath SK, Wilkinson DG, Frischauf AM. PHF2, a novel PHD finger gene located on human chromosome 9q22. Mamm Genome. 1999;10:294–298. doi: 10.1007/s003359900989. [DOI] [PubMed] [Google Scholar]
- Hashimoto H, Horton JR, Zhang X, Bostick M, Jacobsen SE, Cheng X. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008;455:826–829. doi: 10.1038/nature07280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassig CA, Fleischer TC, Billin AN, Schreiber SL, Ayer DE. Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell. 1997;89:341–347. doi: 10.1016/s0092-8674(00)80214-7. [DOI] [PubMed] [Google Scholar]
- Hayakawa T, Ohtani Y, Hayakawa N, Shinmyozu K, Saito M, Ishikawa F, Nakayama J. RBP2 is an MRG15 complex component and down-regulates intra-genic histone H3 lysine 4 methylation. Genes Cells. 2007;12:811–826. doi: 10.1111/j.1365-2443.2007.01089.x. [DOI] [PubMed] [Google Scholar]
- He J, Kallin EM, Tsukada Y, Zhang Y. The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15(Ink4b) Nat Struct Mol Biol. 2008;15:1169–1175. doi: 10.1038/nsmb.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hediger F, Gasser SM. Heterochromatin protein 1: Don’t judge the book by its cover! Curr Opin Genet Dev. 2006;16:143–150. doi: 10.1016/j.gde.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Hirota T, Lipp JJ, Toh BH, Peters JM. Histone H3 serine 10 phosphorylation by Aurora B causes HP1 dissociation from heterochromatin. Nature. 2005;438:1176–1180. doi: 10.1038/nature04254. [DOI] [PubMed] [Google Scholar]
- Hoog C, Schalling M, Grunder-Brundell E, Daneholt B. Analysis of a murine male germ cell-specific transcript that encodes a putative zinc finger protein. Mol Reprod Dev. 1991;30:173–181. doi: 10.1002/mrd.1080300302. [DOI] [PubMed] [Google Scholar]
- Hu Z, Gomes I, Horrigan SK, Kravarusic J, Mar B, Arbieva Z, Chyna B, Fulton N, Edassery S, Raza A, Westbrook CA. A novel nuclear protein, 5qNCA (LOC51780) is a candidate for the myeloid leukemia tumor suppressor gene on chromosome 5 band q31. Oncogene. 2001;20:6946–6954. doi: 10.1038/sj.onc.1204850. [DOI] [PubMed] [Google Scholar]
- Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, Kubicek S, Opravil S, Jenuwein T, Berger SL. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006a;444:629–632. doi: 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science. 2006b;312:748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T, Berger SL. p53 is regulated by the lysine demethylase LSD1. Nature. 2007a;449:105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- Huang Y, Greene E, Murray Stewart T, Goodwin AC, Baylin SB, Woster PM, Casero RA., Jr Inhibition of lysine-specific demethylase 1 by polyamine analogues results in reexpression of aberrantly silenced genes. Proc Natl Acad Sci USA. 2007b;104:8023–8028. doi: 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntsman DG, Chin SF, Muleris M, Batley SJ, Collins VP, Wiedemann LM, Aparicio S, Caldas C. MLL2, the second human homolog of the Drosophila trithorax gene, maps to 19q13.1 and is amplified in solid tumor cell lines. Oncogene. 1999;18:7975–7984. doi: 10.1038/sj.onc.1203291. [DOI] [PubMed] [Google Scholar]
- Ishizaki H, Yano H, Tsuneoka M, Ogasawara S, Akiba J, Nishida N, Kojiro S, Fukahori S, Moriya F, Matsuoka K, Kojiro M. Overexpression of the myc target gene Mina53 in advanced renal cell carcinoma. Pathol Int. 2007;57:672–680. doi: 10.1111/j.1440-1827.2007.02156.x. [DOI] [PubMed] [Google Scholar]
- Ito T. Role of histone modification in chromatin dynamics. J Biochem. 2007;141:609–614. doi: 10.1093/jb/mvm091. [DOI] [PubMed] [Google Scholar]
- Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077–1088. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Jensen LR, Amende M, Gurok U, Moser B, Gimmel V, Tzschach A, Janecke AR, Tariverdian G, Chelly J, Fryns JP, Van Esch H, Kleefstra T, et al. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am J Hum Genet. 2005;76:227–236. doi: 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi AA, Struhl K. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol Cell. 2005;20:971–978. doi: 10.1016/j.molcel.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Kahl P, Gullotti L, Heukamp LC, Wolf S, Friedrichs N, Vorreuther R, Solleder G, Bastian PJ, Ellinger J, Metzger E, Schule R, Buettner R. Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006;66:11341–11347. doi: 10.1158/0008-5472.CAN-06-1570. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, Schuldiner M, Chin K, Punna T, Thompson NJ, Boone C, Emili A, et al. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005;123:593–605. doi: 10.1016/j.cell.2005.10.025. [DOI] [PubMed] [Google Scholar]
- Kim S, Benoiton L, Paik WK. Epsilon-alkyllysinase. Purification and properties of the enzyme. J Biol Chem. 1964;239:3790–3796. [PubMed] [Google Scholar]
- Kim KC, Geng L, Huang S. Inactivation of a histone methyltransferase by mutations in human cancers. Cancer Res. 2003;63:7619–7623. [PubMed] [Google Scholar]
- Kim DH, Villeneuve LM, Morris KV, Rossi JJ. Argonaute-1 directs siRNA-mediated transcriptional gene silencing in human cells. Nat Struct Mol Biol. 2006a;13:793–797. doi: 10.1038/nsmb1142. [DOI] [PubMed] [Google Scholar]
- Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006b;7:397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipreos ET, Pagano M. The F-box protein family. Genome Biol. 2000;1:REVIEWS3002. doi: 10.1186/gb-2000-1-5-reviews3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirmizis A, Santos-Rosa H, Penkett CJ, Singer MA, Vermeulen M, Mann M, Bahler J, Green RD, Kouzarides T. Arginine methylation at histone H3R2 controls deposition of H3K4 trimethylation. Nature. 2007;449:928–932. doi: 10.1038/nature06160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet. 2006a;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Yamane K, Bae Y, Zhang D, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. The transcriptional repressor JHDM3A demethylates trimethyl histone H3 lysine 9 and lysine 36. Nature. 2006b;442:312–316. doi: 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Yan Q, Tothova Z, Yamane K, Erdjument-Bromage H, Tempst P, Gilliland DG, Zhang Y, Kaelin WG., Jr The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell. 2007;128:889–900. doi: 10.1016/j.cell.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Koivisto AM, Ala-Mello S, Lemmela S, Komu HA, Rautio J, Jarvela I. Screening of mutations in the PHF8 gene and identification of a novel mutation in a Finnish family with XLMR and cleft lip/cleft palate. Clin Genet. 2007;72:145–149. doi: 10.1111/j.1399-0004.2007.00836.x. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Shen L, Ahmed S, Boumber Y, Sekido Y, Haddad BR, Issa JP. Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells. PLoS ONE. 2008;3:e2037. doi: 10.1371/journal.pone.0002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg RD, Thomas JO. Chromatin structure; oligomers of the histones. Science. 1974;184:865–868. doi: 10.1126/science.184.4139.865. [DOI] [PubMed] [Google Scholar]
- Kortschak RD, Tucker PW, Saint R. ARID proteins come in from the desert. Trends Biochem Sci. 2000;25:294–299. doi: 10.1016/s0968-0004(00)01597-8. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Koyama-Nasu R, David G, Tanese N. The F-box protein Fbl10 is a novel transcriptional repressor of c-Jun. Nat Cell Biol. 2007;9:1074–1080. doi: 10.1038/ncb1628. [DOI] [PubMed] [Google Scholar]
- Krimpenfort P, Ijpenberg A, Song JY, van der Valk M, Nawijn M, Zevenhoven J, Berns A. p15Ink4b is a critical tumour suppressor in the absence of p16Ink4a. Nature. 2007;448:943–946. doi: 10.1038/nature06084. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Feng Z, Lemieux ME, Faber J, Vempati S, Sinha AU, Xia X, Jesneck J, Bracken AP, Silverman LB, Kutok JL, Kung AL, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14:355–368. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicek S, Jenuwein T. A crack in histone lysine methylation. Cell. 2004;119:903–906. doi: 10.1016/j.cell.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Kurash JK, Lei H, Shen Q, Marston WL, Granda BW, Fan H, Wall D, Li E, Gaudet F. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol Cell. 2008;29:392–400. doi: 10.1016/j.molcel.2007.12.025. [DOI] [PubMed] [Google Scholar]
- Kuratomi K, Yano H, Tsuneoka M, Sakamoto K, Kusukawa J, Kojiro M. Immunohistochemical expression of Mina53 and Ki67 proteins in human primary gingival squamous cell carcinoma. Kurume Med J. 2006;53:71–78. doi: 10.2739/kurumemedj.53.71. [DOI] [PubMed] [Google Scholar]
- Kuzmichev A, Margueron R, Vaquero A, Preissner TS, Scher M, Kirmizis A, Ouyang X, Brockdorff N, Abate-Shen C, Farnham P, Reinberg D. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc Natl Acad Sci USA. 2005;102:1859–1864. doi: 10.1073/pnas.0409875102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Starza R, Gorello P, Rosati R, Riezzo A, Veronese A, Ferrazzi E, Martelli MF, Negrini M, Mecucci C. Cryptic insertion producing two NUP98/NSD1 chimeric transcripts in adult refractory anemia with an excess of blasts. Genes Chromosomes Cancer. 2004;41:395–399. doi: 10.1002/gcc.20103. [DOI] [PubMed] [Google Scholar]
- Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- Lagarou A, Mohd-Sarip A, Moshkin YM, Chalkley GE, Bezstarosti K, Demmers JA, Verrijzer CP. dKDM2 couples histone H2A ubiquitylation to histone H3 demethylation during Polycomb group silencing. Genes Dev. 2008;22:2799–2810. doi: 10.1101/gad.484208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmikuttyamma A, Pastural E, Takahashi N, Sawada K, Sheridan DP, DeCoteau JF, Geyer CR. Bcr-Abl induces autocrine IGF-1 signaling. Oncogene. 2008;27:3831–3844. doi: 10.1038/onc.2008.8. [DOI] [PubMed] [Google Scholar]
- Lal G, Padmanabha L, Smith BJ, Nicholson RM, Howe JR, O’Dorisio MS, Domann FE., Jr RIZ1 is epigenetically inactivated by promoter hypermethylation in thyroid carcinoma. Cancer. 2006;107:2752–2759. doi: 10.1002/cncr.22325. [DOI] [PubMed] [Google Scholar]
- Lan F, Bayliss PE, Rinn JL, Whetstine JR, Wang JK, Chen S, Iwase S, Alpatov R, Issaeva I, Canaani E, Roberts TM, Chang HY, et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature. 2007;449:689–694. doi: 10.1038/nature06192. [DOI] [PubMed] [Google Scholar]
