

Abstract
Chronic myeloid leukemia (CML) is a clonal hematopoietic disorder characterized by the presence of the Philadelphia chromosome which resulted from the reciprocal translocation between chromosomes 9 and 22. The pathogenesis of CML involves the constitutive activation of the BCR-ABL tyrosine kinase, which governs malignant disease by activating multiple signal transduction pathways. The BCR-ABL kinase inhibitor, imatinib, is the front-line treatment for CML, but the emergence of imatinib resistance and other tyrosine kinase inhibitors (TKIs) has called attention for additional resistance mechanisms and has led to the search for alternative drug treatments. In this paper, we discuss our current understanding of mechanisms, related or unrelated to BCR-ABL, which have been shown to account for chemoresistance and treatment failure. We focus on the potential role of the influx and efflux transporters, the inhibitor of apoptosis proteins, and transcription factor-mediated signals as feasible molecular targets to overcome the development of TKIs resistance in CML.
1. Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative disorder that results from the reciprocal translocation of the ABL1 oncogene on chromosome 9 with the breakpoint cluster region (BCR) gene on chromosome 22 [t(9; 22)], leading to the formation of the BCR-ABL oncoprotein. The shortened chromosome 22 formed by this translocation is the Philadelphia (Ph) chromosome. The BCR-ABL fusion oncogene, which is responsible for the pathogenesis of CML, has greatly enhanced ABL1 tyrosine kinase constitutive activity [1]. CML is characterized by a biphasic evolutive course. Most patients are diagnosed in the chronic phase (CML-CP), which is characterized by the absence of symptoms in half of the patients. However, a prominent leukocytosis is frequently observed by routine testing. In the other half of patients, symptoms are common and include splenomegaly, weight loss, lethargy, and anemia [2]. The disease may progress either directly to blast phase (BP) or through an intermediate accelerated phase (AP). The time course for progression to BP is variable and the molecular mechanisms underlying disease progression are extremely complex. BCR-ABL-dependent pathways to blast transformation include an increase in genomic instability, telomere shortening, loss of tumor-suppressor function, and inhibition of tumor suppressors with cell regulatory functions [2, 3].
In order to identify prognostic factors for CML patients, many clinical and biological characteristics have been analyzed. Sokal risk score (based on spleen size, age, platelet count, and peripheral blood blast) is a prognostic factor widely used for prediction of cytogenetic response and of progression-free and overall survival in CML-CP with imatinib as front-line therapy. Other factor predictors for therapy response include OCT-1 activity, ABCB1/P-glycoprotein overexpression and polymorphisms, in vivo measurement of the Crkl phosphorylation, and molecular response [4].
The treatment of CML-CP can be divided into pre-imatinib and post-imatinib era. Prior to the imatinib era, busulphan and interferon-α recombinant [5, 6] were used to control and to prolong CML survival in the CP phase, but allogenic stem-cell transplantation was, and is still, the only therapy with potential for curing CML patients [7]. After the introduction of imatinib, a potent tyrosine kinase inhibitor (TKI), there was a dramatic change in the CML outcome. Imatinib acts by binding to the BCR-ABL protein in the inactive conformation and is unable to bind to the active configuration [8]. The survival rate attributed to imatinib is arguably more elevated than interferon-based therapy [9]. In addition, imatinib is generally well tolerated [10]. Imatinib treatment is associated with high rates of complete cytogenetic and major molecular responses in patients with CML-CP. On the other hand, despite improvements related to survival by using imatinib or other TKIs, CML-BP prognosis remains disappointing [11].
Currently, imatinib is the standard therapy for all CML phases [12–14]. Despite the clinical success with imatinib demonstrating long-term survival for the majority of patients, one-third of patients need an alternative therapy, frequently a second-generation TKI, such as dasatinib and nilotinib. Patients who need second-line therapy include those with imatinib intolerance [10] or mainly primary or acquired imatinib resistance [15, 16].
The most common mechanism of resistance to imatinib is the development of point mutations or amplification of the BCR-ABL gene, which alters the kinase domain (KD) of BCR-ABL and is responsible for imatinib loss of efficacy [17]. KD mutations can be found at any phase of CML. Not all KD mutations are responsible for TKI resistance. However, T315I mutation is generally resistant to all TKIs [18].
BCR-ABL acts with other multiple cellular and genetic events that accumulate progressively to drive the disease into the blast phase. Therefore, additional mechanisms—dependent or independent to BCR-ABL—may also account for resistance to imatinib treatment and result in a poor outcome. In this review, the role of efflux and influx transporters, inhibitor of apoptosis proteins (IAP), and transcription factors as additional mechanisms responsible for chemoresistance in CML will be discussed.
1.1. Efflux and Influx Transporters
The multidrug resistance (MDR) phenotype related to increased expression of efflux pumps, such as ABCB1/P-glycoprotein (Pgp) and ABCG2/breast-cancer-related protein (BCRP), is one of the most studied mechanisms of resistance in CML. More recently, the decrease in influx transporters, such as the organic cation transporter-1 (Oct-1), has also emerged as a mechanism responsible for inefficient drug uptake and consequent treatment failure [16, 19].
1.1.1. ABCB1/P-Glycoprotein
The most common mechanism developed by tumor cells to escape a drug-induced death is displayed in intrinsic or acquired MDR phenotype by the overexpression of the drug-efflux protein ABCB1 [20, 21]. ABCB1, a product of the ABCB1 gene, was first described in 1976 by Juliano and Ling, who observed a cell surface glycoprotein that altered drug permeability in hamster drug-resistant cells. Human cells also express ABCB1 on the cell surface, acting as a drug efflux pump and, consequently, decreasing intracellular drug concentration [22, 23]. Meanwhile, physiological ABCB1 expression has been identified in some tissues, particularly on the membranes of kidney tubules, in the canalicular membranes of hepatocytes, in the gastrointestinal tract, at blood tissue barriers, in the placenta, and in blood cells including CD34+ hematopoietic stem cells, natural killer cells, antigen-presenting dendritic cells (DC), and T and B lymphocytes [24–28]. Its physiological function suggests a protection against potentially toxic compounds and harmful substances found in the blood stream. Studies on ABCB1 knockout mice showed no physiological abnormalities under normal conditions, although these animals display hypersensitivity to drugs and an increase in ABCB1 substrate accumulation [27, 29–31].
Clinical insensitivity to anticancer agents is mainly attributed to an elevated expression of ABCB1, which is related to treatment failure associated with lower remission and survival rates in some types of cancer, including leukemias [32–34]. Meanwhile, gene and protein expressions of ABCB1 are commonly acquired or increased during the course of chemotherapy, which make drug treatment a responsible factor for MDR [35, 36]. Other extrinsic factors may induce MDR by acquisition of ABCB1 expression. Levchenko et al. [37] showed that ABCB1, and, consequently, MDR are transferred by direct membrane contact of tumor cells. Moreover, resistant tumor cells may release membrane microparticles carrying surface ABCB1. The shared microparticles can bind to receptor cells, spread ABCB1 and, consequently, induce MDR phenotype [38].
Even though the ABCB1 efflux functions, other functions for this transporter have been studied. Studies have shown that the resistance induced by ABCB1 is also associated with the inhibition of cell death, and ABCB1 promotes additional protection to caspases-dependent apoptosis, UV radiation, serum starvation condition, and spontaneous apoptosis [39–42]. Recently, our group demonstrated that ABCB1 expression induced by drug treatment promotes resistance to apoptosis in BCR-ABL cells independently of its drug-efflux activity [43].
ABCB1 is related to resistance phenotype in some leukemias and it has been studied in advanced CML. A randomized trial evaluated the relevance of ABCB1 expression in CML patients. The authors observed that the response to cytarabine and daunorubicin was significantly related with both ABCB1 expression and function mainly in the blast phase. For this reason, chemotherapy resistance in CML-BP patients should be considered multifactorial and cannot be associated only with BCR-ABL [44–47]. Our group recently demonstrated that CML patients show high levels of ABCB1 expression independently of CML phases. Nevertheless, we showed that ABCB1 expression is more frequent than multidrug-resistant protein 1 (MRP1) in CML-BP [48].
In vitro data suggest that imatinib is able to induce ABCB1 in sensitive CML cell lines and, as a result, ABCB1 activity may confer resistance to this drug [49–51]. Mahon et al. [52] demonstrated that a multidrug-resistant CML cell line displayed resistance to many drugs including imatinib and the induced overexpression of ABCB1 gene by retroviral transduction in BCR-ABL cell line also leads to imatinib resistance. Moreover, Rumpold et al., [53] showed that a stable silencing of ABCB1 in imatinib-resistant CML cell lines abolished ABCB1-efflux substrates and induced sensibility to imatinib. Regardless of the in vitro data, there is no consistent evidence for this resistance in vivo, although several studies have discussed the role of ABCB1 in imatinib-resistant CML patients [54]. Zong et al. [55] demonstrated that bone marrow mice cells Mdr1a/1b-null transduced with BCR-ABL display a similar response to imatinib, which is related to increased peripheral white blood cells counts and marked hepatosplenomegaly, compared with BCR-ABL-transduced wild-type bone marrow. The authors concluded that the expression of ABCB1 in hematopoietic stem cells does not interfere with imatinib resistance. Another in vivo study revealed that imatinib treatment in CML patients in the accelerated phase induced an increase of ABCB1-positive cells with efflux activity. However, in imatinib-resistant CML patients, the efflux activity was independent of ABCB1 expression, suggesting participation of other ABC transporters [56]. Hatziieremia et al. [57] inhibited ABCB1 using PSC833 in CD34+ cells from CML-CP patients and did not observe imatinib efficiency in eliminating CML cells.
Although these previously described works do not identify the role of ABCB1 in imatinib resistance, studies in polymorphisms of ABCB1 have shown the importance of ABCB1 in CML treatment resistance. Moreover, this kind of study may provide information for the prediction of drug disposition in a specific way and promote better response to imatinib in CML patients [58, 59]. Dulucq et al. [60] analyzed 1236C>T, 2677G>T/A, and 3435C>T ABCB1 single nucleotide polymorphisms (SNPs) in CML patients treated with imatinib. The authors observed that allele G in 2677G>T/A polymorphism was associated with the worst response to imatinib. In a Chinese population, Ni et al. [61] observed more imatinib resistance in patients homozygous for 1236T allele and 3435 TT/CT genotypes.
Studies have suggested that second- and third-generation TKIs can overcome imatinib resistance [62, 63]. There are studies suggesting that nilotinib does not induce resistance in CML cells through ABCB1 overexpression [64]. Nevertheless, Mahon et al. [65] developed nilotinib-resistant CML cell lines and observed that nilotinib is a substrate for ABCB1. Moreover, concomitant overexpression of ABCB1 and BCR-ABL provides nilotinib resistance in CML cells. Studies also revealed the interaction of dasatinib and ABCB1 efflux protein. Giannoudis et al. [66] showed that cell lines BCR-ABL (positive or not) are able to extrude dasatinib through ABCB1 activity. In concordance, Hiwase et al. [67] demonstrated that ABCB1 is able to transport dasatinib from CML cells. These studies show the importance of researching more about ABCB1 expression, function, and inhibition.
An important strategy to try reversing clinical MDR involves modulation or inhibition of ABCB1. The cyclosporine A (CsA) is capable of regulating the efflux function of ABCB1 dependently on its concentration in cancer cells [68, 69]. Some studies in hematological cancer have shown the benefits of CsA on reversing MDR or potentiating drug effects [70]. In a clinical study of our group, we evaluated the effect of CsA on the circumvention of leukemia patients MDR in vitro. Our data showed that combination of CsA and etoposide (VP-16) could induce a good response in ABCB1-positive CML patients [71]. In the same year, we also published a case report showing that the cytotoxic effect of VP-16 was enhanced in combination with CsA in blast cells of CML. Moreover, the patient returned from blast phase to chronic phase [72]. All these studies and others emphasized the importance of reversing the MDR phenotype.
1.1.2. BCRP/ABCG2
Another important efflux pump associated with chemotherapy resistance in CML is BCRP or ABCG2, coded by the gene ABCG2. ABCG2 is a 72-kDa protein composed of 665 amino acids. It has an N-terminal ATP-binding domain (NBF) and a C-terminal transmembrane domain (TMD), a structure half the size and in reverse configuration to most other ABC proteins comprising two NBFs and two TMDs. Because ABCG2 is a half-transporter, it is believed to homodimerize, or possibly oligomerize, in order to function [73].
Fetsch et al., [74] reported high levels of ABCG2 expression in normal placenta, interstitial cells of testes, endocervical cells of uterus, squamous epithelium of cervix, kidney, hepatocytes, pancreas, and small and large intestinal mucosa/epithelial cells. The first reported chemotherapy agent substrate of ABCG2 was mitoxantrone [75]. Other chemotherapeutic substrates include flavopiridol, topotecan, methotrexate, and the TKIs imatinib, gefitinib, and erlotinib [76]. If the amino acid at position 482 is mutated, mitoxantrone transport is more efficient and ABCG2 can additionally transport rhodamine 123 and anthracyclines such as doxorubicin and bisantrene [77, 78].
It was demonstrated that TKI had high-affinity interaction with ABCG2 and that it occurs at submicromolar concentrations [79, 80]. Although other TKIs promote ATPase activity, imatinib was the only one able to inhibit it, suggesting that this drug acts as a modulator agent. In addition, imatinib has promoted the accumulation of a fluorescent substrate inside the cells which reinforced its role as a modulator. Using a different methodology, Houghton et al. [81] demonstrated that overexpression of ABCG2 was not able to confer resistance to imatinib, suggesting that it is not a substrate for this transporter. In addition, imatinib promoted the accumulation of topotecan in functional ABCG2-expressing cell lines, indicating a role of imatinib as a modulator but not as a competitor. Conversely, Ko-143, an ABCG2 specific modulator, could increase the imatinib accumulation in ABCG2-overexpressing cell lines, suggesting its role as a competitor. Interestingly, mitoxantrone accumulation in the same cell lines was increased by the addition of imatinib, suggesting its role as a modulator. These findings suggest that imatinib can be both a substrate and a modulator [82]. Furthermore, Brendel et al. [83] confirmed that ABCG2 expression confers imatinib resistance and reduces imatinib accumulation in K562 cells, effects that are abrogated by the ABCG2 inhibitor fumitremorgin C (FTC). More importantly they observed that differences on imatinib accumulation were only seen when imatinib was used at low concentrations but not at high concentrations. These data support the idea that imatinib may act as a modulator or a substrate depending on the concentration level. However, there is still no consensus on whether imatinib is a substrate or a modulator of ABCG2 transport. Regardless of its role as a substrate or modulator, imatinib interacts with ABCG2, which may have their effectiveness limited by the overexpression of this protein.
