

Abstract
The LIM-only family of proteins comprises four members; two of these (LMO1 and LMO2) are involved in human T-cell leukemia via chromosomal translocations, and LMO2 is a master regulator of hematopoiesis. We have carried out gene targeting of the other members of the LIM-only family, viz., genes Lmo1, Lmo3 and Lmo4, to investigate their role in mouse development. None of these genes has an obligatory role in lymphopoiesis. In addition, while null mutations of Lmo1 or Lmo3 have no discernible phenotype, null mutation of Lmo4 alone causes perinatal lethality due to a severe neural tube defect which occurs in the form of anencephaly or exencephaly. Since the Lmo1 and Lmo3 gene sequences are highly related and have partly overlapping expression domains, we assessed the effect of compound Lmo1/Lmo3 null mutations. Although no anatomical defects were apparent in compound null pups, these animals also die within 24 h of birth, suggesting that a compensation between the related Lmo1 and 3 proteins can occur during embryogenesis to negate the individual loss of these genes. Our results complete the gene targeting of the LIM-only family in mice and suggest that all four members of this family are important in regulators of distinct developmental pathways.
The family of genes encoding LIM-only proteins (the family of LMO genes) comprises four members. The founding member, LMO1, was identified as a transcription unit near the breakpoint of the chromosomal translocation t(11;14)(p15;q11) in human T-cell acute leukemia (2, 4, 24). The gene was subsequently used to identify two other LIM-only genes, subsequently called LMO2 and LMO3 (3, 10), of which LMO2 was also found near a human T-cell acute leukemia chromosomal translocation, also involving chromosome 11p—in this case t(11;14)(p13;q11) (3, 33). Both of the chromosomal translocations involve the T-cell receptor gene TCRδ or TCRα on chromosome 14q11. In addition, the LMO2 gene is activated in human T-cell acute leukemias via a variant chromosomal translocation, t(7;11)(q35;p13), which breaks at a point near LMO2 on 11p13 and within the T-cell receptor β TCRβ chain gene (35). The final member of this family to be identified was LMO4 (14, 22, 32), which is the most distantly related member of the family. The genes do not exist in tandem, indicating that gene duplication occurred early in the evolution of the family (10, 41). While the LMO1 and LMO2 genes encode proteins that are not closely related (Fig. 1), they are both found on the short arm of human chromosome 11. Conversely, LMO1 and LMO3 genes occur on different chromosomes in humans and mice and encode highly related proteins, especially within the LIM domains (Fig. 1). While LMO1 and LMO2 were discovered because they are activated in distinct human T-cell leukemias by chromosomal translocations, no such association is known for LMO3 or LMO4.
FIG. 1.

Alignment of LIM-only family protein sequences. Proteins of the LIM-only protein family are expressed from four genes—LMO1, LMO2, LMO3, and LMO4—in humans and mice. The derived protein sequences (LMO = human; Lmo = mouse) are shown in single-letter code and are aligned to the human LMO1 sequence (stars represent identities). The proteins are broken into the two LIM domains (LIM1 and LIM2), and the Cys, His, and Asp residues involved in zinc binding are underlined. There is a remarkable conservation in amino acid sequence in each family member between species. Conservation between the different proteins is lower, except between LMO1 and LMO3.
Analysis of the derived protein sequences from cDNA clones showed that the LMO1 mRNA encoded a potential zinc finger protein (24) and that LMO proteins comprise two LIM domains (4, 31), which are zinc-binding domains initially found in three proteins called Lin11, Isl-1, and Mec1 (11, 20, 50). An alignment of the LMO proteins of humans and mice is shown in Fig. 1. A LIM domain comprises two zinc finger-like structures related to the GATA DNA-binding domain (28), although there is no evidence of direct, sequence-specific DNA binding by LIM domains. Rather, a protein-protein interaction function has been predicted (31) and demonstrated for the LMO proteins (42, 43, 47, 48). The interaction of the LMO2 protein with the basic helix-loop-helix protein TAL1/SCL, with GATA-1, and with LDB1 in a bipartite DNA-binding complex (27, 43, 47, 48) in erythroid cells indicates a role for LMO2 in controlling expression of target genes, by positive or negative regulation (44, 49). Similarly, the T-cell acute-leukemia-associated complex, which has a bipartite DNA-binding function by interaction of Lmo2 with Tal1 (13), presumably controls target genes in a specific manner dictated by the two DNA-binding arms of the protein complex.
The normal role of Lmo2 in development has been established by gene targeting in mice. These studies showed that Lmo2 is a key regulator of hematopoiesis, being necessary both for primitive, yolk sac erythropoiesis in embryos and in definitive hematopoiesis in adults (49, 54). Furthermore, Lmo2 has a controlling function in vascular formation during embryogenesis, as the null mutation of the gene results in failure of the process of endothelial remodeling (angiogenesis) (53). In this way, Lmo2 has features of a master regulator of cell fate, which is a feature thought to be crucial for its role in tumorigenesis following chromosomal translocations (30). Little is known about the function of the other members of the LIM-only family. Lmo1, Lmo3, and Lmo4 are expressed in the developing mouse brain (5, 6, 10, 14, 17, 22, 32, 37, 46, 51) and there is some overlap in the profiles of expression. The related genes Lmo1 and Lmo3 have a different timing of expression in embryonic brain, with Lmo1 being early and Lmo3 being relatively late (5, 10). Neither gene has distinctive expression in hematopoietic tissue. Lmo4, on the other hand, has prolonged expression in brain and is expressed during hematopoiesis (22).
Since Lmo2 has a distinctive role in development, we have studied possible roles of the other members of the LIM-only family by creating null mutations in mice using homologous recombination. Null mutation of Lmo4 results in embryonic lethality, and this mutation can give rise to exencephaly in homozygous null mice. In the case of Lmo1 and Lmo3, null mutation of either gene alone permits production of viable mice, but perinatal death occurs in pups with compound null mutations in both genes. The cause of death could not be identified, but a neural defect seems likely, as expression of these genes is largely restricted to the brain in mouse embryos.
MATERIALS AND METHODS
Targeting constructs.
Lmo1 and Lmo3 genomic λ clones were isolated from a 129 library in λ2001 (49). Gene targeting into exon 2 of Lmo1 was facilitated by the addition of BamHI adapters to the unique ScaI site and cloning the 1.1-kb pMC1-neopA cassette (55) modified as a BamHI fragment or cloning a 5.1-kb β-galactosidase reporter comprising an encephalomyocarditis virus (EMC) internal ribosome entry site (IRES) linked to the lacZ gene plus the simian virus 40 polyadenylation site followed by the MC1-neopA sequence (clone p1056). This fragment was cloned as a BamHI fragment into the engineered BamHI site of exon 2. The targeting vector comprised a 5.4-kb EcoRI fragment cloned in pBSpt. Targeted clones were detected using a 3′ external probe comprising a 1.5-kb EcoRI-XbaI fragment, which was also used to monitor the transmission of the targeted allele in mice. The Lmo3 targeting strategy employed the introduction of a BamHI site into Lmo3 exon 2 and cloning either pMC1-neopA or the p1056 lacZ reporter. The targeting vector comprised a 6.6-kb BamHI-XhoI fragment cloned in pBSpt. Targeted clones were detected using a 5′ external probe comprising a 1-kb BamHI fragment which was also used to monitor the transmission of the targeted allele in mice. The nomenclature of targeted alleles is summarized in Table 1.
TABLE 1.
