

Abstract
The development of enantioselective synthetic routes to (–)-kinamycin F (9) and (–)-lomaiviticin aglycon (6) is described. The diazotetrahydrobenzo[b]fluorene (diazofluorene) functional group of the targets was prepared by fluoride-mediated coupling of a β-trimethylsilylmethyl-α,β-unsaturated ketone (38) with an oxidized naphthoquinone (19), palladium-catalyzed cyclization (39→37), and diazo transfer (37→53). The D-ring precursors 60 and 68 were prepared from m-cresol and 3-ethylphenol, respectively. Coupling of the β-trimethylsilylmethyl-α,β-unsaturated ketone 60 with the juglone derivative 61, cyclization, and diazo transfer, provided the advanced diazofluorene 63, which was elaborated to (–)-kinamycin F (9) in three steps. The diazofluorene 87 was converted to the C2-symmetric lomaiviticin aglycon precursor 91 by enoxysilane formation and oxidative dimerization with manganese tris(hexafluoroacetylacetonate) (94, 26%). The stereochemical outcome is attributed to the steric bias engendered by the mesityl acetal of 87 and contact ion pairing of the intermediates. The coupling product 91 was deprotected (tert-butylhydrogen peroxide, trifluoroacetic acid–dichloromethane) to form the chain isomer of lomaiviticin aglycon 98, which cyclizes to (–)-lomaiviticin aglycon (6, 39–41% overall). The scope of the fluoride-mediated coupling process is delineated (nine products, average yield = 72%, Table 2); a related enoxysilane quinonylation reaction is also described (10 products, average yield = 77%, Table 1). We establish that dimeric diazofluorenes undergo hydrodediazotization 3-fold faster then related monomeric diazofluorenes (Table 6). The simple diazofluorene 103 is a potent inhibitor of ovarian cancer stem cells (IC50 = 500 nM).
Introduction
Lomaiviticins A–E (1–5, Figure 1) are the most complex members of a family of bacterial metabolites that contain a diazotetrahydrobenzo[b]fluorene (diazofluorene) functional group.1 The lomaiviticins are structurally related to the monomeric diazofluorenes known as the kinamycins (7–10).2 No less than ten distinct kinamycins have been described; kinamycins A (7), C (8), F (9), and the epoxykinamycin FL-120B’ (10),3 are representative. The kinamycins and lomaiviticins are potent antiproliferative and antimicrobial agents, and a large body of data now supports bioreductive activation4 of the diazofluorene as a first step in a cascade leading to cell death.5 It has been proposed that redox cycling of the quinone to produce reactive oxygen species (ROS),5a–d formation of vinyl radical (11),5c,d,e,g,i ortho-quinone methide (12),5c–e,g–i and acylfulvene (13) intermediates by reduction,4a,5k or addition of biological nucleophiles to the diazo substituent (14),5b may contribute to cytotoxicity (Figure 2). The targets of these metabolites are not known; various kinamycins and lomaiviticin A (1) are known to cleave dsDNA in vitro1a,5g,i and in tissue culture5f and a protein target has been implicated in the activity of kinamycin F (9).5j
Figure 1.

Structures of (–)-Lomaiviticins A–E (1–5, respectively), (–)-Lomaiviticin Aglycon (6), (–)-Kinamycins A, C, and F (7–9, respectively), and the Epoxykinamycin (–)-FL-120B’ (10).
Figure 2.
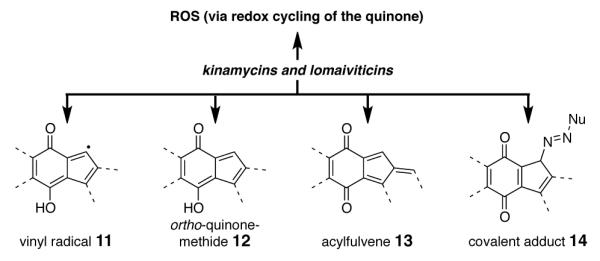
Reactive Intermediates Proposed to Form From the Kinamycins and Lomaiviticins.
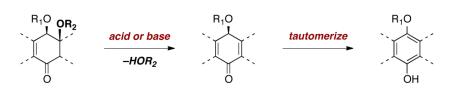
Synthetic planning toward these targets is significantly complicated by the sensitivity of the diazofluorene toward nucleophiles and reducing agents. In addition, the β-alkoxyenone substructure of lomaiviticin A (1) is unstable toward an elimination–aromatization pathway (eq. 1), which limits the range of conditions that are compatible with late-stage intermediates. Moreover, syntheses and installation of the carbohydrate residues of the lomaiviticins, in particular, the β-linked 2,6-dideoxyaminoglycoside residue, is
 |
expected to be challenging. Finally, formation of the central and highly congested bridging carbon–carbon bond in the lomaiviticins constitutes perhaps the most significant obstacle toward their synthesis.
To date, several completed routes to the kinamycins6 and studies toward the lomaiviticins7 have been described. Our own efforts have resulted in an efficient synthesis of (–)-kinamycin F (9),8d the first synthesis of (–)-lomaiviticin aglycon (6),7i and the development of a general and versatile strategy to prepare diazofluorenes of wide structural variability. We have discovered several new reactions during our investigations, have obtained insights into the factors influencing reductive activation of the diazofluorene, and have identified certain diazofluorenes as potent inhibitors of ovarian cancer stem cells. The full details of these investigations are described below.
Results and Discussion
Development of a Convergent and Versatile Route to the Diazofluorene Function
Given the substantial body of evidence implicating the diazofluorene in the biological effects of the kinamycins and lomaiviticins, a versatile strategy to access this functional group was deemed valuable. We targeted diazofluorenes represented by the generalized structure 15 (Scheme 1). Deconstruction of 15 by excision of dinitrogen and a one-carbon synthon, followed by separation of the naphthoquinone and enone residues, formed the hypothetical synthetic precursors 16 and 17. Initial efforts focusing on appending the one-carbon synthon and dinitrogen substituents to the naphthoquinone 16 were unsuccessful.8 Therefore, we pursued a strategy comprising addition of the one-carbon substituent to the enone, followed by bond formation to the quinone, as discussed below.
Scheme 1.
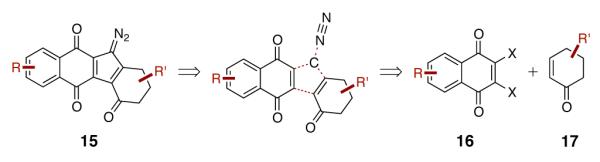
Strategy to Access the Diazofluorene Functional Group.
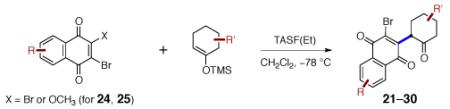
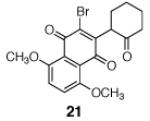
In preliminary studies, we employed the hydrazone 18 (Scheme 2), which was obtained in one step from cyclohex-2-ene-1-one.9 The hydrazone and enoxysilane functions of 18 were envisioned to serve as handles for bond formation to the naphthoquinone. We discovered that activation of 18 with tris-(diethylamino)sulfonium difluorotrimethylsilicate [TASF(Et)]10 in the presence of 5,8-dimethoxy-2,3-dibromonaphthoquinone (19)11 cleanly formed the α-quinonylated product 20 in 73% yield as a single detectable anti diastereomer (1H NMR analysis).
Scheme 2.
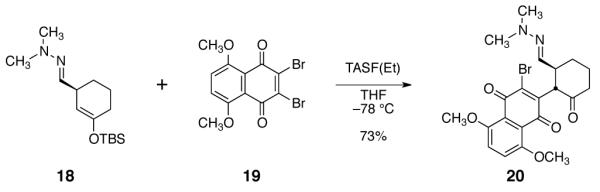
Fluoride-Mediated Coupling of the Enoxysilane 18 with 2,3-Dibromo-5,8-dimethoxynaphthoquinone (19).
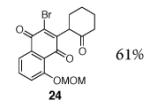
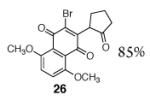
A broad range of enoxysilanes undergo α-quinonylation in high yield (Table 1). In cases where the enoxysilane possesses a β-stereocenter, the coupling products were formed with high anti selectivity (>5:1; entries 2,3,5,7, and 20, 72–85%). Unsymmetrical naphthoquinones couple with regiocontrol, provided that the starting reagent contains a substituent to differentiate the 2- and 3-positions (entries 4, 5, 61%, 76%). Enoxysilanes derived from cyclopentanone (entry 6, 85%), acyclic ketones (entry 9, 82%), and heterocyclic ketones (entry 10, 85%) are also competent nucleophiles. α-Quaternary ketones are formed in good yield (entry 8, 65%). Common methods for the construction of carbon–carbon bonds to quinones include metal-catalyzed cross–coupling reactions,12 lithiation of halogenated hydroquinone ethers,13 and the oxidative addition of boronic acids.14 This fluoride-mediated coupling reaction provides a useful complement to these protocols.
Table 1.
