

Chemical communication among bacteria, termed quorum sensing (QS), is a phenomenon that has attracted considerable interest over the last three decades. In this process, the exchange of small chemical signals enables bacterial populations to act together as an ensemble rather than single organisms.[1] This allows bacteria to achieve functions beneficial to an entire population and promotes coexistence with higher organisms. For example, several bacterial species use QS to regulate biofilm formation, resulting in an increased survival rate due to a higher tolerance to antibiotics.[1] Thus, interference of this communication could lead to improved control of bacterial infections and contaminations in health care settings. Importantly, this approach presents advantages over traditional antimicrobials because of a presumed diminished selective pressure to develop resistance.[2] Therefore, a sound understanding of this communication system at a molecular level could be vital for new anti-microbial therapeutics.
Autoinducer-2 (AI-2) is an important class of QS signals that are produced by many bacteria and are purported to be interspecies signals. Currently, only two AI-2 chemical signals have been characterized, both of which are derived from 4,5-dihydroxy-2,3-pentanedione (DPD, 1), which is produced by the enzyme LuxS.[3] DPD, while at face value seemingly presents itself as a simple, linear five carbon fragment, in aqueous solution DPD exists in a complex equilibrium between both linear (A) and cyclic stereoisomers (B1 and B2, Figure 1a). Indeed, the subsequent hydration of both cyclic forms (C1 and C2) of this highly functionalized molecule is described in almost all reports, and in fact X-ray crystallography has shown that C2 is the isomer recognized by the human pathogen Salmonella typhimurium, the plant symbiont Sinorhizobium meliloti and recently in Yersinia pestis.[4] Crystallographic analysis has also revealed that the active species in Vibrio harveyi is a borate ester (D) in complex with LuxP in 2002.[5] Adding to DPD’s intricacy is that phosphorylation of its primary alcohol by the kinase LsrK occurs in members of the Enterobacteriaceae family (e.g. S. typhimurium and Escherichia coli) to form the species that directly affect gene expression.[6] Thus, the dense oxygenation of DPD grants an interconversion of several signals from a single precursor.
Figure 1.
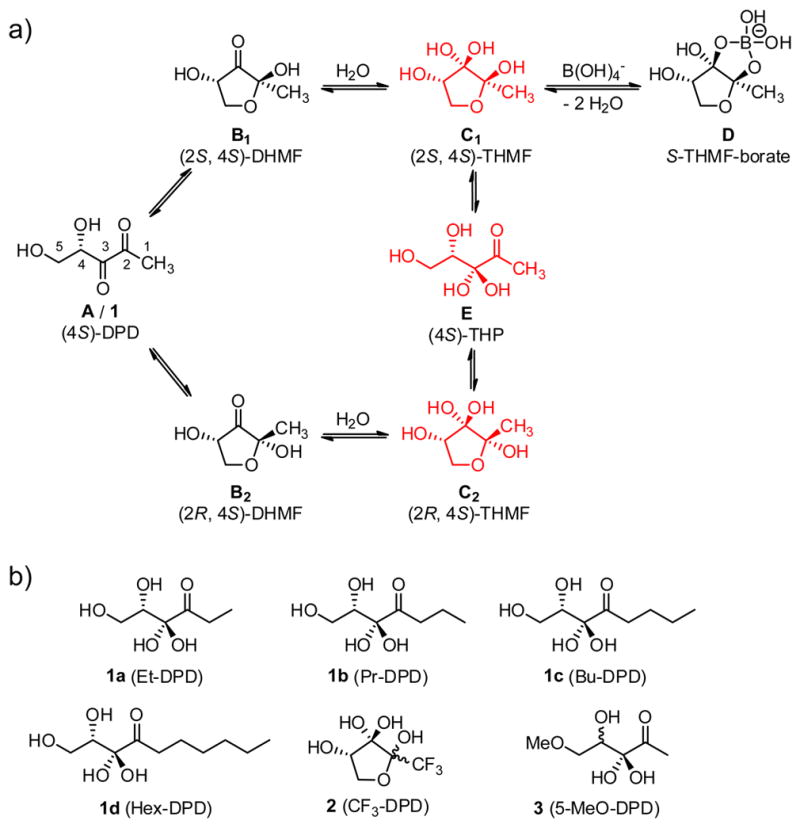
a) The equilibrium of autoinducer-2 (AI-2/1) as typically described in the literature (black color) and the equilibrium identified in this study (red color). The diketone species A is in equilibrium with B1 and B2 which are hydrated to grant C1 and C2. The borate species D was discovered as the active species in V. harveyi. Species E is proven as the major linear structure in this study. b) DPD analogues described in the current study.
