


Abstract
Mutations in immunoglobulin µ-binding protein 2 (Ighmbp2) cause distal spinal muscular atrophy type 1 (DSMA1), an autosomal recessive disease that is clinically characterized by distal limb weakness and respiratory distress. However, despite extensive studies, the mechanism of disease-causing mutations remains elusive. Here we report the crystal structures of the Ighmbp2 helicase core with and without bound RNA. The structures show that the overall fold of Ighmbp2 is very similar to that of Upf1, a key helicase involved in nonsense-mediated mRNA decay. Similar to Upf1, domains 1B and 1C of Ighmbp2 undergo large conformational changes in response to RNA binding, rotating 30° and 10°, respectively. The RNA binding and ATPase activities of Ighmbp2 are further enhanced by the R3H domain, located just downstream of the helicase core. Mapping of the pathogenic mutations of DSMA1 onto the helicase core structure provides a molecular basis for understanding the disease-causing consequences of Ighmbp2 mutations.
INTRODUCTION
Distal spinal muscular atrophy type 1 (DSMA1) is an autosomal recessive disorder resulting from the degeneration of α-motoneurons in the spinal cord. It manifests itself in early childhood and is characterized by early respiratory failure due to diaphragmatic paralysis (1). Although clinical and pathological features are similar to spinal muscular atrophy (SMA), DSMA1 and SMA are genetically distinct. SMA is caused by mutations of the survival of motor neurons (SMN) gene coding for SMN protein, a member of the SMN complex (2–5). DSMA1 is caused by genetic mutations in the Ighmbp2 gene, which encodes immunoglobulin µ-binding protein 2 (Ighmbp2) (6,7). The precise cellular function of Ighmbp2 remains elusive, although it has been implicated in transcription and pre-mRNA processing (8,9). More recently, Ighmbp2 has been reported to associate with ribosomes and tRNAs, suggesting that it is functionally linked to translation (10,11).
Ighmbp2 is a multidomain protein composed of a DNA/RNA helicase domain and an R3H domain and a zinc-finger domain (Figure 1A). Based on sequence homology, Ighmbp2 has been classified as a member of the Upf1-like group within the helicase superfamily 1 (SF1) (12). The other two members of the Upf1-like family of SF1 helicases are Upf1, a key protein involved in nonsense-mediated mRNA decay (13–15), and Senataxin, implicated in transcription regulation (16–18). Biochemical characterization has shown that Ighmbp2 is an ATP-dependent 5′→3′ DNA/RNA helicase (10). Most of the pathogenic mutations in DSMA1 patients occurred in the helicase domain, suggesting that this domain plays a major role in the DSMA1 disease-causing mechanism. Consistent with this notion, the pathogenic mutants of Ighmbp2 exhibited loss of either ATPase or helicase activity or both activities (10), arguing that the biochemical defects of Ighmbp2 may be the primary causes underlying DSMA1.
Figure 1.

Structural overview of hIghmbp2hd and hIghmbp2hd–RNA. (A) Schematic representation of the domain arrangement in Upf1 and Ighmbp2. The helicase core region contains two RecA-like domains: domain 1A (pink) and domain 2A (wheat) and additional regulatory domains: domain 1B (green), domain 1C (cyan) and the ‘stalk’ (orange). Upf1 contains another regulatory domain, the CH domain (gray) located upstream of the helicase region while Ighmbp2 features two more another regulatory domains: a R3H domain (gray) and an AN-1 type zinc-finger domain (gray) downstream of the helicase region. Missense mutations and canonical motifs in Ighmbp2 are labeled. (B) Crystal structures of hIghmbp2hd. (C) Crystal structure of hIghmbp2hd–RNA. The coloring schemes for domains are as in (A). The bound phosphate ion and ssRNA are shown as red sphere and yellow cartoon tube, respectively.
SF1 helicases can be divided into two classes, SF1A and SF1B, on the basis of the direction of translocation, with SF1B further branched to Pif1-like (Pif1, RecD2) and Upf1-like subfamily (Upf1, Ighmbp2, Senataxin) (19). The Upf1-like family differs from the Pif1-like family as it can unwind both DNA and RNA duplexes (20). A great deal is known about the mechanisms of SF1A enzymes (from PcrA, Rep and UvrD) (21–26); however, only two SF1B helicase structures (Upf1 and RecD2) are available. The structures of RecD2 have been determined in three states: the free form, in complex with single-stranded DNA (ssDNA) and in the presence and absence of an ATP analog (27,28). The mechanistic basis for 5′→3′ translocation by the Pif1-like family has been revealed from the structural characterization of the RecD2 enzyme (28). For the understanding of the mechanism by which Upf1-like helicase functions, several crystal structures of Upf1 in different functional states have been determined. These include Upf1 in complex with either phosphate or ADP or the non-hydrolyzable ATP analog, AMPPNP (29), as well as in transition-state with bound single-stranded RNA (ssRNA) (30), but no structure of the RNA-bound Upf1 in the absence of a nucleotide has been reported.
Here we have determined the crystal structure of the helicase core domain of human Ighmbp2 with and without bound ssRNA in the absence of nucleotide. These structures allow a molecular interpretation of a number of pathogenic mutations that cause DSMA1. Moreover, structural comparison combined with biochemical characterization reveal the critical role of the R3H domain in modulation of enzymatic and RNA-binding activities of Ighmbp2.
MATERIALS AND METHODS
Protein expression and purification
The cDNA encoding the helicase domain of human Ighmbp2 (hIghmbp2hd, residues 1–652) was cloned into the pGEX6p-1 vector (GE Healthcare) with a GST tag at the N-terminus. The hIghmbp2hd protein was expressed in Escherichia coli BL21 Rosetta (DE3) pLysS strain (Novagen). Cells were grown in Luria–Bertani medium supplemented with 100 μg/ml ampicillin at 37°C until the optical density at 600 nm (OD600) reached ∼0.6. Protein expression was induced by the addition of 0.4 mM isopropyl-ß-d-1-thiogalactopyranoside and further cell growth was performed at 18°C for 16 h. Cells were harvested by centrifugation at 4000 rpm and resuspended in lysis buffer (25 mM HEPES, 500 mM NaCl, 3 mM DTT, 3 mM MgCl2, pH 7.3) supplemented with 1 mg/ml lysozyme from chicken egg-white (Sigma). Cells were disrupted by sonication on ice and cell debris was cleared by centrifugation at 18 000 rpm. The clarified supernatant was loaded into a column self-packed with Glutathione Sepharose 4B beads (GE Healthcare), pre-equilibrated with the lysis buffer. The column was washed with the lysis buffer and the protein was eluted with elution buffer (25 mM HEPES, 500 mM NaCl, 3 mM DTT, 3 mM MgCl2, 1% w/v reduced glutathione, pH 7.3). The GST tag is then cleaved by incubating with PreScission protease overnight. The protein sample was then further purified by a second glutathione sepharose column and gel filtration using HiPrep Superdex-75 column on an AKTA Xpress system (GE Healthcare) in lysis buffer. hIghmbp2hd was pooled and concentrated to 12 mg/ml. We failed to obtain SeMet-substituted protein as it became insoluble after PreScission protease cleavage (data not shown).
mIghmbp2hd-R3H, a construct containing both helicase and R3H domains (residues 1–786) of mouse Ighmbp2, was cloned and purified in the same way except a Superdex-200 gel filtration column was used in gel filtration.
