

Abstract
Ts65Dn is a mouse model of Down syndrome; a syndrome that results from Chromosome (Chr) 21 trisomy and is associated with congenital defects, cognitive impairment, and ultimately Alzheimer’s Disease. Ts65Dn mice have segmental trisomy for distal mouse Chr 16, a region sharing conserved synteny with human Chr 21. As a result, this strain harbors three copies of over half of the human Chr 21 orthologs. The trisomic segment of Chr 16 is present as a translocation chromosome (Mmu 1716), with breakpoints that have not been defined previously. To molecularly characterize the Chr 16 and Chr 17 breakpoints on the translocation chromosome in Ts65Dn mice, we used a selective enrichment and high-throughput, paired end sequencing approach. Analysis of paired end reads flanking the Chr 16, Chr 17 junction on Mmu1716 and de-novo assembly of the reads directly spanning the junction provided the precise locations of the Chr 16 and Chr 17 breakpoints at 84,351,351 bp and 9,426,822 bp, respectively. These data provide the basis for low cost, highly efficient genotyping of Ts65Dn mice. More importantly, these data provide, for the first time, complete characterization of gene dosage in Ts65Dn mice.
Introduction
Down syndrome (DS) is a congenital condition caused by the presence of three copies (trisomy) of all or part of human Chromosome (Chr) 21. Trisomy 21 (Ts21) remains the most common aneuploidy among live born infants and DS is the most frequent genetically defined cause of mental retardation in humans, with over 350,000 affected individuals in the US population (Down Syndrome Research and Treatment Foundation). Mouse models for DS are a valuable resource for research as they provide a genetically defined, experimentally malleable system that recapitulates many features of human DS. The development of mouse models was predicated on regions of conserved synteny between human Chr 21 and mouse Mmu10, 16 and 17, with the highest proportion of orthologs on Mmu16 (Chr 16). Because trisomy 16 mice do not survive past birth and contain a large number of orthologs from other human chromosomes, they are not a good model for DS. Therefore, a segmental trisomy 16 strain containing the majority of the syntenic region, Ts(1716)65Dn (abbreviated Ts65Dn), was developed in Dr. Muriel Davisson’s laboratory and this model has since become the best characterized and most widely used mouse model for Down syndrome research (Costa et al. 2010; Davisson et al. 1993; Reeves et al. 1995).
Ts65Dn mice carry one product of a reciprocal translocation between Chrs 16 and 17. The translocation was induced through irradiation of male mice of the strain DBA/2J and isolated by selective breeding for translocation products involving Chr 16 (Davisson et al. 1993). Ts65Dn mice are aneuploid and carry the translocation chromosome, Mmu1716 (1716), as a freely segregating, supernumerary chromosome. In male mice, the extra chromosome is associated with abnormal meiotic pairing and infertility (Davisson et al. 2007; Reinholdt et al. 2009), therefore the strain is maintained by breeding through the female germ line, and while there is evidence that the translocation chromosome undergoes recombination during female meiosis, it is likely that recombination frequencies are low due to lack of a true homolog. Indeed, DBA/2J SNPs have been retained on the translocation chromosome, despite over 200 outcrosses to an unrelated F1 strain, B6C3F1, indicating that recombination frequencies are likely suppressed during female meiosis. The discovery of these SNPs has allowed for the recent development of a SNP based genotyping assay (Lorenzi et al. 2010), allowing for a simpler and cheaper pre-screening genotyping methodology to use with quantitative PCR or DNA FISH methods that are typically employed for genotyping these mice (Liu et al. 2003). However this SNP based assay has been shown to have a low percentage of false negative and false positive genotypes due, in part, to its dependence on a segregating allele.
The 1716 chromosome carries ~13.4 Mb of distal Chr 16, from Mrpl39 to Zfp295 fused with the centromere of Chr 17 and up to 10 Mb of Chr 17 sequence just proximal to the centromere (Figure 1). While significant progress towards mapping of the 1716 translocation breakpoint has been made, the sequence of the breakpoint has yet to be defined. Previous work has shown that the Chr 16 breakpoint is between two adjacent genes, Ncam2 (81,624,530 bp; NCBI37/mm9) and Mrpl39 (84,718,526; NCBI37/mm9) and that the Chr 17 breakpoint is between D17Mit19 (4,804,658 bp; NCBI37/mm9) and D17Mit58 (10,489,635 bp; NCBI37/mm9) (Akeson et al. 2001; Kahlem et al. 2004). The Chr 17 breakpoint is distal to 5,988,977 by DNA FISH (Li et al. 2007), indicating that the chromosome 17 region on 1716 is at least ~6.0 Mb and could be as high as 10.4 Mb. Consequently, while the breakpoint region on Chr 16 has been mapped to a relatively short interval (~3 Mb), the Chr 17 breakpoint is less well defined and by extension, the dosage of proximal Chr 17 genes remains unknown. Therefore, the extent to which Chr 17 alleles might contribute to the phenotypes observed in Ts65Dn mice cannot be determined.
Figure 1.