Numerous syntheses of DPD have been described in the last decade following the structural elucidation of AI-2.[7] Forthcoming from these syntheses have been analogues revealing how drastic changes in biological activity occur even with minor scaffold alterations, i.e. C1-alkyl analogues 1a-d (Figure 1b).[8] Notably, several of these studies have focused on the ring dynamics of DPD and the corresponding requirements for signaling activity. For example, CF3-DPD (2) has been reported to exist exclusively in the cyclic form.[9] However, detailed structural analyses of the equilibrium and hydration states of the natural molecule DPD (1) are lacking. Thus, only limited quantitative information has been described for each species of DPD, a problem exacerbated by the fact that mass spectrometry techniques are unable to distinguish between molecules of the same molecular weight.[10] Accordingly, we sought to characterize the important but confounding equilibrium and hydration states of DPD using a variety of NMR techniques. We examined these interactions over a wide range of pH that we reasoned would provide insights into DPD’s stability as well as unique isomers present at physiological pH. Finally, we prepared and analyzed several key analogues enabling logical deduction of previously unrecognized DPD species.
Previous NMR analysis has shown the ratio of the linear to both cyclic forms as 1:4 under strongly acidic conditions.[7b] To provide insight into DPD’s equilibrium at physiological pH, we buffered an aqueous solution of 1 to pH 7 (1 M NaD2PO4/Na2DPO4). Based on the previously assigned 1H NMR spectrum under acidic conditions, we anticipated a straightforward assignment of the signals, however, we observed major changes in both the number and intensities of the methyl group signals (δ = 1.3–2.4 ppm) as well as the signals of the DPD-core (C4–5/δ = 3.5–4.4 ppm), mainly resulting in overlapping peaks. These findings became even more convoluted as we observed another signal change after 24 h, which we ascribed to be the final stage of the equilibrium (Supporting Figure S1). Due to the complexity of the spectra, we were neither able to clearly assign peaks to any species nor integrate single peaks, but we did observe a decrease in intensity of the original peaks. As such, it is evident that the equilibrium of DPD is pH-dependent and that there are previously unaccounted species in equilibrium at physiological pH.
To obtain a more detailed picture of this shift in equilibrium, we titrated DPD with NaOD in D2O (0.1 M) to follow the shifts in equilibrium stepwise. The most significant changes came between pH 4–5, with several new signals appearing in the region of the methyl protons. Importantly, the final 1H NMR spectrum at pH 7 of DPD under these unbuffered conditions is exactly the same as the spectrum in aqueous phosphate solution after 24 h incubation. In addition, we titrated DPD 1 to pH 10 to investigate the stability and the changes under basic conditions. Interestingly, the new methyl peaks around 1.3 ppm are even more dominant under basic conditions and the three major peaks under acidic conditions disappear completely. We next acidified this solution to pH 1 and observed the same 1H NMR spectrum as our starting solution (Supporting Figure S2). Thus, the changes are reversible, which precludes polymerization or decomposition of DPD during the titration, and indicates an unexpected stability of this highly oxygenated molecule over a broad pH range. We thus highlight DPD’s chemical stability in complete contrast to other well documented QS molecules such as acylhomoserine lactones (AHLs), and autoinducing peptides (AIPs), which have inherent susceptibility to hydrolytic degradation.[11] QS signaling molecules stability/instability has been probed previously[11a] and suggests alternate biochemical roles for DPD and its equilibrium species.