It is believed that CML is a clonal disorder originating from the hematopoietic stem cell (HSC). Graham et al. [84] demonstrated that primitive HSCs expressing BCR-ABL in CML-CP patients were resistant to imatinib. This discovery led to the hypothesis that the resistant population could contribute to the failure of treatment with imatinib. Nakanishi et al. [85] studied the interaction of ABCG2 and imatinib on a cell line expressing BCR-ABL. This cell line was resistant to substrates of ABCG2 as well as imatinib, and the resistance was reversed by inhibiting ABCG2. Another interesting finding was that the initial resistance to imatinib caused by ABCG2 was attenuated by the inhibition of BCR-ABL, suggesting that BCR-ABL regulates the expression of ABCG2 at a later stage of transcription.
Some authors have identified the presence of ABCG2 in a particular group of HSC called “side population” (SP) due to its efflux of the fluorochrome Hoechst 33342 and its ability to reconstitute bone marrow in irradiated mice [86]. Afterward, it was demonstrated that ABCG2 was responsible for the SP in mouse and human bone marrow [87, 88]. ABCG2-deficient mice are viable with normal numbers of stem cells. Despite the absence of SP, these data suggest that ABCG2 protein is not necessary for normal hematopoiesis [89]. However, ABCG2 may play a protective role for stem cells, because Zhou et al. [90] demonstrated that stem cells derived from ABCG2-deficient mice were more sensitive to cytotoxic substrates.
Once ABCG2 is expressed in the apical membrane of cells in the epithelium of the small intestine and colon, it is very likely that ABCG2 is involved in the active return of drug entering the intestine. This role would be important in reducing the systemic bioavailability of oral drugs such as imatinib. Studies in ABCG2 knockout mice indicate that ABCG2 and ABCB1 appear to regulate the penetration of imatinib into the brain tissue. Imatinib brain penetration in ABCG2 knockout mice was found to be increased [91].
The SNP 421C>A is responsible for decreased plasma membrane expression of ABCG2, reduced ATPase activity, or decreased drug transport [92–94]. Therefore, the daily imatinib dose for patients with the ABCG2 421C/C genotype might be higher than for those with the 421C/A or 421A/A genotype [95]. Knowledge of the ABCG2 421 genotype could be useful when making dosing decisions aimed at achieving the optimal imatinib exposure.
1.2. SLC22A1/OCT-1 Influx Transporter Protein
Members of the solute carriers (SLCs) superfamily of transporters are known as passive facilitator carriers that allow the passage of solute through the membrane without spending energy [96]. This superfamily is divided into 43 families according to the type of substrate transported and the type of transport. Some families carry specific substrates such as oligopeptides, sugars, phosphatases, or metals, whereas other families are polyspecific, transporting substrates with different sizes and structures [97].
SLC transporters are mostly expressed in the plasma membrane and play a critical role in a variety of physiological cellular processes such as import/export neurotransmitters, nutrients, or metabolites [96]. The family 22 of solute carrier proteins is composed of 12 members mostly of poly-specific transporters. Many members of this family are expressed in the intestine, liver, and kidney, indicating an important role in the absorption and excretion of drugs, xenobiotics, and endogenous compounds that exist as cations at physiological pH. The family is further divided into subgroups according to the substrate and the transport mechanism [96].
A growing number of scientific papers have shown that some chemotherapeutics are substrates for influx transporters. Recently, it was reported that imatinib is transported into the cell, preferably via SLC22A1 (also called OCT-1), and the expression of this transporter is predictive of achieving a complete cytogenetic remission after 6 months of treatment with imatinib [98].
It was reported that the influx of imatinib is temperature dependent, indicating the involvement of an active process of influence. When the cells were incubated with inhibitors of the transporter SLC22A1, the influx of imatinib was significantly reduced [99]. Since then, other studies have been published supporting the hypothesis that imatinib is a substrate for the transporter SLC22A1. White et al. [100] analyzed the activity of SLC22A1 in samples from CML patients before starting treatment with imatinib and compared this with getting a major molecular response at 24 months. In this study, the activity of SLC22A1 was an important determinant of molecular response to imatinib with strong predictive value on the dose. The analysis of SLC22A1 activity before the start of treatment with imatinib was able to identify patients who would need a higher dose of the drug to respond to medical treatment with imatinib.
Besides the SLC22A1 activity, the levels of expression of SLC22A1 may be related to a decreased influx of imatinib. Crossman et al. [101] analyzed the expression of the SLC22A1 gene in samples from CML patients before starting treatment and observed that the expression of SLC22A1 was variable and did not differ significantly from levels found in samples of bone marrow healthy individuals. However, patients who responded to treatment with imatinib had significantly higher levels of expression of the SLC22A1 gene as compared to the group of nonresponders. Despite this and other articles suggesting a direct correlation between SLC22A1 and response to treatment with imatinib, Hu et al. [102] believe that SLC22A1 per se is not able to influence the retention of imatinib, as this drug would be a poor substrate for SLC22A1. On the other hand, Wang et al. [98] suggested that clinical responses to imatinib could be affected by transporters SLC22A1, ABCB1, and ABCG2. Patients with high pretreatment SLC22A1 expression had a higher probability of achieving a cytogenetic response and a superior progression-free and overall survival. The same was not observed when analyzing ABCB1 and ABCG2.
The contribution of the SLC22A1 transporter to the clinical response to imatinib has not yet been elucidated. Therefore, further studies are needed to evaluate the role of this influx transporter in the clinical outcome of imatinib treatment.
2. Inhibitor of Apoptosis Proteins (IAPs)
The IAP family members are characterized by a common baculoviral IAP repeat (BIR) domain [103] and by the ability to block apoptosis through the inhibition of both mitochondrial-dependent and -independent apoptotic pathways [104, 105]. Among IAPs, much attention has been focused on survivin and XIAP due to their potential role as therapeutic targets.
2.1. XIAP
XIAP (X-linked of inhibitor of apoptosis protein) is a singular IAP because it is the only member of the family known to directly inhibit caspases-3, -7, and -9 [106, 107]. XIAP is able to bind their target caspases by a two-site interaction mechanism, which inhibits the apoptotic pathway by blocking the active caspase site or by dissociating the dimer of caspases [108].
There are at least two proteins, Smac/DIABLO [109] and XAF1 (XIAP-associated factor 1) [110], known to interact with XIAP and modulate its antiapoptotic activity, which suggests a significant role of XIAP in the maintenance of the cellular homeostasis [111]. Other relevant XIAP properties are the involvement in copper metabolism [112] and the capacity of self-ubiquitination and of other targets involved or not in the control of the cell death [113], demonstrating its versatility in the cellular physiologic processes. Studies using knockout murine models for XIAP (XIAP/BIRC4 −/−) showed that its absence does not alter caspases-dependent or -independent apoptosis, but increases the expression of other IAPs, possibly as a compensatory mechanism [114].
XIAP is widely expressed in normal tissues [115]; however, its overexpression in cancer is usually associated with an unfavorable prognosis [116–119]. Although it has been demonstrated that the nuclear localization of XIAP is an independent prognostic marker in breast cancer [120], little is known about the expression and subcellular localization relevance of XIAP in CML patient samples.
Increasing evidence demonstrates that treatment of CML cells with chemotherapeutic agents can overcome resistance through negatively regulating XIAP levels. Fang et al. [121] have observed that one of the mechanisms involved in BCR-ABL-positive cells sensitivity to imatinib is XIAP downregulation. Corroborating this data, a study conducted in K562 cells and leukemic blasts obtained from patients with CML in blast crisis showed that apicidin, a histone deacetylase inhibitor, was able to potentiate imatinib effects on apoptosis through XIAP degradation and the release of the proapoptotic protein Smac/DIABLO into cytosol [122]. These events were associated with reduced BCR-Abl protein expression and decreased phosphorylated Akt levels and were caspase dependent [122]. Imatinib-induced apoptosis could also be potentiated when coadministered with ABT-737, a Bcl-2 and Bcl-xL inhibitor [123]. Cotreatment of K562 cells and primary CML samples led to caspase-3 activation and HtrA2/Omi-mediated decreased XIAP levels both in K562 cells and TKI-insensitive CML hematopoietic progenitors [123]. In addition to these findings, treatment of K562 cells with TRAIL led to an apoptosis-resistant phenotype through the upregulation of antiapoptotic proteins, including XIAP [124], further emphasizing its role in chemoresistance in CML.
Many strategies have been used to inhibit both the expression and function of XIAP and resensitize cancer cells to different cytotoxic stimuli [125–127]. One study demonstrated that the downregulation of XIAP expression using antisense oligonucleotides increased the sensitivity to cytotoxic stimuli, inducing apoptosis and decreasing cell viability in the K562 cell line [128]. Recently, the same group showed that the simultaneous inhibition of XIAP and P-glycoprotein in cells that overexpress this efflux pump decreases imatinib resistance [129]. Consistent with this, a recent work published by our group found that cyclosporine-A-mediated Pgp modulation was associated with XIAP inhibition and an increased apoptotic index as a response of resistant CML cells to vincristine [130]. Altogether, these findings point XIAP as an interesting therapeutic target and suggest that combining chemotherapeutic agents with XIAP-targeted therapy seems to represent a promising strategy in CML.
2.2. Survivin
Survivin, another IAP member, is an antiapoptotic protein [131], which also regulates cell division by controlling mitotic spindle checkpoint [132]. Survivin gene generates five different splice variant mRNAs, which encodes different proteins: wild-type survivin, survivin-2B, survivin-3B, survivin-δEx3, and survivin-2α [133]. Compared to wild-type survivin, little prognostic information is known about the functions of alternative splicing forms, which are generally expressed at lower levels than the wild-type survivin. In a recent study, it was found that patients in blast and accelerated phases displayed significantly lower levels of survivin-2B and -δEx3, compared to patients in CML-CP. However, there was no correlation between the isoform expression and clinical parameters or response to imatinib treatment [134].
Undetectable in normal differentiated tissues, survivin is abundantly expressed in all the most common human cancers [131, 135], which makes this protein a potential target for drug discovery and new anticancer interventions. Survivin can also be found in normal tissues characterized by self-renewal and proliferation [136], but its expression is significantly lower than in tumor cells. In CD34+ hematopoietic progenitor stem cells, survivin was found to be expressed and associated with the inhibition of apoptosis [137]. However, a recent report showed that despite survivin being quite expressed in CD34+ cells, its levels are low in more precursor leukemia stem cells [138], indicating that survivin is not an optimal therapeutic target for CML stem cells compartment and suggesting that it may not be the main factor accounting for resistance to targeted therapy in CML [139].
In CML patient samples, several studies have reported that survivin was expressed in the accelerated and blast phases but it was low or undetectable in the chronic phase [4, 140–143], suggesting that survivin may be involved in the pathogenesis of progression from the CML-CP to the CML-BP. In addition, survivin overexpression in CML patients was correlated with the percentage of Ph chromosome positive cells and BCR-ABL expression [142], indicating that it can be regulated by BCR-ABL tyrosine kinase. In fact, Carter et al. [144] demonstrated that BCR-ABL and its downstream effector mitogen-activated protein kinase (MAPK) could target survivin expression at both RNA and protein levels in cells derived from a patient with CML-BP Ph chromosome positive. Survivin downregulation resulted in reduced cell viability in imatinib-sensitive CML cells, but not in imatinib-resistant CML cells or Ph chromosome negative cells, showing that survivin is regulated by the BCR-ABL/MAPK cascade in Ph positive CML. The prognostic importance of survivin in CML was also evaluated in a study from our group, where a correlation between survivin highest levels and high/intermediate Sokal score patients could be observed [145]. In addition, it was reported that survivin overexpression at diagnosis correlated with a low probability to achieve an optimal response to imatinib [134]. These data suggest that survivin may be closely involved in a more aggressive evolution of CML.
Growing evidence suggests that survivin plays an important role in chemoresistance phenotype of human malignancies [146], including CML. It has been demonstrated by our group that treatment of K562 CML cells with imatinib resulted in survivin downregulation and cell death [147]. Consistent with this, imatinib-induced apoptosis was increased when survivin expression was disrupted in BCR-ABL cells, as shown by enhanced cytochrome c release, caspase-9 activity, and BCR-ABL cleavage 199, which indicate that targeting survivin might be a useful tool to sensitize BCR-ABL cells to imatinib. Survivin has also been shown to play a resistant factor to agents other than imatinib in CML cells. In a recent publication, our group showed that survivin overexpression was involved in the resistance to idarubicin, an anthracycline commonly used to treat acute leukemia. On the other hand, idarubicin could induce DNA fragmentation and caspase-mediated apoptosis in K562 cells when survivin levels were down-regulated [148]. In addition, other groups have demonstrated that survivin inhibition is a common mechanism of apoptosis induced in CML cells by different classes of anticancer agents such as aurora kinase inhibitors, histone deacetylase inhibitors [149], microtubule targeting agents (MTAs), and cyclin-dependent kinase (CDK1) inhibitors [150]. Altogether, this amount of data shows that the modulation of survivin expression seems to be an interesting approach to overcome resistance and induce cell death in CML cells.
In recent years, considerable efforts have been made to validate survivin as a new target in cancer therapy. YM-155, a small-molecule inhibitor of survivin, was the first survivin-targeted therapy to be developed and tested in clinical trials. In CML, YM-155 anticancer efficacy has been recently assessed in a preclinical study, where CML-derived cell lines showed great sensitivity to the molecule [151]. This effect has also been demonstrated for sheperdin, which is a novel antagonist of the interaction between hsp90 and survivin, known to be important for stabilizing survivin cytoprotective functions [152]. Although sheperdin did not decrease the viability of phytohemagglutinin-stimulated peripheral blood mononuclear cells or induced organ toxicity in a xenograft acute myeloid leukemia (AML) model, it could inhibit viability in K562 cells and in patient-derived AML peripheral blasts [153], demonstrating that it is a highly selective molecule. Antisurvivin therapies developed, to date, have not revealed major systemic toxicities in animal models and clinical trials and are extremely encouraging. Targeting survivin alone or in conjunction with chemotherapeutic agents has a great potential as a novel therapeutic regimen in CML.