Crystallization
In the initial crystallization screening, 5 mM potassium phosphate was added to the protein. Plate-like crystals were obtained at 15°C in a reservoir solution containing 0.2 M sodium malonate pH 7.0 and 20% (w/v) PEG 3350. Mercury-derivative crystals were obtained by soaking native crystals in the reservoir solution supplemented with 0.1 mM EMTS (ethyl mercury thiosalicylate) for 6 h. Iodide-derivative crystals were obtained by quick soaking the crystals in the reservoir solution containing 500 mM potassium iodide for 5 s.
To crystallize the hIghmbp2hd–RNA complex, a single-stranded A10 RNA was mixed with hIghmbp2hd at a molar ratio of 2:1. Crystals were obtained by hanging drop method with equal volumes of hIghmbp2hd–RNA solution and reservoir solution containing 0.1 M imidazole pH 8.0, 1.0 M sodium citrate at 15°C. All crystals were cryoprotected with 25% ethylene glycol before being flash-cooled in liquid nitrogen.
Data collection and structure determination
Native data sets of the hIghmbp2hd and hIghmbp2hd–RNA complexes were collected at 100K at beamlines ID14-4 and ID23-1 at the European Synchrotron Facility (ESRF, Grenoble, France). The best diffraction volumes of the crystals were defined using diffraction cartography (31). For the hIghmbp2hd–RNA complex crystals the c cell edge was aligned parallel to the spindle using a mini-kappa goniometer (32). This allowed better separation of Bragg spots due to the length of this axis. Mercury and iodide derivative data sets were collected at 100K at beamline 13B1 at the National Synchrotron Radiation Research Centre (NSRRC, Taiwan, R.O.C).
The structure of hIghmbp2hd was determined by the multiple isomorphous replacement with anomalous scattering (MIRAS) method in the space group C2. The data sets were integrated using MOSFLM (33). Scaling of intensities was carried out using SCALA from the CCP4i package (34). Heavy atom search was carried out by Autosol in the PHENIX software suite (35). Three mercury and 17 iodide sites, corresponding to one molecule of hIghmbp2hd in the asymmetric unit, were found. Automatic partial model building was carried out by AutoBuild in the PHENIX software suite (35). The model was built manually using COOT (36). Crystallographic refinement was performed using the PHENIX software suite (35) to a final Rfree of 25.23%. TLS refinement was introduced and a free phosphate group was included at the final stages of refinement.
The crystals of hIghmbp2hd–RNA belong to space group P3121, with two molecules in the asymmetric unit. The data for hIghmbp2hd–RNA were integrated with XDS (37,38), then scaled and reduced using SCALA in CCP4i. The structure was determined by molecular replacement with PHASER (39), using the hIghmbp2hd structure as the search model. A good match for domains 1A and 2A was found. Domain 1B was manually built with the aid of 2Fo–Fc and Fo–Fc maps using COOT. Model refinement was carried out with the PHENIX software suite to a final Rfree of 29.28%. RNA molecules were included in the final stages of refinement. Difference Fourier maps clearly showed the electron density for nine and eight ribonucleotides in molecules A and B, respectively (Supplementary Figure S1). As there is no substantial difference between the two molecules in the ASU (r.m.s.d. of 0.85 Å for all equivalent Cα atoms of hIghmbp2hd), all subsequent analyses are based on molecule A as it is more complete. The stereochemical geometry of the structures was validated using PROCHECK (40). The Ramachandran plot showed that one outlier (residue 580) is in hIghmbp2hd, and three outliers of molecule A (residues 82, 270 and 580) and one outlier of molecule B (residue 580) are in hIghmbp2hd–RNA. All these residues have poorly-defined electron density except for residue 270 which has relatively well defined electron density and is located at a loop region. The statistics for data collection and refinement and the quality of the final models are summarized in Table 1.
Table 1.
Data collection and refinement statistics
| Data collection | hIghmbp2hd | hIghmbp2hd–Hg | hIghmbp2hd–Iodide | hIghmbp2hd–RNA |
|---|---|---|---|---|
| Derivative | – | Mercury | Iodide | – |
| Wavelength (Å) | 0.9795 | 0.99315 | 1.54 | 0.9760 |
| Resolution limit (Å) | 2.5 | 3.0 | 3.0 | 2.85 |
| Space group | C2 | C2 | C2 | P3121 |
| Cell parameters | ||||
| a/b/c (Å) | 116.57/76.72 88.54 | 115.83/76.65/87.29 | 117.67/76.67/87.97 | 87.29/87.29/372.69 |
| α/β/γ (°) | 90/107.32/90 | 90/107.40/90 | 90/107.72/90 | 90/90/120 |
| Unique reflections (N) | 25 840 | 14 640 | 15006 | 39 419 |
| Total reflections (N) | 114 847 | 95 240 | 95 220 | 222 197 |
| I/σ | 16.0 (1.4) | 13.0 (2.2) | 8.4 (1.8) | 21.6 (1.5) |
| Mean (I/σ) | 18.9 (2.3) | 24.4 (3.6) | 26.0 (3.5) | 12.5 (1.9) |
| Completeness (%) | 91.8 (99.7) | 99.9 (99.7) | 99.8 (99.7) | 90.2 (99.5) |
| Rmergea | 0.04 (0.512) | 0.044 (0.341) | 0.065 (0.418) | 0.026 (0.551) |
| Number of derivative sites | – | 3 | 17 | – |
| Refinement statistics | ||||
| Data range (Å) | 63.16–2.50 | 19.97–2.85 | ||
| Used reflections (N) | 23 987 | 38 694 | ||
| Protein residues | 625 | 1222 | ||
| Protein atoms | 4853 | 18 460 | ||
| Solvent molecules | 99 | 140 | ||
| Phosphate molecules | 1 | 2 | ||
| RNA bases | – | 17 | ||
| Rworkb(%) | 19.02 | 20.58 | ||
| Rfreec (%) | 25.23 | 29.28 | ||
| R.m.s.d. | ||||
| Bond length (Å) | 0.008 | 0.009 | ||
| Bond angles (°) | 1.181 | 1.338 | ||
| Ramchandran plot (% residues) | ||||
| Allowed | 98.5 | 97.2 | ||
| Generously allowed | 1.2 | 2.3 | ||
| Disallowedd | 0.2 | 0.5 |
Values in parentheses indicate the specific values in the highest resolution shell.
aRmerge = ∑|Ij − <I>|/∑Ij, where Ij is the intensity of an individual reflection and <I> is the average intensity of that reflection.
bRwork = ∑||Fo| − |Fc||/∑|Fc|, where Fo denotes the observed structure factor amplitude and Fc denotes the structure factor amplitude calculated from the model.
cRfree is as for Rwork but calculated with 5.0% of randomly chosen reflections omitted from the refinement.
dDisallowed: Val580 in hIghmbp2hd and Ser82 (A), Arg270 (A), Val580 (A) and Val580 (B) in hIghmbp2hd–RNA.