Scope of the α-Quinonylation Reaction.a
| entry | product | yield | entry | product | yield |
|---|---|---|---|---|---|
| 1 |
|
84% | 2b |
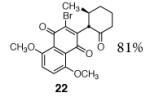
|
81% |
| 3b |
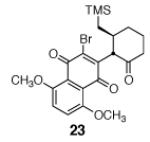
|
86% | 4 |
|
61% |
| 5 |
|
76% | 6 |
|
85% |
| 7 |
|
64% | 8 |
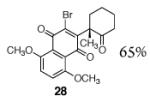
|
65% |
| 9 |
|
82% | 10 |
|
85% |
Conditions: TASF(Et) (1.10 equiv), CH2Cl2, –78 °C.
THF used as solvent.
Synthesis of the quinonylated ketone 20 was encouraging since it contained all of the carbon atoms of our target. We envisioned forming the final ring by intramolecular 1,4-addition of the hydrazone to the naphthoquinone, followed by loss of hydrogen bromide (20→31, Scheme 3). However, under a variety of conditions, the cyclization product 31 could not be formed (UPLC/MS analysis), potentially due to the anti arrangement of the quinone and hydrazone substituents of 20.8
Scheme 3.

Attempted Elaboration of the Hydrazone 20.
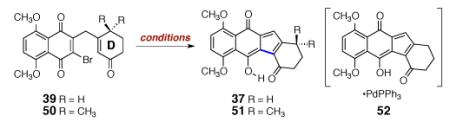
We next targeted the β-methyl-α-β-unsaturated ketone 35 (Scheme 4). We envisioned forming an extended enolate of 35, followed by 5-endo-trig cyclization to construct the tetracycle 36. The precursor 34 was synthesized by copper-catalyzed 1,4-addition of methyl magnesium chloride to cyclohex-2-ene-1-one, trapping with chlorotrimethylsilane, and coupling with 19 (80% overall). Although we were unable to oxidize 34 by several conventional methods, we discovered that thermoloysis of 34 under an atmosphere of dioxygen formed the oxidation product 35 (13% yield). Heating solutions of 35 in the presence of palladium (II) acetate, bis(diphenylphosphino)ferrocene, and potassium carbonate afforded the hydroxyfulvene 37 in 77% yield. Attempts to cyclize 35 using bases alone were unsuccessful.
Scheme 4.
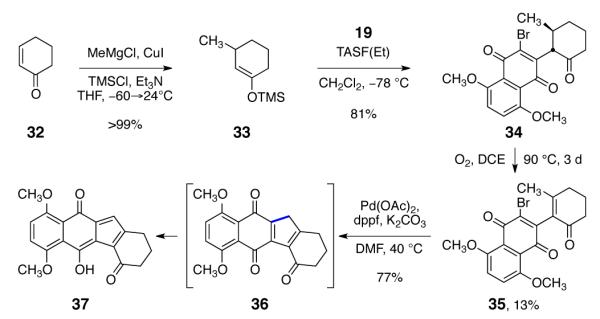
Synthesis of the Tetracycle 37.
The cyclization product 37, and structurally-related intermediates (vide infra), exist exclusively in the hydroxyfulvene (e.g., 37) rather than the keto tautomer (36, Scheme 4). Substituent-dependent ketone–hydroxyfulvene equilibria have been reported for related structures.5h In the present system, the hydroxyfulvene tautomer may be stabilized by intramolecular hydrogen-bonding and π-conjugation of the hydroxy group with the carbonyl substituents. Although we had made substantial progress toward our target, the low efficiency of the oxidation step (34→35) led us to reorganize the sequence of bond-forming events. Three considerations were critical in the development of our successful approach. First, we recognized that reoxidation (e.g., palladium acetate15 or benzeneselenyl chloride–hydrogen peroxide16) of conjugate addition products such as 33 provides a route to β-substituted-α,β-unsaturated ketones that circumvents the difficult oxidation observed above (34→35). Second, the fluoride-mediated quinonylation reaction appeared to be unique in its ability to unite the enone and quinone fragments, and we sought to retain this chemistry. Finally, although not mechanistically well-understood, the palladium-catalyzed cyclization (35→37) provided an efficient method to forge the cyclopentadiene ring of the target.
With these considerations in mind, the β-trimethylsilylmethyl-α,β-unsaturated ketone 38 was synthesized by copper-catalyzed 1,4-addition of trimethylsilylmethyl magnesium chloride to 32, trapping with chlorotrimethylsilane, and reoxidation (76% overall, Scheme 5). TASF(Et)-mediated coupling of 38 with 2,3-dibromo-5,8-dimethoxynaphthoquinone (19) proceeded smoothly, to afford the the ketoquinone product 39 (85%). The observed preference for γ-quinonylation may be steric in origin.
Scheme 5.
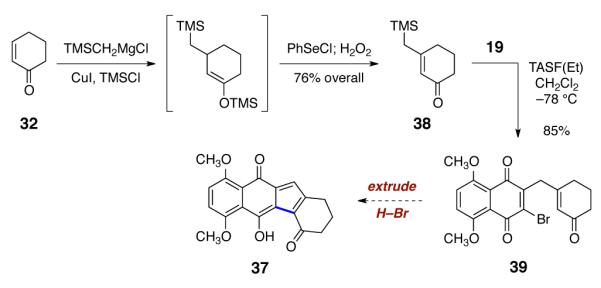
Synthesis of the Ketoquinone 39.
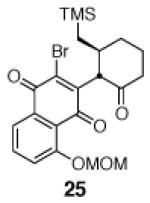
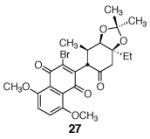
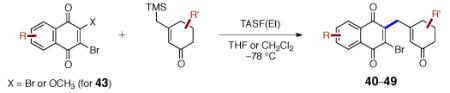
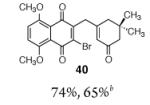
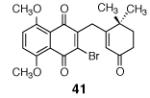
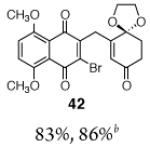
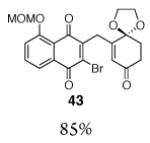
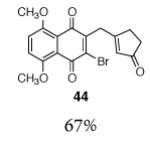
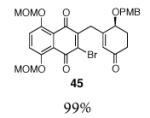
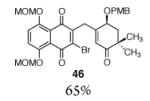
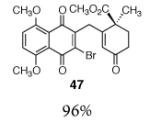
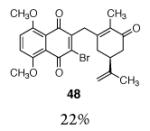
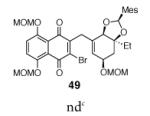
This γ-quinonylation reaction is also relatively general (Table 2). A variety of β-trimethylsilylmethyl-α,β-unsaturated ketones, including those with quaternary carbon centers (entries 1, 2, 7, 8, 57–96%), acetal (entries 3, 4, 83%, 85%, respectively), para-methoxybenzyl ether (entries 6, 7, 99%, 65%, respectively), and ester substituents (entry 8, 96%), couple in high yields. As in our α-quinonylation reaction, unsymmetrical, electronically-biased naphthoquinones react with regiocontrol (entry 4, 85%). 3-(Trimethylsilylmethyl)-cyclopent-2-ene-1-one is also a competent nucleophile for this reaction (entry 5, 67%). Nucleophiles bearing α-substitution, however, couple in diminished yields (entry 9, 22%), and the presence of a ketone substituent appears to be essential to achieve activation of the allylsilane function (entry 10). We have recently found that the bench-stable, commercially available salt tetra-butylammonium difluorotriphenylsilicate (TBAT),17 may be employed in place of TASF(Et) (entries 1, 3).
Table 2.
Scope of the γ-Quinonylation Reaction.a
| entry | product and yield | entry | product and yield |
|---|---|---|---|
| 1 |
|
2 |
|
| 3 |
|
4 |
|
| 5 |
|
6 |
|
| 7 |
|
8 |
|
| 9 |
|
10 |
|
conditions: TASF(Et) (1.1 equiv), CH2Cl2, –78 °C.
TBAT (1.1–1.5 equiv), CH2Cl2, 0 °C.
none detected by 1H NMR analysis of the unpurified reaction mixture.
With the ketoquinone 39 in hand, we investigated methods to forge the final carbon–carbon bond of the diazofluorene (39→37). Surprisingly, the conditions employed for the cyclization of 35 were ineffective (entry 1, Table 3). Further optimization was conducted with stoichiometric palladium to facilitate accurate measurement. Among a variety of ligands and palladium precursors examined, the combination of palladium (II) acetate and triphenylphosphine emerged as the most promising. Using these reagents and cesium carbonate as base, the hydroxyfulvene 37 was isolated in 31% yield (entry 2). Triethylamine (TEA, entry 3) and silver phosphate (entry 4) were less effective. By employing silver carbonate as base, raising the temperature to 80 °C, and using polymer-supported triphenylphosphine (PS-PPh3, to simplify purification), the isolated yield of 37 was increased to 40% (entry 6). The low isolated yield of 37 was attributed to formation of the palladium complex 52, identified by UPLC/MS analysis and accounting for a significant mole fraction of 37 at these high loadings of palladium. Complexes between the structurally related anthracyclines and palladium are known.18 Although we were unable to ascertain the nature of the bonding interactions in 52 (e.g. η5, κ2), attempts to displace the product 37 by addition of excess acetylacetone or bis(diphenylphosphino)ferrocene were unsuccessful. The substrate 50, which bears a quaternary center on the D-ring, formed the cyclization product 51 in 79% yield (entry 7), and a complex with palladium could not be detected during the reaction mixture (UPLC/MS analysis). Using this substrate, we were also able to decrease the loading of palladium to 25 mol% (86% yield of 51, entry 8). In general, cyclization of substrates bearing a quaternary center on the D-ring, including those employed en route to the targets (vide infra), are much more efficient than simple model systems such as 39.