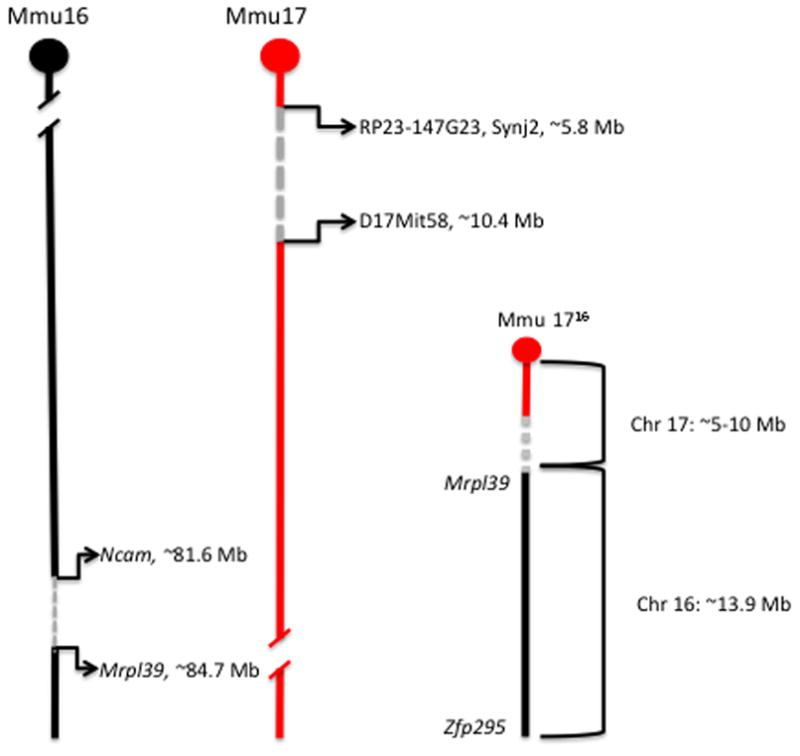
Mapping of the Chr 16 and Chr 17 breakpoints in Ts65Dn. The 1716 chromosome (Mmu 1716) carries ~13.4 Mb of distal Chr 16 (black), from Mrpl39 to Zfp295 fused with the centromere of Chr 17 and up to 10 Mb of Chr 17 sequence just proximal to the centromere (red). The Chr 16 breakpoint is between Ncam2 (81,624,530 bp; NCBI37/mm9) and Mrpl39 (84,718,526; NCBI37/mm9) and the Chr 17 breakpoint is between RP23-147G23, Synj2 (5,988,977 bp; NCBI37/mm9) and D17Mit58 (10,489,635bp; NCBI37/mm9) (Akeson et al. 2001; Kahlem et al. 2004; Li et al. 2007).
To molecularly define the translocation breakpoint in Ts65Dn mice, a targeted re-sequencing approach was used. An analysis of paired end reads spanning the breakpoint and single reads, mapping directly to the breakpoint revealed the precise locations of the Chr 16 and 17 breakpoints, as well the specific sequence of the breakpoint region on the 1716 translocation product. These data provide, for the first time, a complete picture of gene dosage in this important disease model.
Materials and Methods
DNA capture and High Throughput Sequencing
B6EiC3Sn a/A-Ts(1716)65Dn (Ts65Dn, The Jackson Laboratory stock #1924) DNA was obtained from the DNA Resource at The Jackson Laboratory. The HTS libraries were made following Hodges et al., 2009 (Hodges et al. 2009) except that size selection was done using Pippin Prep from Sage Science (http://www.sagescience.com/) with a target size of 400 bp. Twenty ug of the adapter-modified DNA library fragments were hybridized to each of two, custom designed 1M Agilent SureSelect DNA Capture Arrays representing the target sequence: Chr 16: 83,500,000–85,500,000 and Chr 17: 3,500,000–11,250,000. The two arrays contained 1,871,618 tiled probes (3 bp offset) representing 6,168,324 bases of the 9,750,000 Mb target sequence or 63.7% of the target sequence. Regions not represented by probes were repeat sequences. Capture and recovery of enriched genomic DNA was performed as described by the manufacturer. After amplification of the DNA from the arrays, ALINE SizeSelector-1 beads (ALINE Bioscience, Woburn, MA) were used to purify the desired size fragments away from residual primers. The libraries were then quantified using a Thermo Scientific NanoDrop 2000 spectrophotometer, and quality-tested on an Agilent Technologies 2100 Bioanalyzer. Samples were diluted for loading on an Illumina cBot Cluster Generation System which carries out automated solid phase PCR (bridge PCR) on the 8-lane flow cell to create the clustered template DNA fragments for sequencing via the Illumina GA IIx. Nearly 30 million, 76 bp paired end reads were generated.
High Throughput Sequencing Data Analysis
The sequencing data were aligned to the C57BL/6J reference genome (NCBI m37/mm9) using BWA (Burrows Wheeler Alignment) and the alignments were visualized and analyzed using the Integrated Genomics Viewer (Broad Institute, http://www.broadinstitute.org/igv/). Sequencher 4.9 (Gene Codes Corporation) was used for de novo assembly of reads spanning the translocation breakpoint and custom scripts were used to calculate coverage statistics.