3. Transcription Factors
Signal transduction pathways within the cell act by transmitting the extracellular signals to transcription factors, which result in changes in gene expression. However, it is well known that most key signaling pathways are deregulated in cancer, leading to altered expression and function of transcription factors. The constitutive activation of the nuclear factor kappa B (NFκB) [169] and the inactivation of the forkhead box O (FoxO) factors [170] were shown to be important steps in carcinogenic transformation. Therefore, modulating the activity of FoxO and NFκB seems to represent a reasonable therapeutic strategy.
3.1. NFκB
Nuclear Factor κB (NFκB) was discovered in 1986 as a factor in the nucleus of B cells that bind to the enhancer of the kappa light chain of immunoglobulin [171]. It has been shown to be expressed in the cytoplasm of all cell types and, once activated, it translocates to the nucleus, where it regulates the expression of over 200 genes [172]. NFκB is an important transcription factor typically activated by proinflammatory cytokines and other specific stimuli, and is involved in the regulation of a variety of biological responses, such as inflammatory, apoptotic, and immune processes. It achieves this by regulating the expression of proteins such as cytokines, chemokines, adhesion molecules, and the cellular death cascade [173]. The members of NFκB protein family form dimers (usually heterodimers of p50 and p65 subunits) that interact in the cytoplasm with inhibitor of NFκB (IκB) proteins. When IκB is phosphorylated by IκB kinases (IKKs), it is degraded by the ubiquitin-proteasome pathway, liberating NFκB dimers from their inhibition and allowing them to migrate to the nucleus and to activate NFκB target genes [174].
In addition to its function as a central mediator of human immune responses, NFκB plays a major role in activating genes involved in cellular survival, transformation, and oncogenesis. Loss of the normal regulation of NFκB has become apparent as a major contributor to the deregulated growth, resistance to apoptosis, and propensity to metastasize observed in many cancers [175]. The overexpression of p65- or c-Rel-containing dimers can impair apoptosis, whereas the inhibition of NFκB/Rel activity can enhance death induced by TNF-alpha, ionizing radiations, or chemotherapeutic agents in many cell types. Aberrant activation of NFκB/Rel factors contributes to reduce the sensitivity to apoptosis in a vast range of hematologic malignancies. Although alterations in NFκB or IκB genes are documented in some neoplasms, in other cases, dysfunctions in components of the NFκB/Rel-activating signaling pathways or influences of other mutated proteins on NFκB/Rel can be recognized [176]. Constitutively active NFκB has been detected in malignant cells derived from patients with multiple myeloma, AML, ALL, CML, and, most recently, in myelodysplastic syndromes. Targeting NFκB in these hematopoietic malignancies leads to apoptosis, corroborating the role of NFκB in the survival and clonal expansion of malignant cells [174].
The expression of BCR-ABL leads to the activation of NFκB-dependent transcription by causing nuclear translocation of NFκB and by increasing the transactivation function of the RelA/p65 subunit of NFκB. Importantly, this activation is dependent on the tyrosine kinase activity of BCR-ABL that partially requires Ras. It has also been demonstrated that NFκB is required for BCR-ABL-mediated tumorigenicity in nude mice and for transformation of primary bone marrow cells [169]. This activation regulates the transcription of important genes, such as c-myc, which are necessary for the transformation of BCR-ABL+cells, as well as surface molecules, which are necessary for cellular adhesion and interaction, giving advantages for cellular growth [161, 177]. In particular, the constitutive activation of NFκB exists selectively in leukemia stem cells but not in normal HSC [178].
Alterations in NFκB regulation and in the signaling pathways that control its activities are involved in cancer progression, as well as in the treatment resistance during chemo- and radiotherapy. NFκB blocking can stop the proliferation of tumor cells or cause the tumor cells to become more sensitive to antitumor agents. This way, drugs that are capable of suppressing NFκB activation have important therapeutic potential in the carcinogenesis inhibition [179]. Several studies have demonstrated that the expression of BCR-ABL kinase activity in CML cell lines leads to a constitutive activation of NFκB through IKKβ downstream of BCR-ABL and the suppression of NFκB activation by the expression of IκBα blocked BCR-ABL-dependent xenograft tumor formation [161–163].
Cilloni et al. [161] demonstrated that a selective inhibitor of the IκB kinase (IKK) was capable of reducing NFκB binding activity and proliferation, followed by induction of apoptosis in CML cell lines sensitive and resistant to imatinib, as well as in bone marrow cells from sensitive and resistant CML patients. Corroborating with these data, Duncan et al. [162] demonstrated that a selective IKKβ inhibitor strongly suppressed growth and viability and induced cell death of cell lines expressing either wild-type or mutant versions of BCR-ABL, including the T315I mutation. Following the same rationale, Lounnas et al. [163] used another IKKβ inhibitor to block NFκB pathway capable of reducing cell survival and inducing apoptosis of imatinib-sensitive and imatinib-resistant cell lines. This work also demonstrated that cells from patients with T315I mutation appeared sensitive to NFκB inhibition in terms of proliferation. Furthermore, in vivo experiments resulted in a significant regression of the tumors after the administration of the IKKβ-inhibitor in nude mice injected with BCR-ABL wild-type and T315I mutant cells. Taken together, these results indicate that NFκB/IKK is essential for BCR-ABL—induced cell growth and survival and that the kinase IKKβ represents an attractive therapeutic target in CML.
Among these compounds acting as NFκB inhibitors, proteasome inhibitors have been widely used. Recently, it has been shown that BCR-ABL induces the activity of the proteasome, supporting the idea of using the proteasome as a suitable target for BCR-ABL-expressing cells [180]. The proteasome inhibition results in the accumulation of IκB in the cytoplasm, leading to inhibition of NFκB translocation to the nucleus. The most used proteasome inhibitor is bortezomib/Velcade/PS341, inhibitor of the chymotrypsin-like activity of the β5 subunit of the proteasome. In several studies, proteasome inhibition induced proliferation arrest and apoptosis in imatinib-resistant cells, providing a rationale for the use of this drug in the subset of patients resistant to imatinib [164, 165]. Hu et al. [165] showed the combined effect of bortezomib and imatinib in CML. The combinatory regimens in CML murine models significantly reduced disseminated disease, decreased tumor growth, and induced apoptosis in tumor sections. In this work, the combination of bortezomib and imatinib repressed the DNA-binding activity of NFκB. Albero et al. [164] demonstrated that bortezomib reduces proliferation and survival of Bcr-Abl- expressing cells, regardless of their sensitivity to imatinib, and including the highly resistant mutant T315I. In both studies, bortezomib inhibited proteasomal degradation of IκB, leading to its accumulation. Taken together, these results suggest that an approach combining imatinib and proteasome inhibitors can be a therapeutic strategy in reducing relapse and overcoming imatinib resistance by inactivating the NFκB pathway.
3.2. FoxO
FoxO transcription factors belong to the forkhead family of proteins, which are characterized by a conserved DNA-binding domain termed forkhead box (Fox) [181]. The FoxO class contains four members: FoxO1, FoxO3a, FoxO4, and FoxO6, whose expression can be found in a variety of different tissues [182]. FoxO proteins are implicated in crucial cellular functions including cell cycle regulation, stress response, glucose metabolism, and apoptosis [183]. Accumulating evidence suggests that FoxO act as tumor suppressors, inhibiting tumor growth by the activation of genes such as Bim, FasL, p27kip, cyclin D, GADD45a, glucose-9-phosphatase, and manganese dismutase [184]. Except for FoxO6, which is constitutively nuclear [185], phosphorylation by kinases, mainly Akt, ERK, (IκB kinase) IKK, and serum and glucocorticoid-regulated kinase (SGK), regulates FoxO nuclear/cytoplasmic shuttling [186], leading to its nuclear exclusion, retention in the cytoplasm, and subsequent proteasome degradation and inactivation [187]. FoxO transcription factors can also be regulated by other posttranslational modifications such as acetylation, methylation, ubiquitination, and glycosylation [188].
Because BCR/ABL activity requires an activated PI3K/Akt pathway [189] and the inactivation of FoxO transcription factors was shown to be essential for tumorigenesis and resistance to treatment [190], the activation of FoxO by chemotherapeutic drugs seems to be a great strategy to overcome resistance [191]. Komatsu et al. [157] showed that BCR-ABL-positive cells have FoxO3a in a constitutively phosphorylated status and p27/kip1 downregulated. In agreement, exposure of CML cells to imatinib inhibited FoxO3a phosphorylation and induced p27/kip1 expression and G0/G1 arrest, blocking cell cycle progression. Essafi et al. [156] also showed that BCR-ABL inhibition induced by imatinib in CML cells resulted in FoxO3a activation. As a consequence, the induction of the FoxO3a-direct transcriptional target Bim was observed concomitantly with increased apoptosis. More recently, it was demonstrated that BCR-ABL-mediated FoxO3a inactivation was proteasome dependent [160]. Bortezomib treatment was able to restore FoxO3a expression, sensitize BCR-ABL T315I expressing cells to apoptosis, and inhibit CML-like disease in leukemic mice [160]. Regulation of FoxO3a expression affects the expression not only of Bim and p27/kip1, but also of cyclin D [155]. Imatinib-mediated inhibition of BCR-ABL represses cyclin D4 expression, upon FoxO3a activation and binding to cyclin D4 promoter. However, this effect can be prevented after FoxO3a silencing, indicating that FoxO3a is a key signaling molecule for BCR-ABL pathway and a relevant factor for apoptosis and cell cycle arrest in CML cells [155]. Imatinib can also exert its antileukemic effects through the concomitant activation of FoxO3a and the down-regulation of the inhibitor of DNA binding 1 (Id1) in K562 cells [159]. This study demonstrated that Id1 promoter is transcriptionally inhibited by FoxO3a, leading to differentiation of BCR-ABL transformed cells [159], suggesting that Id1 is essential for maintaining the leukemia phenotype. Moreover, experimental data suggest that FoxO3a activation can overcome imatinib resistance by increasing tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression and by inducing apoptosis [158], further emphasizing the importance of the FoxO pathway in determining drug sensitivity.
Although a great amount of evidence demonstrates that FoxO3a functions as a downstream factor for TKI-induced apoptosis, recent data suggest that FoxO3a has a crucial role in maintenance of CML stem cells. In a recent study, it was demonstrated that FoxO3a deficiency is associated with a decreased ability of leukemia-initiating cells (LICs) to provoke CML in FoxO3a−/− mice [192]. Moreover, in CML stem cells, FoxO3a is predominantly nuclear and plays a resistant factor against TKI therapy [192]. Corroborating these data, the transcription factor Bcl-6 was identified as a target for the FoxO family, responsible for CML stem cells' self-renewal, repression of p53, leukemia initiation and resistance to TKI treatment [193]. As previously discussed [194], these findings reflect a “stem cell paradox” and may explain, in part, why CML stem cells persist after TKI treatment. The mechanisms and implications of these unexpected results regarding differential FoxO dynamics in CML stem cells still remain to be elucidated.
In conclusion, various findings have found that the activation of FoxO3a and its downstream genes are of clinical importance in diverse anticancer therapeutics, including in CML treatment. Different from p53 [17], FoxO mutation has not yet been found in human cancer, favoring FoxO targeted therapy. Clinical drugs which activate FoxO transcription factors can be used in combination with therapeutic agents for sensitizing CML malignant cells to therapy.
4. Molecular Interactions in Chemoresistance
Growing evidence has demonstrated that the development of the MDR phenotype arises as a result of a complex network involving multiple cellular and molecular mechanisms. It is a multifactorial process rather than a consequence of a single and isolated mechanism (Figure 1). As the problem of drug resistance cannot be solved by circumventing only an individual protein, many efforts have been made in order to target diverse mechanisms and enhance cell sensitivity to antineoplastic therapy (Table 1).
Figure 1.
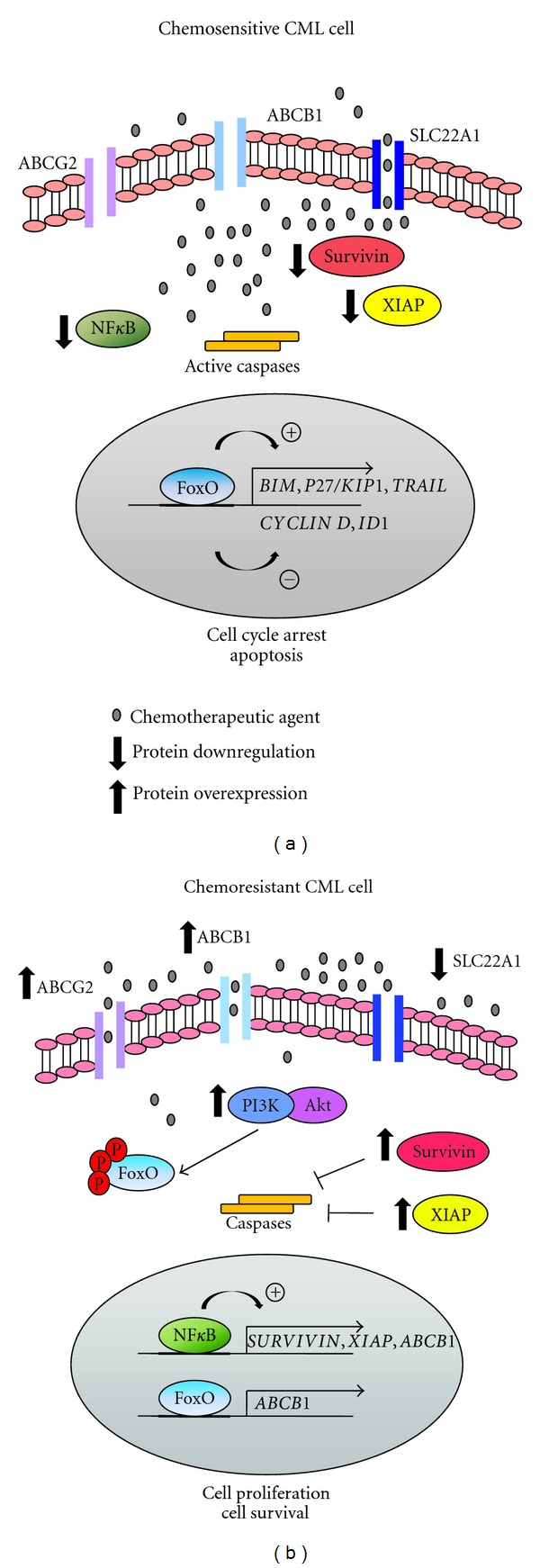
Molecular interactions in chemoresistance. Chemoresistant chronic myeloid leukemia (CML) cells display a multifactorial resistance phenotype characterized by deregulation of diverse signaling pathways which may act in concert or individually to prevent chemotherapy sensitivity (b). Resistant cells display constitutively active nuclear expression of NFκB which contributes to stimulate transcription of the inhibitor of apoptosis proteins (IAPs) survivin and XIAP and also the efflux drug transporter ABCB1. The transcription factor FoxO3a, which usually acts as an apoptosis mediator, may also lead to enhanced ABCB1 transcription when chronically activated. In addition, chemoresistant CML cells display an overexpression of the efflux pump ABCG2 and reduced levels of the influx drug transporter SLC22A1. By contrast, many chemotherapeutic agents may overcome resistance and sensitize cells to apoptosis by modulating these pathways (a). Drug-mediated down-regulation of NFκB, survivin, XIAP, and ABCB1 is associated with increased apoptotic levels, emphasizing their role as resistance factors. In addition, chemotherapy-induced FoxO3a activation results in cell cycle arrest and apoptosis by up-regulating BIM, P27/KIP1, and TRAIL and inhibiting CYCLIN D and ID1 genes.