Fluorescence anisotropy
Fluorescence anisotropy measurements were performed with single-stranded U15 RNA labeled at the 5′-end with 6-carboxy-fluorescein (6-FAM) (Metabion International AG). 100 µl reactions were carried out in a 96-well plate at 25°C and measured using Safire II fluorescent plate reader (Tecan). 100 nM of the labeled oligonucleotide was incubated with increasing protein concentration at ambient temperature for 30 min prior to measurements, in a buffer containing 20 mM HEPES, 2 mM MgCl2, 2 mM DTT, 100 mM NaCl, 10% glycerol and pH 7.4. The excitation and emission wavelengths were 470 and 535 nm, respectively. All anisotropy measurements were performed in triplicate. Each titration point was measured using three reads with an integration time of 40 µs. Dissociation constants (Kd) values were determined by fitting the experimental data to a binding equation describing a single-site binding model (shown below) using Prism software 4.0 (GraphPad).
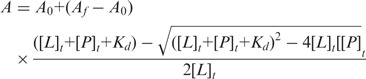 |
where A is the anisotropy measured, A0 is the initial anisotropy in the absence of protein, Af is the final anisotropy, [P] is the protein concentration, [L] is the labeled RNA concentration
In vitro ATPase assay
The ATPase assay was performed in 50 μl reaction volume containing 40 pmol of purified Ighmbp2 proteins (hIghmbp2hd or mIghmbp2hd-R3H) in 50 mM MES (pH 6.0), 50 mM KAc, 2.5 mM Mg(Ac)2, 5 μg of 15-mer poly(A), 30 μM cold ATP and 0.3 μCi [γ32P]ATP. Upon incubation at 37°C for 1 h, 5 μl of 500 mM EDTA was added to terminate the reaction. In order to identify the ATPase hydrolysis products, a standard reaction with the same substrates and conditions was performed using 0.1 U of Calf Intestine Phosphatase (CIP) at 37°C for 6 min. Aliquots (10 μl) of the reaction products were analyzed by thin-layer chromatography on a polyethyleneimine-cellulose plate (Sigma) and developing it in 0.3 M K2HPO4 (pH 7.6).
RESULTS
Overall structure
The crystal structure of the helicase domain of human Ighmbp2 (residues 1–652, designated as hIghmbp2hd) was solved by the MIRAS method using mercury and iodide derivatives to a resolution of 2.5 Å. Residues 73–75, 120–126, 310–315, 503–507 and 649–652 were not visible in the electron density map and assumed to be disordered in the structure. The helicase core of Ighmbp2 has essentially the same fold as that of the Upf1 protein (29,30), which contains four domains: two RecA-like domains (domains 1A and 2A) with two subdomains 1B and 1C inserted into domain 1A (Figure 1B). Domains 1A (residues 159–270 and 347–440) and 2A (residues 441–648) share the α–β fold with a central seven-stranded parallel β-sheet surrounded by α helices, resembling the RecA-like fold seen in all SF1 and SF2 helicases (20,41). Domain 1B (residues 34–141) consisting of a short α-helix and a β-barrel, is connected to domain 1A by two α-helices (residues 3–33 and 142–158) also referred to as the ‘stalk’ (30). Domain 1C (residues 271–346) is inserted between the Walker A and Walker B motifs and forms four helices situated above domain 1A (Figure 1B). The canonical motifs I, II, III, V and VI known from other SF1 helicases to be involved in ATP binding [reviewed in (20)] and motifs III and Va known to coordinate between NTP and nucleic acid-binding sites (19), are present in the equivalent positions in hIghmbp2hd (Figure 1).
To gain insights into how the helicase interacts with nucleic acid, we solved the crystal structure of hIghmbp2hd in complex with a ssRNA (thereafter referred to as hIghmbp2hd–RNA). Residues 32–36, 94–97, 121–124, 308–320, 502–506 and 649–652 were disordered in the structure. Although a 10-nt ssRNA (A10) was used in crystallization, we only observed nine adenine bases in the hIghmbp2hd–RNA complex. For simplicity, we have numbered nucleotides A1 to A9 in a 5′→3′ direction. The ssRNA binds in an extended conformation with the 5′-end at domain 2A and the 3′-end pointing towards the channel between domains 1B and 1C (Figure 1C). Both the structures of hIghmbp2hd and hIghmbp2hd–RNA contain a phosphate situated in the nucleotide-binding cleft between domains 1A and 2A.
RNA recognition
The ssRNA binds to a composite RNA-binding channel formed by domains 1A, 2A, 1B and 1C (Figure 1C). The backbone phosphates of ssRNA mainly contact domains 1A and 2A with the adenine bases pointing either to the solvent region or towards domain 1B. Domains 1B and 1C directly interact with the 3′-end of RNA, similar to that observed in the Upf1-RNA-bound structure with the presence of the CH domain (30). Protein–RNA interactions were mediated mainly by a combination of hydrogen bonds and electrostatic interactions as depicted in Figure 2. Briefly, A1 is well ordered by stacking its adenine base with the side chain of His411. The adenine bases A1 to A3 are in parallel orientation to each other at an average distance of ∼3.7 Å, a common feature of base stacking in ssRNA (42). As the adenine base of A3 is perpendicular to that of A4, it protrudes outwards causing a bend in the RNA backbone between A3 and A4. The side chain of residue Gln507 forms hydrogen bonding with the O2′ atom of the ribose sugar of base A3 while the O2′ atom of the ribose sugar of A4 interacts with the side chains of Asp565 and Arg596. The hydroxyl group of residue Thr407 forms a hydrogen bond with the N3 atom of the nitrogenous base of A5 which in turn forms base stacking interactions with A6 to A8. Motif Ia interacts with the phosphate backbone of A7 via the side chain of Asn245. Residues Thr351 and Thr353 from motif Ic interact with the phosphate backbone of A7 and A8, respectively. A second bend of the RNA backbone is observed whereby A9, located ∼10 Å from A8, flips away from the base stacking configuration. Arg270 makes two important contacts with the RNA at this position. First, its NH2 group interacts with the N7 atom of the adenine base of A8. Second, its NH1 group forms electrostatic interactions with the O1P atom of the phosphodiester backbone of A9. Since only residues Gln507, Asp565 and Arg596 interact with the ribose 2′-OH groups of A3 and A4 (Figure 2) through polar interactions, the ability of Ighmbp2 to unwind both DNA and RNA duplexes (10) could be explained by the lack of specific recognition of 2′-OH group of the sugar ribose ring that renders Ighmbp2 unspecific to DNA or RNA.
Figure 2.

Protein–RNA interactions. (A) Stereo view showing the ssRNA-binding site in hIghmbp2hd–RNA. The bound ssRNA and the residues involved protein–RNA interactions are shown in ball-stick models. The color scheme is as in Figure 1. (B) Schematic representation of the contacts between the hIghmbp2hd protein and RNA.