Because of the overlapping signals in the 1H NMR spectra, we next turned to 13C NMR spectroscopy with isotopically labeled DPD derivatives, 13CH3-DPD 4[7b] as well as the 1,2-bis-13C-labeled compound 5 (Figure 2). As anticipated, both labeled derivatives presented the same pH dependent behavior as natural DPD as viewed by 1H NMR spectra (Figure 2a/Supporting Figure S3). However, the presence of the 13C isotope resulted in altered splitting patterns of each methyl group in the 1H NMR spectra (1JCH = 127.9–129.1 Hz and 2JCH = 5.0–6.1 Hz). Nevertheless, the inclusion of these two isotopically labeled compounds allowed us to focus on the two carbon atoms in 13C NMR, C1 and C2, that undergo significant changes (vide supra) based on hydration or cyclization. Analysis of the labeled compounds indeed revealed that the 13C NMR peaks are of better resolution than the proton signals and we observed for the methyl carbons three major (20.0, 20.6 and 25.2 ppm) and four minor peaks at acidic pH. Basic titration resulted in a decrease in signal intensity of the major peaks and the appearance of new peaks, clearly separated from the original signals, at 23.1 ppm. However, these new peaks overlapped with each other indicating similar structures of the new species. From the 13C NMR spectrum of the monolabeled compound 4 we can surmise that 8–9 different DPD species are in equilibrium at physiological pH (Supporting Figure S4). To quantify each labeled signal, we determined the relaxation time for the 13C NMR nuclei, a technique rarely used in literature for small molecules but made possible with the multiple 13C isotopes. Thus, the overlapping signals at 23.1 ppm were quantified to be 1.5–2 times more abundant at pH 7 than the three originally identified major peaks (20.0, 20.5 and 25.2 ppm). Surprisingly, quantification revealed that the ratio of the original peaks is constant over the whole pH range with an excess of 4.3–4.6 of both cyclic species to the linear species. This clearly shows that the abundance of the original species is reduced with higher pH, but that the linear/cyclic equilibrium is not influenced by the new species at physiological pH.
Figure 2.

a) 1H NMR spectra illustrating the pH dependency of 13C labeled DPD 4. b) Relevant 13C NMR spectra for the base titration of bis-13C labeled DPD 5 (Full spectrum in/Supporting Figure S3). The different DPD species are assigned into 3 groups I, II, III, while 13C isotopes are indicated with an asterisk (*).
The additional 13C label incorporated within compound 5, at position C2 is important because multiple ring closing equilibria are taking place at this position. In addition, hydration and dehydration processes could be followed at this position, which again are readily seen via 13C NMR spectroscopy (Figure 2b). Importantly, this compound allows direct assignments of each methyl group to the corresponding quaternary signal of position C2 using the C–C coupling constants. We have identified three major species present that we have labeled as I, II, and III as seen in Figure 2b (Group II consists of both closed species).
The signal at 211 ppm corresponds to a linear species of DPD containing a carbonyl group and can be assigned to the methyl group of species I at 25.2 ppm with 1JCC = 42 Hz (Figure 2b). In addition, we could assign both cyclic forms of DPD at 20.0 and 20.5 ppm to the quaternary carbon signals at 104.0 and 104.5 ppm with a higher coupling constant (1JCC = 47–48 Hz/group II). The tendency of higher coupling constants for the more rigid cyclic forms compared with the linear form is in accordance with literature values.[12] Yet, the newly observed peaks of group III have an even higher coupling constant of 1JCC= 51 Hz. These large values provide a glimpse that other cyclic forms exist; however, these NMR spectra did not serve to fully dissect their exact structure.
We observe only one major signal at 211 ppm corresponding to the C2 carbonyl group of the linear species and we do not observe any shift during the titration. Consequently, we can exclude any hydration/dehydration processes occurring at C3 and this position is readily hydrated in aqueous solution upon deprotection. These findings are in agreement with NMR studies that describe a high tendency of monohydration of α-diketones in aqueous solutions of up to 70 %;[13] however, we believe that the electron withdrawing effects of the adjacent alcohol also serve to increase the ratio of monohydration of the diketone in DPD. These observations allow us to assign the signals of species I to (4S)-3,3,4,5-tetrahydroxypentan-2-one [(4S)-THP/structure E], Figure 1a. The corresponding species C1 and C2 are in equilibrium with E and this can be observed in NOESY and ROESY spectra (Supporting Figure S5). However, we are not able to fully exclude the linear diketone A and both unhydrated cyclic forms B1 and B2 (Figure 1a), but here we postulate that they are of minor importance in aqueous solutions. This is also in agreement with the calculated equilibrium constant of the hydration of this molecule, which favors hydration, as well as the equilibrium suggested in another study.[7b, 14]
Our NMR structural analysis of DPD has opened up an entirely new series of equilibrium structures previously not thought to be important. However, we wished to further probe the chemical basis of these equilibrating chemical partners. Therefore, we synthesized two derivatives as model systems for the cyclic and the linear species of DPD (Figure 3). As our model for the cyclized form we examined the CF3 analogue of DPD (2); interestingly, this derivative is the most active agonist described to date and is only present in the hydrated form at the C3 position.[9] In our analysis of 2, we observed the equilibrium of CF3-DPD 2 to be highly pH dependent (Figure 3a/Supporting Figure S6). Thus, the 1H NMR signal changes for C4 and C5 are very similar to the cyclic isomers of natural DPD again showing complete hydration, while the spectra of 2 and DPD parallel each other and reemphasize that the equilibrium of the hydrated species E with C1 and C2 are important.