Table 1.
Anticancer drugs sensitize CML cells by targeting IAPs, drug transporters, NFκB and FoxO proteins.
| Drug or therapy | Protein(s) targeted | Signaling pathways affected |
|---|---|---|
| Imatinib, idarubicin | Survivin | Imatinib and idarubicin inhibited viability and induced apoptosis in cells derived from a Ph+ patient in blast crisis and K562 cells, respectively, through survivin downregulation [144]. |
| Imatinib | Survivin | Enhanced imatinib-mediated apoptosis by modulating reactive oxygen species [147] and using antisense oligonucleotide or dominant-negative survivin [154] in CML cell lines. |
| Microtubule stabilizing agents and flavopiridol vorinostat, MK0457 | Survivin | The combination of microtubule stabilizing agents and the cyclin-dependent kinase inhibitor flavopiridol [149] as well as the cotreatment with vorinostat and the aurora kinase inhibitor [155] led to survivin inhibition and increased apoptosis levels in K562 cells. |
| Sheperdin | Survivin | The survivin inhibitor molecule showed great toxicity against CML and AML cells, with no decrease in viability of phytohemagglutinin-stimulated peripheral blood mononuclear cells [153]. |
| Imatinib | FoxO3a | Imatinib-mediated BCR-ABL inhibition resulted in FoxO3a activation, induction of Bim [156], p27/kip1 [157] and tumor-necrosis-factor-related apoptosis-inducing ligand (TRAIL) [158], repression of cyclin D4 expression [156] and inhibitor of DNA binding 1 (Id1) [159], and consequent increased apoptosis in CML cell lines. |
| Bortezomib | FoxO3a | Bortezomib treatment was able to restore FoxO3a expression, sensitize imatinib-resistant T315I expressing cells to apoptosis, and inhibit CML-like disease in leukemic mice [160]. |
| IKKB inhibitors | NFκB | The IKKB inhibitors led to the induction of apoptosis in cell lines (K562 and KCL) and bone marrow cells sensitive and resistant to imatinib [161], induced cell death in cell lines BaF3 BCR-ABL wild-type or mutant, including T315I mutation [162], suppressed proliferation of cells from patients with T315I mutation and in vivo experiments resulted in a regression of the tumors in nude mice [163]. |
| Bortezomib | NFκB | Bortezomib reduced proliferation and survival of BCR-ABL-expressing cells, regardless of their sensitivity to imatinib and including the mutant T315I [164], and the combinatory effect with imatinib in CML led to reduced disseminated disease, decreased tumor growth and induced apoptosis in tumor sections [165]. |
| Vincristine | ABCB1 and survivin | Overexpression of ABCB1 and survivin were associated with low apoptosis index induced by vincristine treatment [43]. |
| LQB-118 | ABCB1, survivin and XIAP | LQB-118 overcome resistance phenotype through ABCB1, survivin and XIAP downregulation [166]. |
| Imatinib and nilotinib | SLC22A1, ABCB1 and ABCG2 | K562 cells displayed upregulated levels of SLC22A1, ABCB1, and ABCG2 genes, after exposure to increasing concentrations of imatinib and nilotinib, respectively [167]. |
| Imatinib | SLC22A1, ABCB1 and ABCG2 | Chronic exposure to imatinib increased ABCB1 and ABCG2 at the protein and gene levels, but SLC22A1 expression remained unaltered [168]. |
| Imatinib and vincristine | XIAP and ABCB1 | Simultaneous inhibition of XIAP and ABCB1 in cells that overexpress this efflux pump decreases the resistance to imatinib [129] and vincristine [130]. |
| Imatinib, apicidin and EBT-737 | XIAP | Imatinib-induced apoptosis was found to be associated with XIAP downregulation [121] and could be potentiated when combined with apicidin [122] and EBT-737 [123] in K562 cells and CML progenitors. |
| Etoposide and doxorubicin | XIAP | The downregulation of XIAP expression with antisense oligonucleotides increased apoptosis and enhanced the effects of doxorubicin in K562 cells [128]. |
AML: acute myeloid leukemia, CML: chronic myeloid leukemia; IAPs: inhibitor apoptosis proteins.
Wang et al. [98] had suggested that clinical responses to imatinib treatment could be affected by transporters SLC22A1, ABCB1, and ABCG2; however, a recent work showed no significant differences between ABCB1, ABCG2, and SLC22A1 genotypes and imatinib plasma or intracellular concentrations [195].
These data indicate that other transporters may be crucial for determining imatinib intracellular and plasma concentrations in CML patients. By contrast, in experiments using in vitro models of acquired resistance, K562 cells displayed upregulated levels of ABCB1 and ABCG2 genes, after exposure to increasing concentrations of imatinib [167, 168], which would imply the involvement of these transporters in resistance to TKIs [168]. However, different from the ABCG2 inhibitor, the ABCB1 inhibitor was able to restore imatinib sensitivity, indicating that only ABCB1 is essential for the development of acquired resistance in CML. Regarding the expression of SLC22A1 gene, contradictory data show that K562 resistant-cells had an increased [167] or similar [168] expression compared to their parental ones. Another work has demonstrated that imatinib and nilotinib are capable of inhibiting ABCB1 and ABCG2 and may overcome resistance, despite high levels of these transporters [63].
Current studies have proposed the role of IAPs in MDR phenotype promotion in association with ABCB1 expression [196]. Recently, we evaluated the resistance induced by the overexpression of both ABCB1 and survivin proteins [43]. In this work, we showed that K562 cells (ABCB1-negative) progressively became resistant to vincristine treatment by simultaneous overexpression of ABCB1 and survivin. We also showed that ABCB1 promoted resistance to cell death independently of its membrane expression. Besides that, we could observe that ABCB1 and survivin colocalize in the cytoplasmatic compartment, suggesting a common regulatory pathway of apoptosis resistance control [43]. In another work, we observed that both ABCB1 and survivin protein expressions are associated in CML patients [145]. We could establish a positive correlation between ABCB1 and survivin expression, but not with ABCB1 activity in samples from late-phase CML-CP patients. These data suggest that ABCB1 and survivin may act in promoting resistance in CML patients and, thus, reinforce the hypothesis that ABCB1 is able to induce resistance independently of its activity function [145]. As discussed above, CML patients usually develop imatinib resistance, and, therefore, new treatment approaches are necessary to overcome CML resistance. Netto et al. [197] showed that a new compound named LQB-118 was effective against leukemia cell lines with low toxicity to peripheral blood cells. Recently, we evaluated the effect of LQB-118 on CML cell lines and observed that this compound was able to induce apoptosis in both sensitive and resistant CML cells [166]. Moreover, cells treated with LQB-118 also presented decreased levels of survivin, XIAP, and ABCB1 expression. We also analyzed the LQB-118 effect in CML patient samples and observed that this compound was effective in inducing apoptosis in patients displaying the MDR phenotype [166]. Corroborating these data, Seca et al. [129] showed that the simultaneous inhibition of XIAP and ABCB1 in cells overexpressing ABCB1 could decrease imatinib resistance.
Recent studies reported that ABCB1 expression can be regulated by the NFκB transcription factor in hepatocytes and in drug-resistant cells. Moreover, the inhibition of NFκB activity sensitizes resistant colon cancer cells through a decreased ABCB1 expression, providing a link between NFκB and resistance to chemotherapy through the regulation of human ABCB1 gene expression [198]. In CML, Assef et al. [51] demonstrated that the resistance to imatinib exhibited in multidrug-resistant human leukemic K562 cells mediated by ABCB1 was reversed by the blockade of the NFκB pathway using a specific NFκB inhibitor [51]. Moreover, experimental evidence demonstrated the enhanced binding of NFκB to the promoter region of ABCB1 after K562 treatment with doxorubicin [199], further confirming the regulation of ABCB1 by NFκB in the promotion of chemoresistance. In accordance to that, FoxO3a may also interact with ABCB1 gene and decrease cell sensitivity. Some reports have postulated that chronic induction of Foxo3a expression and nuclear localization may activate mechanisms of resistance in CML cells. By using doxorubicin-sensitive and resistant K562 CML cells, Hui et al. [200, 201] have demonstrated that resistance to doxorubicin is associated with increased activity of PI3K/Akt, through a mechanism of feedback and with the ABCB1 gene induction. In contrast, it was recently demonstrated that FoxO3a is able to inhibit survivin expression while inducing cell death in melanoma [202] and neuroblastoma-derived cell lines [203]. Moreover, FoxO3a and FoxO1 were able to physically interact and inhibit survivin promoter, confirming the interaction between FoxO transcription factors and the antiapoptotic protein survivin [204]. However, the interaction between survivin and FoxO proteins, and its role in imatinib sensitivity, has not been investigated yet in CML-derived cells.
Survivin can also be targeted by NFκB [205], although it remains unclear how this interaction occurs. It was reported that inhibitors of the NFκB pathway, such as the natural compounds triptolide [206] and berbamine [207], have been shown to induce apoptosis in CML imatinib-resistant cells by down-regulating survivin levels. XIAP is another identified NFκB target, which is also implicated in modulating NFκB activation, through a feedback loop mechanism, in response to DNA damage and bacterial infection [208]. Studies suggest that XIAP recruits TAK1 in order to achieve NFκB activation and can mediate NFκB activation by promoting degradation of COMMD1, a negative regulator of NFκB [208]. As survivin, the interaction of XIAP and NFκB in CML remains unclear.
5. Conclusions
Although the introduction of imatinib and other TKIs in CML therapy has brought improvements in survival, CML prognosis still remains unfavorable for a group of patients. In addition to mutations found in the BCR-ABL gene, which alter the BCR-ABL kinase domain, there are currently identified secondary mechanisms of TKIs resistance. Multiple factors, such as inhibition of apoptotic signaling pathways, reduction in drug accumulation, and alterations in transcription factors, are known to contribute to the development of MDR and treatment failure in CML. These mechanisms usually act in concert in a multifactorial resistance context and play their role independent of or downstream BCR-ABL tyrosine kinase. Because the inhibition of only one mechanism is not effective enough to overcome clinical TKIs resistance, suppressing simultaneously several proteins must be required to increase the efficacy of the treatment in CML patients. Several questions remain to be answered to understand the interplay between these modes of resistance. For instance, how these proteins interact with each other to promote resistance and which one must be completely suppressed to antagonize malignancy? Regardless, what we know is that chemoresistance in CML is a multifactorial phenomenon and targeting these molecules seems to represent an interesting and feasible approach to overcome the development of TKIs-resistance in CML.
Conflict of Interests
The authors declare that they have no conflict of interests.
Acknowledgments
This study was supported by research grants from CNPq, FAPERJ, INCT para Controle do Câncer, CNPq 573806/2008-0, FAPERJ EE26/170.026/2008, Programa de Oncobiologia (UFRJ/Fundação do Câncer), and FAPERJ-PPSUS.
Abbreviations
- ALL:
Acute lymphoid leukemia
- AML:
Acute myeloid leukemia
- BCR-ABL:
Breakpoint cluster region/V-abl Abelson murine leukemia viral oncogene homolog 1
- BCRP:
Breast-cancer-related protein
- BIR:
Baculoviral IAP repeat
- BP:
Blast phase of chronic myeloid leukemia
- CDK1:
Cyclin-dependent kinase 1
- CLL:
Chronic lymphoid leukemia
- CML:
Chronic myeloid leukemia
- FOX:
Forkhead box
- HSC:
Hematopoietic stem cell
- IAP:
Inhibitor of apoptosis proteins
- Id1:
Inhibitor of DNA binding 1
- IκB:
Inhibitor of NFκB
- IKK:
IκB kinase
- KD:
Kinase domain
- LIC:
Leukemia initiating cells
- MAPK:
Mitogen-activated protein kinase
- MDR:
Multidrug resistance
- MRP1:
Multidrug resistance protein 1
- MTA:
Microtubule targeting agents
- NFκB:
Nuclear factor kappa B
- OCT-1:
Organic cation transporter-1
- Pgp:
P-glycoprotein
- Ph:
Philadelphia
- SGK:
Serum and glucocorticoid-regulated kinase
- Si-RNA:
Small interfering RNA
- SP:
Side population
- TKI:
Tyrosine kinase inhibitors
- TRAIL:
Tumor-necrosis-factor-related apoptosis-inducing ligand
- XIAP:
X-linked of inhibitor of apoptosis protein.