RNA binding induced conformational changes
Superposition of domains 1A and 2A of hIghmbp2hd and hIghmbp2hd–RNA showed little change in the positions of Cα atoms (r.m.s.d. of 0.87 Å, Figure 3A), indicating that the relative orientations of domains 1A and 2A are largely conserved in the presence and absence of RNA. In contrast to this observation, large conformational changes are observed in domains 1B and 1C. When RNA is bound, domain 1B rotates ∼30° away from domain 1 C such that it bends towards domain 2A to form a channel (diameter of ∼15 Å and length ∼50 Å) that could accommodate a single stranded but not a duplex nucleic acid (Figure 3A). This marked conformational change widens the channel between domains 1B and 1C where the 3′-end of ssRNA is situated. The opening of the channel between 1B and 1C also requires the outwards tilting of domain 1C by 10°. In addition to these inter-domain movements, a prominent intra-domain conformational change occurs, which involves the reorganization of a loop (residues 264–273 in domain 1A, located just before domain 1C). This results in reorientation of the side chain of Arg270 from the nucleotide-binding pocket to the RNA-binding pocket (between bases A8 and A9) upon ssRNA binding (Figure 3B). These conformational changes demonstrate the conformational flexibility of Ighmbp2, similar to that observed in Upf1 (30).
Figure 3.

Conformational changes of hIghmbp2hd upon RNA binding. (A) Stereo view showing the superposition of hIghmbp2hd–RNA (pink) with hIghmbp2hd (green). Phosphate ions are shown as sticks and the bound ssRNA is shown in yellow cartoon. The arrows indicate the movement of domain 1B and 1C in the RNA-bound state compared to the RNA-free state. (B) Conformational change of a loop region (residues 264–273) and the reorientation of residue Arg270 in response to ssRNA binding. The position of the phosphate ions indicates the location of the nucleotide-binding site.
Structural comparison of Ighmbp2 with Upf1
Superposition of the structure of hIghmbp2hd (Figure 4A) with that of hUpf1ΔCH-PO4− (Figure 4B) showed that these two structures are very similar. The best fitting lies in domains 1A and 2A and the largest change in orientation occurs in domain 1B. Domain 1B of hIghmbp2hd interacts with domain 1C but has no contacts with domain 2A. In contrast, domain 1B in hUpf1ΔCH-PO4− has no contacts with domain 1C but interacts with domain 2A.
Figure 4.

Structural comparison of Ighmbp2 with Upf1. The ribbon diagrams are drawn with domains 1A (pink) and 2A (wheat) in the same orientation. The bound phosphate ion and nucleotide are shown in gray sticks. ssRNA is show in yellow sticks. The coloring scheme for domains are as in Figure 1. (A) hIghmbp2hd. (B) hUpf1ΔCH-PO4− (PDB code: 2GK7). (C) hIghmbp2hd–RNA. (D) hUpf1ΔCH-RNA-ADP:AlF4− (PDB code 2XZO). (E) Yeast Upf1-RNA-ADP:AlF4− (PDB code: 2XZL).
Comparison of hIghmbp2hd–RNA (Figure 4C) with the RNA-bound Upf1 structures (30) showed that the conformation of domain 1B in Ighmbp2 is drastically different from those in Upf1 in response to RNA binding. In Ighmbp2, upon RNA binding, domain 1B rotates ∼30° towards domain 2A, therefore forming a composite RNA-binding channel together with domains 1A, 2A and 1C (Figure 4C versus A). In contrast, upon RNA binding, domain 1B in the structure of human Upf1ΔCH-RNA-ADP:AlF4− rotates ∼35° and moves away from the helicase core, thereby relieving its steric hindrance imposed on domain 2A but making no contacts with RNA (30) (Figures 4D versus B). In addition to this conformational change, domain 1B in the structure of yeast Upf1-RNA-ADP:AlF4− shifts by ∼20° toward domain 1C, thereby narrowing the channel between them (Figure 4E versus D). In contrast to the large conformational change of domain 1B, domain 1C in hIghmbp2hd–RNA tilts only 10° away from domain 1B with respect to its position in the RNA-free structure, whereas no conformational change of domain 1C has been observed in the Upf1 structures with and without RNA (Figures 3A and 4).
Another notable difference lies in the RNA-binding sites. In the structure of hUpf1ΔCH-RNA-ADP:AlF4− (Figure 4D), domains 1A and 2A form an extended surface to bind RNA while domain 1B changes from an inhibitory conformation seen in hUpf1ΔCH-PO4− (Figure 4B) to a conformation that neither hinders nor promotes RNA binding. Consequently, Upf1 lacking the CH domain (human Upf1ΔCH-RNA-ADP:AlF4−) could only bind to a ssRNA of 6 nt and domains 1B and 1C have no contribution to RNA binding. In the presence of CH domain (yeast Upf1-RNA-ADP:AlF4−), domain 1B is pushed towards domain 1C and the RNA-binding channel of Upf1 is extended to accommodate eight bases with domains 1B and 1C directly interacting with RNA (30) (Figure 4E). In this conformation, Upf1 binds more tightly to the 3′-end of the RNA, ‘clamping’ it and reduces its helicase activity (30). Ighmbp2 lacks the CH domain but has a R3H domain and a zinc-finger domain located downstream of the helicase core domain. Without these two accessory domains, a 9-nt ssRNA is bound to Ighmbp2hd in the absence of a nucleotide (Figure 4C) because all four domains contribute to the formation of the composite RNA-binding channel. One simple explanation for the ability of Ighmbp2hd to bind longer RNA without the contribution of the accessory domain is that domain 1B in Ighmbp2hd is not in an inhibitory conformation (no contact with domain 2A but interacts with domain 1C), and a simple 30° rotation coupled with the outward movement of domain 1C is sufficient to allow RNA binding. Given that helicases make use of accessory domains for their specific and regulatory functions and that the R3H domain has been proposed to bind nucleic acids (9), it is envisaged that the R3H domain might have a role in regulating Ighmbp2’s RNA-binding and catalytic activities.
R3H domain has a regulatory role on Ighmbp2
The R3H domain is a conserved sequence motif characterized by an invariant arginine residue and a highly conserved histidine residue that are separated by three residues (43). The solution structure of the R3H domain from human Ighmbp2 (PDB:1MSZ) showed that the R3H domain consists of three-stranded anti-parallel β-sheet and two α-helices and has a positively charged surface (44). To examine the functional role of R3H domain in Ighmbp2 protein, we expressed and purified the protein fragment composed of the helicase core and the R3H domain from mouse mIghmbp2 (mIghmbp2hd-R3H) since the human protein containing the corresponding regions degraded severely during purification (data not shown). To investigate the contribution of R3H domain to RNA binding, we carried out fluorescence anisotropy experiments to determine the binding affinities of the proteins (hIghmbp2hd and mIghmbp2hd-R3H) to RNA. In these experiments, the tightest RNA affinity was observed with the mIghmbp2hd-R3H in the absence of any nucleotides with a Kd value of 107±29 nM (Figure 5A). In comparison, hIghmbp2hd interacted ∼10-fold less tightly with a Kd value of 1077±130 nM (Figure 5A). These strikingly different Kd values strongly suggest that R3H domains have a role in promoting nucleic acid binding, consistent with the results from Fukita and coworkers (9). Similar to Upf1, both hIghmbp2hd and mIghmbp2hd-R3H showed higher RNA-binding affinity in the absence of a nucleotide (Figure 5A) (30,45,46). Although the presence of nucleotides did lower RNA-binding affinity, it was not significant (Figure 5A). The difference in RNA binding could be due to conformational changes associated with nucleotide binding. Hence, the variation in the RNA-binding affinities of Ighmbp2 is mainly contributed by the R3H domain.