Table 3.
Optimization of the Palladium-Mediated Cyclization Reaction.a
| entry | subs. | solvent | mol% Pd |
ligand | base | T (°C) |
yield |
|---|---|---|---|---|---|---|---|
| 1 | 39 | DMF | 40 | dppf | K2CO3 | 50 | <5% |
| 2 | 39 | THF | 80 | PPh3 | Cs2CO3 | 50 | 31% |
| 3 | 39 | THF | 100 | PPh3 | TEA | 50 | <5% |
| 4 | 39 | THF | 100 | PPh3 | Ag3PO4 | 50 | dec. |
| 5 | 39 | PhCH3 | 75 | PS-PPh3 | Ag2CO3 | 80 | 22% |
| 6 | 39 | PhCH3 | 100 | PS-PPh3 | Ag2CO3 | 80 | 40% |
| 7 | 50 | PhCH3 | 100 | PS-PPh3 | Ag2CO3 | 80 | 79% |
| 8 | 50 | PhCH3 | 25 | PS-PPh3 | Ag2CO3 | 80 | 86% |
Conditions: 1–2 equiv of ligand (with respect to palladium) and 2–3 equivalents of base (with respect to substrate) were employed.
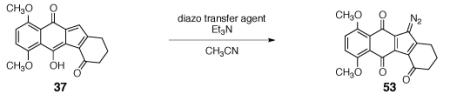
Diazo transfer to the hydroxyfulvene 37 was all that was required to complete the synthesis of the diazofluorene. Historically, an early application of para-toluenesulfonyl azide (p-TsN3), by Doering,19 was in the synthesis of diazocyclopentadiene itself from lithium cyclopentadienide. The conjugate base of 37 possesses five inequivalent cyclopentadienide carbon atoms, but only addition to the desired position generates a stable diazo adduct. Exposure of the hydroxyfulvene 37 to triethylamine and common diazotransfer agents, such as methanesulfonylazide (MSA),20 p-acetamidobenzenesulfonyl azide (p-ABSA), 21 or p-TsN3,19 afforded low yields of the diazofluorene product 53 (entries 1–3, Table 4, 4–20%). Use of the more electrophilic imidazole-1-sulfonylazide hydrochloride (ISA)22 formed the diazofluorene 53 in 74% yield in less than one hour at 0 °C (entry 4). Trifluoromethanesulfonyl azide (TfN3)23 and nonaflyl azide24 were slightly more effective (81%, 85%, entries 5, 6 respectively). The latter is preferred due to its increased thermal stability and lower volatility.
Table 4.
Diazo Transfer to the Hydroxyfulvene 37.a
| entry | diazo transfer agent (equiv) | t (h) | T (°C) | yield |
|---|---|---|---|---|
| 1 | MSA (7.5) | 4 | 24 | 4% |
| 2 | p-ABSA (5.0) | 1.5 | 24 | 20% |
| 3 | p-TsN3 (7.5) | 4 | 24 | 18% |
| 4 | ISA (2.5) | 0.3 | 0 | 74% |
| 5 | TfN3 (2.5) | 0.3 | 24 | 81% |
| 6 | CF3(CF2)3SO2N3 (2.5) | 0.3 | 0 | 85% |
Conditions: diazo transfer reagent (2.5–7.5 equiv), TEA (5.0–15 equiv), CH3CN.
The chemistry outlined above has allowed us to prepare a large variety of monomeric diazofluorenes. The antiproliferative activity of many of these compounds has been evaluated, and certain synthetic diazofluorenes have emerged as potent inhibitors of cancer stem cells (vide infra). Of greatest significance, these studies laid the foundation for syntheses of (–)-kinamycin F (9) and (–)- lomaiviticin aglycon (6), as described in detail below.
Syntheses of the Enone Fragments
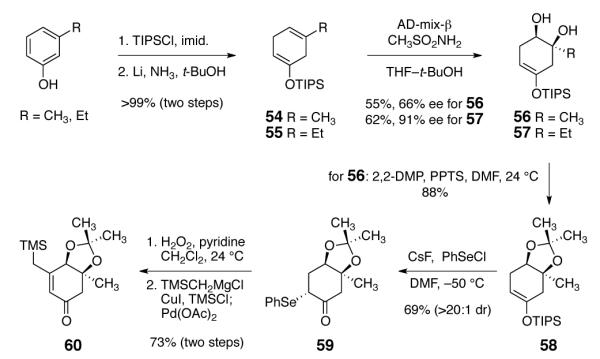
Our route to the diazofluorene defined the enone fragments required for syntheses of kinamycins and lomaiviticins, and these were prepared by the routes outlined in Scheme 6. Beginning with m-cresol (kinamycin series), silyl ether formation (tri-isopropylsilyl chloride, imidazole) and Birch reduction (lithium, ammonia) formed the cyclohexadiene 54 (99%, two steps). Regio- and stereoselective asymmetric dihydroxylation25 then generated the vicinal diol 56 in 66% ee (55% yield). The use of the tri-isopropylsilyl protecting group was critical to the success of this step; presumably, its steric bulk prevents entry of the enoxysilane into the binding pocket of cinchona alkaloid–osmium complex, allowing selective oxidation of the less reactive alkene. The modest enantiomeric excess observed in this transformation was attributed to the negligible steric requirements of the methyl substituent, which renders discrimination of the faces of the alkene difficult. No attempt to optimize the enantiomeric excess was made, as we found that subsequent intermediates were highly crystalline and could be resolved to 97% ee.
Scheme 6.
Syntheses of the Enone 60.
The diol 56 was then transformed to the acetonide 58 by stirring with 2,2-dimethoxypropane and pyridinium para-toluenesulfonate (PPTS, 88%). Treatment of 58 with phenylselenium chloride formed the α-selenyl ketone 59 (69%, >20:1 mixture of diastereomers). Finally, oxidation of 59 (hydrogen peroxide, pyridine) and thermal elimination of the resulting selenyl oxide formed an enone (not shown) that was homologated by copper-catalyzed 1,4-addition of trimethylsilyl methylmagnesium chloride, trapping with chloro trimethylsilane, and reoxidation (60, 73% over two steps).
In the lomaiviticin series, silylation and Birch reduction of 3-ethylphenol provided the diene 55 (>99%). Asymmetric dihydroxylation of 55 formed the diol 57 (62%, 91% ee). Derivatives of 57 were used in our lomaiviticin studies (vide infra).
Completion of (–)-Kinamycin F (9)
To complete the synthesis of (–)-kinamycin F (9), a suitable naphthoquinone coupling partner, biased toward addition at the C-3 position, was required.8 Toward this end, O-(methoxymethyl)-2-bromo-3-methoxyjuglone (61) was prepared by heating a methanolic solution of O-(methoxymethyl)-2,3-dibromo-juglone and sodium carbonate (96%).8 Fluoride-mediated coupling with the β-trimethylsilylmethyl-α,β-unsaturated ketone 60 proceeded with complete regioselectivity, forming the C-3 substituted product 62 in 79% yield (Scheme 7). The cyclization and diazotransfer steps proceeded smoothly, as in our model system, to form the diazofluorene 63 (65%, two steps). To complete the synthesis we needed to develop a method for the stereocontrolled conversion of the ketone function of 62 to a trans-1,2-diol. Site-selectivity in the reduction step was a serious concern, as an earlier study by Feldman and Eastman5c,d had established the diazofluorene as highly reactive toward reducing agents. To control selectivity, we pursued an α-oxidation, directed reduction sequence. Treatment of the diazofluorene 63 with tri-isopropylsilyl trifluoromethanesulfonate (TIPSOTf) and di-isopropylethylamine (DIPEA) formed an enoxysilane (not shown) that was exposed to an excess of dimethyldioxirane in a mixture of methylene chloride and methanol at –40 °C. Under these conditions, the free α-hydroxy ketone 65 was obtained, presumably formed by in situ methanolysis of the silyloxy epoxide 64. The stereoselectivity of this oxidation [verified by successful conversion of 65 to (–)-kinamycin F (9)] is consistent with faster addition of the dioxirane to the convex face of the cis-fused 6-5 system. The α-hydroxyl group of 65 was then used to control selectivity in the reduction step.
Scheme 7.

Completion of the Synthesis of (–)-Kinamycin F (9).