References
- 1.Goldman JM, Melo JV. Mechanisms of disease: chronic myeloid leukemia—advances in biology and new approaches to treatment. New England Journal of Medicine. 2003;349(15):1451–1464. doi: 10.1056/NEJMra020777. [DOI] [PubMed] [Google Scholar]
- 2.Perrotti D, Jamieson C, Goldman J, Skorski T. Chronic myeloid leukemia: mechanisms of blastic transformation. Journal of Clinical Investigation. 2010;120(7):2254–2264. doi: 10.1172/JCI41246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nature Reviews Cancer. 2007;7(6):441–453. doi: 10.1038/nrc2147. [DOI] [PubMed] [Google Scholar]
- 4.Hernández-Boluda JC, Bellosillo B, Vela MC, Colomer D, Alvarez-Larrán A, Cervantes F. Survivin expression in the progression of chronic myeloid leukemia: a sequential study in 16 patients. Leukemia and Lymphoma. 2005;46(5):717–722. doi: 10.1080/10428190500052131. [DOI] [PubMed] [Google Scholar]
- 5.Hehlmann R, Heimpel H, Hasford J, et al. Randomized comparison of busulfan and hydroxyurea in chronic myelogenous leukemia: prolongation of survival by hydroxyurea. Blood. 1993;82(2):398–407. [PubMed] [Google Scholar]
- 6.Bonifazi F, de Vivo A, Rosti G, et al. Chronic myeloid leukemia and interferon-α: a study of complete cytogenetic responders. Blood. 2001;98(10):3074–3081. doi: 10.1182/blood.v98.10.3074. [DOI] [PubMed] [Google Scholar]
- 7.Gratwohl A, Brand R, Apperley J, et al. Allogeneic hematopoietic stem cell transplantation for chronic myeloid leukemia in Europe 2006: transplant activity, long-term data and current results. An analysis by the chronic leukemia working party of the European Group for Blood and Marrow Transplantation (EBMT) Haematologica. 2006;91(4):513–521. [PubMed] [Google Scholar]
- 8.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289(5486):1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 9.Kantarjian HM, Cortes JE, O’Brien S, et al. Long-term survival benefit and improved complete cytogenetic and molecular response rates with imatinib mesylate in Philadelphia chromosome-positive chronic-phase chronic myeloid leukemia after failure of interferon-α . Blood. 2004;104(7):1979–1988. doi: 10.1182/blood-2004-02-0711. [DOI] [PubMed] [Google Scholar]
- 10.Pinilla-Ibarz J, Cortes J, Mauro MJ. Intolerance to tyrosine kinase inhibitors in chronic myeloid leukemia. Cancer. 2011;117(4):688–697. doi: 10.1002/cncr.25648. [DOI] [PubMed] [Google Scholar]
- 11.Baccarani M, Saglio G, Goldman J, et al. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2006;108(6):1809–1820. doi: 10.1182/blood-2006-02-005686. [DOI] [PubMed] [Google Scholar]
- 12.Saglio G, Kantarjian H, Holyoake T, Ranganathan A, Cortés JE. Proceedings of the third global workshop on chronic myeloid leukemia. Clinical Lymphoma, Myeloma and Leukemia. 2010;10(6):443–451. doi: 10.3816/CLML.2010.n.077. [DOI] [PubMed] [Google Scholar]
- 13.Breccia M, Efficace F, Alimena G. Imatinib treatment in chronic myelogenous leukemia: what have we learned so far? Cancer Letters. 2011;300(2):115–121. doi: 10.1016/j.canlet.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 14.Eiring AM, Khorashad JS, Morley K, Deininger MW. Advances in the treatment of chronic myeloid leukemia. BMC Medicine. 2011;9, article 99 doi: 10.1186/1741-7015-9-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melo JV, Chuah C. Resistance to imatinib mesylate in chronic myeloid leukaemia. Cancer Letters. 2007;249(2):121–132. doi: 10.1016/j.canlet.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 16.Bixby D, Talpaz M. Mechanisms of resistance to tyrosine kinase inhibitors in chronic myeloid leukemia and recent therapeutic strategies to overcome resistance. Hematology. 2009:461–476. doi: 10.1182/asheducation-2009.1.461. [DOI] [PubMed] [Google Scholar]
- 17.Quintás-Cardama A, Cortes J. Molecular biology of BCR-ABL1-positive chronic myeloid leukemia. Blood. 2009;113(8):1619–1630. doi: 10.1182/blood-2008-03-144790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roychowdhury S, Talpaz M. Managing resistance in chronic myeloid leukemia. Blood Reviews. 2011;25(6):279–290. doi: 10.1016/j.blre.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 19.Eechoute K, Sparreboom A, Burger H, et al. Drug transporters and imatinib treatment: Implications for clinical practice. Clinical Cancer Research. 2011;17(3):406–415. doi: 10.1158/1078-0432.CCR-10-2250. [DOI] [PubMed] [Google Scholar]
- 20.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annual Review of Pharmacology and Toxicology. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 21.Kruh GD. Introduction to resistance to anticancer agents. Oncogene. 2003;22(47):7262–7264. doi: 10.1038/sj.onc.1206932. [DOI] [PubMed] [Google Scholar]
- 22.Kartner N, Evernden-Porelle D, Bradley G, Ling V. Detection of P-glycoprotein in multidrug-resistant cell lines by monoclonal antibodies. Nature. 1985;316(6031):820–823. doi: 10.1038/316820a0. [DOI] [PubMed] [Google Scholar]
- 23.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nature Reviews Cancer. 2002;2(1):48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 24.Chaudhary PM, Roninson IB. Expression and activity of P-glycoprotein, a multidrug efflux pump, in human hematopoietic stem cells. Cell. 1991;66(1):85–94. doi: 10.1016/0092-8674(91)90141-k. [DOI] [PubMed] [Google Scholar]
- 25.Klimecki WT, Futscher BW, Grogan TM, Dalton WS. P-glycoprotein expression and function in circulating blood cells from normal volunteers. Blood. 1994;83(9):2451–2458. [PubMed] [Google Scholar]
- 26.Schinkel AH, Mayer U, Wagenaar E, et al. Normal viability and altered pharmacokinetics in mice lacking MDR1-type (drug-transporting) P-glycoproteins. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(8):4028–4033. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fromm MF. Importance of P-glycoprotein at blood-tissue barriers. Trends in Pharmacological Sciences. 2004;25(8):423–429. doi: 10.1016/j.tips.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 28.Luurtsema G, Verbeek J, Lubberink M, et al. Carbon-11 labeled tracers for in vivo imaging of P-glycoprotein function: kinetics, advantages and disadvantages. Current Topics in Medicinal Chemistry. 2010;10(17):1820–1833. doi: 10.2174/156802610792928013. [DOI] [PubMed] [Google Scholar]
- 29.Lown KS, Mayo RR, Leichtman AB, et al. Role of intestinal P-glycoprotein (MDR1) in interpatient variation in the oral bioavailability of cyclosporine. Clinical Pharmacology and Therapeutics. 1997;62(3):248–260. doi: 10.1016/S0009-9236(97)90027-8. [DOI] [PubMed] [Google Scholar]
- 30.Schinkel AH. P-glycoprotein, a gatekeeper in the blood-brain barrier. Advanced Drug Delivery Reviews. 1999;36(2-3):179–194. doi: 10.1016/s0169-409x(98)00085-4. [DOI] [PubMed] [Google Scholar]
- 31.Sarkadi B, Homolya L, Szakács G, Váradi A. Human multidrug resistance ABCB and ABCG transporters: participation in a chemoimmunity defense system. Physiological Reviews. 2006;86(4):1179–1236. doi: 10.1152/physrev.00037.2005. [DOI] [PubMed] [Google Scholar]
- 32.Svoboda-Beusan I, Kusec R, Bendelja K, et al. The relevance of multidrug resistance-associated P-glycoprotein expression in the treatment response of B-cell chronic lymphocytic leukemia. Haematologica. 2000;85(12):1261–1267. [PubMed] [Google Scholar]
- 33.Ohsawa M, Ikura Y, Fukushima H, et al. Immunohistochemical expression of multidrug resistance proteins as a predictor of poor response to chemotherapy and prognosis in patients with nodal diffuse large B-cell lymphoma. Oncology. 2005;68(4–6):422–431. doi: 10.1159/000086984. [DOI] [PubMed] [Google Scholar]
- 34.Lourenço JJ, Maia RC, Scheiner MAM, Vasconcelos FC, Moreira MAM. Genomic variation at the MDR1 promoter and P-glycoprotein expression and activity in AML patients. Leukemia Research. 2008;32(6):976–979. doi: 10.1016/j.leukres.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 35.Grogan TM, Spier CM, Salmon SE, et al. P-glycoprotein expression in human plasma cell myeloma: correlation with prior chemotherapy. Blood. 1993;81(2):490–495. [PubMed] [Google Scholar]
- 36.Ambudkar SV, Sauna ZE, Gottesman MM, Szakacs G. A novel way to spread drug resistance in tumor cells: functional intercellular transfer of P-glycoprotein (ABCB1) Trends in Pharmacological Sciences. 2005;26(8):385–387. doi: 10.1016/j.tips.2005.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levchenko A, Mehta BM, Niu X, et al. Intercellular transfer of P-glycoprotein mediates acquired multidrug resistance in tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(6):1933–1938. doi: 10.1073/pnas.0401851102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bebawy M, Combes V, Lee E, et al. Membrane microparticles mediate transfer of P-glycoprotein to drug sensitive cancer cells. Leukemia. 2009;23(9):1643–1649. doi: 10.1038/leu.2009.76. [DOI] [PubMed] [Google Scholar]
- 39.Robinson LJ, Roberts WK, Ling TT, Lamming D, Sternberg SS, Roepe PD. Human MDR 1 Protein overexpression delays the apoptotic cascade in chinese hamster ovary fibroblasts. Biochemistry. 1997;36(37):11169–11178. doi: 10.1021/bi9627830. [DOI] [PubMed] [Google Scholar]
- 40.Smyth MJ, Krasovskis E, Sutton VR, Johnstone RW. The drug efflux protein, P-glycoprotein, additionally protects drug-resistant tumor cells from multiple forms of caspase-dependent apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(12):7024–7029. doi: 10.1073/pnas.95.12.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnstone RW, Cretney E, Smyth MJ. P-glycoprotein protects leukemia cells against caspase-dependent, but not caspase-independent, cell death. Blood. 1999;93(3):1075–1085. [PubMed] [Google Scholar]
- 42.Pallis M, Turzanski J, Grundy M, Seedhouse C, Russell N. Resistance to spontaneous apoptosis in acute myeloid leukaemia blasts is associated with P-glycoprotein expression and function, but not with the presence of FLT3 internal tandem duplications. British Journal of Haematology. 2003;120(6):1009–1016. doi: 10.1046/j.1365-2141.2003.04210.x. [DOI] [PubMed] [Google Scholar]
- 43.Souza PS, Vasconcelos FC, de Souza Reis FR, De Moraes GN, Maia RC. P-glycoprotein and survivin simultaneously regulate vincristine-induced apoptosis in chronic myeloid leukemia cells. International Journal of Oncology. 2011;39(4):925–933. doi: 10.3892/ijo.2011.1103. [DOI] [PubMed] [Google Scholar]
- 44.Kuwazuru Y, Yoshimura A, Hanada S, et al. Expression of the multidrug transporter, P-glycoprotein, in chronic myelogenous leukaemia cells in blast crisis. British Journal of Haematology. 1990;74(1):24–29. doi: 10.1111/j.1365-2141.1990.tb02533.x. [DOI] [PubMed] [Google Scholar]
- 45.Stavrovskaya A, Turkina A, Sedyakhina N, et al. Prognostic value of P-glycoprotein and leukocyte differentiation antigens in chronic meloid leukemia. Leukemia and Lymphoma. 1998;28(5-6):469–482. doi: 10.3109/10428199809058354. [DOI] [PubMed] [Google Scholar]
- 46.List AF, Kopecky KJ, Willman CL, et al. Cyclosporine inhibition of P-glycoprotein in chronic myeloid leukemia blast phase. Blood. 2002;100(5):1910–1912. [PubMed] [Google Scholar]
- 47.Weisberg E, Griffin JD. Mechanism of resistance to the ABL tyrosine kinase inhibitor STI571 in BCR/ABL-transformed hematopoietic cell lines. Blood. 2000;95(11):3498–3505. [PubMed] [Google Scholar]
- 48.Vasconcelos FC, Silva KL, Souza PS, et al. Variation of MDR proteins expression and activity levels according to clinical status and evolution of CML patients. Cytometry, B. 2011;80(3):158–166. doi: 10.1002/cyto.b.20580. [DOI] [PubMed] [Google Scholar]
- 49.Mahon FX, Deininger MW, Schultheis B, et al. Selection and characterization of BCR-ABL positive cell lines with differential sensitivity to the signal transduction inhibitor STI571: diverse mechanisms of resistance. Blood. 2000;96:1070–1079. [PubMed] [Google Scholar]
- 50.Widmer N, Rumpold H, Untergasser G, Fayet A, Buclin T, Decosterd LA. Resistance reversal by RNAi silencing of MDR1 in CML cells associated with increase in imatinib intracellular levels. Leukemia. 2007;21(7):1561–1562. doi: 10.1038/sj.leu.2404671. [DOI] [PubMed] [Google Scholar]
- 51.Assef Y, Rubio F, Coló G, del Mónaco S, Costas MA, Kotsias BA. Imatinib resistance in multidrug-resistant K562 human leukemic cells. Leukemia Research. 2009;33(5):710–716. doi: 10.1016/j.leukres.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 52.Mahon FX, Belloc F, Lagarde V, et al. MDR1 gene overexpression confers resistance to imatinib mesylate in leukemia cell line models. Blood. 2003;101(6):2368–2373. doi: 10.1182/blood.V101.6.2368. [DOI] [PubMed] [Google Scholar]
- 53.Rumpold H, Wolf AM, Gruenewald K, Gastl G, Gunsilius E, Wolf D. RNAi-mediated knockdown of P-glycoprotein using a transposon-based vector system durably restores imatinib sensitivity in imatinib-resistant CML cell lines. Experimental Hematology. 2005;33(7):767–775. doi: 10.1016/j.exphem.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 54.Illmer T, Schaich M, Platzbecker U, et al. P-glycoprotein-mediated drug efflux is a resistance mechanism of chronic myelogenous leukemia cells to treatment with imatinib mesylate. Leukemia. 2004;18(3):401–408. doi: 10.1038/sj.leu.2403257. [DOI] [PubMed] [Google Scholar]