Figure 5.

RNA-binding and ATPase activities of Ighmbp2. (A) Quantitative measurements of RNA-binding affinities of hIghmbp2hd and mIghmbp2hd-R3H in solution by fluorescence anisotropy. Dissociation constants (KD) values were determined by fitting the experimental data to a binding equation describing a single-site binding model. The KD values and their corresponding errors are the mean and standard deviation of three independent experiments. (B) ATPase activities of hIghmbp2hd and hIghmbp2hd-R3H in the absence or presence of ssRNA. The position of phosphate was determined by treating the substrate with CIP.
We hypothesize that tighter RNA binding will lead to enhanced ATPase activity as Ighmbp2 is a nucleic acid-dependent ATPase (10). ATPase assays in the absence and presence of nucleic acids as shown in Figure 5B (lanes 3–6) indicated that ATP hydrolysis is stimulated by the presence of ssRNA. Importantly, mIghmbp2hd-R3H exhibits 4-fold stronger ATPase activity compared to hIghmbp2hd in the presence of ssRNA (Figure 5B, lanes 4 and 6), suggesting R3H domain stimulates ATPase activity through enhancing RNA binding. Altogether, our results reveal the synergy between the R3H domain and the helicase domain as demonstrated by enhanced RNA binding which in turn potentiates its ATPase activity.
Structural and functional placement of DSMA1 missense mutations on Ighmbp2
Of all the disease-causing missense-mutations in IGHMBP2, which are located within or adjacent to the hecliase domain of Ighmbp2 (7,47–52), only nine representative mutants have been biochemically characterized (10). We mapped these nine missense mutants onto the hIghmbp2hd–RNA structure (Figure 6A) and classified them according to their locations: residing in or close to the nucleotide-binding pocket (class I), in RNA-binding channel (class II) and in neither ligand-binding sites (class III).
Figure 6.

Structural roles of residues associated with disease-causing missense mutations in SMARD1. (A) Disease-causing missense mutations in DSMA1 are mapped in the structure of hIghmbp2hd–RNA with the Cα atoms of the mutated residues shown as red spheres. AMPPNP (in stick model) is modeled by superposition of the structure of hIghmbp2hd–RNA with that of human Upf1ΔCH-AMPPNP (PBD code: 2GJK). (B) Class I mutations residing in or close to the nucleotide-binding pocket. (C) Class II mutations located in the RNA-binding channel. (D) and (E) Class III mutations located far away from either the nucleotide-binding pocket or the RNA-binding channel.
Class I mutants comprise Q96R, T221A, H445P and R603H. Gln196 is the invariant residue in the Q motif, a motif previously proposed to be unique to the DEAD box family of helicases to control ATP binding and hydrolysis (53,54). When AMPPNP from the Upf1 structure is superimposed onto Ighmbp2 nucleotide-binding pocket, residue Gln196 is shown to make a Van der Waals (VDW) contact with the adenine base of AMPPNP with the shortest distance of 3.6 Å (Figure 6B). The mutation of Gln196 to an arginine would result in a longer side chain protruding into the nucleotide-binding pocket. This would prevent ATP binding, thereby leading to loss of the ATPase activity.
Residue Thr221 is part of the Walker A sequence in motif I and could form hydrogen bonds with the α- and β-phosphate groups of modeled AMPPNP (Figure 6B) and also coordinate the catalytic magnesium ion. Motif I is well characterized to be important for nucleotide binding and hydrolysis (55,56). Hence mutation of Thr221 to alanine will inevitably impact the ATPase activity.
Residue His445 is well conserved in mouse, human and hamster Ighmbp2. It is located at one end of motif IIIa which is the loop linking domains 1A and 2A (Figure 6A and B). Since the movement of motor domains 1A and 2A relative to each other in response to ATP binding/hydrolysis is critical for the SF1 helicases [reviewed in (20)], this loop might act as a hinge to regulate the conformational change between domains 1A and 2A. Mutation of His445 to a proline would reduce the flexibility of this loop, thus restraining the movement of motor domains 1A and 2A and affect the ATPase activity.
Arg603 lies in motif VI (Figure 5), which has been shown to function in ATP hydrolysis and RNA binding (55,57). From the hIghmbp2hd–RNA structure, Arg603 forms electrostatic interactions with the γ-phosphate of AMPPNP. The equivalent residue in PcrA has been proposed as an ‘arginine finger’ to stabilize the transition state (21,55). Moreover, it was reported that mutating the corresponding conserved arginine residue in eIF4A would abolish its RNA binding and ATPase and helicase activities (57). In hIghmbp2, mutation of Arg603 to histidine would render this residue unable to interact with the nucleotide, hence affecting the ATPase activity of the enzyme.
The Class II mutants consist of D565N and N583I. Asp565 lies in motif V, which is a flexible loop in domain 2A (Figure 6C). Although the function of motif V is less well studied, there is evidence showing that that it is involved in RNA binding (56) and the translocation activity of hepatitis C virus NS3 helicase (58). The hIghmbp2hd–RNA structure shows that the side-chain of Asp565 contacts the ribose O2′ of A4 via VDW interactions (Figures 2 and 6C) and forms a hydrogen bond with Arg596, which in turn contacts the ribose group of A4. Residues Asp565 and Arg596 correspond to Asp827 and Arg858 in human Upf1, which show similar interactions with the RNA backbone (30). Substitution of Asp565 by Asn would not extensively change the interactions of this residue with the RNA backbone, therefore not affecting the RNA binding to Ighmbp2. Asn583 is located near the 5′-end of the RNA-binding channel but Asn583 does not directly interact with the bound RNA (Figure 6C). We observed that Asn583 is surrounded by several basic residues Arg581, Lys585 and Arg584. Mutation of Asn583 to an isoleucine would place a hydrophobic residue into a positively-charged groove formed by these three residues, thus affecting the local charge distribution and reducing RNA binding.
Class III mutants include C241R, E382K and T493I. Cys241 is located at the central β-sheet of domain 1A and surrounded by several hydrophobic residues (Leu239, Val348, Phe369, Val372, Ile374 and Pro388) (Figure 6D). Therefore, mutation of Cys241 to a positively-charged arginine would most likely disrupt the central β-sheet, hence destabilizing domain 1A.
Residue Glu382 lies on the helix just before motif II. It forms a hydrogen bond with Trp386 in the same helix and also forms a salt-bridge interaction with Arg425 in a neighboring helix, all in domain 1A (Figure 6E). The mutation of Glu382 to a lysine (which has a longer and positively charged side chain) would result in charge-charge repulsion with Arg425 and also could disrupt the hydrogen bond formed between Glu382 and Trp386. Thus it is likely that this mutation would reduce the stability of domain 1A.