- Latres E, Malumbres M, Sotillo R, Martin J, Ortega S, Martin-Caballero J, Flores JM, Cordon-Cardo C, Barbacid M. Limited overlapping roles of P15(INK4b) and P18(INK4c) cell cycle inhibitors in proliferation and tumorigenesis. EMBO J. 2000;19:3496–3506. doi: 10.1093/emboj/19.13.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnier F, Holbert S, Ronce N, Faravelli F, Lenzner S, Schwartz CE, Lespinasse J, Van Esch H, Lacombe D, Goizet C, Phan-Dinh Tuy F, van Bokhoven H, et al. Mutations in PHF8 are associated with X linked mental retardation and cleft lip/cleft palate. J Med Genet. 2005;42:780–786. doi: 10.1136/jmg.2004.029439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DY, Teyssier C, Strahl BD, Stallcup MR. Role of protein methylation in regulation of transcription. Endocr Rev. 2005a;26:147–170. doi: 10.1210/er.2004-0008. [DOI] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005b;437:432–435. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Schmidt DM, McCafferty DG, Shiekhattar R. Histone H3 lysine 4 demethylation is a target of nonselective antidepressive medications. Chem Biol. 2006;13:563–567. doi: 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Lee JS, Shukla A, Schneider J, Swanson SK, Washburn MP, Florens L, Bhaumik SR, Shilatifard A. Histone Crosstalk between H2B Monoubiquitination and H3 Methylation Mediated by COMPASS. Cell. 2007;131:1084–1096. doi: 10.1016/j.cell.2007.09.046. [DOI] [PubMed] [Google Scholar]
- Lee J, Thompson JR, Botuyan MV, Mer G. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat Struct Mol Biol. 2008;15:109–111. doi: 10.1038/nsmb1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Cao R, Wang M, Myers MP, Zhang Y, Xu RM. Structure of a Bmi-1-Ring1B polycomb group ubiquitin ligase complex. J Biol Chem. 2006;281:20643–20649. doi: 10.1074/jbc.M602461200. [DOI] [PubMed] [Google Scholar]
- Linnoila RI, Zhao B, DeMayo JL, Nelkin BD, Baylin SB, DeMayo FJ, Ball DW. Constitutive achaete-scute homologue-1 promotes airway dysplasia and lung neuroendocrine tumors in transgenic mice. Cancer Res. 2000;60:4005–4009. [PubMed] [Google Scholar]
- Loh YH, Zhang W, Chen X, George J, Ng HH. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 2007;21:2545–2557. doi: 10.1101/gad.1588207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Bigas N, Kisiel TA, Dewaal DC, Holmes KB, Volkert TL, Gupta S, Love J, Murray HL, Young RA, Benevolenskaya EV. Genome-wide analysis of the H3K4 histone demethylase RBP2 reveals a transcriptional program controlling differentiation. Mol Cell. 2008;31:520–530. doi: 10.1016/j.molcel.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PJ, Sundquist K, Baeckstrom D, Poulsom R, Hanby A, Meier-Ewert S, Jones T, Mitchell M, Pitha-Rowe P, Freemont P, Taylor-Papadimitriou J. A novel gene (PLU-1) containing highly conserved putative DNA/chromatin binding motifs is specifically up-regulated in breast cancer. J Biol Chem. 1999;274:15633–15645. doi: 10.1074/jbc.274.22.15633. [DOI] [PubMed] [Google Scholar]
- Lubitz S, Glaser S, Schaft J, Stewart AF, Anastassiadis K. Increased apoptosis and skewed differentiation in mouse embryonic stem cells lacking the histone methyltransferase Mll2. Mol Biol Cell. 2007;18:2356–2366. doi: 10.1091/mbc.E06-11-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Majumder S, Liu Y, Ford OH, 3rd, Mohler JL, Whang YE. Involvement of arginine methyltransferase CARM1 in androgen receptor function and prostate cancer cell viability. Prostate. 2006;66:1292–1301. doi: 10.1002/pros.20438. [DOI] [PubMed] [Google Scholar]
- Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, Reinberg D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell. 2008;32:503–518. doi: 10.1016/j.molcel.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews AG, Kuo AJ, Ramon-Maiques S, Han S, Champagne KS, Ivanov D, Gallardo M, Carney D, Cheung P, Ciccone DN, Walter KL, Utz PJ, et al. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature. 2007;450:1106–1110. doi: 10.1038/nature06431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- Metzger E, Yin N, Wissmann M, Kunowska N, Fischer K, Friedrichs N, Patnaik D, Higgins JM, Potier N, Scheidtmann KH, Buettner R, Schule R. Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation. Nat Cell Biol. 2008;10:53–60. doi: 10.1038/ncb1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- Mohd-Sarip A, Cleard F, Mishra RK, Karch F, Verrijzer CP. Synergistic recognition of an epigenetic DNA element by Pleiohomeotic and a Polycomb core complex. Genes Dev. 2005;19:1755–1760. doi: 10.1101/gad.347005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohd-Sarip A, van der Knaap JA, Wyman C, Kanaar R, Schedl P, Verrijzer CP. Architecture of a polycomb nucleoprotein complex. Mol Cell. 2006;24:91–100. doi: 10.1016/j.molcel.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Mohd-Sarip A, Venturini F, Chalkley GE, Verrijzer CP. Pleiohomeotic can link polycomb to DNA and mediate transcriptional repression. Mol Cell Biol. 2002;22:7473–7483. doi: 10.1128/MCB.22.21.7473-7483.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan P, Westbrook TF, Ottinger M, Pavlova N, Chang B, Macia E, Shi YJ, Barretina J, Liu J, Howley PM, Elledge SJ, Shi Y. CDYL bridges REST and histone methyltransferases for gene repression and suppression of cellular transformation. Mol Cell. 2008;32:718–726. doi: 10.1016/j.molcel.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrasov M, Klymenko T, Fraterman S, Papp B, Oktaba K, Kocher T, Cohen A, Stunnenberg HG, Wilm M, Muller J. Pcl-PRC2 is needed to generate high levels of H3-K27 trimethylation at Polycomb target genes. EMBO J. 2007;26:4078–4088. doi: 10.1038/sj.emboj.7601837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CJ, Santos-Rosa H, Kouzarides T. Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell. 2006;126:905–916. doi: 10.1016/j.cell.2006.07.026. [DOI] [PubMed] [Google Scholar]