Nomenclature of targeted allelesa
| Allele nomenclature | Selectable marker | Gene targeting strategy |
|---|---|---|
| Lmo1− | Neomycin | neo insertion |
| Lmo1Z | Neomycin | IRES-lacZ insertion |
| Lmo3− | Neomycin | neo insertion |
| Lmo3Z | Neomycin | IRES-lacZ insertion |
| Lmo4− | Neomycin | neo insertion |
| Lmo4-GFP | Hygromycin | GFP knockin |
The various Lmo gene targeted alleles used in the study comprise either (i) gene inactivation by insertion of a gene encoding a selectable marker (either neomycin [neo] or hygromycin [hyg] genes) to create the Lmo− allele, (ii) inactivation using insertion of an IRES-lacZ cassette including a neo gene to create the LmoZ allele, or (iii) knockin of the GFP gene in frame with the Lmo4 coding sequence (together with the hyg selectable marker) to create the Lmo4-GFP allele. Compound Lmo1 and Lmo3 genotypes comprising combinations of alleles are designated +/− (a wild-type and a neo insertion allele), Z/+ (IRES-lacZ insertion and a wild-type allele), and Z/Z (both alleles have IRES-lacZ insertions).
The restriction map of the mouse Lmo4 gene was determined from 129 genomic λ clones (41). A 9.8-kb XhoI Lmo4 genomic DNA fragment containing exon 1b to exon 4 was isolated from the λ phage genomic clone and subcloned into pBluescript. The neomycin resistance gene (neo) was cloned as a 1.1-kb BamHI neo fragment from pMC1-neopA (55) into the blunted EagI site of Lmo4 exon 2, producing a truncated gene which encodes only the first 15 amino acids of Lmo4. The herpes simplex viral thymidine kinase (tk) gene was inserted as a 1.8-kb blunted XbaI fragment from plasmid MC1-tk (38). The Lmo4-GFP knockin clone was made by cloning the hygromycin resistance gene as a 1.7-kb blunted ClaI-BglII fragment from pPGK-hyg (1, 21) containing hyg into the filled-in AflII site of pEGFP-N1 (Clontech) to create pEGFP-hyg. A 2.7-kb blunted BamHI-EcoRV fragment from pEGFP-hyg was inserted into the blunted EagI site of Lmo4 exon 2. The herpes simplex virus tk gene was inserted to allow for negative selection.
Gene targeting in ES cells and generation of mutant mice.
Embryonic stem (ES) cell lines used were E14 for Lmo1 and Lmo3 or CCB for Lmo4 targeting. Cells were grown on neomycin- or hygromycin-resistant feeders in the presence of leukemia inhibitory factor as described previously (49). DNA transfection was carried out by electroporation, and clones were selected by resistance to G418 (400 μg/ml) or hygromycin (300 μg/ml). Targeted clones were confirmed by filter hybridization and injected in donor blastocysts for generation of chimeric mice. Male chimeras were bred with C57BL/6 females, and germ line transmission was assessed by filter hybridization of tail biopsy specimen DNA. Filter hybridization (36) was carried out as described previously (23). Restriction enzyme-digested DNA was fractionated on 0.8 or 1% agarose and transferred to nylon membranes following denaturation. The filters were hybridized with randomly 32P-labeled DNA probes (9), and specific signal was detected by autoradiography.
Analysis of mice.
Anesthetized animals were transcardially perfused with 4% paraformaldehyde (PFA) in phosphate-buffered saline. Brains were removed, postfixed for 15 min in 4% PFA on ice, and equilibrated overnight at 4°C in phosphate-buffered saline-30% sucrose. Sections (40 μm thick) were cut on a sliding microtome and incubated free-floating in X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) at 37°C (19). After X-Gal staining, sections were mounted on slides, counterstained with neutral red, and coverslipped with DePeX. RNA analysis was carried out by filter hybridization using glyoxal-treated RNA (39). Total cellular RNA (10 μg per lane) was fractionated on 1.4% agarose and transferred to nylon membranes which were hybridized with randomly 32P-labeled DNA probes (9), and specific signal was detected by autoradiography. Flow cytometry was carried out using antibodies detecting specific hematopoietic lineage markers (purchased from Pharmingen). Mice were screened for populations expressing Sca-1, Mac-1, Gr-1, B220, Thy-1, CD3, CD4, CD8, CD44, and Ter119.
RESULTS
Effect of targeted mutation of the Lmo1 or Lmo3 gene.
Two kinds of gene targeting constructs were made with Lmo1 and Lmo3 genes, namely, insertion into exons 2 of neo or lacZ genes, in which the latter was expressed from an EMC-IRES (Fig. 2A and B). Targeted ES cells were generated from the four constructs, and these were injected into blastocysts to make chimeras and germ line carriers of the null alleles (Fig. 2C and D). Identical results were obtained with neo- or lacZ-targeted (herein shown as Z) ES cells in each case. No effect on viability, longevity, or fertility could be observed in either Lmo1 or Lmo3 homozygous null mice, and offspring of heterozygous pairs were produced with the expected Mendelian ratios (Table 2 shows genotype data of pups at weaning from Lmo1 and Lmo3 neomycin targeting). Histology of major tissues such as lung, heart, kidney, thymus, spleen, bone marrow, and pancreas did not reveal any noticeable differences between wild-type, heterozygous, or homozygous mice (J. Anderson and T. H. Rabbitts, data not shown). The expression of Lmo1 and Lmo3 has been found mainly in brain of the developing mouse embryos (10, 17); Lmo1 expression is highest at early stages, and little expression is seen by in situ hybridization of RNA by about embryonic day 19 (E19) (10). Lmo3 on the other hand, is expressed to a much lower extent at early embryonic stages and is quite widely expressed in the brain and spinal cord by E19 (10, 17). Examination of brains from Lmo1−/− or Lmo3−/− mice did not reveal any differences from brains of wild-type mice (S. Hunt and T. H. Rabbitts, data not shown). The expression of β-galactosidase was in this respect a useful marker of Lmo-expressing cells, giving a guide both to overall architecture and to specific structural features. Lmo3 is the more widely expressed of the two genes just before birth, as illustrated in Fig. 3, which shows the distribution of β-galactosidase expressed from the Lmo3-lacZ allele in either heterozygous (Lmo3+/Z) or homozygous (Lmo3Z/Z) states combined with a heterozygous Lmo1+/− background. Expression is evident in cerebral cortex, hippocampus, cerebellum, and brain stem, in keeping with the in situ-hybridization data. No differences appear to occur between mice heterozygous for Lmo3 and those homozygous for Lmo3. We conclude that there is no detectable phenotypic change resulting from null mutation of either Lmo1 or Lmo3.
FIG. 2.

Targeted disruption of the Lmo1 and Lmo3 genes. (A) Restriction maps of wild-type Lmo1, neo replacement targeting vector, and lacZ knockin targeting vector. The MC1-neo-pA cassette or an IRES-lacZ reporter was inserted at the ScaI site of Lmo1 exon 2. (B) Restriction maps of wild-type Lmo3, neo replacement targeting vector, and the lacZ knockin targeting vector. The MC1-neo-pA cassette or an IRES-lacZ reporter cassette was inserted into a BamHI site mutagenized into the Lmo3 exon 2. (C and D) Detection of targeted Lmo1 (C) or Lmo3 (D) loci was carried out by filter hybridization using the indicated probes. The Lmo1 targeted allele was detected as a 7.6-kb BamHI fragment, and the Lmo3 targeted allele was detected as a 7.8-kb EcoRI fragment. The results shown were obtained using DNA from pups of a cross between (Lmo1Z/Z; Lmo3+/−) and (Lmo1Z/+; Lmo3−/−) mice. The litter of nine pups at P0 included two pups with the compound null genotype (Lmo1Z/Z; Lmo3−/−) (pups 8 and 9).