Figure 3.

a) 1H NMR of model compound 2 in comparison with 13C-DPD (4) at pH 1 and pH 7. b) 13C NMR of model compound 3 at pH 1 and pH 7. c) The proposed mechanism for the reaction of the monoketone species with 1,2-phenylenediamine.
We synthesized the methylated primary alcohol (3) as our linear model compound, which is unable to engage in any ring closing events. The titration of 5-MeO-DPD in the range of pH 1–7 indicates the presence of a single major species over the entire pH range as observed by 1H NMR (Supporting Figure S7), Only one new minor species of less than 5 % was observed and the 13C NMR clearly shows the presence of only one carbonyl moiety (Figure 3b). This linear structure thus houses a singular ketone, further strengthening our analysis that the linear form of DPD is species E in aqueous solution (Figure 1a). However, seemingly contradictory the diketone A has always been implicated as the major linear species of DPD in solution, largely because addition of 1,2-phenylenediamine grants the corresponding quinoxaline.[3b, 7a, 7b] To test this, we treated 5-MeO-DPD 3 with an aqueous solution of 1,2-phenylenediamine and found complete conversion (Supporting Figure S7). To reconcile this finding we now submit that the reaction likely occurs via rapid dehydration of position C3 after the conversion of the C2 carbonyl to the corresponding imine, thus obviating the requirement of the α-diketone (Figure 3c).
While the preceding studies were focused on the equilibrium of the natural DPD signal, we also sought to understand the equilibria of DPD analogues and their capability of modulating QS (1a-d). In total, these compounds exhibit variable biological activity in V. harveyi and S. typhimurium reporter assays. In V. harveyi, all act only as synergistic activators of QS in the presence of DPD.[8] However, in S. typhimurium the biological activity varies based on the simple addition of methylene groups: ethyl-DPD (1a) is a weak agonist, whereas propyl-DPD (1b), butyl-DPD (1c), and hexyl-DPD (1d) are potent antagonists.[8a, 8b]
In stark contrast to DPD, which exhibits a ratio of both cyclic forms to the single linear form of 4.3:1, the ratios of cyclic to linear isomers of the alkyl-DPD analogues were about 1:1 at pH 1.5 (Figure 4). Only the ethyl derivative 1a has a ratio slightly above 1, whereas it is less than 1 for the three analogues with the longer alkyl chain (propyl, butyl and hexyl, 1b-d). This drastic change in equilibrium is surprising given the only weakly electron-donating capability of the additional methylene groups. However, even these minor changes appear sufficient to alter the electrophilicity of the C2 carbonyl.
Figure 4.