- Ng HH, Feng Q, Wang H, Erdjument-Bromage H, Tempst P, Zhang Y, Struhl K. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 2002;16:1518–1527. doi: 10.1101/gad.1001502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SS, Kavanagh KL, McDonough MA, Butler D, Pilka ES, Lienard BM, Bray JE, Savitsky P, Gileadi O, von Delft F, Rose NR, Offer J, et al. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature. 2007;448:87–91. doi: 10.1038/nature05971. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Rice JC, Sarma K, Erdjument-Bromage H, Werner J, Wang Y, Chuikov S, Valenzuela P, Tempst P, Steward R, Lis JT, Allis CD, et al. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol Cell. 2002;9:1201–1213. doi: 10.1016/s1097-2765(02)00548-8. [DOI] [PubMed] [Google Scholar]
- Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, Zhang Y. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Okada Y, Scott G, Ray MK, Mishina Y, Zhang Y. Histone demethylase JHDM2A is critical for Tnp1 and Prm1 transcription and spermatogenesis. Nature. 2007;450:119–123. doi: 10.1038/nature06236. [DOI] [PubMed] [Google Scholar]
- Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, Cheng X, Bestor TH. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozer A, Bruick RK. Non-heme dioxygenases: Cellular sensors and regulators jelly rolled into one? Nat Chem Biol. 2007;3:144–153. doi: 10.1038/nchembio863. [DOI] [PubMed] [Google Scholar]
- Paik WK, Kim S. Enzymatic demethylation of calf thymus histones. Biochem Biophys Res Commun. 1973;51:781–788. doi: 10.1016/0006-291x(73)91383-1. [DOI] [PubMed] [Google Scholar]
- Paik WK, Kim S. Epsilon-alkyllysinase. New assay method, purification, and biological significance. Arch Biochem Biophys. 1974;165:369–378. doi: 10.1016/0003-9861(74)90175-1. [DOI] [PubMed] [Google Scholar]
- Pal S, Baiocchi RA, Byrd JC, Grever MR, Jacob ST, Sif S. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 2007;26:3558–3569. doi: 10.1038/sj.emboj.7601794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, Jonsdottir GA, Stewart R, Thomson JA. Whole-genome analysis of histone H3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell. 2007;1:299–312. doi: 10.1016/j.stem.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Pasini D, Hansen KH, Christensen J, Agger K, Cloos PA, Helin K. Coordinated regulation of transcriptional repression by the RBP2 H3K4 demethylase and Polycomb-Repressive Complex 2. Genes Dev. 2008;22:1345–1355. doi: 10.1101/gad.470008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perillo B, Ombra MN, Bertoni A, Cuozzo C, Sacchetti S, Sasso A, Chiariotti L, Malorni A, Abbondanza C, Avvedimento EV. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science. 2008;319:202–206. doi: 10.1126/science.1147674. [DOI] [PubMed] [Google Scholar]
- Pfau R, Tzatsos A, Kampranis SC, Serebrennikova OB, Bear SE, Tsichlis PN. Members of a family of JmjC domain-containing oncoproteins immortalize embryonic fibroblasts via a JmjC domain-dependent process. Proc Natl Acad Sci USA. 2008;105:1907–1912. doi: 10.1073/pnas.0711865105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao GH, Piao WH, He Y, Zhang HH, Wang GQ, Piao Z. Hypermethylation of RIZ1 tumor suppressor gene is involved in the early tumorigenesis of hepatocellular carcinoma. Histol Histopathol. 2008;23:1171–1175. doi: 10.14670/HH-23.1171. [DOI] [PubMed] [Google Scholar]
- Pietersen AM, van Lohuizen M. Stem cell regulation by polycomb repressors: Postponing commitment. Curr Opin Cell Biol. 2008;20:201–207. doi: 10.1016/j.ceb.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Pollard PJ, Loenarz C, Mole DR, McDonough MA, Gleadle JM, Schofield CJ, Ratcliffe PJ. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1alpha. Biochem J. 2008;416:387–394. doi: 10.1042/BJ20081238. [DOI] [PubMed] [Google Scholar]
- Polytarchou C, Pfau R, Hatziapostolou M, Tsichlis PN. The JmjC domain histone demethylase Ndy1 regulates redox homeostasis and protects cells from oxidative stress. Mol Cell Biol. 2008;28:7451–7464. doi: 10.1128/MCB.00688-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothof J, van Haaften G, Thijssen K, Kamath RS, Fraser AG, Ahringer J, Plasterk RH, Tijsterman M. Identification of genes that protect the C. elegans genome against mutations by genome-wide RNAi. Genes Dev. 2003;17:443–448. doi: 10.1101/gad.1060703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian C, Zhang Q, Li S, Zeng L, Walsh MJ, Zhou MM. Structure and chromosomal DNA binding of the SWIRM domain. Nat Struct Mol Biol. 2005;12:1078–1085. doi: 10.1038/nsmb1022. [DOI] [PubMed] [Google Scholar]
- Qiao Y, Liu X, Harvard C, Hildebrand MJ, Rajcan-Separovic E, Holden JJ, Lewis ME. Autism-associated familial microdeletion of Xp11.22. Clin Genet. 2008;74:134–144. doi: 10.1111/j.1399-0004.2008.01028.x. [DOI] [PubMed] [Google Scholar]
- Raman JD, Mongan NP, Tickoo SK, Boorjian SA, Scherr DS, Gudas LJ. Increased expression of the polycomb group gene, EZH2, in transitional cell carcinoma of the bladder. Clin Cancer Res. 2005;11:8570–8576. doi: 10.1158/1078-0432.CCR-05-1047. [DOI] [PubMed] [Google Scholar]
- Rosati R, La Starza R, Veronese A, Aventin A, Schwienbacher C, Vallespi T, Negrini M, Martelli MF, Mecucci C. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15) Blood. 2002;99:3857–3860. doi: 10.1182/blood.v99.10.3857. [DOI] [PubMed] [Google Scholar]
- Rowley JD. Rearrangements involving chromosome band 11Q23 in acute leukaemia. Semin Cancer Biol. 1993;4:377–385. [PubMed] [Google Scholar]