Mutant T493I is in a loop region, away from the nucleotide-binding site and is not located in proximity to any helicase motifs (Figure 6A). In addition, the side chain of Thr493 does not make significant contacts with neighboring residues. Based on our current structures, it is difficult to predict how the T493I mutation would affect the conformation and functionality of Ighmbp2.
The mutational effects predicted based on structural analysis are consistent with the previous biochemical data (10). All class I mutants (Q196R, T221A, H445P and R603H) and one of the class II mutants (N583I) and two of the class III mutants (C241R, E382K) exhibited defective ATPase and helicase activities (10). Interestingly, class II mutant D565N retains the ability to bind nucleic acid and has an ATPase activity similar to the wild type but does lose its helicase activity. As unwinding of a nucleic acid duplex requires translocation, the defective helicase activity of this mutant is probably due to its inability to translocate along the RNA substrate. The only mutant with minimal effect on Ighmbp2’s catalytic activities is T493I in class III. It has been reported that the T493I mutant has severely reduced steady-state protein level in DSMA1 patient cell lines (50). Therefore, the pathogenicity of this mutant is most likely due to an effect on Ighmbp2 protein levels, rather than its enzymatic activity. Taken together, our structural data rationalized the biochemical defects observed for the disease-causing mutations of DSMA1 and strengthen the notion that the loss of the helicase activity of Ighmbp2 is the primary pathological basis of DSMA1.
DISCUSSION
Previous studies have shown that the Ighmbp2 protein belongs to the SF1B helicase family based on its sequence alignment and the ability to unwind both RNA and DNA duplex in the 5′→3′ direction (10). Indeed, our crystal structure of hIghmbp2hd reveals that the helicase domain of Ighmbp2 is structurally similar to that of Upf1 and both have similar nucleotide and nucleic acid-binding sites. Comparison of the RNA-free and the RNA-bound states of hIghmbp2hd demonstrates that ssRNA binding induces conformational changes in domains 1B and 1C. The importance of these domains was also demonstrated by the Upf1 helicase, whereby conformation changes are observed in domain 1B upon nucleic acid binding (30) and deletion of domain 1C abolished nucleic acid binding (29). These observations indicate that members of the Upf1-like family have a similar domain fold and their mechanistic actions could be very similar.
Many helicases have N-terminal or C-terminal accessory domains. These domains usually have defined folds and can influence the specific function of the helicase. For instance, the N-terminal cysteine–histidine-rich domain (CH-domain, residues 115–275) of Upf1 have been shown to play a regulatory role on the helicase activity of Upf1 by modulating RNA-binding kinetics and the length of the RNA-binding channel through the interaction with Upf2 (30,59). In comparison, Ighmbp2 has an accessory C-terminal R3H domain and an AN1-type zinc-finger motif. Previous biochemical results have argued that the R3H domain of Ighmbp2 is crucial to RNA binding (9). It is clear from our data that although the helicase domain alone was able to bind RNA and has basal ATPase activity, these activities are augmented by the R3H domain. Given that the CH domain of Upf1 extends the RNA-binding channel and enables it to bind longer RNA, the R3H might act in analogy to the CH domain of Upf1 to extend the RNA-binding channel in Ighmbp2, therefore increasing RNA binding. Consistent with this notion, it has been shown that the R3H domain alone is not sufficient for high-affinity RNA binding and that the presence of both the helicase and R3H domains would enhance RNA binding (9). This would suggest that the helicase and R3H domains work in a cooperative manner to enhance RNA binding.
The structure of the Ighmbp2 helicase provides an opportunity to address how genetic defects in patients with DSMA1 impact the molecular function of the protein. Available information indicates specific mutations do not correlate with the severity of the clinical features (47,60). Our structural information, together with the previous biochemical data (10), indicate that seven out of the nine representative missense mutations affect ATPase activity, either through disrupting ATP binding/hydrolysis or through reducing the structural stability of the helicase motors. The two exceptional cases are the mutations T493I and D565N, which do not affect the ATPase activity of the enzyme. Instead, the mutation T493I causes neuronal degeneration through reducing the intracellular Ighmbp2 protein levels while the D565N mutation probably affects the ability of Ighmbp2 translocation along the bound RNA, therefore leading to its enzymatic defect. Our structure-based functional annotation of disease mutations in Ighmbp2 can facilitate further experimental studies to explore the pathology of SMARD1.
There has been an increasing interest in the study of the senataxin protein encoded by the human SETX gene, known to be involved in the neurodegenerative diseases ALS4 and AOA2 (16,17,61,62). Senataxin has a C-terminal helicase domain homologous to the helicase domains of Ighmbp2 and Upf1. Mutations in the helicase domain of the SETX gene show defects in transcription, RNA processing and DNA damage repair (17,62,63). Recently, Senataxin has been elucitated to function as an helicase that resolves RNA/DNA hybrids so that Xrn2-dependent termination can occur (18). Although senataxin and Ighmbp2 share a homologous helicase domain, the mutations in the helicase domain of senataxin and the disease phenotypes generated by these mutations are different from those of Ighmbp2, suggesting that these two proteins have different and specific cellular functions, presumably determined by their accessory domains outside the helicase domains. Clearly, more studies are required to reveal the exact molecular function of Ighmbp2. These studies, in conjunction with those on senataxin, could help to understand the patho-mechanisms of DSMA1, ALS4 and AOA2.
ACCESSION NUMBERS
The coordinates and structure-factor amplitudes of hIghmbp2hd and hIghmbp2hd–RNA have been deposited in the Protein Data Bank with accession codes 4B3F and 4B3G, respectively.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–2.
FUNDING
The Biomedical Research Council of A*STAR (Agency for Science, Technology and Research, Singapore). Funding for open access charge: Agency for Science, Technology and Research (A*STAR), Singapore.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank the beamline scientists at ID14-4 and ID23-1 (ESRF, France) and 13B1 (NSRRC, Taiwan R.O.C.) for assistance and access to synchrotron radiation facilities.