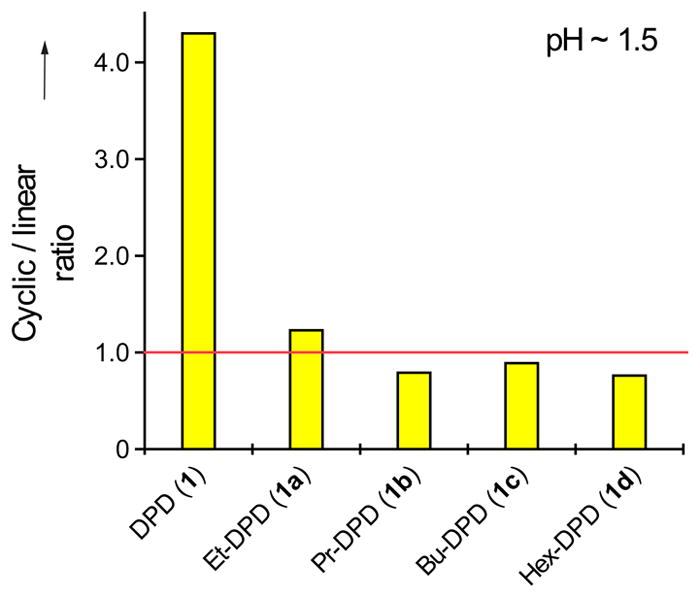
1H NMR quantification of the cyclic/linear ratio of DPD (1) and the C1-alkyl DPD analogues (1a-d) at around pH 1.5. Shown are the values for both cyclic species C1 and C2 over species E.
We adjusted the pH of these solutions to 7 and also observed the appearance of new peaks in a manner similar to that observed with DPD (Supporting Figure S8). Because of the complexity of the spectra, we were unable to rigorously characterize the equilibria of 1a-d at pH 7. However, as we observed constant values of 4.3:1 for the ratio of the equilibrium of species C1 and C2 with species E within DPD (1), we posit that the corresponding ratios of the C1-alkyl derivatives 1a-d at pH 1.5 (Figure 4) are also similar over the whole investigated pH range.
According to our NMR observations for DPD, we postulate that the lower presence of cyclic forms may explain the lack of signaling activity of these analogues in V. harveyi, as a boron complexed cyclic isomer of DPD must bind to the receptor protein LuxP to initiate the QS cascade inside the cell.[5a] In S. typhimurium, on the other hand, activity does not depend on the cyclic DPD signal or such species to bind its receptor, and the linear forms of these analogues may enter the cell, where they can then activate or antagonize gene expression.[8b]
In summary, detailed NMR analysis has provided us with better understanding of the structural diversity that DPD possesses at physiological pH. Our results allow us to conclude that the linear species of DPD is present as the monohydrate E [(4S)-THP], which is in equilibrium with both hydrated forms C1 and C2 at pH 7. The species previously postulated to be major components of the DPD equilibrium described species A, B1 and B2 were not observed and are at best only minor species. Furthermore, the presence of the multitude of closed DPD isomers at physiological pH, coupled with the ability for DPD to complex anions as observed with [B(OH)4]−, highlights the complexity of AI-2 based signaling in that any of these isomers could potentially mediate bacterial communication. This study suggests that the lexicon of AI-2 communication may be significantly larger than appreciated.
Supplementary Material
Acknowledgments
This work was supported by The Skaggs Institute for Chemical Biology and the National Institute of Health (AI077644) and by a postdoctoral fellowship from the German Academic Exchange Service (DAAD) to DG. We also thank Dr. Laura Pasternack for helpful discussions about the NMR studies.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Costerton JW, Stewart PS, Greenberg EP. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]; b) Ng WL, Bassler BL. Annu Rev Genet. 2009;43:197–222. doi: 10.1146/annurev-genet-102108-134304. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lowery CA, Dickerson TJ, Janda KD. Chem Soc Rev. 2008;37:1337–1346. doi: 10.1039/b702781h. [DOI] [PubMed] [Google Scholar]; d) Galloway WRJD, Hodgkinson JT, Bowden SD, Welch M, Spring DR. Chem Rev. 2011;111:28–67. doi: 10.1021/cr100109t. [DOI] [PubMed] [Google Scholar]
- 2.Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ. Nat Rev Micro. 2008;6:17–27. doi: 10.1038/nrmicro1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Surette MG, Miller MB, Bassler BL. Proc Natl Acad Sci U S A. 1999;96:1639–1644. doi: 10.1073/pnas.96.4.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhu JG, Hu XB, Dizin E, Pei DH. J Am Chem Soc. 2003;125:13379–13381. doi: 10.1021/ja0369663. [DOI] [PubMed] [Google Scholar]