We considered several reagents known to effect the directed reduction of α- and β-hydroxy ketones. This reaction is most commonly accomplished with tetramethylammonium triacetoxyborohydride,26 but the use of acetic acid as cosolvent was undesirable. We employed borane•tetrahydrofuran complex, as this reagent had been shown to effect the directed reduction of α-hydroxy ketones, though the stereoselectivity for simple cycloalkanones was low.27 To implement the reduction, the α-hydroxyketone 65 was dissolved in tetrahydrofuran and cooled to – 78 °C. Borane•tetrahydrofuran complex (1 equiv) was added, and the resulting mixture was warmed to –20 °C. Under these conditions the trans-1,2-diol 67 was formed in 58% yield. Minor amounts (<5%) of quinone reduction products were detected by UPLC/MS analysis, suggesting that the reduction proceeds via the in situ generated borinite ester 66. To complete the synthesis, the diol 67 was deprotected by treatment with hydrochloric acid in methanol at low temperature, to provide (–)-kinamycin F (9) in 65% yield.
Synthesis of (–)-Lomaiviticin Aglycon (6)
With a route to the kinamycins established, we focused on the preparation of (–)-lomaiviticin aglycon (6). When we began our studies we hypothesized that, given the existence of the kinamycins, nature may synthesize lomaiviticins A (1) and B (2) by oxidative coupling of monomeric diazofluorenes (see Supporting Information for a working mechanism for the putative biosynthetic dimerization). Accordingly, we considered several synthetic methods to effect this transformation. Direct oxidative coupling of cycloalkanones,28 oxidative coupling of ketone, ester, and amide enolates,29 and single electron oxidation of enoxysilane30 or enamine31 derivatives all had precedent. Two considerations led us to focus on enoxysilane coupling pathways. First, in our kinamycin work, we discovered that β-oxygenated enoxysilanes (Scheme 7) derived from protected diazofluorenes could be generated in near quantitative yield. Second, we expected that β-elimination would be problematic using enolate-based methods.
In a model study, we found that enoxysilanes derived from simple diazofluorenes could be oxidatively coupled in good yield.8 This motivated us to study this transformation in more detail using a fully-functionalized substrate, and the diazofluorene 71 was prepared by the sequence outlined in Scheme 8. Oxidation15 of the enoxysilane 57 formed the enone 68 (92%). Treatment of the enone 68 with mesitylaldehyde dimethyl acetal32 in the presence of PPTS as catalyst formed the acetal 69 (12:1 mixture of endo and exo isomers, 80%). Homologation, fluoride-mediated coupling with 5,8-bis(methoxymethyl)-2,3-dibromonaphthoquinone (70), cyclization, and diazo transfer provided the endo-mesityl diazofluorene 71 in 37% overall yield. Treatment of the endo-mesityl diazofluorene 71 with p-toluenesulfonic acid in methanol formed the (–)-monomeric lomaiviticin aglycon (72, 55%).33 Alternatively, treatment of 71 with TBSOTf and DIPEA formed an enoxysilane (not shown) that was oxidatively coupled (ceric ammonium nitrate) to provide the C2-symmetric dimeric product 73 (42%), as well as the monomeric quinone 74 (8%). In these experiments, only a single dimeric product was formed, as determined by 1H NMR and UPLC/MS analysis of the unpurified reaction mixture. Although the C2-symmetry of the product was evident by NMR analysis, this element of symmetry also prevented rigorous assignment of relative stereochemistry. To obtain evidence for stereochemistry, we deprotected the dimer 73 by heating with 20% trifluoroacetic acid in methanol at 50 °C. Under these strongly acidic conditions, the starting material was converted to the aglycon 75 (41%). This compound was completely characterized by multidimensional NMR analysis, and these data revealed the presence of the C-1/1’ carbonyl substituents, providing evidence that ring closure to form the hemiketal structure of lomaiviticin B (2) had not occurred. Based on this result, and our kinamycin studies, we assigned the stereochemistry of 75 as the unnatural anti, anti-configuration. In retrospect, the formation of the anti, anti-diastereomer 73 is not surprising; the concave topology of the molecule and the steric congestion introduced by the mesityl protecting groups both favor bond formation to the α-face of the monomer 71.
Scheme 8.

Synthesis and Dimerization of the Bis(methoxymethyl)-endo-mesityldiazofluorene 71.
Aromatization issues notwithstanding, among several strategies considered to arrive at the natural syn, syn-configuration, double-epimerization of the anti, anti-aglycon 75 seemed the most straightforward. An extensive battery of conditions were evaluated with the goal of enolizing the ketone functional groups within 75. We hypothesized that even if the relative energies of the diastereomers were similar, cyclization to form lomaiviticin aglycon (6) should drive the equilibrium forward. Unfortunately, however, the anti, anti-aglycon 75 exhibited limited stability toward nucleophiles and bases. Under forcing acidic conditions, 75 was remarkably inert. For example, no deuterium incorporation was observed in the α-positions of 75 after heating with trifluoroacetic acid (20% v/v) in methanol-d4 for 7 min at 120 °C under microwave irradiation (1H NMR analysis).
We then turned our attention toward epimerization of the protected anti, anti-dimer 73. In this case, hemiketal formation is not possible, but we expected that protonation of an enol(ate) from the convex face to afford the desired syn, syn-stereochemistry would be kinetically favored. Although this protected intermediate was significantly more robust, activation of the α-protons was not possible. This may be due to the increased steric bulk about these protons originating from the mesityl acetal protecting groups. After several months of focused experimentation, our epimerization studies were abandoned.
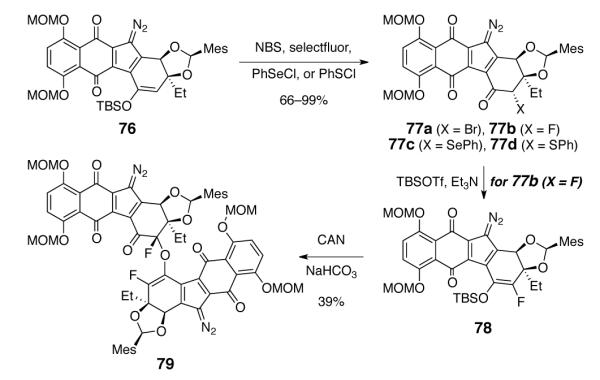
We next explored the installation of a removable functional group at the α-position of the diazofluorene (Scheme 9). In this approach, the stereochemistry of the resulting dimer would be ultimately controlled by removal of this functional group, rather than the dimerization step. Several α-functionalized diazofluorenes were prepared from the enoxysilane 76, including the α-bromo, α-fluoro, α-phenylselenyl, and α-phenylsulfenyl ketones (77a–d, respectively). However, with the exception of the α-fluoroketone derivative 77b, all attempts to generate α-substituted enoxysilanes from these functionalized diazofluorenes were unsuccessful. Although conditions for the reductive cleavage of α-fluoroketones are not widespread,34 we attempted to advance the fluorinated product 77b. Treatment of 77b with TBSOTf and TEA formed the enoxysilane 78. Exposure to CAN and sodium bicarbonate formed a single C1-symmetric dimeric product 79 (39%). The presence of extensive oxidation and fluorine substituents about the newly-formed bond made complete characterization of 79 difficult, but all NMR data are consistent with carbon–oxygen bond formation (as shown), rather than the expected carbon–carbon coupling product.
Scheme 9.
Synthesis and Reactions of α-Functionalized Diazofluorenes 77.
Although we were cognizant of the reactivity of the diazofluorene toward reducing agents,5c,d,g,h, we prepared8 the α-phenylselenyl and α, β-epoxy ketones 80 and 81 and examined the possibility of conducting a reductive coupling reaction by formation of an α-keto radical (Figure 3). Unfortunately, under a variety of reducing conditions8 extensive decomposition of the substrates was observed. We also attempted the direct oxidative coupling of the monomeric lomaiviticin aglycon (72). The flexibility introduced by removal of the cyclic protecting group was anticipated to allow access to the natural syn, syn-stereochemistry. However, efforts to dimerize 72, directly or in the presence of air or added oxidants [CAN, bis(trifluoroacetoxy)iodobenzene, potassium ferricyanide], were unsuccessful. In some instances oxidation to a putative diquinone (UPLC/MS and NMR analysis, not shown) was observed, but this could not be manipulated to a dimeric product.
Figure 3.
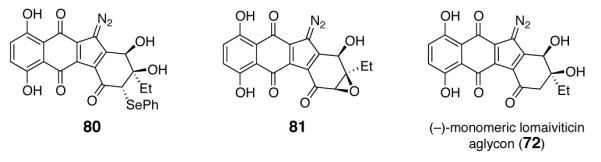
Structures of the α-Phenylselenyldiazofluorene 80, the α, β-Epoxydiazofluorene 81, and the Monomeric Lomaiviticin Aglycon (72).
The extensive studies described above served to define the stability profile of synthetic diazofluorenes: while they were reactive toward reducing agents and nucleophiles, they were robust toward strong oxidants. With this in mind, we prepared the C-3-deoxydiazofluorene 83 (Scheme 10) because we believed that removal of the C-3 oxygen substituent would bias the system for oxidative coupling anti to the ethyl substituent, to provide the natural relative configuration. Stereoretentive oxidation of the C-3/3′ C–H bonds of the product would then provide lomaiviticin aglycon (6). This strategy found encouraging precedent in the work of Wender and Baran, who had demonstrated that highly selective C–H hydroxylations can be effected in complex settings using dioxirane-based reagents. 35
Scheme 10.