- 55.Zong Y, Zhou S, Sorrentino BP. Loss of P-glycoprotein expression in hematopoietic stem cells does not improve responses to imatinib in a murine model of chronic myelogenous leukemia. Leukemia. 2005;19(9):1590–1596. doi: 10.1038/sj.leu.2403853. [DOI] [PubMed] [Google Scholar]
- 56.Stromskaya TP, Rybalkina EY, Kruglov SS, et al. Role of P-glycoprotein in evolution of populations of chronic myeloid leukemia cells treated with imatinib. Biochemistry. 2008;73(1):29–37. doi: 10.1134/s0006297908010045. [DOI] [PubMed] [Google Scholar]
- 57.Hatziieremia S, Jordanides NE, Holyoake TL, Mountford JC, Jørgensen HG. Inhibition of MDR1 does not sensitize primitive chronic myeloid leukemia CD34+ cells to imatinib. Experimental Hematology. 2009;37(6):692–700. doi: 10.1016/j.exphem.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 58.Gurney H, Wong M, Balleine RL, et al. Imatinib disposition and ABCB1 (MDR1, P-glycoprotein) genotype. Clinical Pharmacology and Therapeutics. 2007;82(1):33–40. doi: 10.1038/sj.clpt.6100201. [DOI] [PubMed] [Google Scholar]
- 59.Kimchi-Sarfaty C, Oh JM, Kim IW, et al. A "silent" polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315(5811):525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 60.Dulucq S, Bouchet S, Turcq B, et al. Multidrug resistance gene (MDR1) polymorphisms are associated with major molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood. 2008;112(5):2024–2027. doi: 10.1182/blood-2008-03-147744. [DOI] [PubMed] [Google Scholar]
- 61.Ni LN, Li JY, Miao KR, et al. Multidrug resistance gene (MDR1) polymorphisms correlate with imatinib response in chronic myeloid leukemia. Medical Oncology. 2011;28(1):265–269. doi: 10.1007/s12032-010-9456-9. [DOI] [PubMed] [Google Scholar]
- 62.Bhatia R, Holtz M, Niu N, et al. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood. 2003;101(12):4701–4707. doi: 10.1182/blood-2002-09-2780. [DOI] [PubMed] [Google Scholar]
- 63.Dohse M, Scharenberg C, Shukla S, et al. Comparison of ATP-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib, nilotinib, and dasatinib. Drug Metabolism and Disposition. 2010;38(8):1371–1380. doi: 10.1124/dmd.109.031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hegedus C, Özvegy-Laczka C, Apáti AÁ, et al. Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: implications for altered anti-cancer effects and pharmacological properties. British Journal of Pharmacology. 2009;158(4):1153–1164. doi: 10.1111/j.1476-5381.2009.00383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mahon FX, Hayette S, Lagarde V, et al. Evidence that resistance to nilotinib may be due to BCR-ABL, Pgp, or Src kinase overexpression. Cancer Research. 2008;68(23):9809–9816. doi: 10.1158/0008-5472.CAN-08-1008. [DOI] [PubMed] [Google Scholar]
- 66.Giannoudis A, Davies A, Lucas CM, Harris RJ, Pirmohamed M, Clark RE. Effective dasatinib uptake may occur without human organic cation transporter 1 (hOCT1): implications for the treatment of imatinib-resistant chronic myeloid leukemia. Blood. 2008;112(8):3348–3354. doi: 10.1182/blood-2007-10-116236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hiwase DK, Saunders V, Hewett D, et al. Dasatinib cellular uptake and efflux in chronic myeloid leukemia cells: therapeutic implications. Clinical Cancer Research. 2008;14(12):3881–3888. doi: 10.1158/1078-0432.CCR-07-5095. [DOI] [PubMed] [Google Scholar]
- 68.Bartlett NL, Lum BL, Fisher GA, et al. Phase I trial of doxorubicin with cyclosporine as a modulator of multidrug resistance. Journal of Clinical Oncology. 1994;12(4):835–842. doi: 10.1200/JCO.1994.12.4.835. [DOI] [PubMed] [Google Scholar]
- 69.Garrigues A, Nugier J, Orlowski S, Ezan E. A high-throughput screening microplate test for the interaction of drugs with P-glycoprotein. Analytical Biochemistry. 2002;305(1):106–114. doi: 10.1006/abio.2002.5650. [DOI] [PubMed] [Google Scholar]
- 70.Frydrych I, Mlejnek P, Dolezel P. Cyclosporin A sensitises Bcr-Abl positive cells to imatinib mesylate independently of P-glycoprotein expression. Toxicology in Vitro. 2009;23(8):1482–1490. doi: 10.1016/j.tiv.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 71.Maia RC, Carriço MK, Klumb CENP, et al. Clinical approach to circumvention of multidrug resistance in refractory leukemic patients: association of cyclosporin A with etoposide. Journal of Experimental and Clinical Cancer Research. 1997;16(4):419–424. [PubMed] [Google Scholar]
- 72.Maia RC, Noronha H, Vasconcelos FC, Rumjanek VM. Interaction of cyclosporin A and etoposide. Clinical and in vitro assessment in blast phase of chronic myeloid leukaemia. Clinical and Laboratory Haematology. 1997;19(3):215–217. [PubMed] [Google Scholar]
- 73.Ni Z, Mark ME, Cai X, Mao Q. Fluorescence resonance energy transfer (FRET) analysis demonstrates dimer/oligomer formation of the human breast cancer resistance protein (BCRP/ABCG2) in intact cells. International Journal of Biochemistry and Molecular Biology. 2010;1(1):1–11. [PMC free article] [PubMed] [Google Scholar]
- 74.Fetsch PA, Abati A, Litman T, et al. Localization of the ABCG2 mitoxantrone resistance-associated protein in normal tissues. Cancer Letters. 2006;235(1):84–92. doi: 10.1016/j.canlet.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 75.Nakagawa M, Schneider E, Dixon KH, et al. Reduced intracellular drug accumulation in the absence of P-glycoprotein (MDR1) overexpression in mitoxantrone-resistant human MCF-7 breast cancer cells. Cancer Research. 1992;52(22):6175–6181. [PubMed] [Google Scholar]
- 76.Lemos C, Jansen G, Peters GJ. Drug transporters: recent advances concerning BCRP and tyrosine kinase inhibitors. British Journal of Cancer. 2008;98(5):857–862. doi: 10.1038/sj.bjc.6604213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Honjo Y, Hrycyna CA, Yan QW, et al. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Research. 2001;61(18):6635–6639. [PubMed] [Google Scholar]
- 78.Miwa M, Tsukahara S, Ishikawa E, Asada S, Imai Y, Sugimoto Y. Single amino acid substitutions in the transmembrane domains of breast cancer resistance protein (BCRP) alter cross resistance patterns in transfectants. International Journal of Cancer. 2003;107(5):757–763. doi: 10.1002/ijc.11484. [DOI] [PubMed] [Google Scholar]
- 79.Özvegy-Laczka C, Hegedus T, Várady G, et al. High-affinity interaction of tyrosine kinase inhibitors with the ABCG2 multidrug transporter. Molecular Pharmacology. 2004;65(6):1485–1495. doi: 10.1124/mol.65.6.1485. [DOI] [PubMed] [Google Scholar]
- 80.Shukla S, Sauna ZE, Ambudkar SV. Evidence for the interaction of imatinib at the transport-substrate site(s) of the multidrug-resistance-linked ABC drug transporters ABCB1 (P-glycoprotein) and ABCG2. Leukemia. 2008;22(2):445–447. doi: 10.1038/sj.leu.2404897. [DOI] [PubMed] [Google Scholar]
- 81.Houghton PJ, Germain GS, Harwood FC, et al. Imatinib mesylate is a potent inhibitor of the ABCG2 (BCRP) transporter and reverses resistance to topotecan and SN-38 in vitro. Cancer Research. 2004;64(7):2333–2337. doi: 10.1158/0008-5472.can-03-3344. [DOI] [PubMed] [Google Scholar]
- 82.Burger H, van Tol H, Boersma AWM, et al. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood. 2004;104(9):2940–2942. doi: 10.1182/blood-2004-04-1398. [DOI] [PubMed] [Google Scholar]
- 83.Brendel C, Scharenberg C, Dohse M, et al. Imatinib mesylate and nilotinib (AMN107) exhibit high-affinity interaction with ABCG2 on primitive hematopoietic stem cells. Leukemia. 2007;21(6):1267–1275. doi: 10.1038/sj.leu.2404638. [DOI] [PubMed] [Google Scholar]
- 84.Graham SM, Jørgensen HG, Allan E, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319–325. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- 85.Nakanishi T, Shiozawa K, Hassel BA, Ross DD. Complex interaction of BCRP/ABCG2 and imatinib in BCR-ABL-expressing cells: BCRP-mediated resistance to imatinib is attenuated by imatinib-induced reduction of BCRP expression. Blood. 2006;108(2):678–684. doi: 10.1182/blood-2005-10-4020. [DOI] [PubMed] [Google Scholar]
- 86.Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. Journal of Experimental Medicine. 1996;183(4):1797–1806. doi: 10.1084/jem.183.4.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhou S, Schuetz JD, Bunting KD, et al. The ABC transporter BCRP1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nature Medicine. 2001;7(9):1028–1034. doi: 10.1038/nm0901-1028. [DOI] [PubMed] [Google Scholar]
- 88.Scharenberg CW, Harkey MA, Torok-Storb B. The ABCG2 transporter is an efficient hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood. 2002;99(2):507–512. doi: 10.1182/blood.v99.2.507. [DOI] [PubMed] [Google Scholar]
- 89.Jonker JW, Buitelaar M, Wagenaar E, et al. The breast cancer resistance protein protects against a major chlorophyll-derived dietary phototoxin and protoporphyria. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(24):15649–15654. doi: 10.1073/pnas.202607599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhou S, Morris JJ, Barnes Y, Lan L, Schuetz JD, Sorrentino BP. Bcrp1 gene expression is required for normal numbers of side population stem cells in mice, and confers relative protection to mitoxantrone in hematopoietic cells in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(19):12339–12344. doi: 10.1073/pnas.192276999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Breedveld P, Pluim D, Cipriani G, et al. The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Research. 2005;65(7):2577–2582. doi: 10.1158/0008-5472.CAN-04-2416. [DOI] [PubMed] [Google Scholar]
- 92.Imai Y, Nakane M, Kage K, et al. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and low-level drug resistance. Molecular Cancer Therapeutics. 2002;1(8):611–616. [PubMed] [Google Scholar]
- 93.Tamura A, Watanabe M, Saito H, et al. Functional validation of the genetic polymorphisms of human ATP-binding cassette (ABC) transporter ABCG2: identification of alleles that are defective in porphyrin transport. Molecular Pharmacology. 2006;70(1):287–296. doi: 10.1124/mol.106.023556. [DOI] [PubMed] [Google Scholar]
- 94.Mizuarai S, Aozasa N, Kotani H. Single nucleotide polymorphisms result in impaired membrane localization and reduced atpase activity in multidrug transporter ABCG2. International Journal of Cancer. 2004;109(2):238–246. doi: 10.1002/ijc.11669. [DOI] [PubMed] [Google Scholar]
- 95.Takahashi N, Miura M. Therapeutic drug monitoring of imatinib for chronic myeloid leukemia patients in the chronic phase. Pharmacology. 2011;87(5-6):241–248. doi: 10.1159/000324900. [DOI] [PubMed] [Google Scholar]
- 96.Koepsell H, Lips K, Volk C. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharmaceutical Research. 2007;24(7):1227–1251. doi: 10.1007/s11095-007-9254-z. [DOI] [PubMed] [Google Scholar]
- 97.Hediger MA, Romero MF, Peng JB, Rolfs A, Takanaga H, Bruford EA. The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteins. Pflugers Archiv European Journal of Physiology. 2004;447(5):465–468. doi: 10.1007/s00424-003-1192-y. [DOI] [PubMed] [Google Scholar]
- 98.Wang L, Giannoudis A, Lane S, Williamson P, Pirmohamed M, Clark RE. Expression of the uptake drug transporter hOCT1 is an important clinical determinant of the response to imatinib in chronic myeloid leukemia. Clinical Pharmacology and Therapeutics. 2008;83(2):258–264. doi: 10.1038/sj.clpt.6100268. [DOI] [PubMed] [Google Scholar]
- 99.Thomas J, Wang L, Clark RE, Pirmohamed M. Active transport of imatinib into and out of cells: implications for drug resistance. Blood. 2004;104(12):3739–3745. doi: 10.1182/blood-2003-12-4276. [DOI] [PubMed] [Google Scholar]
- 100.White DL, Saunders VA, Dang P, et al. Most CML patients who have a suboptimal response to imatinib have low OCT-1 activity: higher doses of imatinib may overcome the negative impact of low OCT-1 activity. Blood. 2007;110(12):4064–4072. doi: 10.1182/blood-2007-06-093617. [DOI] [PubMed] [Google Scholar]
- 101.Crossman LC, Druker BJ, Deininger MWN, Pirmohamed M, Wang L, Clark RE. hOCT 1 and resistance to imatinib. Blood. 2005;106(3):1133–1134. doi: 10.1182/blood-2005-02-0694. [DOI] [PubMed] [Google Scholar]
- 102.Hu S, Franke RM, Filipski KK, et al. Interaction of imatinib with human organic ion carriers. Clinical Cancer Research. 2008;14(10):3141–3148. doi: 10.1158/1078-0432.CCR-07-4913. [DOI] [PubMed] [Google Scholar]
- 103.Crook NE, Clem RJ, Miller LK. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. Journal of Virology. 1993;67(4):2168–2174. doi: 10.1128/jvi.67.4.2168-2174.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Deveraux QL, Reed JC. IAP family proteins—suppressors of apoptosis. Genes and Development. 1999;13(3):239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 105.Wrzesień-Kuś A, Smolewski P, Sobczak-Pluta A, Wierzbowska A, Robak T. The inhibitor of apoptosis protein family and its antagonists in acute leukemias. Apoptosis. 2004;9(6):705–715. doi: 10.1023/B:APPT.0000045788.61012.b2. [DOI] [PubMed] [Google Scholar]