TABLE 2.
Genotypes of litters from interbreedings of Lmo1 and Lmo3 mutant micea
| Parents | No. of pups (total) | No. (%) of pups with genotype
|
||
|---|---|---|---|---|
| Wild type (+/+) | Heterozygous (+/−) | Homozygous (−/−) | ||
| Lmo1+/− × Lmo1+/− | 57 | 19 (33) | 26 (46) | 12 (21) |
| Lmo3+/− × Lmo3+/− | 50 | 15 (30) | 25 (50) | 10 (20) |
Genotypes of mice from various interbreeding regimens for Lmo1 and Lmo3 mutant mice were determined by filter hybridization of tail biopsy specimen DNA. The numbers shown exemplify the genotypes of pups from about postnatal day 28 (P28) from a cross between Lmo1+/− heterozygous mice or between Lmo3+/− heterozygous mice.
FIG. 3.

Macroscopic β-galactosidase expression in brains from Lmo3 mutant mice. (Lmo1+/−; Lmo3Z/+) mice were interbred with (Lmo1+/−; Lmo3Z/Z) mice, and offspring were sacrificed at E19. The brains were dissected and whole mount stained with X-Gal to reveal β-galactosidase expression. Simultaneously, the genotypes of the mice were established using filter hybridization. The whole mount photographs show comparisons of the indicated genotypes of Lmo3 heterozygous (A) and homozygous (B) mouse brains. Abbreviations: ob, olfactory bulb; cc, cerebral cortex; th, thalamus; mb, midbrain; bs, brain stem; sc, spinal cord.
Postnatal death in Lmo1/Lmo3 compound null mutant mice.
The derived protein sequences of Lmo1 and Lmo3 show extremely high homology (Fig. 1), and considering their overlapping patterns of expression in the developing mouse brain (10, 17), it seemed possible that complementation of Lmo1-Lmo3 protein activity could explain the viability of the null mutant mice. Interbreeding was undertaken to generate mice with the various possible compound genotypes (Table 3). The most striking observation was that no mice with a compound null phenotype were found in any weaned litters; this included crosses with both neo replacement and lacZ knockins; i.e., no mice with any of the following genotypes survived to weaning: (Lmo1−/−; Lmo3−/−), (Lmo1−/−; Lmo3Z/Z), (Lmo1Z/Z; Lmo3−/−), or (Lmo1Z/Z; Lmo3Z/Z). Heterozygous Lmo1 or Lmo3 mice with the reciprocal null mutation were viable. Thus, the compound (Lmo1; Lmo3) null mutation was lethal.
TABLE 3.
Genotypes of litters resulting from (Lmo1Z/Z; Lmo3+/−) × (Lmo1Z/+; Lmo3−/−) interbreedinga
| Age | No. of pups (total) | Genotype of pups | No. (%) of pups with genotype |
|---|---|---|---|
| P0 | 36 | Lmo1Z/Z; Lmo3−/− | 8 (22) |
| Lmo1Z/Z; Lmo3+/− | 12 (33) | ||
| Lmo1Z/+; Lmo3−/− | 12 (33) | ||
| Lmo1Z/+; Lmo3+/− | 4 (11) | ||
| P2 | 28 | Lmo1Z/Z; Lmo3+/− | 12 (43) |
| Lmo1Z/+; Lmo3−/− | 12 (43) | ||
| Lmo1Z/+; Lmo3+/− | 4 (14) | ||
| Lmo1Z/Z; Lmo3−/− | 0 (0) |
Genotypes of mice from the indicated interbreeding regimen were determined by filter hybridization of tail biopsy specimen DNA. The numbers shown exemplify the genotypes of pups from a cross of compound-genotype mice [(Lmo1Z/Z; Lmo3+/−) × (Lmo1Z/+; Lmo3−/−)]. Note that a similar outcome of P0-P2 compound homozygous pups was obtained when either lacZ knockin or neo replacement was used for either Lmo1 or Lmo3 mutant mice.
We further analyzed the lethality by examining newborn pups from (Lmo1Z/Z; Lmo3+/−) mice crossed with (Lmo1Z/+; Lmo3−/−) mice and found that immediately following birth, compound null mice (Lmo1Z/Z; Lmo3−/−) were alive but died shortly thereafter. Genotyping data from a typical postnatal day 0 (P0) litter of such a cross is shown in Fig. 2, using either Lmo1 (Fig. 2C) or Lmo3 (Fig. 2D) probes. This litter had nine pups, and two of these (pups 8 and 9) had the compound genotype. These pups died within a few hours of birth. Typically, compound genotype pups were able to feed as judged by milk in their stomachs but did not thrive and we observed that these mice die usually within 24 h of birth. For instance, a group of litters comprising 36 mice analyzed at P0 showed eight (Lmo1Z/Z; Lmo3−/−) pups but the same litters analyzed 48 h later had no surviving (Lmo1Z/Z; Lmo3−/−) pups (Table 3). In separate analyses, one (Lmo1Z/Z; Lmo3−/−) pup survived for 18 days (until P18), but its growth was very stunted.
The compound Lmo1/Lmo3 null mutation affects viability within the first few hours after birth but the cause of death is currently obscure. Histological analysis of major organs including heart, lung, kidney, spleen, thymus, and pancreas showed no obvious difference between compound null mice and controls, and flow cytometric analysis of lineage marker distribution of hematopoietic cells showed no variance for normal patterns (data not shown). As the major expression of Lmo1 and Lmo3 is in developing brain and spinal cord, the anatomy of these structures was examined, but no gross morphological change was observed. The lacZ knockin provided a simple reporter for Lmo1 or Lmo3 expression, and brains taken at P0 from mutant mice were sectioned and subjected to histochemical analysis. All the main structures could be observed in compound Lmo1/Lmo3 null brains. Sections of brains from P0 mice were studied after X-Gal staining, and representatives are shown in Fig. 4 from a (Lmo1−/−; Lmo3Z/Z) null compound. These studies did not reveal any specific differences between null and control brains or any defects to which the cause of death of the compound genotype mice could be attributed.
FIG. 4.

Histological comparison of brain morphology in compound null Lmo1 or Lmo3 mutant mice. Sections of various brain regions from (Lmo1−/−; Lmo3Z/Z) (designated compound null) or (Lmo1+/−; Lmo3Z/+) (designated control) mice are compared. Brains were removed from mice transcardially perfused with 4% PFA at P0, and sections were made on a sliding microtome. These were stained free-floating with X-Gal, mounted, and counterstained with neutral red. Regions of brain illustrated in sections shown are abbreviated as follows: hp, hippocampus; s, subiculum; ec, entorhinal cortex; oc, olfactory cortex; gp, globus pallidus; am, amygdala; CA1, CA3 of hippocampus.
Targeted mutation of the Lmo4 gene.