REFERENCES
- 1.Mellins RB. Pulmonary physiotherapy in the pediatric age group. Am. Rev. Respir. Dis. 1974;110:137–142. doi: 10.1164/arrd.1974.110.6P2.137. [DOI] [PubMed] [Google Scholar]
- 2.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 3.Gubitz AK, Feng W, Dreyfuss G. The SMN complex. Exp. Cell Res. 2004;296:51–56. doi: 10.1016/j.yexcr.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 4.Paushkin S, Gubitz AK, Massenet S, Dreyfuss G. The SMN complex, an assemblyosome of ribonucleoproteins. Curr. Opin. Cell Biol. 2002;14:305–312. doi: 10.1016/s0955-0674(02)00332-0. [DOI] [PubMed] [Google Scholar]
- 5.Meister G, Eggert C, Fischer U. SMN-mediated assembly of RNPs: a complex story. Trends Cell. Biol. 2002;12:472–478. doi: 10.1016/s0962-8924(02)02371-1. [DOI] [PubMed] [Google Scholar]
- 6.Kaindl AM, Guenther UP, Rudnik-Schoneborn S, Varon R, Zerres K, Schuelke M, Hubner C, von Au K. Spinal muscular atrophy with respiratory distress type 1 (SMARD1) J. Child Neurol. 2008;23:199–204. doi: 10.1177/0883073807310989. [DOI] [PubMed] [Google Scholar]
- 7.Grohmann K, Schuelke M, Diers A, Hoffmann K, Lucke B, Adams C, Bertini E, Leonhardt-Horti H, Muntoni F, Ouvrier R, et al. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat. Genet. 2001;29:75–77. doi: 10.1038/ng703. [DOI] [PubMed] [Google Scholar]
- 8.Molnar GM, Crozat A, Kraeft SK, Dou QP, Chen LB, Pardee AB. Association of the mammalian helicase MAH with the pre-mRNA splicing complex. Proc. Natl Acad. Sci. USA. 1997;94:7831–7836. doi: 10.1073/pnas.94.15.7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukita Y, Mizuta TR, Shirozu M, Ozawa K, Shimizu A, Honjo T. The human S mu bp-2, a DNA-binding protein specific to the single-stranded guanine-rich sequence related to the immunoglobulin mu chain switch region. J. Biol. Chem. 1993;268:17463–17470. [PubMed] [Google Scholar]
- 10.Guenther UP, Handoko L, Laggerbauer B, Jablonka S, Chari A, Alzheimer M, Ohmer J, Plottner O, Gehring N, Sickmann A, et al. IGHMBP2 is a ribosome-associated helicase inactive in the neuromuscular disorder distal SMA type 1 (DSMA1) Hum. Mol. Genet. 2009;18:1288–1300. doi: 10.1093/hmg/ddp028. [DOI] [PubMed] [Google Scholar]
- 11.de Planell-Saguer M, Schroeder DG, Rodicio MC, Cox GA, Mourelatos Z. Biochemical and genetic evidence for a role of IGHMBP2 in the translational machinery. Hum. Mol. Genet. 2009;18:2115–2126. doi: 10.1093/hmg/ddp134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jankowsky E. RNA helicases at work: binding and rearranging. Trends Biochem. Sci. 2011;36:19–29. doi: 10.1016/j.tibs.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhattacharya A, Czaplinski K, Trifillis P, He F, Jacobson A, Peltz SW. Characterization of the biochemical properties of the human Upf1 gene product that is involved in nonsense-mediated mRNA decay. RNA. 2000;6:1226–1235. doi: 10.1017/s1355838200000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Czaplinski K, Weng Y, Hagan KW, Peltz SW. Purification and characterization of the Upf1 protein: a factor involved in translation and mRNA degradation. RNA. 1995;1:610–623. [PMC free article] [PubMed] [Google Scholar]
- 15.Sun X, Perlick HA, Dietz HC, Maquat LE. A mutated human homologue to yeast Upf1 protein has a dominant-negative effect on the decay of nonsense-containing mRNAs in mammalian cells. Proc. Natl Acad. Sci. USA. 1998;95:10009–10014. doi: 10.1073/pnas.95.17.10009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen YZ, Hashemi SH, Anderson SK, Huang Y, Moreira MC, Lynch DR, Glass IA, Chance PF, Bennett CL. Senataxin, the yeast Sen1p orthologue: characterization of a unique protein in which recessive mutations cause ataxia and dominant mutations cause motor neuron disease. Neurobiol. Dis. 2006;23:97–108. doi: 10.1016/j.nbd.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 17.Suraweera A, Lim Y, Woods R, Birrell GW, Nasim T, Becherel OJ, Lavin MF. Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum. Mol. Genet. 2009;18: 3384–3396. doi: 10.1093/hmg/ddp278. [DOI] [PubMed] [Google Scholar]
- 18.Skourti-Stathaki K, Proudfoot NJ, Gromak N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell. 2011;42:794–805. doi: 10.1016/j.molcel.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fairman-Williams ME, Guenther UP, Jankowsky E. SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol. 2010;20:313–324. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 21.Velankar SS, Soultanas P, Dillingham MS, Subramanya HS, Wigley DB. Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell. 1999;97:75–84. doi: 10.1016/s0092-8674(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 22.Korolev S, Hsieh J, Gauss GH, Lohman TM, Waksman G. Major domain swiveling revealed by the crystal structures of complexes of E. coli Rep helicase bound to single-stranded DNA and ADP. Cell. 1997;90:635–647. doi: 10.1016/s0092-8674(00)80525-5. [DOI] [PubMed] [Google Scholar]
- 23.Subramanya HS, Bird LE, Brannigan JA, Wigley DB. Crystal structure of a DExx box DNA helicase. Nature. 1996;384:379–383. doi: 10.1038/384379a0. [DOI] [PubMed] [Google Scholar]
- 24.Tomko EJ, Fischer CJ, Niedziela-Majka A, Lohman TM. A nonuniform stepping mechanism for E. coli UvrD monomer translocation along single-stranded DNA. Mol. Cell. 2007;26:335–347. doi: 10.1016/j.molcel.2007.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dillingham MS, Soultanas P, Wiley P, Webb MR, Wigley DB. Defining the roles of individual residues in the single-stranded DNA binding site of PcrA helicase. Proc. Natl Acad. Sci. USA. 2001;98:8381–8387. doi: 10.1073/pnas.131009598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soultanas P, Dillingham MS, Wiley P, Webb MR, Wigley DB. Uncoupling DNA translocation and helicase activity in PcrA: direct evidence for an active mechanism. EMBO J. 2000;19:3799–3810. doi: 10.1093/emboj/19.14.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saikrishnan K, Griffiths SP, Cook N, Court R, Wigley DB. DNA binding to RecD: role of the 1B domain in SF1B helicase activity. EMBO J. 2008;27:2222–2229. doi: 10.1038/emboj.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saikrishnan K, Powell B, Cook NJ, Webb MR, Wigley DB. Mechanistic basis of 5′-3′ translocation in SF1B helicases. Cell. 2009;137:849–859. doi: 10.1016/j.cell.2009.03.036. [DOI] [PubMed] [Google Scholar]
- 29.Cheng Z, Muhlrad D, Lim MK, Parker R, Song H. Structural and functional insights into the human Upf1 helicase core. EMBO J. 2007;26:253–264. doi: 10.1038/sj.emboj.7601464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chakrabarti S, Jayachandran U, Bonneau F, Fiorini F, Basquin C, Domcke S, Le Hir H, Conti E. Molecular mechanisms for the RNA-dependent ATPase activity of Upf1 and its regulation by Upf2. Mol. Cell. 2011;41:693–703. doi: 10.1016/j.molcel.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 31.Bowler MW, Guijarro M, Petitdemange S, Baker I, Svensson O, Burghammer M, Mueller-Dieckmann C, Gordon EJ, Flot D, McSweeney SM, et al. Diffraction cartography: applying microbeams to macromolecular crystallography sample evaluation and data collection. Acta Crystallogr. D Biol. Crystallogr. 2010;66:855–864. doi: 10.1107/S0907444910019591. [DOI] [PubMed] [Google Scholar]