- Rozovskaia T, Tillib S, Smith S, Sedkov Y, Rozenblatt-Rosen O, Petruk S, Yano T, Nakamura T, Ben-Simchon L, Gildea J, Croce CM, Shearn A, et al. Trithorax and ASH1 interact directly and associate with the trithorax group-responsive bxd region of the Ultrabithorax promoter. Mol Cell Biol. 1999;19:6441–6447. doi: 10.1128/mcb.19.9.6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez C, Sanchez I, Demmers JA, Rodriguez P, Strouboulis J, Vidal M. Proteomics analysis of Ring1B/Rnf2 interactors identifies a novel complex with the Fbxl10/Jhdm1B histone demethylase and the Bcl6 interacting corepressor. Mol Cell Proteomics. 2007;6:820–834. doi: 10.1074/mcp.M600275-MCP200. [DOI] [PubMed] [Google Scholar]
- Santi DV, Garrett CE, Barr PJ. On the mechanism of inhibition of DNA-cytosine methyltransferases by cytosine analogs. Cell. 1983;33:9–10. doi: 10.1016/0092-8674(83)90327-6. [DOI] [PubMed] [Google Scholar]
- Santos C, Rodriguez-Revenga L, Madrigal I, Badenas C, Pineda M, Mila M. A novel mutation in JARID1C gene associated with mental retardation. Eur J Hum Genet. 2006;14:583–586. doi: 10.1038/sj.ejhg.5201608. [DOI] [PubMed] [Google Scholar]
- Saramaki OR, Tammela TL, Martikainen PM, Vessella RL, Visakorpi T. The gene for polycomb group protein enhancer of zeste homolog 2 (EZH2) is amplified in late-stage prostate cancer. Genes Chromosomes Cancer. 2006;45:639–645. doi: 10.1002/gcc.20327. [DOI] [PubMed] [Google Scholar]
- Sarma K, Margueron R, Ivanov A, Pirrotta V, Reinberg D. Ezh2 requires PHF1 to efficiently catalyze H3 lysine 27 trimethylation in vivo. Mol Cell Biol. 2008;28:2718–2731. doi: 10.1128/MCB.02017-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt DM, McCafferty DG. Trans-2-Phenylcyclopropylamine is a mechanism-based inactivator of the histone demethylase LSD1. Biochemistry. 2007;46:4408–4416. doi: 10.1021/bi0618621. [DOI] [PubMed] [Google Scholar]
- Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004;18:1251–1262. doi: 10.1101/gad.300704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128:735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Schurter BT, Koh SS, Chen D, Bunick GJ, Harp JM, Hanson BL, Henschen-Edman A, Mackay DR, Stallcup MR, Aswad DW. Methylation of histone H3 by coactivator-associated arginine methyltransferase 1. Biochemistry. 2001;40:5747–5756. doi: 10.1021/bi002631b. [DOI] [PubMed] [Google Scholar]
- Schwartz YB, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet. 2007;8:9–22. doi: 10.1038/nrg1981. [DOI] [PubMed] [Google Scholar]
- Scibetta AG, Santangelo S, Coleman J, Hall D, Chaplin T, Copier J, Catchpole S, Burchell J, Taylor-Papadimitriou J. Functional analysis of the transcription repressor PLU-1/JARID1B. Mol Cell Biol. 2007;27:7220–7235. doi: 10.1128/MCB.00274-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoumanne A, Chen X. The lysine-specific demethylase 1 is required for cell proliferation in both p53-dependent and -independent manners. J Biol Chem. 2007;282:15471–15475. doi: 10.1074/jbc.M701023200. [DOI] [PubMed] [Google Scholar]
- Scoumanne A, Chen X. Protein methylation: A new mechanism of p53 tumor suppressor regulation. Histol Histopathol. 2008;23:1143–1149. doi: 10.14670/hh-23.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secombe J, Eisenman RN. The function and regulation of the JARID1 family of histone H3 lysine 4 demethylases: The Myc connection. Cell Cycle. 2007;6:1324–1328. doi: 10.4161/cc.6.11.4269. [DOI] [PubMed] [Google Scholar]
- Sellers WR, Loda M. The EZH2 polycomb transcriptional repressor—A marker or mover of metastatic prostate cancer? Cancer Cell. 2002;2:349–350. doi: 10.1016/s1535-6108(02)00187-3. [DOI] [PubMed] [Google Scholar]
- Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413:86–91. doi: 10.1038/35092592. [DOI] [PubMed] [Google Scholar]
- Shi Y, Whetstine JR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell. 2007;25:1–14. doi: 10.1016/j.molcel.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Shi Y, Sawada J, Sui G, Affarel B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y, Shi Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell. 2005;19:857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Shi X, Hong T, Walter KL, Ewalt M, Michishita E, Hung T, Carney D, Pena P, Lan F, Kaadige MR, Lacoste N, Cayrou C, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–99. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A. Chromatin modifications by methylation and ubiquitination: Implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- Shin S, Janknecht R. Diversity within the JMJD2 histone demethylase family. Biochem Biophys Res Commun. 2007;353:973–977. doi: 10.1016/j.bbrc.2006.12.147. [DOI] [PubMed] [Google Scholar]
- Shirato H, Ogawa S, Nakajima K, Inagawa M, Kojima M, Tachibana M, Shinkai Y, Takeuchi T. A Jumonji (Jarid2) protein complex represses cyclin D1 expression by methylation of histone H3-K9. J Biol Chem. 2009;284:733–739. doi: 10.1074/jbc.M804994200. [DOI] [PubMed] [Google Scholar]
- Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647:21–29. doi: 10.1016/j.mrfmmm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Sims RJ, 3rd, Millhouse S, Chen CF, Lewis BA, Erdjument-Bromage H, Tempst P, Manley JL, Reinberg D. Recognition of trimethylated histone H3 lysine 4 facilitates the recruitment of transcription postinitiation factors and pre-mRNA splicing. Mol Cell. 2007;28:665–676. doi: 10.1016/j.molcel.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer MS, Kahana A, Wolf AJ, Meisinger LL, Peterson SE, Goggin C, Mahowald M, Gottschling DE. Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics. 1998;150:613–632. doi: 10.1093/genetics/150.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha SM, Singh RD, Alam ND, Roy AD, Roychoudhury SD, Panda CD. Alterations in candidate genes PHF2, FANCC, PTCH1 and XPA at chromosomal 9q22.3 region: Pathological significance in early- and late-onset breast carcinoma. Mol Cancer. 2008;7:84. doi: 10.1186/1476-4598-7-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smothers JF, Henikoff S. The hinge and chromo shadow domain impart distinct targeting of HP1-like proteins. Mol Cell Biol. 2001;21:2555–2569. doi: 10.1128/MCB.21.7.2555-2569.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6:846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- Spivakov M, Fisher AG. Epigenetic signatures of stem-cell identity. Nat Rev Genet. 2007;8:263–271. doi: 10.1038/nrg2046. [DOI] [PubMed] [Google Scholar]
- Stallcup MR, Chen D, Koh SS, Ma H, Lee YH, Li H, Schurter BT, Aswad DW. Co-operation between protein-acetylating and protein-methylating co-activators in transcriptional activation. Biochem Soc Trans. 2000;28:415–418. [PubMed] [Google Scholar]
- Stavropoulos P, Blobel G, Hoelz A. Crystal structure and mechanism of human lysine-specific demethylase-1. Nat Struct Mol Biol. 2006;13:626–632. doi: 10.1038/nsmb1113. [DOI] [PubMed] [Google Scholar]
- Steele-Perkins G, Fang W, Yang XH, Van Gele M, Carling T, Gu J, Buyse IM, Fletcher JA, Liu J, Bronson R, Chadwick RB, de la Chapelle A, et al. Tumor formation and inactivation of RIZ1, an Rb-binding member of a nuclear protein-methyltransferase superfamily. Genes Dev. 2001;15:2250–2262. doi: 10.1101/gad.870101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA, Blobel GA, Vakoc CR. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008;28:2825–2839. doi: 10.1128/MCB.02076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Briggs SD, Brame CJ, Caldwell JA, Koh SS, Ma H, Cook RG, Shabanowitz J, Hunt DF, Stallcup MR, Allis CD. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr Biol. 2001;11:996–1000. doi: 10.1016/s0960-9822(01)00294-9. [DOI] [PubMed] [Google Scholar]
- Subramanian K, Jia D, Kapoor-Vazirani P, Powell DR, Collins RE, Sharma D, Peng J, Cheng X, Vertino PM. Regulation of estrogen receptor alpha by the SET7 lysine methyltransferase. Mol Cell. 2008;30:336–347. doi: 10.1016/j.molcel.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surani MA, Hayashi K, Hajkova P. Genetic and epigenetic regulators of pluripotency. Cell. 2007;128:747–762. doi: 10.1016/j.cell.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Shen H, Akagi K, Morse HC, Malley JD, Naiman DQ, Jenkins NA, Copeland NG. New genes involved in cancer identified by retroviral tagging. Nat Genet. 2002;32:166–174. doi: 10.1038/ng949. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Minehata K, Akagi K, Jenkins NA, Copeland NG. Tumor suppressor gene identification using retroviral insertional mutagenesis in Blm-deficient mice. EMBO J. 2006;25:3422–3431. doi: 10.1038/sj.emboj.7601215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki C, Takahashi K, Hayama S, Ishikawa N, Kato T, Ito T, Tsuchiya E, Nakamura Y, Daigo Y. Identification of Myc-associated protein with JmjC domain as a novel therapeutic target oncogene for lung cancer. Mol Cancer Ther. 2007;6:542–551. doi: 10.1158/1535-7163.MCT-06-0659. [DOI] [PubMed] [Google Scholar]
- Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, Shinkai Y. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002;16:1779–1791. doi: 10.1101/gad.989402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Mei P, Fang R, Leonor T, Rutenberg M, Shimizu F, Li J, Rao A, Shi Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature. 2007;447:601–605. doi: 10.1038/nature05823. [DOI] [PubMed] [Google Scholar]
- Tan K, Shaw AL, Madsen B, Jensen K, Taylor-Papadimitriou J, Freemont PS. Human PLU-1 Has transcriptional repression properties and interacts with the developmental transcription factors BF-1 and PAX9. J Biol Chem. 2003;278:20507–20513. doi: 10.1074/jbc.M301994200. [DOI] [PubMed] [Google Scholar]
- Tatton-Brown K, Douglas J, Coleman K, Baujat G, Cole TR, Das S, Horn D, Hughes HE, Temple IK, Faravelli F, Waggoner D, Turkmen S, et al. Genotype-phenotype associations in Sotos syndrome: An analysis of 266 individuals with NSD1 aberrations. Am J Hum Genet. 2005;77:193–204. doi: 10.1086/432082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenney K, Shilatifard A. A COMPASS in the voyage of defining the role of trithorax/MLL-containing complexes: Linking leukemogensis to covalent modifications of chromatin. J Cell Biochem. 2005;95:429–436. doi: 10.1002/jcb.20421. [DOI] [PubMed] [Google Scholar]
- Teye K, Tsuneoka M, Arima N, Koda Y, Nakamura Y, Ueta Y, Shirouzu K, Kimura H. Increased expression of a Myc target gene Mina53 in human colon cancer. Am J Pathol. 2004;164:205–216. doi: 10.1016/S0002-9440(10)63111-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teye K, Arima N, Nakamura Y, Sakamoto K, Sueoka E, Kimura H, Tsuneoka M. Expression of Myc target gene mina53 in subtypes of human lymphoma. Oncol Rep. 2007;18:841–848. [PubMed] [Google Scholar]
- Tong JK, Hassig CA, Schnitzler GR, Kingston RE, Schreiber SL. Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature. 1998;395:917–921. doi: 10.1038/27699. [DOI] [PubMed] [Google Scholar]