While Lmo1 and Lmo3 are highly conserved, Lmo4 is the least conserved of the LIM-only (LMO) family (Fig. 1). Lmo4 has a broad expression pattern (6, 14, 22, 45, 46, 51), unlike Lmo1 and Lmo3. The Lmo4 gene was inactivated by homologous recombination in mouse ES cells. Two targeting vectors were used, one to insert the neomycin (neo) resistance gene in Lmo4 exon 2 (Fig. 5A) and the other to create an in-frame fusion of the amino-terminal end of Lmo4 (at exon2) with green fluorescent protein (GFP) (Fig. 5B). Two independent clones were derived (D1 and D8) with a single Lmo4 allele disrupted by the neo gene. Chimeric mice were obtained from both clones, and germ line transmission was obtained. Representative genotyping of mice at P0 is shown in Fig. 5C, which illustrates that both heterozygous (+/−) and homozygous (−/−) null pups were obtained. Similar results were obtained with both Lmo4 targeted clones. Clone D8 was used for a second targeting event using the GFP vector, and this generated cells in which homologous recombination occurred either in the wild-type Lmo4 allele to give −/− ES cells (Lmo4−/−, comprising one allele mutated with neo and the other by GFP insertion) or in the neo-targeted allele to give GFP knockin heterozygous cells (Lmo4+/− GFP-hyg) (Fig. 5D).
FIG. 5.

Targeted disruption of the Lmo4 gene. (A) Restriction maps of the Lmo4 gene and the BamHI linearized targeting vector plm4-neo-tk, together with the predicted structure of the targeted Lmo4 locus following a homologous recombination event. The black boxes represent coding exons of Lmo4. The expected lengths of the restriction fragments diagnostic for homologous recombination are indicated by double-headed arrows. (B) Restriction maps of the Lmo4 gene and the BamHI-linearized targeting vector plm4GFP-hyg-tk and the predicted structure of the targeted Lmo4 locus following a homologous recombination event are shown. The expected lengths of the restriction fragments diagnostic for homologous recombination are indicated by double-headed arrows. Abbreviations for restriction sites: B, BamHI; S, SacI; X, XhoI; E, EagI. (C) Genotype analysis of mice at P0 derived from an Lmo4+/− × Lmo4+/− intercross. Representative DNA samples from each genotype—i.e., wild-type (wt), heterozygous Lmo4+/−, and homozygous Lmo4 null−/− mice—were digested with SacI, and filter hybridization with the 3′ probe was performed. Wild-type and targeted alleles are 3.8 and 5 kb, respectively. BamHI digestion of the same samples yields fragments of 6.6 (wild-type allele) and 7.9 (targeted allele) kb when hybridized with the 5′ probe which confirms the genotypes. (D) The ES cell line D8 (Lmo4+/−) was subjected to a second round of gene targeting with the linearized vector plm4GFP-hyg-tk. DNA samples from representative ES cell clones of each genotype—Lmo4+/− neo (i.e., the original Lmo4+/− clone with neo inserted into exon 2), Lmo4+/− GFP-hyg (i.e., clone with the original neo mutant allele “re-targeted” by plm4GFP-hyg-tk), and Lmo4−/−—were digested with BamHI and hybridized with the 5′ probe. This yields fragments of 6.6 (wild-type allele) and 7.9 (neo targeted allele) kb in Lmo4+/− neo cells, fragments of 6.6 and 9.3 kb (GFP-hyg targeted allele) in Lmo4+/− GFP-hyg cells, and fragments of 7.9 and 9.3 kb in Lmo4−/− cells. Hybridizing SacI-digested DNA with a neo probe showed only the 5-kb band in Lmo4+/− neo and Lmo4−/− ES cell clones, while rehybridizing the BamHI-digested DNA with the hyg probe yielded only the 7.9-kb GFP-hyg targeted band. Control DNA from the wild-type (wt) ES cell line is also shown. Wild-type, neo targeted, and GFP-hyg targeted alleles are indicated by bands of 6.6, 7.9, and 9.3 kb, respectively.
Neonatal lethality in Lmo4 homozygotes.
Heterozygous germ line carrier mice were obtained from both neo and GFP knockin ES cells and did not display any overt phenotype. The effect of obtaining null mutant offspring was studied. When genotypes were analyzed at weaning, we found no evidence of homozygous mouse survival, whereas heterozygous survivors occurred at the expected frequency (Table 4). Overall, the lethality of Lmo4 null mice is 100% at P28. In addition, homozygous mice could be detected throughout embryogenesis, and identical data were obtained from both targeted ES cell derivatives. As the Lmo4 null mutation appeared to be lethal to embryos, offspring from Lmo4+/− heterozygous crosses were genotyped at progressive time points to establish when lethality was occurring (Table 4). Embryos analyzed at day E9.5 of gestation showed a Mendelian distribution of Lmo4+/+, Lmo4+/−, and Lmo4−/− embryos. At embryonic stages E15.5 to E18.5, although Lmo4−/− embryos were found, they were present at a frequency (∼12%) lower than the expected Mendelian ratio (Table 4). Therefore, about 50% of Lmo4−/− embryos died after embryonic stage E9.5, and the remaining 50% were born dead.
TABLE 4.
Genotypes of litters from intercrosses of Lmo4 heterozygous micea
| Age | No. of pups | No. (%) of pups with genotype
|
||
|---|---|---|---|---|
| Wild type (+/+) | Heterozygous (+/−) | Homozygous (−/−) | ||
| E9.5 | 28 | 9 (32) | 13 (46) | 6 (22) |
| E10.5 | 16 | 3 (19) | 11 (69) | 2 (12) |
| E15.5 | 19 | 5 (26) | 11 (58) | 3 (16) |
| E16.5 | 13 | 3 (33) | 9 (69) | 1 (7) |
| E17.5 | 22 | 5 (23) | 14 (64) | 3 (13) |
| E18.5 | 10 | 4 (40) | 4 (40) | 2 (20) |
| P28 | 151 | 40 (26) | 111 (74) | 0 (0) |
Heterozygous Lmo4 null mutant mice (neo insertion) were bred, and the genotypes of embryos or pups were determined by filter hybridization of yolk sac or tail DNA from the indicated number of progeny at embryonic stages E9.5 to E10.5 and E15.5 to E18.5 and at P28.
Failure of neural tube closure in Lmo4 null mutant embryos.
When Lmo4−/− pups survive to birth, they are born dead. These pups were smaller than their littermates, and about 30% have a physically distinctive phenotype, namely, the cranium is completely absent and the brain is heavily malformed (Fig. 6A), which is reminiscent of the condition of anencephaly, a severe cranial neural tube developmental defect leading to absence of the cranium and exposure and malformation of the brain (7). Spina bifida is another form of neural tube defect (7) but was not found in these mice (Fig. 6B, dorsal view). The facial appearance of Lmo4−/− pups was also different from that of their littermates in that the nasal part was more broad and protruded (Fig. 6A) and the skin was almost white in color, possibly due to depigmentation of the skin (Fig. 6A). All internal organs were present and grossly normal. Not all dead-born Lmo4−/− pups had anencephaly, and some had an appearance indistinguishable from that of their +/− littermates. Therefore, the penetrance of this anencephalic phenotype is incomplete.
FIG. 6.