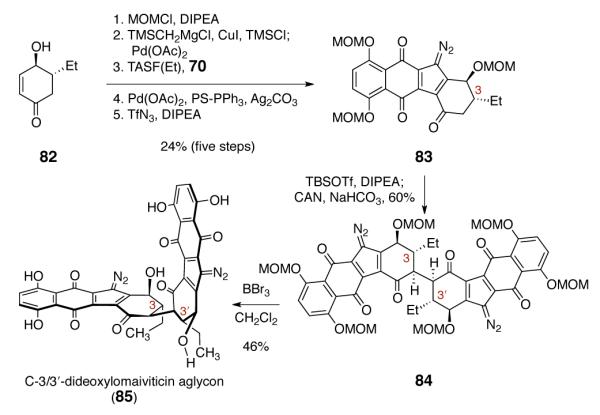
Synthesis of C-3/3′-Dideoxylomaiviticin Aglycon 85.
The synthesis of the protected C-3/3′-dideoxydimer 84 began with the ketone 82.36 Protection of the secondary alcohol (chloromethyl methyl ether, DIPEA) and elaboration as before provided the C-3-deoxydiazofluorene 83 (24% over five steps). Enoxysilane formation (TBSOTf, DIPEA), followed by addition of CAN, formed the oxidative coupling product 84 in 60% yield as a single detectable diastereomer. The relative stereochemistry of 84 was confirmed by observation of a W-plane coupling between the C-2 and C-4 hydrogen atoms. Deprotection of 84 (boron tribromide, dichloromethane) formed C-3/3′-dideoxylomaiviticin aglycon 85 (46%). Unfortunately, neither 84 nor 85 was reactive toward dimethyldioxirane or trifluorodimethyldioxirane, perhaps due to the steric congestion about the targeted C–H bonds.
At this juncture two invariable trends dominated our synthetic planning. First, single electron oxidation of monomeric enoxysilanes was the only method that provided dimeric products. Second, modification of the stereochemistry following the dimerization appeared unachievable. These considerations led us to focus on the synthesis of substrates that might be compatible with the enoxysilane coupling protocol and also provide the natural stereochemistry on dimerization. We considered the use of acyclic protecting groups on the vicinal diol function, however intermediates bearing acyclic alkyl or silyl ether protecting groups were less stable toward elimination of the β-oxygen substituent. The limited stability of the diol 68 prevented installation of smaller (unactivated) cyclic protecting groups, such as formaldehyde acetal.
Inspection of molecular models, and later, computational studies8 clearly revealed the origins of the steric bias in the dimerization of the endo-mesityl diazofluorene 71. In the energy-minimized structure,8 the mesityl substituent protrudes into the cavity of the 6-5 system. Approach from the α-face, to afford the undesired anti, anti-stereochemistry is kinetically favored. In considering ways to invert this bias, we recognized that epimerization of the mesityl substituent, to the exo orientation, might promote bond formation to the desired (concave) face of the enoxysilane. The exo-mesityl diazofluorene 87 was prepared by a modification of our route to the endo-isomer 71 (Scheme 11). Under conditions developed for protection of the diol 68, the endo-acetal is the kinetically-favored product. Warming of the reaction mixture to 50 °C promotes epimerization of the acetal, to form a 1:1 mixture of endo and exo diastereomers (86). Resubjection of the diastereomerically pure exo isomer (obtained by careful purification on ca. 25 mg scales) formed a 1:0.8 mixture of exo:endo isomers, suggesting they are nearly equal in energies. In practice, the mixture of endo and exo acetals 68 (and subsequent intermediates) were advanced through the diazo transfer step, at which point the endo- and exo-diazofluorenes 71 and 87 were isolated separately.
Scheme 11.
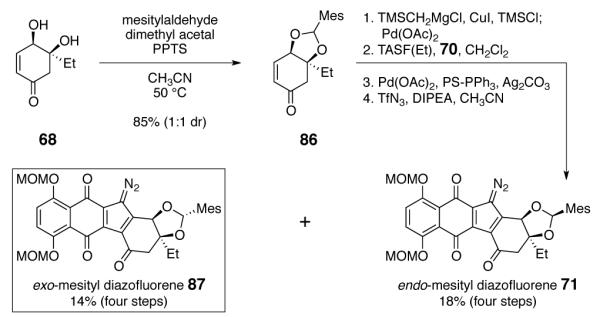
Synthesis of the exo-Mesityl Diazofluorene 87.
Ultimately, the diazofluorene 87 was successfully advanced to lomaiviticin aglycon (6) by a sequence comprising enoxysilane formation, oxidative dimerization, and deprotection (Scheme 12). Among a large number of oxidants evaluated for their ability to dimerize enoxysilanes derived from 87, only two, ceric ammonium nitrate (CAN) and manganese hexafluoroacetylacetonate (94),37, 38 were effective [oxidation potentials (vs. Fc) = 0.88 and 0.9 for CAN39 and 94,38 respectively]. Up to six isolable products are obtained in these transformations: the undesired anti, anti and syn, anti dimers 90 and 92, respectively, the desired syn, syn-dimer 91, the aromatized–oxidized monomer 74, and the α-nitrodiazofluorene 93 (when CAN was employed).
Scheme 12.

Products Identified in the Oxidative Coupling of the exo-Mesityldiazofluorene 87.
A detailed evaluation of the oxidative dimerization step has revealed that the product distribution, efficiency, and stereoselectivity of the transformation are dependent upon enoxysilane structure (silicon substituents), choice of oxidant, and solvent. Selected experiments that provide an overall representation of the factors influencing this reaction are shown in Table 5. Thus, treatment of the tert-butyldimethylsilyl derivative 88 with ceric ammonium nitrate afforded a 24% yield of the undesired anti, anti-diastereomer 90, 14% of the α-nitrodiazofluorene 93, and extensive amounts of unidentified decomposition products (entry 1). Within the limits of UPLC/MS analysis, no additional dimeric products were formed. Oxidation of 88 with manganese tris(hexafluoroacetylacetonate) (94) resulted in aromatization (74, 28%) and production of a large amount of unidentified decomposition products; less than 5% of the anti, anti-diastereomer 90 was formed (entry 2). Oxidation of the trimethylsilyl derivative 89 was much more efficient using 94, and the undesired anti, anti-dimer 90 was obtained in 26% yield when dichloromethane was employed as solvent. Encouragingly, oxidation of 89 with CAN formed the three dimeric products 90–92 in 17%, 6%, and 10% yield, respectively, as well as the aromatized monomer 74 (8%) and the diazofluorene 87 (8%, entry 4). By employing the trimethylenoxysilane 89, manganese tris(hexafluoroacetylacetonate) (94) as oxidant, and benzene as solvent, the desired syn, syn coupling product 91 was obtained as the major product (26%, entry 5). Smaller amounts of the undesired anti, anti-coupling product 90 (12%) were also isolated, and only trace quantities of the syn, anti-dimer 92 could be detected under these conditions (UPLC/MS analysis). These latter conditions were reproducibly executed on 100-mg scales.
Table 5.
Dimerization of the exo-Mesityldiazofluorene 87.a
| entry | R3b | oxidant | solvent | 90 | 91 | 92 | 74 | 87 |
|---|---|---|---|---|---|---|---|---|
| 1c,d | 88 | CAN | CH3CN | 24% | nde | nd | nd | 3% |
| 2 f | 88 | 94 | PhH | <5% | nd | nd | 28% | 5% |
| 3 g | 89 | 94 | CH2Cl2 | 26% | nd | <5% | 15% | <5% |
| 4 b | 89 | CAN | CH3CN | 17% | 6% | 10% | 8% | 8% |
| 5 f | 89 | 94 | PhH | 12% | 26% | <5% | 15% | 36% |
Isolated yields.
88: R3 = t-Bu(CH3)2; 89: R3 = (CH3)3.
CAN (2.0 equiv), sodium bicarbonate (20 equiv), CH3CN, –35 °C.
The α-nitrodiazofluorene 93 was obtained in 14% yield.
none detected.
Mn(hfacac)3 (94, 1.20 equiv), PhH, 24 °C.
Mn(hfacac)3 (94, 1.20 equiv), CH2Cl2, 24 °C.
The complexities of radical cation reactions40 and low mass balances in entries 1–3 of Table 5 caution against overinterpretation of these results. However, the enhanced anti, anti-stereoselectivity in the dimerization of 89 using manganese tris(hexafluoroacetylacetonate) (94) in benzene (entry 5) compared to dichloromethane (entry 3) may be due to formation of a tight ion pair in which the manganese anion is associated with the convex face of the radical cation.8 Kochi has shown that ion pairs formed from enoxysilanes are closely associated in non-polar solvents.41 This association may direct bond formation to the more hindered (concave) face of the substrate. Under identical conditions, oxidation of the trimethylenoxysilane derived from the endo-isomer 71 provides the anti, anti-dimer 73 exclusively (38%),8 suggesting that the shielding of the endo-mesityl substituent overrides the steric encumbrance of the manganese anion.