- Torres-Padilla ME, Parfitt DE, Kouzarides T, Zernicka-Goetz M. Histone arginine methylation regulates pluripotency in the early mouse embryo. Nature. 2007;445:214–218. doi: 10.1038/nature05458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremethick DJ. Higher-order structures of chromatin: The elusive 30 nm fiber. Cell. 2007;128:651–654. doi: 10.1016/j.cell.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, Sedgwick B. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature. 2002;419:174–178. doi: 10.1038/nature00908. [DOI] [PubMed] [Google Scholar]
- Trewick SC, McLaughlin PJ, Allshire RC. Methylation: Lost in hydroxylation? EMBO Rep. 2005;6:315–320. doi: 10.1038/sj.embor.7400379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojer P, Reinberg D. Histone lysine demethylases and their impact on epigenetics. Cell. 2006;125:213–217. doi: 10.1016/j.cell.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Trojer P, Zhang J, Yonezawa M, Schmidt A, Zheng H, Jenuwein T, Reinberg D. Dynamic histone H1 isotype 4 methylation and demethylation by histone lysine methyltransferase G9a/KMT1C and the jumonji domain-containing JMJD2/KDM4 proteins. J Biol Chem. 2009;284:8395–8405. doi: 10.1074/jbc.M807818200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WW, Nguyen TT, Shi Y, Barton MC. p53-targeted LSD1 functions in repression of chromatin structure and transcription in vivo. Mol Cell Biol. 2008;28:5139–5146. doi: 10.1128/MCB.00287-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsichlis PN, Lazo PA. Virus-host interactions and the pathogenesis of murine and human oncogenic retroviruses. Curr Top Microbiol Immunol. 1991;171:95–171. doi: 10.1007/978-3-642-76524-7_5. [DOI] [PubMed] [Google Scholar]
- Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Tsuneoka M, Koda Y, Soejima M, Teye K, Kimura H. A novel myc target gene, mina53, that is involved in cell proliferation. J Biol Chem. 2002;277:35450–35459. doi: 10.1074/jbc.M204458200. [DOI] [PubMed] [Google Scholar]
- Tsuneoka M, Fujita H, Arima N, Teye K, Okamura T, Inutsuka H, Koda Y, Shirouzu K, Kimura H. Mina53 as a potential prognostic factor for esophageal squamous cell carcinoma. Clin Cancer Res. 2004;10:7347–7356. doi: 10.1158/1078-0432.CCR-03-0543. [DOI] [PubMed] [Google Scholar]
- Tu S, Teng YC, Yuan C, Wu YT, Chan MY, Cheng AN, Lin PH, Juan LJ, Tsai MD. The ARID domain of the H3K4 demethylase RBP2 binds to a DNA CCGCCC motif. Nat Struct Mol Biol. 2008;15:419–421. doi: 10.1038/nsmb.1400. [DOI] [PubMed] [Google Scholar]
- Tzatsos A, Pfau R, Kampranis SC, Tsichlis PN. Ndy1/KDM2B immortalizes mouse embryonic fibroblasts by repressing the Ink4a/Arf locus. Proc Natl Acad Sci USA. 2009;106:2641–2646. doi: 10.1073/pnas.0813139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzschach A, Lenzner S, Moser B, Reinhardt R, Chelly J, Fryns JP, Kleefstra T, Raynaud M, Turner G, Ropers HH, Kuss A, Jensen LR. Novel JARID1C/SMCX mutations in patients with X-linked mental retardation. Hum Mutat. 2006;27:389. doi: 10.1002/humu.9420. [DOI] [PubMed] [Google Scholar]
- Uren AG, Kool J, Matentzoglu K, de Ridder J, Mattison J, van Uitert M, Lagcher W, Sie D, Tanger E, Cox T, Reinders M, Hubbard TJ, et al. Large-scale mutagenesis in p19(ARF)- and p53-deficient mice identifies cancer genes and their collaborative networks. Cell. 2008;133:727–741. doi: 10.1016/j.cell.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakoc CR, Mandat SA, Olenchock BA, Blobel GA. Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol Cell. 2005;19:381–391. doi: 10.1016/j.molcel.2005.06.011. [DOI] [PubMed] [Google Scholar]
- van Leeuwen F, Gafken PR, Gottschling DE. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002;109:745–756. doi: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- van Oevelen C, Wang J, Asp P, Yan Q, Kaelin WG, Jr, Kluger Y, Dynlacht BD. A role for mammalian Sin3 in permanent gene silencing. Mol Cell. 2008;32:359–370. doi: 10.1016/j.molcel.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA, Chinnaiyan AM. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeq B, Laxman B, Cao X, Jing X, Ramnarayanan K, Brenner JC, Yu J, et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008;322:1695–1699. doi: 10.1126/science.1165395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M, Mulder KW, Denissov S, Pijnappel WW, van Schaik FM, Varier RA, Baltissen MP, Stunnenberg HG, Mann M, Timmers HT. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- Wang H, Huang ZQ, Xia L, Feng Q, Erdjument-Bromage H, Strahl BD, Briggs SD, Allis CD, Wong J, Tempst P, Zhang Y. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science. 2001;293:853–857. doi: 10.1126/science.1060781. [DOI] [PubMed] [Google Scholar]
- Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004a;431:873–878. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, Sonbuchner LS, McDonald CH, Cook RG, Dou Y, Roeder RG, Clarke S, et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science. 2004b;306:279–283. doi: 10.1126/science.1101400. [DOI] [PubMed] [Google Scholar]
- Wang GG, Cai L, Pasillas MP, Kamps MP. NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat Cell Biol. 2007;9:804–812. doi: 10.1038/ncb1608. [DOI] [PubMed] [Google Scholar]