- 106.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388(6639):300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 107.Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Reports. 2006;7(10):988–994. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Scott FL, Denault JB, Riedl SJ, Shin H, Renatus M, Salvesen GS. XIAP inhibits caspase-3 and -7 using two binding sites: evolutionary conserved mechanism of IAPs. EMBO Journal. 2005;24(3):645–655. doi: 10.1038/sj.emboj.7600544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Verhagen AM, Ekert PG, Pakusch M, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102(1):43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 110.Liston P, Fong WG, Kelly NL, et al. Identification of XAF1 as an antagonist of XIAP anti-Caspase activity. Nature Cell Biology. 2001;3(2):128–133. doi: 10.1038/35055027. [DOI] [PubMed] [Google Scholar]
- 111.Holcik M, Korneluk RG. XIAP, the guardian angel. Nature Reviews Molecular Cell Biology. 2001;2(7):550–556. doi: 10.1038/35080103. [DOI] [PubMed] [Google Scholar]
- 112.Mufti AR, Burstein E, Duckett CS. XIAP: cell death regulation meets copper homeostasis. Archives of Biochemistry and Biophysics. 2007;463(2):168–174. doi: 10.1016/j.abb.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Galbán S, Duckett CS. XIAP as a ubiquitin ligase in cellular signaling. Cell Death and Differentiation. 2010;17(1):54–60. doi: 10.1038/cdd.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Harlin H, Reffey SB, Duckett CS, Lindsten T, Thompson CB. Characterization of XIAP-deficient mice. Molecular and Cellular Biology. 2001;21(10):3604–3608. doi: 10.1128/MCB.21.10.3604-3608.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vischioni B, van der Valk P, Span SW, Kruyt FAE, Rodriguez JA, Giaccone G. Expression and localization of inhibitor of apoptosis proteins in normal human tissues. Human Pathology. 2006;37(1):78–86. doi: 10.1016/j.humpath.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 116.Liu SS, Tsang BK, Cheung AN, et al. Anti-apoptotic proteins, apoptotic and proliferative parameters and their prognostic significance in cervical carcinoma. European Journal of Cancer. 2001;37(9):1104–1110. doi: 10.1016/s0959-8049(01)00085-5. [DOI] [PubMed] [Google Scholar]
- 117.Carter BZ, Kornblau SM, Tsao T, et al. Caspase-independent cell death in AML: caspase inhibition in vitro with pan-caspase inhibitors or in vivo by XIAP or Survivin does not affect cell survival or prognosis. Blood. 2003;102(12):4179–4186. doi: 10.1182/blood-2003-03-0960. [DOI] [PubMed] [Google Scholar]
- 118.Tamm I, Richter S, Scholz F, et al. Xiap expression correlates with monocytic differentiation in adult de novo AML: impact on prognosis. Hematology Journal. 2004;5(6):489–495. doi: 10.1038/sj.thj.6200549. [DOI] [PubMed] [Google Scholar]
- 119.Tamm I, Richter S, Oltersdorf D, et al. High expression levels of x-linked inhibitor of apoptosis protein and survivin correlate with poor overall survival in childhood de novo acute myeloid leukemia. Clinical Cancer Research. 2004;10(11):3737–3744. doi: 10.1158/1078-0432.CCR-03-0642. [DOI] [PubMed] [Google Scholar]
- 120.Zhang Y, Zhu J, Tang Y, et al. X-linked inhibitor of apoptosis positive nuclear labeling: a new independent prognostic biomarker of breast invasive ductal carcinoma. Diagnostic Pathology. 2011;6(1, article 49) doi: 10.1186/1746-1596-6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fang G, Kim CN, Perkins CL, et al. CGP57148B (STI-571) induces differentiation and apoptosis and sensitizes Bcr-Abl-positive human leukemia cells to apoptosis due to antileukemic drugs. Blood. 2000;96(6):2246–2253. [PubMed] [Google Scholar]
- 122.Kim JS, Jeung HK, Cheong JW, et al. Apicidin potentiates the imatinib-induced apoptosis of Bcr-Abl-positive human leukaemia cells by enhancing the activation of mitochondria-dependent caspase cascades. British Journal of Haematology. 2004;124(2):166–178. doi: 10.1046/j.1365-2141.2003.04759.x. [DOI] [PubMed] [Google Scholar]
- 123.Airiau K, Mahon F-X, Josselin M, Jeanneteau M, Turcq B, Belloc F. ABT-737 increases tyrosine kinase inhibitor-induced apoptosis in chronic myeloid leukemia cells through XIAP downregulation and sensitizes CD34+ CD38− population to imatinib. doi: 10.1016/j.exphem.2012.01.004. Experimental Hematology. In press. [DOI] [PubMed] [Google Scholar]
- 124.Hao XS, Hao JH, Liu FT, Newland AC, Jia L. Potential mechanisms of leukemia cell resistance to TRAIL-induced apopotosis. Apoptosis. 2003;8(6):601–607. doi: 10.1023/A:1026131425204. [DOI] [PubMed] [Google Scholar]
- 125.McManus DC, Lefebvre CA, Cherton-Horvat G, et al. Loss of XIAP protein expression by RNAi and antisense approaches sensitizes cancer cells to functionally diverse chemotherapeutics. Oncogene. 2004;23(49):8105–8117. doi: 10.1038/sj.onc.1207967. [DOI] [PubMed] [Google Scholar]
- 126.Schimmer AD, Dalili S, Batey RA, Riedl SJ. Targeting XIAP for the treatment of malignancy. Cell Death and Differentiation. 2006;13(2):179–188. doi: 10.1038/sj.cdd.4401826. [DOI] [PubMed] [Google Scholar]
- 127.Schimmer AD, Estey EH, Borthakur G, et al. Phase I/II trial of AEG35156 X-linked inhibitor of apoptosis protein antisense oligonucleotide combined with idarubicin and cytarabine in patients with relapsed or primary refractory acute myeloid leukemia. Journal of Clinical Oncology. 2009;27(28):4741–4746. doi: 10.1200/JCO.2009.21.8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lima RT, Martins LM, Guimarães JE, Sambade C, Vasconcelos MH. Chemosensitization effects of XIAP downregulation in K562 leukemia cells. Journal of Chemotherapy. 2006;18(1):98–102. doi: 10.1179/joc.2006.18.1.98. [DOI] [PubMed] [Google Scholar]
- 129.Seca H, Lima RT, Guimarães JE, Vasconcelos MH. Simultaneous targeting of P-gp and XIAP with siRNAs increases sensitivity of P-gp overexpressing CML cells to imatinib. Hematology. 2011;16(2):100–108. doi: 10.1179/102453311X12940641877803. [DOI] [PubMed] [Google Scholar]
- 130.de Souza PS, da Cunha Vasconcelos F, Silva LFR, Maia RC. Cyclosporine A enables vincristine-induced apoptosis during reversal of multidrug resistance phenotype in chronic myeloid leukemia cells. doi: 10.1007/s13277-012-0323-5. Tumor Biology. In press. [DOI] [PubMed] [Google Scholar]
- 131.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nature Medicine. 1997;3(8):917–921. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 132.Li F, Ambrosini G, Chu EY, et al. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396(6711):580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 133.Li F, Ling X. Survivin study: an update of “what is the next wave?”. Journal of Cellular Physiology. 2006;208(3):476–486. doi: 10.1002/jcp.20634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Speletas M, Argentou N, Karanikas V, et al. Survivin isoform expression patterns in CML patients correlate with resistance to imatinib and progression, but do not trigger cytolytic responses. Clinical Immunology. 2011;139(2):155–163. doi: 10.1016/j.clim.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 135.Altieri DC. Validating survivin as a cancer therapeutic target. Nature Reviews Cancer. 2003;3(1):46–54. doi: 10.1038/nrc968. [DOI] [PubMed] [Google Scholar]
- 136.Fukuda S, Pelus LM. Survivin, a cancer target with an emerging role in normal adult tissues. Molecular Cancer Therapeutics. 2006;5(5):1087–1098. doi: 10.1158/1535-7163.MCT-05-0375. [DOI] [PubMed] [Google Scholar]
- 137.Fukuda S, Pelus LM. Regulation of the inhibitor-of-apoptosis family member survivin in normal cord blood and bone marrow CD34+ cells by hematopoietic growth factors: Implication of survivin expression in normal hematopoiesis. Blood. 2001;98(7):2091–2100. doi: 10.1182/blood.v98.7.2091. [DOI] [PubMed] [Google Scholar]
- 138.Gerber JM, Qin L, Kowalski J, et al. Characterization of chronic myeloid leukemia stem cells. American Journal of Hematology. 2011;86(1):31–37. doi: 10.1002/ajh.21915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Helgason GV, Young GAR, Holyoake TL. Targeting chronic myeloid leukemia stem cells. Current Hematologic Malignancy Reports. 2010;5(2):81–87. doi: 10.1007/s11899-010-0043-0. [DOI] [PubMed] [Google Scholar]
- 140.Mori A, Wada H, Nishimura Y, Okamoto T, Takemoto Y, Kakishita E. Expression of the antiapoptosis gene survivin in human leukemia. International Journal of Hematology. 2002;75(2):161–165. doi: 10.1007/BF02982021. [DOI] [PubMed] [Google Scholar]
- 141.Badran A, Yoshida A, Yujiwano, et al. Expression of the antiapoptotic gene survivin in chronic myeloid leukemia. Anticancer Research. 2003;23(1):589–592. [PubMed] [Google Scholar]
- 142.Conte E, Stagno F, Guglielmo P, Scuto A, Consoli C, Messina A. Survivin expression in chronic myeloid leukemia. Cancer Letters. 2005;225(1):105–110. doi: 10.1016/j.canlet.2004.10.034. [DOI] [PubMed] [Google Scholar]
- 143.Zhara M, Mourad H, Farouk G, Elbatch M, Ezzat S, Sami W. Molecular detection of survivin expression, antiapoptotic gene, and other prognostic markers, how they are correlated and how it could be of prognostic value in chronic myeloid leukemia patient. The Egyptian Journal of Immunology. 2007;14(2):51–62. [PubMed] [Google Scholar]
- 144.Carter BZ, Mak DH, Schober WD, et al. Regulation of survivin expression through BCR-Abl/MAPK cascade: targeting survivin overcomes imatinib resistance and increases imatinib sensitivity in imatinib-responsive CML cells. Blood. 2006;107(4):1555–1563. doi: 10.1182/blood-2004-12-4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Reis FRDS, Vasconcelos FDC, Pereira DL, Moellman-Coelho A, Silva KL, Maia RC. Survivin and P-glycoprotein are associated and highly expressed in late phase chronic myeloid leukemia. Oncology Reports. 2011;26(2):471–478. doi: 10.3892/or.2011.1296. [DOI] [PubMed] [Google Scholar]
- 146.Yamamoto H, Ngan CY, Monden M. Cancer cells survive with survivin. Cancer Science. 2008;99(9):1709–1714. doi: 10.1111/j.1349-7006.2008.00870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kwee JK, Luque DG, Ferreira ACDS, et al. Modulation of reactive oxygen species by antioxidants in chronic myeloid leukemia cells enhances imatinib sensitivity through survivin downregulation. Anti-Cancer Drugs. 2008;19(10):975–981. doi: 10.1097/CAD.0b013e3283140c6f. [DOI] [PubMed] [Google Scholar]
- 148.Nestal de Moraes G, Silva KL, Vasconcelos FDC, Maia RC. Survivin overexpression correlates with an apoptosis-resistant phenotype in chronic myeloid leukemia cells. Oncology Reports. 2011;25(6):1613–1619. doi: 10.3892/or.2011.1224. [DOI] [PubMed] [Google Scholar]
- 149.Fiskus W, Wang Y, Joshi R, et al. Cotreatment with vorinostat enhances activity of MK-0457 (VX-680) against acute and chronic myelogenous leukemia cells. Clinical Cancer Research. 2008;14(19):6106–6115. doi: 10.1158/1078-0432.CCR-08-0721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bright SA, Campiani G, Deininger MW, Lawler M, Williams DC, Zisterer DM. Sequential treatment with flavopiridol synergistically enhances pyrrolo-1,5-benzoxazepine-induced apoptosis in human chronic myeloid leukaemia cells including those resistant to imatinib treatment. Biochemical Pharmacology. 2010;80(1):31–38. doi: 10.1016/j.bcp.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 151.Nakahara T, Kita A, Yamanaka K, et al. Broad spectrum and potent antitumor activities of YM155, a novel small-molecule survivin suppressant, in a wide variety of human cancer cell lines and xenograft models. Cancer Science. 2011;102(3):614–621. doi: 10.1111/j.1349-7006.2010.01834.x. [DOI] [PubMed] [Google Scholar]
- 152.Fortugno P, Wall NR, Giodini A, et al. Survivin exists in immunochemically distinct subcellular pools and is involved in spindle microtubule function. Journal of Cell Science. 2002;115(3):575–585. doi: 10.1242/jcs.115.3.575. [DOI] [PubMed] [Google Scholar]
- 153.Gyurkocza B, Plescia J, Raskett CM, et al. Antileukemic activity of shepherdin and molecular diversity of Hsp90 inhibitors. Journal of the National Cancer Institute. 2006;98(15):1068–1077. doi: 10.1093/jnci/djj300. [DOI] [PubMed] [Google Scholar]
- 154.Wang Z, Sampath J, Fukuda S, Pelus LM. Disruption of the inhibitor of apoptosis protein survivin sensitizes BCR-ABL-positive cells to STI571-induced apoptosis. Cancer Research. 2005;65(18):8224–8232. doi: 10.1158/0008-5472.CAN-05-0303. [DOI] [PubMed] [Google Scholar]
- 155.de Mattos SF, Essafi A, Soeiro I, et al. FOXO3a and BCR-ABL regulate cyclin D2 transcription through a STAT5/BCL6-dependent mechanism. Molecular and Cellular Biology. 2004;24(22):10058–10071. doi: 10.1128/MCB.24.22.10058-10071.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Essafi A, Fernández De Mattos S, Hassen YAM, et al. Direct transcriptional regulation of Bim by FOXO3a mediates STI571-induced apoptosis in Bcr-Abl-expressing cells. Oncogene. 2005;24(14):2317–2329. doi: 10.1038/sj.onc.1208421. [DOI] [PubMed] [Google Scholar]
- 157.Komatsu N, Watanabe T, Uchida M, et al. A member of forkhead transcription factor FKHRL1 is a downstream effector of STI571-induced cell cycle arrest in BCR-ABL-expressing cells. Journal of Biological Chemistry. 2003;278(8):6411–6419. doi: 10.1074/jbc.M211562200. [DOI] [PubMed] [Google Scholar]