- 32.Brockhauser S, White KI, McCarthy AA, Ravelli RB. Translation calibration of inverse-kappa goniometers in macromolecular crystallography. Acta Crystallogr. A. 2011;67:219–228. doi: 10.1107/S0108767311004831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leslie AG. The integration of macromolecular diffraction data. Acta Crystallogr. D Biol. Crystallogr. 2006;62:48–57. doi: 10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- 34.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 35.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Kabsch W. Automatic indexing of rotation diffraction patterns. J. Appl. Crystallogr. 1988;21:67–72. [Google Scholar]
- 38.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 1993;26:795–800. [Google Scholar]
- 39.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283. [Google Scholar]
- 41.Singleton MR, Wigley DB. Modularity and specialization in superfamily 1 and 2 helicases. J. Bacteriol. 2002;184:1819–1826. doi: 10.1128/JB.184.7.1819-1826.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Šponer J, Hobza P. Molecular interactions of nucleic acid bases. A review of quantum-chemical studies. Collect. Czech. Chem. Commun. 2003;68:2231–2282. [Google Scholar]
- 43.Grishin NV. The R3H motif: a domain that binds single-stranded nucleic acids. Trends Biochem. Sci. 1998;23:329–330. doi: 10.1016/s0968-0004(98)01258-4. [DOI] [PubMed] [Google Scholar]
- 44.Liepinsh E, Leonchiks A, Sharipo A, Guignard L, Otting G. Solution structure of the R3H domain from human Smubp-2. J. Mol. Biol. 2003;326:217–223. doi: 10.1016/s0022-2836(02)01381-5. [DOI] [PubMed] [Google Scholar]
- 45.Weng Y, Czaplinski K, Peltz SW. ATP is a cofactor of the Upf1 protein that modulates its translation termination and RNA binding activities. RNA. 1998;4:205–214. [PMC free article] [PubMed] [Google Scholar]
- 46.Chamieh H, Ballut L, Bonneau F, Le Hir H. NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat. Struct. Mol. Biol. 2008;15:85–93. doi: 10.1038/nsmb1330. [DOI] [PubMed] [Google Scholar]
- 47.Grohmann K, Varon R, Stolz P, Schuelke M, Janetzki C, Bertini E, Bushby K, Muntoni F, Ouvrier R, Van Maldergem L, et al. Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1) Ann. Neurol. 2003;54:719–724. doi: 10.1002/ana.10755. [DOI] [PubMed] [Google Scholar]
- 48.Guenther UP, Varon R, Schlicke M, Dutrannoy V, Volk A, Hubner C, von Au K, Schuelke M. Clinical and mutational profile in spinal muscular atrophy with respiratory distress (SMARD): defining novel phenotypes through hierarchical cluster analysis. Hum. Mutat. 2007;28:808–815. doi: 10.1002/humu.20525. [DOI] [PubMed] [Google Scholar]
- 49.Maystadt I, Zarhrate M, Landrieu P, Boespflug-Tanguy O, Sukno S, Collignon P, Melki J, Verellen-Dumoulin C, Munnich A, Viollet L. Allelic heterogeneity of SMARD1 at the IGHMBP2 locus. Hum. Mutat. 2004;23:525–526. doi: 10.1002/humu.9241. [DOI] [PubMed] [Google Scholar]
- 50.Guenther UP, Handoko L, Varon R, Stephani U, Tsao CY, Mendell JR, Lutzkendorf S, Hubner C, von Au K, Jablonka S, et al. Clinical variability in distal spinal muscular atrophy type 1 (DSMA1): determination of steady-state IGHMBP2 protein levels in five patients with infantile and juvenile disease. J. Mol. Med. 2008;87:31–41. doi: 10.1007/s00109-008-0402-7. [DOI] [PubMed] [Google Scholar]
- 51.Pierson TM, Tart G, Adams D, Toro C, Golas G, Tifft C, Gahl W. Infantile-onset spinal muscular atrophy with respiratory distress-1 diagnosed in a 20-year-old man. Neuromuscul. Disord. 2011;21:353–355. doi: 10.1016/j.nmd.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Giannini A, Pinto AM, Rossetti G, Prandi E, Tiziano D, Brahe C, Nardocci N. Respiratory failure in infants due to spinal muscular atrophy with respiratory distress type 1. Intensive Care Med. 2006;32:1851–1855. doi: 10.1007/s00134-006-0346-8. [DOI] [PubMed] [Google Scholar]
- 53.Tanner NK, Cordin O, Banroques J, Doere M, Linder P. The Q motif: a newly identified motif in DEAD box helicases may regulate ATP binding and hydrolysis. Mol. Cell. 2003;11:127–138. doi: 10.1016/s1097-2765(03)00006-6. [DOI] [PubMed] [Google Scholar]
- 54.Cordin O, Tanner NK, Doere M, Linder P, Banroques J. The newly discovered Q motif of DEAD-box RNA helicases regulates RNA-binding and helicase activity. EMBO J. 2004;23:2478–2487. doi: 10.1038/sj.emboj.7600272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Caruthers JM, McKay DB. Helicase structure and mechanism. Curr. Opin. Struct. Biol. 2002;12:123–133. doi: 10.1016/s0959-440x(02)00298-1. [DOI] [PubMed] [Google Scholar]
- 56.Cordin O, Banroques J, Tanner NK, Linder P. The DEAD-box protein family of RNA helicases. Gene. 2006;367:17–37. doi: 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 57.Pause A, Methot N, Sonenberg N. The HRIGRXXR region of the DEAD box RNA helicase eukaryotic translation initiation factor 4A is required for RNA binding and ATP hydrolysis. Mol. Cell. Biol. 1993;13:6789–6798. doi: 10.1128/mcb.13.11.6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gu M, Rice CM. Three conformational snapshots of the hepatitis C virus NS3 helicase reveal a ratchet translocation mechanism. Proc. Natl Acad. Sci. USA. 2010;107:521–528. doi: 10.1073/pnas.0913380107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clerici M, Mourao A, Gutsche I, Gehring NH, Hentze MW, Kulozik A, Kadlec J, Sattler M, Cusack S. Unusual bipartite mode of interaction between the nonsense-mediated decay factors, UPF1 and UPF2. EMBO J. 2009;28:2293–2306. doi: 10.1038/emboj.2009.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Joseph S, Robb SA, Mohammed S, Lillis S, Simonds A, Manzur AY, Walter S, Wraige E. Interfamilial phenotypic heterogeneity in SMARD1. Neuromuscul. Disord. 2009;19:193–195. doi: 10.1016/j.nmd.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 61.Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, Dierick I, Abel A, Kennerson ML, Rabin BA, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) Am. J. Hum. Genet. 2004;74:1128–1135. doi: 10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moreira MC, Klur S, Watanabe M, Nemeth AH, Le Ber I, Moniz JC, Tranchant C, Aubourg P, Tazir M, Schols L, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat. Genet. 2004;36:225–227. doi: 10.1038/ng1303. [DOI] [PubMed] [Google Scholar]
- 63.Suraweera A, Becherel OJ, Chen P, Rundle N, Woods R, Nakamura J, Gatei M, Criscuolo C, Filla A, Chessa L, et al. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J. Cell Biol. 2007;177:969–979. doi: 10.1083/jcb.200701042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.