Neural tube closure defect in Lmo4 homozygous null mutant mice. Timed matings between heterozygous Lmo4 mice were terminated at P0 (A), E18.5 (B), or E9.5 (C and D), and pups were genotyped by filter hybridization using the 3′ probe. (A to D) Whole-mount photographs are shown. (A) Lateral view of a homozygous Lmo4−/− dead-born pup, compared to a heterozygous Lmo4+/− live-born littermate. The Lmo4−/− pup exhibits anencephaly in which the cranium and the brain are absent. The facial features are also markedly abnormal compared to those of the Lmo4+/− pup. (B) Lateral and dorsal views comparing an E18.5 homozygous Lmo4−/− embryo with a heterozygous Lmo4+/− littermate. The Lmo4−/− embryo exhibits exencephaly in which the cranium is absent and the is brain exposed. The nasal part of the Lmo4−/− embryo is broader, and the ears are located at a much lower position. (C and D) Posterior views of an E9.5 Lmo4−/− embryo show that the cranial neural tube remains open at the position of the anterior neuropore (arrowhead). The same views taken from a wild-type littermate reveal that the cranial neural tube has closed by E9.5. (E and F) Histology of dorsal telecephalon of homozygous or heterozygous Lmo4 embryos at E18.5. Coronal section through the head, showing complete loss of brain structure, necrosis, and loss of tissue (skin and mesenchyme) overlying the dorsal part of the brain in an Lmo4 homozygous embryo (E) compared with a heterozygous littermate (F). The brain stem in Lmo4 homozygous embryos was intact and apparently normal. Abbreviations: e, epidermis; cg, cortical grey matter; bg, basal ganglia. The sections were stained with neutral red. Scale bar: 100 μm.
Between E15.5 and E18.5, Lmo4−/− embryos were found at a lower frequency than the expected Mendelian distribution (Table 4). About half of the Lmo4−/− embryos exhibited the cranial neural tube defect exencephaly, in which there is a defect in the cranium and the brain is exposed (Fig. 6B). This is consistent with the findings in the Lmo4−/− dead-born pups. The developmental consequences of the failure of neural tube closure were studied with histological analysis of Lmo4−/− and Lmo4+/− littermates. The normal architecture of the cerebral cortex was lost in Lmo4−/− embryos (compare Fig. 6E and F, showing a section of an Lmo4+/− embryo) with the brain showing widespread necrosis, and thus it is not possible, for instance, to distinguish areas within the cortex. The anencephaly condition seen in the dead-born pups might be due to the fact that the friable brains were worn off during the process of labor. Moreover, like the dead-born pups, the exencephalic embryos also had very white skin and abnormal facial appearances compared to that of their littermates (Fig. 6B). This indicates that Lmo4 is required for the proper development of the neural tube.
The penetrance of this neural tube defect in Lmo4−/− embryos is only about 40 to 50%. Since the Lmo4 gene in the homozygous null animals is interrupted by inserting a neo gene in its exon 2, which encodes the first LIM domain, it could be possible that an alternative splicing variant of the Lmo4 transcript consisting of the 5′ untranslated region and exons 3, 4, and 5 is formed (41). This splicing variant, if formed and translated, would contain the second LIM domain of the protein, which might be sufficient for a degree of function. RNA filter hybridization (Fig. 7) was used to check if the nonexencephalic Lmo4−/− embryos have such a splicing variant of Lmo4. Total RNA was isolated from wild-type, Lmo4+/−, exencephalic Lmo4−/−, or nonexencephalic Lmo4−/− whole embryos and assayed by filter hybridization using a probe containing Lmo4 cDNA sequences 3′ to the neo insertion site. By contrast to the wild-type and Lmo4+/− embryos, both Lmo4−/− embryos had no detectable Lmo4 transcript, suggesting that no alternative Lmo4 splicing variant occurs in the nonexencephalic Lmo4−/− embryo (Fig. 5).
FIG. 7.
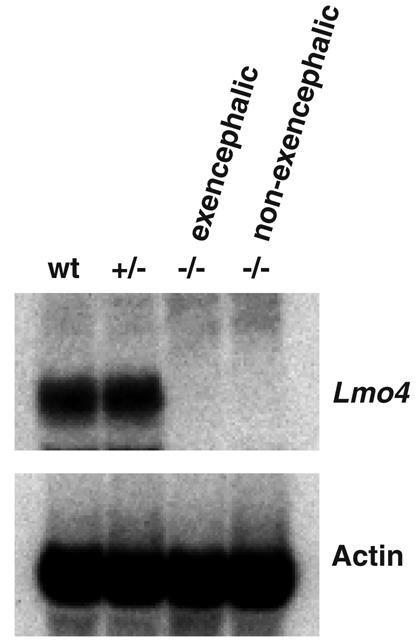
Filter hybridization of RNA from Lmo4 mutant embryos. Total RNA was extracted from E12.5 embryos with normal and mutant phenotypes, fractionated on a 1.4% agarose gel, transferred to a nylon membrane, and hybridized to a probe containing Lmo4 cDNA sequences 3′ to the neo insertion site in exon 2 (see Fig. 5). RNA loading was monitored by reprobing the filter with a mouse β-actin cDNA. RNA of representatives from wild-type (wt), Lmo4+/−, and Lmo4−/− exencephalic or nonexencephalic embryos.
The mouse neural tube forms during E8 to E10 of gestation (15). The neural folds, comprised of neuroectoderm (the future brain and spinal cord tissue), the neural crest, and underlying mesenchyme, fuse to form the neural tube, and if this fails to fuse, it results in exencephaly. The neural tubes were therefore examined in day E9.5 and E10.5 embryos derived from Lmo4+/− crosses. Eight Lmo4−/− embryos were found among 44 embryos examined (Table 4). In three of these Lmo4−/− embryos, the cranial neural tubes failed to close, whereas those of all Lmo4+/− and wild-type embryos had completed closure (Fig. 5). There was no other significant difference between the Lmo4−/− embryos and the Lmo4+/− or wild-type embryos between E9.5 and E10.5. The percentage of Lmo4−/− embryos at E9.5 and E10.5 that failed to have neural tube closure (38%) was very similar to that of embryos at late stages of gestation which exhibited exencephaly (44%). This suggests that the loss of Lmo4−/− embryos during development does not preferentially involve the exencephalic ones.
Intact hematopoiesis in Lmo4 mutant mice.
Various internal organs were taken from E18.5 Lmo4−/− and wild-type embryos for histological analyses. Microscopic examination of tissue sections of skin, heart, lung, liver, spleen, thymus, and kidney of the Lmo4−/− embryos showed no abnormalities. As mouse spleen at embryonic stage E18.5 is still a major hematopoietic organ and Lmo4−/− mice die at birth, examination of spleen at E18.5 was used to determine if hematopoiesis in Lmo4−/− embryos is grossly intact. Cells at different stages of maturation were seen for both erythroid and myeloid lineages. Lymphocytes and megakaryocytes were also present. Therefore, no obvious hematopoietic anomaly was found in Lmo4−/− embryos. Histological sections of the thymus were also examined, and no marked difference in cellularity was observed between the thymus of Lmo4−/− embryos and that of the wild-type mice, and the cortex-medulla demarcation and the general architecture of the Lmo4−/− thymus were well preserved. Fluorescence-activated cell sorter analysis of the lymphocytes from E18.5 Lmo4−/− embryos did not reveal any abnormalities. To ensure that normal T- and B-cell development could occur in adult mice, we examined ES-cell-derived lymphopoiesis in Rag1 null mice, which lack the ability to develop mature lymphocytes (25). Injecting Lmo4−/Lmo4−/− null ES cells into Rag1 null blastocysts and using GFP as a marker of Lmo4 expression resulted in adult mice with a normal lymphocyte differentiation pattern derived from ES cells, indicating that lymphopoiesis is not altered by the Lmo4 null mutation (W. Swat and F. W. Alt, data not shown).