The final obstacle involved deprotection of the syn, syn-dimer 91. While the anti, anti-dimeric products 73 and 90 were stable toward strongly acidic conditions (vide supra; see Supporting Information for deprotection of 90), the syn, syn-dimer 91 decomposed under mildly acidic conditions. We attribute the heightened reactivity of 91 to the anti arrangement of the α-proton and β-oxygen substituents, which facilitates β-elimination. After many experiments that resulted in unidentifiable decomposition products, a useful lead was obtained. Treatment of 91 with excess trifluoroacetic acid in wet dichloromethane at –35 °C resulted in rapid formation of the mono mesityl-protected aglycon 95 (UPLC/MS analysis), which slowly converted to the aromatized aglycon 97 (Scheme 13A). We reasoned that the aromatized product 97 was forming by elimination of the β-oxocarbenium ion 96. To prevent this, we conducted the deprotection in the presence of a large excess of tert-butyl hydroperoxide (Scheme 13B).42 LC/MS analysis revealed that 95 accumulated under these conditions as well, but now neutralization of the reaction mixture at low temperature (–78 °C) followed by extractive workup provided mixtures of the chain isomer of lomaiviticin aglycon 98 as well as the expected ring isomer 6 (98:6 = 8–2:1), rather than the dehydrated product 97. Presumably, under these conditions, the oxocarbenium ion 96 is trapped by the peroxide nucleophile before it has a chance to eliminate.
Scheme 13.

A. Deprotection of the syn,syn-Dimer 91 (in the Absence of TBHP). B. Deprotection of the syn, syn-Dimer 91 (in the Presence of TBHP). C. Selected 1H and NOE NMR Data for the Mono Mesityl-protected Aglycon 95.
The chain isomer 98 was characterized by multidimensional NMR analysis, which established the presence of the C-1 carbonyl substituent. Attempted separation of this mixture by preparative thin-layer chromatography resulted in cyclization to form the ring isomer 6 exclusively (39–41% overall).
The aglycon 6 was fully characterized by NMR, IR, and HRMS analysis and all data are consistent with the structure shown. However, we observed some differences in NMR chemical shifts between the bis(hemiketal) core of 6 and those of lomaiviticin B (2). In particular, the H-2 and C-2 resonances of 6 are observed at 3.41 and 62.1 ppm, respectively (30% DMF-d7–CD3OD), while those of lomaiviticin B (2) are reported to resonate at 2.64 and 44.0 ppm, respectively (CD3OD; the aglycon 6 is sparingly soluble in CD3OD, which prohibited acquisition of spectroscopic data in this solvent).1a The C-2 and H-2 resonances of lomaiviticin aglycon 6 are in good agreement with a model system prepared by Nicolaou and co-workers (δ H-2 = 2.87, δ C-2 = 60.4, C6D6)7a and NMR spectroscopic data for the chain isomer 98 matched closely those of lomaiviticin A (1). To provide further evidence for connectivity and stereochemistry in 4, we isolated the monomesityl aglycon 95 (Scheme 13A) by arresting the deprotection of 91 prematurely. The 1H and 13C resonances within the bis(cyclohexenone) substructure of 95 were resolved in CDCl3, and were assigned by multidimensional NMR analysis, providing confirmation of connectivity. An NOE between H-3′ and H-1′ was observed, providing further confirmation of the syn, syn-stereochemistry of the dimerization product 91 (Scheme 13C).
The data above left little doubt as to the structure of synthetic lomaiviticin aglycon 6. We recognized, however, that natural lomaiviticin B (2) was obtained as its bis(trifluoroacetate) salt, following a final HPLC purification step.1a To probe for potential effects of charge on chemical shifts, lomaiviticin B (2) was obtained by partial hydrolysis of lomaiviticin A1b (1, trifluoroacetic acid, aqueous methanol, 45 °C, 82%).8 The H-2 and C-2 resonances (as well as all other resonances) of the bis(trifluoroacetate) salt of lomaiviticin B (2) obtained in this way were in exact agreement with literature values. The free base of 2 was obtained by a basic wash. The chemical shifts of H-2 and C-2 of the free base form were in better agreement with (–)-lomaiviticin aglycon (6, free base of 2: δ H-2 = 3.64, δ C-2 = 71.3, CD3OD). Thus, the chemical shift discrepancies observed between (–)-lomaiviticin aglycon (6) and natural lomaiviticin B (2) are attributed to the charged state of 2 when it was obtained from the producing organism.1a
Hydrodediazotization Studies
A growing body of data suggests the cytotoxic effects of the kinamycins and lomaiviticins are mediated by a reducing co-factor,5 and the dimeric and monomeric diazofluorene derivatives prepared above provide an opportunity to probe substituents effects on the redox activity of the diazofluorene. We evaluated the rate of reduction of the unnatural anti, anti-aglycon 75, (–)-lomaiviticin aglycon (6), the monomeric lomaiviticin aglycon 72, and (–)-kinamycin C (8) by dithiothreitol (1 equiv) in methanol at 37 °C (Table 6). The anti,anti-dimer 75 was studied because it contains the flexibility and conjugated ketone substituents found within lomaiviticin A (1). Under these conditions, the anti, anti-dimer 75 and the monomeric lomaiviticin aglycon (72) undergo hydrodediazotization to form the hydroxyfulvenes 99 and 100, respectively, and more slowly, the products of methanolysis (101 and 102).5k Interestingly, these data reveal that the rate of reduction of the anti,anti-dimer 75 is ca. 3-fold faster than that of the monomeric lomaiviticin aglycon (72). Perhaps more strikingly, under otherwise identical conditions, neither (–)-lomaiviticin aglycon (6) nor (–)-kinamycin C (8) formed detectable levels of reduction products (UPLC/MS analysis). Collectively, these data suggest that the ketone function of lomaiviticin A (1) raises the oxidation potential of the diazofluorene, allowing for facile electron transfer. Additionally, a stabilizing donor–acceptor interaction between the hydroxyfulvene and the remaining diazofluorene may further promote reductive activation of dimers such as 75, and lomaiviticin A (1). Thus, though lomaiviticins A (1) and B (2) share a good deal of homology, the additional flexibility present in the structure of lomaiviticin A (1) is suggested to enhance the rate of reduction of the metabolite.
Table 6.
Reduction–Solvolysis of 75 and 72.a
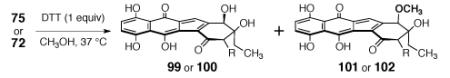
| time (h) | 75 | 99 | 101 | 72 | 100 | 102 |
|---|---|---|---|---|---|---|
| 0 | 100% | 0% | 0% | 100% | 0% | 0% |
| 1 | 62% | 38% | 0% | 89% | 10% | 1% |
| 10 | 22% | 78% | 0% | 76% | 11% | 13% |
% composition was determined by UPLC/MS analysis.
Identification of Simple Diazofluorenes as Potent Inhibitors of Ovarian Cancer Stem Cells
In parallel with our synthetic studies we have conducted structure–function studies of synthetic diazofluorenes. One aspect of this research has focused on evaluating the activity of our synthetic analogs against cancer stem cells (CSCs), a small population of cells within tumors that are highly tumorigenic and chemoresistant. CSCs have been identified in many different cancers, including ovarian cancer.43
We have found that the diazofluorene 103 (Figure 4) potently inhibits the growth of cultured ovarian cancer stem cells (IC50 = 500 nM). This compound is readily-prepared (500 mg of 103 was synthesized in less than one week in our laboratory).8 Although the development of 103 as a clinical agent is of interest, of equal or greater significance are identification of the biological mechanisms underlying the cytotoxic effects of 103 in this cell line, an avenue of research we are currently pursuing.
Figure 4.
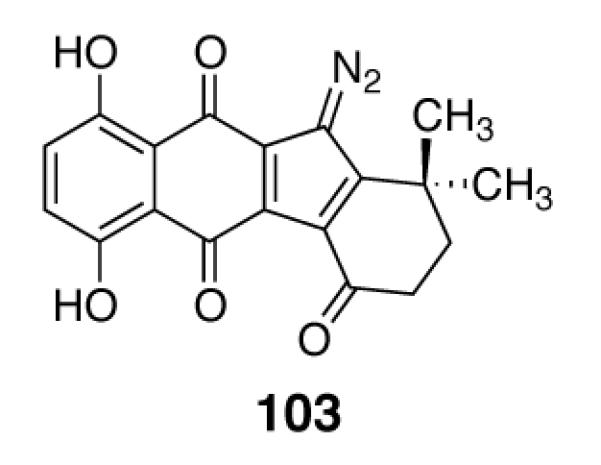
Structure of the Anti-Cancer Stem Cell Agent 103.