- Wang L, Pal S, Sif S. Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol Cell Biol. 2008a;28:6262–6277. doi: 10.1128/MCB.00923-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SC, Oelze B, Schumacher A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS ONE. 2008b;3:e2698. doi: 10.1371/journal.pone.0002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Hevi S, Kurash JK, Lei H, Gay F, Bajko J, Su H, Sun W, Chang H, Xu G, Gaudet F, Li E, et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet. 2009;41:125–129. doi: 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- Wellmann S, Bettkober M, Zelmer A, Seeger K, Faigle M, Eltzschig HK, Buhrer C. Hypoxia upregulates the histone demethylase JMJD1Avia HIF-1. Biochem Biophys Res Commun. 2008;372:892–897. doi: 10.1016/j.bbrc.2008.05.150. [DOI] [PubMed] [Google Scholar]
- Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- Wilsker D, Patsialou A, Dallas PB, Moran E. ARID proteins: A diverse family of DNA binding proteins implicated in the control of cell growth, differentiation, and development. Cell Growth Differ. 2002;13:95–106. [PubMed] [Google Scholar]
- Wissmann M, Yin N, Muller JM, Greschik H, Fodor BD, Jenuwein T, Vogler C, Schneider R, Gunther T, Buettner R, Metzger E, Schule R. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol. 2007;9:347–353. doi: 10.1038/ncb1546. [DOI] [PubMed] [Google Scholar]
- Wysocka J, Allis CD, Coonrod S. Histone arginine methylation and its dynamic regulation. Front Biosci. 2006a;11:344–355. doi: 10.2741/1802. [DOI] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, Wu C, Allis CD. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006b;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Zhu Z, Han G, Ye X, Xu B, Peng Z, Ma Y, Yu Y, Lin H, Chen AP, Chen CD. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc Natl Acad Sci USA. 2007;104:19226–19231. doi: 10.1073/pnas.0700735104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Jing C, Kelly G, Walker PA, Muskett FW, Frenkiel TA, Martin SR, Sarma K, Reinberg D, Gamblin SJ, Wilson JR. Specificity and mechanism of the histone methyltransferase Pr-Set7. Genes Dev. 2005;19:1444–1454. doi: 10.1101/gad.1315905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi H, Deguchi K, Aono A, Tani Y, Kishimoto T, Komori T. Growth disturbance in fetal liver hematopoiesis of Mll-mutant mice. Blood. 1998;92:108–117. [PubMed] [Google Scholar]
- Yamagishi T, Hirose S, Kondo T. Secondary DNA structure formation for Hoxb9 promoter and identification of its specific binding protein. Nucleic Acids Res. 2008;36:1965–1975. doi: 10.1093/nar/gkm1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell. 2006;125:483–495. doi: 10.1016/j.cell.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Yamane K, Tateishi K, Klose RJ, Fang J, Fabrizio LA, Erdjument-Bromage H, Taylor-Papadimitriou J, Tempst P, Zhang Y. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol Cell. 2007;25:801–812. doi: 10.1016/j.molcel.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Yang ZQ, Imoto I, Fukuda Y, Pimkhaokham A, Shimada Y, Imamura M, Sugano S, Nakamura Y, Inazawa J. Identification of a novel gene, GASC1, within an amplicon at 9p23–24 frequently detected in esophageal cancer cell lines. Cancer Res. 2000;60:4735–4739. [PubMed] [Google Scholar]
- Yang ZQ, Imoto I, Pimkhaokham A, Shimada Y, Sasaki K, Oka M, Inazawa J. A novel amplicon at 9p23–24 in squamous cell carcinoma of the esophagus that lies proximal to GASC1 and harbors NFIB. Jpn J Cancer Res. 2001;92:423–428. doi: 10.1111/j.1349-7006.2001.tb01112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Gocke CB, Luo X, Borek D, Tomchick DR, Machius M, Otwinowski Z, Yu H. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol Cell. 2006;23:377–387. doi: 10.1016/j.molcel.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Yoon KA, Park S, Hwangbo B, Shin HD, Cheong HS, Shin HR, Lee JS. Genetic polymorphisms in the Rb-binding zinc finger gene RIZ and the risk of lung cancer. Carcinogenesis. 2007;28:1971–1977. doi: 10.1093/carcin/bgm156. [DOI] [PubMed] [Google Scholar]
- You A, Tong JK, Grozinger CM, Schreiber SL. CoREST is an integral component of the CoREST-human histone deacetylase complex. Proc Natl Acad Sci USA. 2001;98:1454–1458. doi: 10.1073/pnas.98.4.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu BD, Hanson RD, Hess JL, Horning SE, Korsmeyer SJ. MLL, a mammalian trithorax-group gene, functions as a transcriptional maintenance factor in morphogenesis. Proc Natl Acad Sci USA. 1998;95:10632–10636. doi: 10.1073/pnas.95.18.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger J, Stark A, Kellis M, Hong JW, Nechaev S, Adelman K, Levine M, Young RA. RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat Genet. 2007;39:1512–1516. doi: 10.1038/ng.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Reinberg D. Transcription regulation by histone methylation: Interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15:2343–2360. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Hu CM, Yuan YS, He CH, Zhao Q, Liu NZ. Expression of Mina53 and its significance in gastric carcinoma. Int J Biol Markers. 2008;23:83–88. doi: 10.1177/172460080802300204. [DOI] [PubMed] [Google Scholar]
- Zhao XD, Han X, Chew JL, Liu J, Chiu KP, Choo A, Orlov YL, Sung WK, Shahab A, Kuznetsov VA, Bourque G, Oh S, et al. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell. 2007;1:286–298. doi: 10.1016/j.stem.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Liu C, Ge Z, Fang X, Zhang X, Straat K, Bjorkholm M, Xu D. Lysine-specific demethylase 1 (LSD1) Is required for the transcriptional repression of the telomerase reverse transcriptase (hTERT) gene. PLoS ONE. 2008;3:e1446. doi: 10.1371/journal.pone.0001446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet. 2007;39:61–69. doi: 10.1038/ng1929. [DOI] [PubMed] [Google Scholar]