DISCUSSION
The family of genes encoding LIM-only proteins comprises four members (Lmo1, Lmo2, Lmo3, and Lmo4). No other candidate LMO/Lmo genes have appeared in hybridization searches (unpublished data), and there is no evidence of further LMO/Lmo genes in the human genome sequence; thus, with the experiments described in this work, inactivation of all members of this gene family has now been achieved (Table 5).
TABLE 5.
Summary of effects of null mutation of the Lmo family genes
| Targeted gene(s) | Effect of null mutation on viability | Tissue affected | Other defects |
|---|---|---|---|
| Lmo1 | None | None detectable | |
| Lmo2 | Death at E9.5 | Failure of primitive erythropoiesis | Failure of adult hematopoiesis, failure of angiogenesis (assessed in chimeras) |
| Lmo3 | None | None detectable | |
| Lmo1 + Lmo3 | Immediate postnatal death | Unspecified central nervous system abnormalities | |
| Lmo4 | Perinatal death | Neural tube closure defect |
Lmo4 is necessary for neural tube development.
Most Lmo4 null mice died in utero, and about one-third exhibited a severe neural tube defect in the form of exencephaly. The cause of death for the nonexencephalic Lmo4−/− embryos, however, remains obscure. In situ-hybridization studies have revealed that Lmo4 is highly expressed in neural crest cells of the neural fold (16, 22), which is consistent with a neural tube defect in the Lmo4 null embryos. Additional roles in neuritogenesis (46) and in sensory neurons (6) are possible, and these may impinge on the phenotype of the Lmo4 null mutation. A variety of cell types are descendants of neural crest cells, such as the melanocytes and neuroendocrine cells (56). The facial phenotype may also indicate a problem in neural crest cells, which are known to contribute to facial bone and cartilage. The exencephalic Lmo4−/− animals have very pale skin, and this might also be due to defects in melanocytes in the skin causing depigmentation, although further experiments are required to clarify this point. It is of interest that the penetrance of the exencephaly phenotype is incomplete. This may be attributable to the mixed genetic backgrounds (129/Sv × C57BL/6) of the mice generated in this experiment. Moreover, the mechanisms leading to neural tube defect are complex, and the genes involved are heterogeneous, as exemplified by the various genetic mutant mice that manifest the same neural tube defect (15). Further, there may be a number of modifier genes contributing to development of the overt phenotype (26). There may be alternative compensatory pathways to ensure proper neural tube closure. It is also interesting that penetrance of exencephaly in a number of genetic mouse mutants, including p53 mice, is also incomplete (8, 18, 34, 40)
Complementary and noncomplementary functions of Lmo1, Lmo3, and Lmo4 genes in central nervous system development.
The expression of Lmo1 and Lmo3 is highly restricted and is largely found in the developing nervous system and, in the case of Lmo3, persists in adult brain and spinal cord (5, 10, 12, 17). Lmo4 has more-widespread expression (6, 14, 22, 32, 46). The consequence of creating a null mutation in the Lmo4 gene is more profound than that of creating a null mutation in either Lmo1 or Lmo3, as a high incidence of neural tube closure failure is found in Lmo4 null mice. While the Lmo1-Lmo3 compound null mice die soon after birth, no obvious cause of death could be determined. As Lmo3 expression seems restricted to neural tissue, at least through embryogenesis, the defect must presumably occur in neural tissue itself. The complementary profiles of Lmo1 and Lmo3 gene expression and the overlapping timing of expression suggest that Lmo3 fulfills the role of Lmo1 in Lmo1 null mice, which facilitates the viability of these animals. The high sequence similarity in the derived Lmo1 and Lmo3 proteins, especially in the LIM domains (Fig. 1), makes such a molecular complementation seem possible. In compound Lmo1-Lmo3 null mice, loss of both genes results in a lethal phenotype, and therefore complementarity is restricted to the two related genes.
The LIM-only proteins are developmental regulators.
A normal role for each member of the LIM-only family in development is now established (Table 5). Lmo2 is an essential regulator of primitive and definitive hematopoiesis as well as angiogenesis (52-54). The Lmo2 role in hematopoiesis appears unique among the LIM-only proteins, as null mutation of the other three members does not visibly affect the differentiation and distribution of cell types in the hematopoietic compartment, although a role in the function of individual cell types cannot be ruled out. Rather, the Lmo1, Lmo3, and Lmo4 genes are implicated in the control of neural development. In each case, the absence of the gene results in perinatal lethality, with a discernible phenotype in the case of Lmo4 null mutations, i.e., exencephaly or an inferred neural defect in the case of Lmo1 plus Lmo3 mutations.
These developmental regulators can be involved in cancer pathogenesis. Two of the members, LMO1 and LMO2, are involved in distinct T-cell leukemias by association with chromosomal translocation breakpoints (29) but neither LMO3 nor LMO4 has been found at chromosomal translocation breakpoints. However, LMO4 was first recognized as an auto-antigen in breast tumors (32), where it is overexpressed and has a function in differentiation of mammary epithelium (45, 51). The subversion of developmental regulators after chromosomal translocations have occurred in somatic cells has been highlighted as the principle consequence of such abnormal chromosomes in acute leukemias and sarcomas, and the aberrant use of these and other transcription factors in cancer is likely to be a common feature.
Acknowledgments
This work was supported by the Medical Research Council. E.T. was the recipient of a fellowship of the Croucher Foundation, L.F. was the recipient of a Leukemia Research Fund fellowship, I.L. was the recipient of a fellowship from the National Foundation for Cancer Research, and A.W. was the recipient of an MRC Clinical Research Fellowship.
We are indebted to W. Swat and F. W. Alt (Howard Hughes Medical Institute, Children's Hospital, Boston, Mass.) for conducting the Rag complementation experiments.