Conclusion
We have described the evolution of enantioselective synthetic routes to (–)-kinamycin F (9) and (–)-lomaiviticin aglycon (6). Our synthetic routes were enabled by the development of scalable, efficient, and enantioselective syntheses of the kinamycin and lomaiviticin D-ring precursors 60 and 68, and implementation of a convergent, three-step strategy for construction of the diazofluorene functional group. Use of substrate control to effect the transformation of the ketone 65 to the trans-1,2-diol 67, in the presence of the redox-sensitive diazofluorene, allowed for a short synthesis of (–)-kinamycin F (9, 13 steps from m-cresol).
Extensive investigations into the oxidative coupling of monomeric diazofluorenes such as 87 has allowed for a short (11-step) synthesis of (–)-lomaiviticin aglycon (6). These studies have revealed a complex relationship between substrate structure and reaction conditions, and stereochemical outcome and product distributions. Ultimately, conditions were found to form a precursor to lomaiviticin aglycon (6) in good yield (26–33%), and this was successfully advanced to the target.
Several new reactions, including a novel γ-quinonylation of β-trimethylsilyl- α- β-unsaturated ketones, and a new α-quinonylation of enoxysilanes, were developed. In addition, we have described the application of nonaflylazide as a storable, bench-stable surrogate for the volatile and highly reactive reagent trifluoromethanesulfonyl azide. We have also described the first use of manganese tris(hexafluoroacetylacetonate) (94) in the oxidative coupling of enoxysilanes. This reagent possesses physical properties (high solubility in nonpolar solvents and a coordinatively-saturated metal center) that are complementary to more common oxidants (such as CAN), and this reagent may find applications in settings beyond those described here. We have obtained evidence that the flexible, dimeric structure of lomaiviticin A (1) may enhance the rate of reductive activation of the metabolite, and we have identified simple diazofluorenes (103) as potent inhibitors of ovarian cancer stem cells.
The results reported herein lay the foundation for synthesis of lomaiviticin A (1) itself. We have delineated efficient routes to advanced monomeric diazofluorenes and established their stability and reactivity profiles. The successful oxidative coupling of these systems provides encouragement that the dimerization of fully functionalized (glycosylated) monomeric diazofluorenes may be possible.
Supplementary Material
Acknowledgement
Partial financial support from the American Cancer Society, the National Institute of General Medical Sciences (R01GM090000), the National Science Foundation (Graduate Research Fellowship to C. M. W.), the Searle Scholar Program, and Yale University is gratefully acknowledged. We thank Dr. Gil Mor and Dr. Ayesha Alvero for determining the activity of 103 against ovarian CSCs and Dr. Eric Paulson of the Yale Chemical and Biophysical Instrumentation Center for valuable assistance with NMR. We acknowledge the Developmental Therapeutics Program of the National Cancer Institute for in vivo toxicity analysis of 103. S.B.H. is a fellow of the David and Lucile Packard and the Alfred P. Sloan Foundations, and is a Camille Dreyfus Teacher–Scholar.
Footnotes
Supporting Information Available: Experimental procedures and detailed characterization data of all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1(a).He H, Ding WD, Bernan VS, Richardson AD, Ireland CM, Greenstein M, Ellestad GA, Carter GT. J. Am. Chem. Soc. 2001;123:5362. doi: 10.1021/ja010129o. Woo CM, Beizer NE, Janso JE, Herzon SB. J. Am. Chem. Soc. Article ASAP, DOI: 10.1021/ja3074984. . For reviews, see: Marco-Contelles J, Molina MT. Curr. Org. Chem. 2003;7:1433. Arya DP. Top. Heterocycl. Chem. 2006;2:129. Nawrat CC, Moody CJ. Nat. Prod. Rep. 2011;28:1426. doi: 10.1039/c1np00031d. Herzon SB, Woo CM. Nat. Prod. Rep. 2012;29:87. doi: 10.1039/c1np00052g.
- 2(a).Ito S, Matsuya T, Ōmura S, Otani M, Nakagawa A. J. Antibiot. 1970;23:315. doi: 10.7164/antibiotics.23.315. [DOI] [PubMed] [Google Scholar]; (b) Hata T, Ōmura S, Iwai Y, Nakagawa A, Otani M. J. Antibiot. 1971;24:353. doi: 10.7164/antibiotics.24.353. [DOI] [PubMed] [Google Scholar]; (c) Ōmura S, Nakagawa A, Yamada H, Hata T, Furusaki A, Watanabe T. Chem. Pharm. Bull. 1971;19:2428. doi: 10.1248/cpb.21.931. [DOI] [PubMed] [Google Scholar]; (d) Furusaki A, Matsui M, Watanabe T, Ōmura S, Nakagawa A, Hata T. Isr. J. Chem. 1972;10:173. [Google Scholar]; (e) Cone MC, Seaton PJ, Halley KA, Gould SJ. J. Antibiot. 1989;42:179. doi: 10.7164/antibiotics.42.179. [DOI] [PubMed] [Google Scholar]; (f) Seaton PJ, Gould SJ. J. Antibiot. 1989;42:189. doi: 10.7164/antibiotics.42.189. [DOI] [PubMed] [Google Scholar]
- 3(a).Lin H-C, Chang S-C, Wang N-L, Chang L-R. J. Antibiot. 1994;47:675. doi: 10.7164/antibiotics.47.675. [DOI] [PubMed] [Google Scholar]; (b) Lin H-C, Chang S-C, Wang N-L, Chang L-R. J. Antibiot. 1994;47:681. doi: 10.7164/antibiotics.47.675. [DOI] [PubMed] [Google Scholar]
- 4(a).Moore HW. Science. 1977;197:527. doi: 10.1126/science.877572. [DOI] [PubMed] [Google Scholar]; (b) Rockwell S, Sartorelli AC, Tomasz M, Kennedy KA. Canc. Metastasis Rev. 1993;12:165. doi: 10.1007/BF00689808. [DOI] [PubMed] [Google Scholar]
- 5(a).Arya DP, Jebaratnam DJ. J. Org. Chem. 1995;60:3268. [Google Scholar]; (b) Laufer RS, Dmitrienko GI. J. Am. Chem. Soc. 2002;124:1854. doi: 10.1021/ja0167809. [DOI] [PubMed] [Google Scholar]; (c) Feldman KS, Eastman KJ. J. Am. Chem. Soc. 2005;127:15344. doi: 10.1021/ja053880w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Feldman KS, Eastman KJ. J. Am. Chem. Soc. 2006;128:12562. doi: 10.1021/ja0642616. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zeng W, Ballard TE, Tkachenko AG, Burns VA, Feldheim DL, Melander C. Bioorg. Med. Chem. Lett. 2006;16:5148. doi: 10.1016/j.bmcl.2006.07.024. [DOI] [PubMed] [Google Scholar]; (f) O’Hara KA, Wu X, Patel D, Liang H, Yalowich JC, Chen N, Goodfellow V, Adedayo O, Dmitrienko GI, Hasinoff BB. Free Radic. Biol. Med. 2007;43:1132. doi: 10.1016/j.freeradbiomed.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ballard TE, Melander C. Tetrahedron Lett. 2008;49:3157. [Google Scholar]; (h) Khdour O, Skibo EB. Org. Biomol. Chem. 2009;7:2140. doi: 10.1039/b903844b. [DOI] [PubMed] [Google Scholar]; (i) Heinecke CL, Melander C. Tetrahedron Lett. 2010;51:1455. [Google Scholar]; (j) O’Hara KA, Dmitrienko GI, Hasinoff BB. Chem. Biol. Interact. 2010;184:396. doi: 10.1016/j.cbi.2010.01.013. [DOI] [PubMed] [Google Scholar]; (k) Mulcahy SP, Woo CM, Ding WD, Ellestad GA, Herzon SB. Chem. Sci. 2012;3:1070. [Google Scholar]
- 6(a).Lei X, Porco JA. J. Am. Chem. Soc. 2006;128:14790. doi: 10.1021/ja066621v. Kumamoto T, Kitani Y, Tsuchiya H, Yamaguchi K, Seki H, Ishikawa T. Tetrahedron. 2007;63:5189. Nicolaou KC, Li H, Nold AL, Pappo D, Lenzen A. J. Am. Chem. Soc. 2007;129:10356. doi: 10.1021/ja074297d. Woo CM, Lu L, Gholap SL, Smith DR, Herzon SB. J. Am. Chem. Soc. 2010;132:2540. doi: 10.1021/ja910769j. Scully SS, Porco JA. Angew. Chem., Int. Ed. 2011;50:9722. doi: 10.1002/anie.201104504. For studies toward the kinamycins, see: Chen N, Carriere MB, Laufer RS, Taylor NJ, Dmitrienko GI. Org. Lett. 2008;10:381. doi: 10.1021/ol702650u. Scully SS, Porco JA. Org. Lett. 2012;14:2646. doi: 10.1021/ol3010563.