- 158.Kikuchi S, Nagai T, Kunitama M, Kirito K, Ozawa K, Komatsu N. Active FKHRL1 overcomes imatinib resistance in chronic myelogenous leukemia-derived cell lines via the production of tumor necrosis factor-related apoptosis-inducing ligand. Cancer Science. 2007;98(12):1949–1958. doi: 10.1111/j.1349-7006.2007.00623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Birkenkamp KU, Essafi A, van der Vos KE, et al. FOXO3a induces differentiation of Bcr-Abl-transformed cells through transcriptional down-regulation of Id1. Journal of Biological Chemistry. 2007;282(4):2211–2220. doi: 10.1074/jbc.M606669200. [DOI] [PubMed] [Google Scholar]
- 160.Jagani Z, Song K, Kutok JL, et al. Proteasome inhibition causes regression of leukemia and abrogates BCR-ABL-induced evasion of apoptosis in part through regulation of forkhead tumor suppressors. Cancer Research. 2009;69(16):6546–6555. doi: 10.1158/0008-5472.CAN-09-0605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cilloni D, Messa F, Arruga F, et al. The NF-κB pathway blockade by the IKK inhibitor PS1145 can overcome Imatinib resistance. Leukemia. 2006;20(1):61–67. doi: 10.1038/sj.leu.2403998. [DOI] [PubMed] [Google Scholar]
- 162.Duncan EA, Goetz CA, Stein SJ, et al. IκB kinase β inhibition induces cell death in Imatinib-resistant and T315I Dasatinib-resistant BCR-ABL+ cells. Molecular Cancer Therapeutics. 2008;7(2):391–397. doi: 10.1158/1535-7163.MCT-07-0305. [DOI] [PubMed] [Google Scholar]
- 163.Lounnas N, Frelin C, Gonthier N, et al. NF-κB inhibition triggers death of imatinib-sensitive and imatinib-resistant chronic myeloid leukemia cells including T315I Bcr-Abl mutants. International Journal of Cancer. 2009;125(2):308–317. doi: 10.1002/ijc.24294. [DOI] [PubMed] [Google Scholar]
- 164.Albero MP, Vaquer JM, Andreu EJ, et al. Bortezomib decreases Rb phosphorylation and induces caspase-dependent apoptosis in Imatinib-sensitive and-resistant Bcr-Abl1-expressing cells. Oncogene. 2010;29(22):3276–3286. doi: 10.1038/onc.2010.81. [DOI] [PubMed] [Google Scholar]
- 165.Hu Z, Pan XF, Wu FQ, et al. Synergy between proteasome inhibitors and imatinib mesylate in chronic myeloid leukemia. Plos ONE. 2009;4(7) doi: 10.1371/journal.pone.0006257. Article ID e6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Maia RC, Vasconcelos FC, de Sá Bacelar T, et al. LQB-118, a pterocarpanquinone structurally related to lapachol [2-hydroxy-3-(3-methyl-2-butenyl)-1,4-naphthoquinone]: a novel class of agent with high apoptotic effect in chronic myeloid leukemia cells. Investigational New Drugs. 2011;29(6):1143–1155. doi: 10.1007/s10637-010-9453-z. [DOI] [PubMed] [Google Scholar]
- 167.Gromicho M, Dinis J, Magalhães M, et al. Development of imatinib and dasatinib resistance: dynamics of expression of drug transporters ABCB1, ABCC1, ABCG2, MVP, and SLC22A1. Leukemia and Lymphoma. 2011;52(10):1980–1990. doi: 10.3109/10428194.2011.584005. [DOI] [PubMed] [Google Scholar]
- 168.Hirayama C, Watanabe H, Nakashima R, et al. Constitutive overexpression of P-glycoprotein, rather than breast cancer resistance protein or organic cation transporter 1, contributes to acquisition of imatinib-resistance in K562 cells. Pharmaceutical Research. 2008;25(4):827–835. doi: 10.1007/s11095-007-9376-3. [DOI] [PubMed] [Google Scholar]
- 169.Fuchs O. Transcription factor NF-κB inhibitors as single therapeutic agents or in combination with classical chemotherapeutic agents for the treatment of hematologic malignancies. Current Molecular Pharmacology. 2010;3(3):98–122. doi: 10.2174/1874467211003030098. [DOI] [PubMed] [Google Scholar]
- 170.Arden KC. Multiple roles of FOXO transcription factors in mammalian cells point to multiple roles in cancer. Experimental Gerontology. 2006;41(8):709–717. doi: 10.1016/j.exger.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 171.Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46(5):705–716. doi: 10.1016/0092-8674(86)90346-6. [DOI] [PubMed] [Google Scholar]
- 172.Okamoto T, Sanda T, Asamitsu K. Nk-κb signaling and carcinogenesis. Current Pharmaceutical Design. 2007;13(5):447–462. doi: 10.2174/138161207780162944. [DOI] [PubMed] [Google Scholar]
- 173.Mankan AK, Lawless MW, Gray SG, Kelleher D, McManus R. NF-κB regulation: the nuclear response. Journal of Cellular and Molecular Medicine. 2009;13(4):631–643. doi: 10.1111/j.1582-4934.2009.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Braun T, Carvalho G, Fabre C, Grosjean J, Fenaux P, Kroemer G. Targeting NF-κB in hematologic malignancies. Cell Death and Differentiation. 2006;13(5):748–758. doi: 10.1038/sj.cdd.4401874. [DOI] [PubMed] [Google Scholar]
- 175.Lu T, Stark GR. Cytokine overexpression and constitutive NFκb in cancer. Cell Cycle. 2004;3(9):1114–1117. [PubMed] [Google Scholar]
- 176.Turco MC, Romano MF, Petrella A, Bisogni R, Tassone P, Venuta S. NF-κB/Rel-mediated regulation of apoptosis in hematologic malignancies and normal hematopoietic progenitors. Leukemia. 2004;18(1):11–17. doi: 10.1038/sj.leu.2403171. [DOI] [PubMed] [Google Scholar]
- 177.Cilloni D, Martinelli G, Messa F, Baccarani M, Saglio G. Nuclear factor κB as a target for new drug development in myeloid malignancies. Haematologica. 2007;92(9):1224–1229. doi: 10.3324/haematol.11199. [DOI] [PubMed] [Google Scholar]
- 178.Lu Z, Jin Y, Chen C, Li J, Cao Q, Pan J. Pristimerin induces apoptosis in imatinib-resistant chronic myelogenous leukemia cells harboring T315I mutation by blocking NF-κB signaling and depleting Bcr-Abl. Molecular Cancer. 2010;9, article 112 doi: 10.1186/1476-4598-9-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Luqman S, Pezzuto JM. NFκB: a promising target for natural products in cancer chemoprevention. Phytotherapy Research. 2010;24(7):949–963. doi: 10.1002/ptr.3171. [DOI] [PubMed] [Google Scholar]
- 180.Crawford LJ, Windrum P, Magill L, et al. Proteasome proteolytic profile is linked to Bcr-Abl expression. Experimental Hematology. 2009;37(3):357–366. doi: 10.1016/j.exphem.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 181.Kaestner KH, Knöchel W, Martínez DE. Unified nomenclature for the winged helix/forkhead transcription factors. Genes and Development. 2000;14(2):142–146. [PubMed] [Google Scholar]
- 182.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24(50):7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 183.Tran H, Brunet A, Griffith EC, Greenberg ME. The many forks in FOXO’s road. Science’s STKE. 2003;2003(172):p. RE5. doi: 10.1126/stke.2003.172.re5. [DOI] [PubMed] [Google Scholar]
- 184.Yang JY, Hung MC. A new fork for clinical application: targeting forkhead transcription factors in cancer. Clinical Cancer Research. 2009;15(3):752–757. doi: 10.1158/1078-0432.CCR-08-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.van der Heide LP, Jacobs FM, Burbach JP, Hoekman MFM, Smidt MP. FOXO6 transcriptional activity is regulated by Thr26 and Ser184, independent of nucleo-cytoplasmic shuttling. Biochemical Journal. 2005;391(3):623–629. doi: 10.1042/BJ20050525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.van der Heide LP, Hoekman MFM, Smidt MP. The ins and outs of FOXO shuttling: mechanisms of FOXO translocation and transcriptional regulation. Biochemical Journal. 2004;380(2):297–309. doi: 10.1042/BJ20040167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Huang H, Tindall DJ. Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochimica et Biophysica Acta. 2011;1813(11):1961–1964. doi: 10.1016/j.bbamcr.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zhao Y, Wang Y, Zhu W-G. Applications of post-translational modifications of FOXO family proteins in biological functions. Journal of Molecular Cell Biology. 2011;3(5):276–282. doi: 10.1093/jmcb/mjr013. [DOI] [PubMed] [Google Scholar]
- 189.Skorski T, Bellacosa A, Nieborowska-Skorska M, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. EMBO Journal. 1997;16(20):6151–6161. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Yang JY, Hung MC. Deciphering the role of forkhead transcription factors in cancer therapy. Current Drug Targets. 2011;12(9):1284–1290. doi: 10.2174/138945011796150299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Jagani Z, Singh A, Khosravi-Far R. FOXO tumor suppressors and BCR-ABL-induced leukemia: a matter of evasion of apoptosis. Biochimica et Biophysica Acta. 2008;1785(1):63–84. doi: 10.1016/j.bbcan.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Naka K, Hoshii T, Muraguchi T, et al. TGF-β-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463(7281):676–680. doi: 10.1038/nature08734. [DOI] [PubMed] [Google Scholar]
- 193.Hurtz C, Hatzi K, Cerchietti L, et al. BCL6-mediated repression of p53 is critical for leukemia stem cell survival in chronic myeloid leukemia. Journal of Experimental Medicine. 2011;208(11):2163–2174. doi: 10.1084/jem.20110304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Naka K, Hoshii T, Hirao A. Novel therapeutic approach to eradicate tyrosine kinase inhibitor resistant chronic myeloid leukemia stem cells. Cancer Science. 2010;101(7):1577–1581. doi: 10.1111/j.1349-7006.2010.01584.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Nambu T, Hamada A, Nakashima R, et al. Association of SLCO1B3 polymorphism with intracellular accumulation of imatinib in leukocytes in patients with chronic myeloid leukemia. Biological and Pharmaceutical Bulletin. 2011;34(1):114–119. doi: 10.1248/bpb.34.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Liu F, Liu S, He S, Xie Z, Zu X, Jiang Y. Survivin transcription is associated with P-glycoprotein/MDR1 overexpression in the multidrug resistance of MCF-7 breast cancer cells. Oncology Reports. 2010;23(5):1469–1475. doi: 10.3892/or_00000786. [DOI] [PubMed] [Google Scholar]
- 197.Netto CD, da Silva AJM, Salustiano EJS, et al. New Pterocarpanquinones: synthesis, antineoplasic activity on cultured human malignant cell lines and TNF-α modulation in human PBMC cells. Bioorganic and Medicinal Chemistry. 2010;18(4):1610–1616. doi: 10.1016/j.bmc.2009.12.073. [DOI] [PubMed] [Google Scholar]
- 198.Bentires-Alj M, Barbu V, Fillet M, et al. NF-κB transcription factor induces drug resistance through MDR1 expression in cancer cells. Oncogene. 2003;22(1):90–97. doi: 10.1038/sj.onc.1206056. [DOI] [PubMed] [Google Scholar]
- 199.Shen H, Xu W, Chen Q, Wu Z, Tang H, Wang F. Tetrandrine prevents acquired drug resistance of K562 cells through inhibition of MDR1 gene transcription. Journal of Cancer Research and Clinical Oncology. 2010;136(5):659–665. doi: 10.1007/s00432-009-0704-3. [DOI] [PubMed] [Google Scholar]
- 200.Hui RCY, Gomes AR, Constantinidou D, et al. The forkhead transcription factor FOXO3a increases phosphoinositide-3 kinase/Akt activity in drug-resistant leukemic cells through induction of PIK3CA expression. Molecular and Cellular Biology. 2008;28(19):5886–5898. doi: 10.1128/MCB.01265-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hui RCY, Francis RE, Guest SK, et al. Doxorubicin activates FOXO3a to induce the expression of multidrug resistance gene ABCB1 (MDR1) in K562 leukemic cells. Molecular Cancer Therapeutics. 2008;7(3):670–678. doi: 10.1158/1535-7163.MCT-07-0397. [DOI] [PubMed] [Google Scholar]
- 202.Hilmi C, Larribere L, Deckert M, et al. Involvement of FKHRL1 in melanoma cell survival and death. Pigment Cell and Melanoma Research. 2008;21(2):139–146. doi: 10.1111/j.1755-148X.2008.00440.x. [DOI] [PubMed] [Google Scholar]
- 203.Obexer P, Hagenbuchner J, Unterkircher T, et al. Repression of BIRC5/survivin by FOXO3/FKHRL1 sensitizes human neuroblastoma cells to DNA damage-induced apoptosis. Molecular Biology of the Cell. 2009;20(7):2041–2048. doi: 10.1091/mbc.E08-07-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Guha M, Plescia J, Leav I, Li J, Languino LR, Altieri DC. Endogenous tumor suppression mediated by PTEN involves survivin gene silencing. Cancer Research. 2009;69(12):4954–4958. doi: 10.1158/0008-5472.CAN-09-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Tracey L, Pérez-Rosado A, Artiga MJ, et al. Expression of the NF-κB targets BCL2 and BIRC5/Survivin characterizes small B-cell and aggressive B-cell lymphomas, respectively. Journal of Pathology. 2005;206(2):123–134. doi: 10.1002/path.1768. [DOI] [PubMed] [Google Scholar]
- 206.Shi X, Jin Y, Cheng C, et al. Triptolide inhibits BCR-ABL transcription and induces apoptosis in STI571-resistant chronic myelogenous leukemia cells harboring T315I mutation. Clinical Cancer Research. 2009;15(5):1686–1697. doi: 10.1158/1078-0432.CCR-08-2141. [DOI] [PubMed] [Google Scholar]
- 207.Wei YL, Liang Y, Xu L, Zhao XY. The antiproliferation effect of berbamine on K562 resistant cells by inhibiting NF-κB pathway. Anatomical Record. 2009;292(7):945–950. doi: 10.1002/ar.20924. [DOI] [PubMed] [Google Scholar]
- 208.Gyrd-Hansen M, Meier P. IAPs: from caspase inhibitors to modulators of NF-κB, inflammation and cancer. Nature Reviews Cancer. 2010;10(8):561–574. doi: 10.1038/nrc2889. [DOI] [PubMed] [Google Scholar]