- 4.a) Pereira CS, McAuley JR, Taga ME, Xavier KB, Miller ST. Mol Microbiol. 2008;70:1223–1235. doi: 10.1111/j.1365-2958.2008.06477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miller ST, Xavier KB, Campagna SR, Taga ME, Semmelhack MF, Bassler BL, Hughson FM. Mol Cell. 2004;15:677–687. doi: 10.1016/j.molcel.2004.07.020. [DOI] [PubMed] [Google Scholar]; c) Kavanaugh JS, Gakhar L, Horswill AR. Acta Crystallogr, Sect F: Struct Biol Cryst Commun. 2011;67:1501–1505. doi: 10.1107/S1744309111042953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Chen X, Schauder S, Potier N, Van Dorsselaer A, Pelczer I, Bassler BL, Hughson FM. Nature. 2002;415:545–549. doi: 10.1038/415545a. [DOI] [PubMed] [Google Scholar]; b) Xue X, et al. Angew Chem, Int Ed. 2011;50:9852–9856. doi: 10.1002/anie.201103130. [DOI] [PubMed] [Google Scholar]
- 6.Xavier KB, Bassler BL. J Bacteriol. 2005;187:238–248. doi: 10.1128/JB.187.1.238-248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Meijler MM, Hom LG, Kaufmann GF, McKenzie KM, Sun C, Moss JA, Matsushita M, Janda KD. Angew Chem, Int Ed. 2004;43:2106–2108. doi: 10.1002/anie.200353150. [DOI] [PubMed] [Google Scholar]; b) Semmelhack MF, Campagna SR, Federle MJ, Bassler BL. Org Lett. 2005;7:569–572. doi: 10.1021/ol047695j. [DOI] [PubMed] [Google Scholar]; c) De Keersmaecker SCJ, et al. J Biol Chem. 2005;280:19563–19568. doi: 10.1074/jbc.M412660200. [DOI] [PubMed] [Google Scholar]
- 8.a) Lowery CA, Park J, Kaufmann GF, Janda KD. J Am Chem Soc. 2008;130:9200–9201. doi: 10.1021/ja802353j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Roy V, Smith JAI, Wang J, Stewart JE, Bentley WE, Sintim HO. J Am Chem Soc. 2010;132:11141–11150. doi: 10.1021/ja102587w. [DOI] [PubMed] [Google Scholar]; c) Ganin H, Tang X, Meijler MM. Bioorg Med Chem Lett. 2009;19:3941–3944. doi: 10.1016/j.bmcl.2009.03.163. [DOI] [PubMed] [Google Scholar]
- 9.Frezza M, Balestrino D, Soulere L, Reverchon S, Queneau Y, Forestier C, Doutheau A. Eur J Org Chem. 2006:4731–4736. [Google Scholar]
- 10.a) Campagna SR, Gooding JR, May AL. Anal Chem. 2009;81:6374–6381. doi: 10.1021/ac900824j. [DOI] [PubMed] [Google Scholar]; b) Gooding JR, May AL, Hilliard KR, Campagna SR. Biochemistry. 2010;49:5621–5623. doi: 10.1021/bi1001163. [DOI] [PubMed] [Google Scholar]
- 11.a) Kaufmann GF, et al. Proc Natl Acad Sci U S A. 2005;102:309–314. doi: 10.1073/pnas.0408639102. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Horswill AR, Stoodley P, Stewart PS, Parsek MR. Anal Bioanal Chem. 2007;387:371–380. doi: 10.1007/s00216-006-0720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Park J, et al. Chem Biol. 2007;14:1119–1127. doi: 10.1016/j.chembiol.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Bose B, et al. J Am Chem Soc. 1998;120:11158–11173. [Google Scholar]; b) Bose-Basu B, Klepach T, Bondo G, Bondo PB, Zhang W, Carmichael I, Serianni AS. J Org Chem. 2007;72:7511–7522. doi: 10.1021/jo0706776. [DOI] [PubMed] [Google Scholar]
- 13.a) Lissi E, Abuin E, Espinoza C, Rubio MA. Langmuir. 1994;10:1071–1074. [Google Scholar]; b) Segretario JP, Sleszynski N, Partch RE, Zuman P, Horak V. J Org Chem. 1986;51:5393–5396. [Google Scholar]
- 14.Tsuchikama K, Lowery CA, Janda KD. J Org Chem. 2011;76:6981–6989. doi: 10.1021/jo200882k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.