- 7(a).Nicolaou KC, Denton RM, Lenzen A, Edmonds DJ, Li A, Milburn RR, Harrison ST. Angew. Chem., Int. Ed. 2006;45:2076. doi: 10.1002/anie.200504466. [DOI] [PubMed] [Google Scholar]; (b) Krygowski ES, Murphy-Benenato K, Shair MD. Angew. Chem., Int. Ed. 2008;47:1680. doi: 10.1002/anie.200704830. [DOI] [PubMed] [Google Scholar]; () Morris WJ, Shair MD. Org. Lett. 2008;11:9. doi: 10.1021/ol8022006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang W, Baranczak A, Sulikowski GA. Org. Lett. 2008;10:1939. doi: 10.1021/ol800460a. [DOI] [PubMed] [Google Scholar]; (d) Gholap SL, Woo CM, Ravikumar PC, Herzon SB. Org. Lett. 2009;11:4322. doi: 10.1021/ol901710b. [DOI] [PubMed] [Google Scholar]; (e) Nicolaou KC, Nold AL, Li H. Angew. Chem., Int. Ed. 2009;48:5860. doi: 10.1002/anie.200902509. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Lee HG, Ahn JY, Lee AS, Shair MD. Chem. –Eur. J. 2010;16:13058. doi: 10.1002/chem.201002157. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Morris WJ, Shair MD. Tetrahedron Lett. 2010;51:4310. doi: 10.1016/j.tetlet.2010.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Herzon SB, Lu L, Woo CM, Gholap SL. J. Am. Chem. Soc. 2011;133:7260. doi: 10.1021/ja200034b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Baranczak A, Sulikowski GA. Org. Lett. 2012;14:1027. doi: 10.1021/ol203390w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.See Supporting Information.
- 9.Díez E, Fernández R, Gasch C, Lassaletta JM, Llera JM, Martín-Zamora E, Vázquez J. J. Org. Chem. 1997;62:5144. [Google Scholar]
- 10(a).Middleton WJ. Tris(substituted amino)sulfonium Salts. 3,940,402 U.S. Patent. 1976; (b) Noyori R, Nishida I, Sakata J. Tetrahedron Lett. 1980;21:2085. [Google Scholar]; (c) Noyori R, Nishida I, Sakata J, Nishizawa M. J. Am. Chem. Soc. 1980;102:1223. [Google Scholar]
- 11.Huot R, Brassard P. Can. J. Chem. 1974;52:838. [Google Scholar]
- 12(a).Tamayo N, Echavarren AM, Paredes MC. J. Org. Chem. 1991;56:6488. [Google Scholar]; (b) Liebeskind LS, Riesinger SW. J. Org. Chem. 1993;58:408. [Google Scholar]
- 13.Tietze LF, Gericke KM, Schuberth I. Eur. J. Org. Chem. 2007;2007:4563. doi: 10.1039/b700838d. [DOI] [PubMed] [Google Scholar]
- 14.Fujiwara Y, Domingo V, Seiple IB, Gianatassio R, Del Bel M, Baran PS. J. Am. Chem. Soc. 2011;133:3292. doi: 10.1021/ja111152z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15(a).Ito Y, Hirao T, Saegusa T. J. Org. Chem. 1978;43:1011. [Google Scholar]; (b) Larock RC, Hightower TR, Kraus GA, Hahn P, Zheng D. Tetrahedron Lett. 1995;36:2423. [Google Scholar]
- 16(a).Reich HJ, Reich IL, Renga JM. J. Am. Chem. Soc. 1973;95:5813. [Google Scholar]; (b) Sharpless KB, Launer RF, Teranishi AY. J. Am. Chem. Soc. 1973;95:6137. [Google Scholar]
- 17.Pilcher AS, DeShong P. J. Org. Chem. 1996;61:6901. doi: 10.1021/jo960922d. [DOI] [PubMed] [Google Scholar]
- 18(a).Fiallo MML, Garnier-Suillerot A. Biochemistry. 1986;25:924. doi: 10.1021/bi00352a028. [DOI] [PubMed] [Google Scholar]; (b) Fiallo MML, Garnier-Suillerot A. Inorg. Chim. Acta. 1987;137:119. [Google Scholar]
- 19.Doering W. v. E., DePuy CH. J. Am. Chem. Soc. 1953;75:5955. [Google Scholar]
- 20.Taber DF, Ruckle RE, Hennessy MJ. J. Org. Chem. 1986;51:4077. [Google Scholar]
- 21.Baum JS, Shook BC, Davies HML, Smith HD. Synth. Commun. 1987;17:1709. [Google Scholar]
- 22.Goddard-Borger ED, Stick RV. Org. Lett. 2007;9:3797. doi: 10.1021/ol701581g. [DOI] [PubMed] [Google Scholar]
- 23.Cavender CJ, Shiner VJ. J. Org. Chem. 1972;37:3567. [Google Scholar]
- 24.Zhu S-Z. J. Chem. Soc., Perkin Trans. 1. 1994:2077. [Google Scholar]
- 25.Kolb HC, VanNieuwenhze MS, Sharpless KB. Chem. Rev. 1994;94:2483. [Google Scholar]
- 26.Evans DA, Chapman KT, Carreira EM. J. Am. Chem. Soc. 1988;110:3560. [Google Scholar]
- 27.Brown HC, Vogel FGM. Liebigs Ann. Chem. 1978:695. [Google Scholar]
- 28.Hawkins EGE, Large R. J. Chem. Soc., Perkin Trans. 1. 1974:280. [Google Scholar]
- 29(a).Ito Y, Konoike T, Harada T, Saegusa T. J. Am. Chem. Soc. 1977;99:1487. [Google Scholar]; (b) Baran PS, DeMartino MP. Angew. Chem., Int. Ed. 2006;45:7083. doi: 10.1002/anie.200603024. [DOI] [PubMed] [Google Scholar]; (c) DeMartino MP, Chen K, Baran PS. J. Am. Chem. Soc. 2008;130:11546. doi: 10.1021/ja804159y. [DOI] [PubMed] [Google Scholar]
- 30(a).Baciocchi E, Casu A, Ruzziconi R. Tetrahedron Lett. 1989;30:3707. [Google Scholar]; (b) Avetta CT, Konkol LC, Taylor CN, Dugan KC, Stern CL, Thomson R. J. Org. Lett. 2008;10:5621. doi: 10.1021/ol802516z. [DOI] [PubMed] [Google Scholar]
- 31.Narasaka K, Okauchi T, Tanaka K, Murakami M. Chem. Lett. 1992;21:2099. [Google Scholar]
- 32.Ji N, O’Dowd H, Rosen BM, Myers AG. J. Am. Chem. Soc. 2006;128:14825. doi: 10.1021/ja0662467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.This compound was previously prepared by Nicolaou and co-workers (ref. 7a), and was previously named “the monomeric unit of lomaiviticin aglycon”. Spectroscopic data for synthetic 72 were in agreement with the material synthesized by Nicolaou and co-workers.
- 34(a).Surya Prakash GK, Hu J, Olah GA. J. Fluorine Chem. 2001;112:355. [Google Scholar]; (b) Hata H, Kobayashi T, Amii H, Uneyama K, Welch JT. Tetrahedron Lett. 2002;43:6099. [Google Scholar]
- 35(a).Wender PA, Hilinski MK, Mayweg AVW. Org. Lett. 2004;7:79. doi: 10.1021/ol047859w. [DOI] [PubMed] [Google Scholar]; (b) Chen K, Baran PS. Nature. 2009;459:824. doi: 10.1038/nature08043. [DOI] [PubMed] [Google Scholar]
- 36.Ohnemüller UK, Nising CF, Encinas A, Bräse S. Synthesis. 2007;2007:2175. [Google Scholar]
- 37.Evans S, Hamnett A, Orchard AF, Lloyd DR. Faraday Discuss. Chem. Soc. 1972;54:227. [Google Scholar]
- 38.Bryant JR, Taves JE, Mayer JM. Inorg. Chem. 2002;41:2769. doi: 10.1021/ic025541z. [DOI] [PubMed] [Google Scholar]
- 39.Connelly NG, Geiger WE. Chem. Rev. 1996;96:877. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- 40.Schmittel M, Burghart A. Angew. Chem., Int. Ed. 1997;36:2550. [Google Scholar]
- 41.Bockman TM, Kochi JK. J. Chem. Soc., Perkin Trans. 2. 1996:1633. [Google Scholar]
- 42.Myers AG, Fundy MAM, Lindstrom PA., Jr. Tetrahedron Lett. 1988;29:5609. [Google Scholar]
- 43(a).Zhou JB, Zhang Y. Expet. Opin. Drug. Discov. 2009;4:741. doi: 10.1517/17460440903002059. [DOI] [PubMed] [Google Scholar]; (b) Alvero AB, Chen R, Fu H-H, Montagna M, Schwartz PE, Rutherford T, Silasi D-A, Steffensen KD, Waldstrom M, Visintin I, Mor G. Cell Cycle. 2009;8:158. doi: 10.4161/cc.8.1.7533. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bapat SA. Reproduction. 2010;140:33. doi: 10.1530/REP-09-0389. [DOI] [PubMed] [Google Scholar]; (d) Wei X, Dombkowski D, Meirelles K, Pieretti-Vanmarcke R, Szotek PP, Chang HL, Preffer FI, Mueller PR, Teixeira J, MacLaughlin DT, Donahoe PK. Proc. Natl. Acad. Sci. U.S.A. 2010;107:18874. doi: 10.1073/pnas.1012667107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.