REFERENCES
- 1.Adra, C. N., P. H. Boer, and M. W. McNurney. 1987. Cloning and expression of the mouse pgk-1 gene and the nucleotide sequence of its promoter. Gene 60:65-74. [DOI] [PubMed] [Google Scholar]
- 2.Boehm, T., R. Baer, I. Lavenir, A. Forster, J. J. Waters, E. Nacheva, and T. H. Rabbitts. 1988. The mechanism of chromosomal translocation t(11;14) involving the T-cell receptor Cδ locus on human chromosome 14q11 and a transcribed region of chromosome 11p15. EMBO J. 7:385-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boehm, T., L. Foroni, Y. Kaneko, M. P. Perutz, and T. H. Rabbitts. 1991. The rhombotin family of cysteine-rich LIM-domain oncogenes: distinct members are involved in T-cell translocations to human chromosomes 11p15 and 11p13. Proc. Natl. Acad. Sci. USA 88:4367-4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boehm, T., L. Foroni, M. Kennedy, and T. H. Rabbitts. 1990. The rhombotin gene belongs to a class of transcriptional regulators with a potential novel protein dimerisation motif. Oncogene 5:1103-1105. [PubMed] [Google Scholar]
- 5.Boehm, T., M.-G. Spillantini, M. V. Sofroniew, M. A. Surani, and T. H. Rabbitts. 1991. Developmentally regulated and tissue-specific expression of mRNAs encoding the two alternative forms of the LIM domain oncogene rhombotin: evidence for thymus expression. Oncogene 6:695-703. [PubMed] [Google Scholar]
- 6.Chen, H. H., J. W. Yip, A. F. Stewart, and E. Frank. 2002. Differential expression of a transcription regulatory factor, the LIM domain only 4 protein Lmo4, in muscle sensory neurons. Development 129:4879-4889. [DOI] [PubMed] [Google Scholar]
- 7.Copp, A. J., F. A. Brook, J. P. Estibeiro, A. S. Shum, and D. L. Cockcroft. 1990. The embryonic development of mammalian neural tube defects. Prog. Neurobiol. 35:363-403. [DOI] [PubMed] [Google Scholar]
- 8.Epstein, D. J., M. Vekemans, and P. Gros. 1991. splotch (Sp2H), a mutation affecting development of mouse neural tube, shows a deletion within the paired homeodomain of Pax-3. Cell 67:767-774. [DOI] [PubMed] [Google Scholar]
- 9.Feinberg, A. P., and B. A. Vogelstein. 1983. A technique for radiolabelling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem. 132:6-13. [DOI] [PubMed] [Google Scholar]
- 10.Foroni, L., T. Boehm, L. White, A. Forster, P. Sherrington, X. B. Liao, C. I. Brannan, N. A. Jenkins, N. G. Copeland, and T. H. Rabbitts. 1992. The rhombotin gene family encode related LIM-domain proteins whose differing expression suggests multiple roles in mouse development. J. Mol. Biol. 226:747-761. [DOI] [PubMed] [Google Scholar]
- 11.Freyd, G., S. K. Kim, and H. R. Horvitz. 1990. Novel cysteine-rich motif and homeodomain in the product of the Caenorhabditis elegans cell lineage gene lin-11. Nature 344:876-879. [DOI] [PubMed] [Google Scholar]
- 12.Greenberg, J. M., T. Boehm, M. V. Sofroniew, R. J. Keynes, S. C. Barton, M. L. Norris, M. A. Surani, M.-G. Spillantini, and T. H. Rabbitts. 1990. Segmental and developmental regulation of a presumptive T-cell oncogene in the central nervous system. Nature 344:158-160. [DOI] [PubMed] [Google Scholar]
- 13.Grutz, G., K. Bucher, I. Lavenir, R. Larson, T. Larson, and T. H. Rabbitts. 1998. The oncogenic T cell LIM-protein Lmo2 forms part of a DNA-binding complex specifically in immature T cells. EMBO J. 17:4594-4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grutz, G., A. Forster, and T. H. Rabbitts. 1998. Identification of the LMO4 gene encoding an interaction partner of the LIM-binding protein LDB1/NLI1: a candidate for displacement by LMO proteins in T cell acute leukaemia. Oncogene 17:2799-2803. [DOI] [PubMed] [Google Scholar]
- 15.Harris, M. J., and D. M. Juriloff. 1999. Mini-review: toward understanding mechanisms of genetic neural tube defects in mice. Teratology 60:292-305. [DOI] [PubMed] [Google Scholar]
- 16.Hermanson, O., T. M. Sugihara, and B. Andersen. 1999. Expression of LMO-4 in the central nervous system of the embryonic and adult mouse. Cell. Mol. Biol. (Noisy-Le-Grand) 45:677-686. [PubMed] [Google Scholar]
- 17.Hinks, G. L., B. Shah, S. J. French, L. S. Campos, K. Staley, J. Hughes, and M. V. Sofroniew. 1997. Expression of LIM protein genes Lmo1, Lmo2, and Lmo3 in adult mouse hippocampus and other forebrain regions: differential regulation by seizure activity. J. Neurosci. 17:5549-5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hollander, M. C., M. S. Sheikh, D. V. Bulavin, K. Lundgren, L. Augeri-Henmueller, R. Shehee, T. A. Molinaro, K. E. Kim, E. Tolosa, J. D. Ashwell, M. P. Rosenberg, Q. Zhan, P. M. Fernandez-Salguero, W. F. Morgan, C. X. Deng, and A. J. Fornace, Jr. 1999. Genomic instability in Gadd45a-deficient mice. Nat. Genet. 23:176-184. [DOI] [PubMed] [Google Scholar]
- 19.Jones, A., S. Bahn, A. L. Grant, M. Kohler, and W. Wisden. 1996. Characterization of a cerebellar granule cell-specific gene encoding the gamma-aminobutyric acid type A receptor alpha 6 subunit. J. Neurochem. 67:907-916. [DOI] [PubMed] [Google Scholar]
- 20.Karlsson, O., S. Thor, T. Norberg, H. Ohlsson, and T. Edlund. 1990. Insulin gene enhancer binding protein Isl-1 is a member of a novel class of proteins containing both a homeo- and a cys-his domain. Nature 344:879-882. [DOI] [PubMed] [Google Scholar]
- 21.Kaster, K. R., S. G. Burgett, R. N. Rao, and T. D. Ingolia. 1983. Analysis of a bacterial hygromycin B resistance gene by transcriptional and translational fusions and by DNA sequencing. Nucleic Acids Res. 11:6895-6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kenny, D. A., L. W. Jurata, Y. Saga, and G. N. Gill. 1998. Identification and characterization of LMO4, an LMO gene with a novel pattern of expression during embryogenesis. Proc. Natl. Acad. Sci. USA 95:11257-11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LeFranc, M.-P., A. Forster, R. Baer, M. A. Stinson, and T. H. Rabbitts. 1986. Diversity and rearrangement of the human T cell rearranging γ genes: nine germ-line variable genes belonging to two subgroups. Cell 45:237-246. [DOI] [PubMed] [Google Scholar]
- 24.McGuire, E. A., R. D. Hockett, K. M. Pollock, M. F. Bartholdi, S. J. O'Brien, and S. J. Korsmeyer. 1989. The t(11;14)(p15;q11) in a T-cell acute lymphoblastic leukemia cell line activates multiple transcripts, including Ttg-1, a gene encoding a potential zinc finger protein. Mol. Cell. Biol. 9:2124-2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mombaerts, P., J. Iacomini, R. S. Johnson, K. Herrup, S. Tonegawa, and V. E. Papaioannou. 1992. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68:869-877. [DOI] [PubMed] [Google Scholar]
- 26.Neumann, P. E., W. N. Frankel, V. A. Letts, J. M. Coffin, A. J. Copp, and M. Bernfield. 1994. Multifactorial inheritance of neural tube defects: localization of the major gene and recognition of modifiers in ct mutant mice. Nat. Genet. 6:357-362. [DOI] [PubMed] [Google Scholar]
- 27.Osada, H., G. Grutz, H. Axelson, A. Forster, and T. H. Rabbitts. 1995. Association of erythroid transcription factors: complexes involving the LIM protein RBTN2 and the zinc-finger protein GATA1. Proc. Natl. Acad. Sci. USA 92:9585-9589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perez-Alvarado, G. C., C. Miles, J. W. Michelsen, H. A. Louis, D. R. Winge, M. C. Beckerle, and M. F. Summers. 1994. Structure of the carboxy-terminal LIM domain from the cysteine rich protein CRP. Nat. Struct. Biol. 1:388-398. [DOI] [PubMed] [Google Scholar]
- 29.Rabbitts, T. H. 1998. LMO T-cell translocation oncogenes typify genes activated by chromosomal translocations that alter transcription and developmental processes. Genes Dev. 12:2651-2657. [DOI] [PubMed] [Google Scholar]
- 30.Rabbitts, T. H. 1991. Translocations, master genes, and differences between the origins of acute and chronic leukemias. Cell 67:641-644. [DOI] [PubMed] [Google Scholar]
- 31.Rabbitts, T. H., and T. Boehm. 1990. LIM domains. Nature 346:418. [DOI] [PubMed] [Google Scholar]
- 32.Racevskis, J., A. Dill, J. A. Sparano, and H. Ruan. 1999. Molecular cloning of LMO4, a new human LIM domain gene. Biochim. Biophys. Acta 1445:148-153. [DOI] [PubMed] [Google Scholar]
- 33.Royer-Pokora, B., U. Loos, and W.-D. Ludwig. 1991. TTG-2, a new gene encoding a cysteine-rich protein with the LIM motif, is overexpressed in acute T-cell leukaemia with the t(11;14)(p13;q11). Oncogene 6:1887-1893. [PubMed] [Google Scholar]
- 34.Sah, V. P., L. D. Attardi, G. J. Mulligan, B. O. Williams, R. T. Bronson, and T. Jacks. 1995. A subset of p53-deficient embryos exhibit exencephaly. Nat. Genet. 10:175-180. [DOI] [PubMed] [Google Scholar]
- 35.Sanchez-Garcia, I., Y. Kaneko, R. Gonzalez-Sarmiento, K. Campbell, L. White, T. Boehm, and T. H. Rabbitts. 1991. A study of chromosome 11p13 translocations involving TCRβ and TCRδ in human T cell leukaemia. Oncogene 6:577-582. [PubMed] [Google Scholar]
- 36.Southern, E. M. 1975. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 98:503-517. [DOI] [PubMed] [Google Scholar]
- 37.Sugihara, T. M., I. Bach, C. Kioussi, M. G. Rosenfeld, and B. Andersen. 1998. Mouse deformed epidermal autoregulatory factor 1 recruits a LIM domain factor, LMO-4, and CLIM coregulators. Proc. Natl. Acad. Sci. USA 95:15418-15423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thomas, K. R., and M. R. Capecchi. 1987. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51:503-512. [DOI] [PubMed] [Google Scholar]
- 39.Thomas, P. S. 1980. Hybridization of denatured RNA and small DNA fragments transferred to nitrocellulose. Proc. Natl. Acad. Sci. USA 77:5201-5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tozawa, R., S. Ishibashi, J. Osuga, H. Yagyu, T. Oka, Z. Chen, K. Ohashi, S. Perrey, F. Shionoiri, N. Yahagi, K. Harada, T. Gotoda, Y. Yazaki, and N. Yamada. 1999. Embryonic lethality and defective neural tube closure in mice lacking squalene synthase. J. Biol. Chem. 274:30843-30848. [DOI] [PubMed] [Google Scholar]
- 41.Tse, E., G. Grutz, A. A. Garner, Y. Ramsey, N. P. Carter, N. Copeland, D. J. Gilbert, N. A. Jenkins, A. Agulnick, A. Forster, and T. H. Rabbitts. 1999. Characterisation of the Lmo4 gene encoding a LIM-only protein: genomic organisation and comparative chromosomal mapping. Mamm. Genome 10:1089-1094. [DOI] [PubMed] [Google Scholar]
- 42.Valge-Archer, V., A. Forster, and T. H. Rabbitts. 1998. The LMO1 and LDB1 proteins interact in human T cell acute leukaemia with the chromosomal translocation t(11;14)(p15;q11). Oncogene 17:3199-3202. [DOI] [PubMed] [Google Scholar]
- 43.Valge-Archer, V. E., H. Osada, A. J. Warren, A. Forster, J. Li, R. Baer, and T. H. Rabbitts. 1994. The LIM protein RBTN2 and the bHLH protein TAL1 are present in a complex in erythroid cells. Proc. Natl. Acad. Sci. USA 91:8617-8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Visvader, J. E., X. Mao, Y. Fujiwara, K. Hahm, and S. H. Orkin. 1997. The LIM-domain binding protein Ldb1 and its partner LMO2 act as negative regulators of erythroid differentiation. Proc. Natl. Acad. Sci. USA 94:13707-13712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Visvader, J. E., D. Venter, K. Hahm, M. Santamaria, E. Y. Sum, L. O'Reilly, D. White, R. Williams, J. Armes, and G. J. Lindeman. 2001. The LIM domain gene LMO4 inhibits differentiation of mammary epithelial cells in vitro and is overexpressed in breast cancer. Proc. Natl. Acad. Sci. USA 98:14452-14457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vu, D., P. Marin, C. Walzer, M. M. Cathieni, E. N. Bianchi, F. Saidji, G. Leuba, C. Bouras, and A. Savioz. 2003. Transcription regulator LMO4 interferes with neuritogenesis in human SH-SY5Y neuroblastoma cells. Brain Res. Mol. Brain Res. 115:93-103. [DOI] [PubMed] [Google Scholar]
- 47.Wadman, I., J. Li, R. O. Bash, A. Forster, H. Osada, T. H. Rabbitts, and R. Baer. 1994. Specific in vivo association between the bHLH and LIM proteins implicated in human T cell leukaemia. EMBO J. 13:4831-4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wadman, I. A., H. Osada, G. G. Grutz, A. D. Agulnick, H. Westphal, A. Forster, and T. H. Rabbitts. 1997. The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and Ldb1/NLI proteins. EMBO J. 16:3145-3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Warren, A. J., W. H. Colledge, M. B. L. Carlton, M. J. Evans, A. J. H. Smith, and T. H. Rabbitts. 1994. The oncogenic cysteine-rich LIM domain protein rbtn2 is essential for erythroid development. Cell 78:45-58. [DOI] [PubMed] [Google Scholar]
- 50.Way, J. C., and M. Chalfie. 1988. mec-3, a homeobox-containing gene that specifies differentiation of the touch receptor neurons in C. elegans. Cell 54:5-16. [DOI] [PubMed] [Google Scholar]
- 51.Wittlin, S., E. Y. Sum, N. K. Jonas, G. J. Lindeman, and J. E. Visvader. 2003. Two promoters within the human LMO4 gene contribute to its overexpression in breast cancer cells. Genomics 82:280-287. [DOI] [PubMed] [Google Scholar]
- 52.Yamada, Y., R. Pannell, A. Forster, and T. H. Rabbitts. 2002. The LIM-domain protein Lmo2 is a key regulator of tumour anogiogenesis: a new anti-angiogenesis drug target. Oncogene 21:1309-1315. [DOI] [PubMed] [Google Scholar]
- 53.Yamada, Y., R. Pannell, A. Forster, and T. H. Rabbitts. 2000. The oncogenic LIM-only transcription factor Lmo2 regulates angiogenesis but not vasculogenesis. Proc. Natl. Acad. Sci. USA 97:320-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamada, Y., A. W. Warren, C. Dobson, A. Forster, R. Pannell, and T. H. Rabbitts. 1998. The T cell leukaemia LIM protein Lmo2 is necessary for adult mouse haematopoiesis. Proc. Natl. Acad. Sci. USA 95:3890-3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yenofsky, R. L., M. Fine, and J. W. Pellow. 1990. A mutant neomycin phosphotransferase II gene reduces the resistance of transformants to antibiotic selection pressure. Proc. Natl. Acad. Sci. USA 87:3435-3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ziller, C., M. Fauquet, C. Kalcheim, J. Smith, and N. M. Le Douarin. 1987. Cell lineages in peripheral nervous system ontogeny: medium-induced modulation of neuronal phenotypic expression in neural crest cell cultures. Dev. Biol. 120:101-111. [DOI] [PubMed] [Google Scholar]