Breakpoint PCR
Primers spanning the translocation breakpoint were designed using Primer3 software (Broad Institute). The breakpoint primer sequences were Chr17fwd_5′-GTGGCAAGAGACTCAAATTCAAC-3′ and Chr16rev_5′-TGGCTTATTATTATCAGGGCATTT-3′. These primers amplify a ~275 bp product from junction site of the 1716 translocation product. Positive control primers were IMR8545_5′-AAAGTCGCTCTGAGTTGTTAT-3′ and IMR8546_5′-GGAGCGGGAGAAATGGATATG-3′, which amplify a 600 bp product from the Rosa locus. In a 50 μl total reaction, all 4 primers were used at a final concentration of 0.4 μM each. PCR cycling conditions were 94°C – 2min, 94°C – 45sec, 55°C – 45sec, 72°C –1min for 40 cycles, followed by a final 7 min. extension at 72°C – 7min. PCR products were separated on a 1.5% agarose gel. For the purposes of validation, 209 samples that were previously genotyped by either by quantitative PCR (Liu et al. 2003) or DNA FISH as being either positive, negative or inconclusive for the segmental trisomy were genotyped by breakpoint PCR. Among this set of samples were samples from Ts65Dn mice and 2 derivative strains, B6EiC3Sn.BLiA-Ts(1716)65Dn/DnJ (The Jackson Laboratory, stock #5252) and B6EiC3Sn-Rb(12.Ts171665Dn)2Cje/CjeDn (The Jackson Laboratory, stock #4850). Additionally, 113 samples derived from Ts65Dn mice and genotyped using the SNP based genotyping prescreen (Lorenzi et al 2010) and FISH confirmation (Moore et al. 1999).
Fluorescent in situ hybridization
Bacterial artificial chromosomes (BACs) containing genomic segments from Chr 16 and Chr 17 that were closely located near the breakpoints (within 100 kb) but did not span the breakpoints were selected: RP23-325G3 (Mmu16 coordinates: 84,076,476–84,273,785), RP23-18M23 (Mmu16; 84,404,243–84,614,960), RP23-67A9 (Mmu17; 9,097,314–9,356,527) and RP23-178A3 (Mmu17: 9,503,820–9,692,678)(Figure 4A). Clones were labeled by incorporating Aqua or Orange-dUTP (Abbott) by nick translation (Rigby et al. 1977). Combinations of a Mmu16 and Mmu17 probe were denatured and hybridized overnight to slides containing metaphase spreads prepared from peripheral blood samples (Akeson and Davisson 2001) in hybridization buffer containing 2X saline-sodium citrate (SSC) buffer, 10% dextran sulfate, 50% formamide and 1 mg/ml mouse Cot-1 DNA at 37C. Slides were washed in 1.5X SSC/50% formamide at 25C and then 1.5X SSC, each for 2 minutes at 25C. Slides were visualized using an Olympus BX51 epifluorescent microscope equipped with the appropriate Vysis filter cubes.
Figure 4.
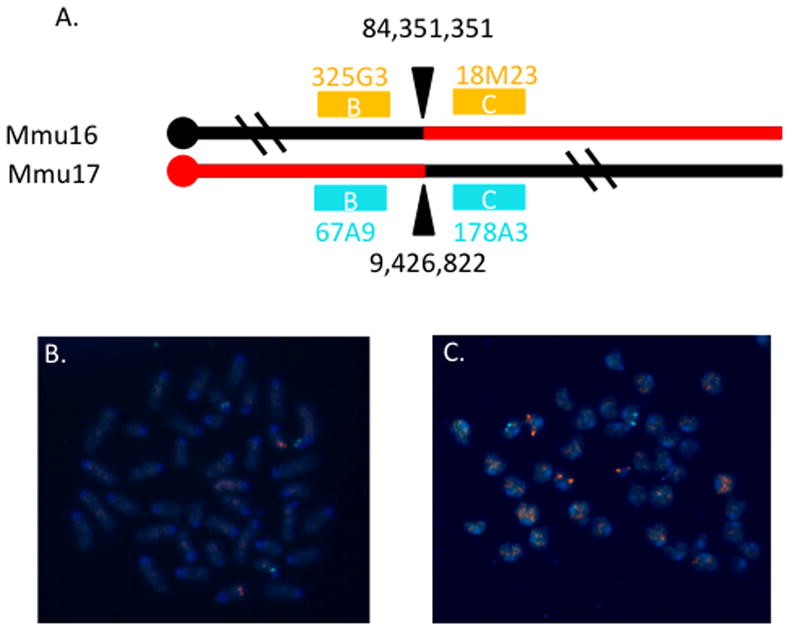
FISH confirmation of the breakpoint. A. Cartoon of breakpoint and BAC probes used for FISH. The breakpoints are designated by arrowheads and genome coordinates for each chromosome. The Ts65Dn marker chromosome contains proximal Chr 17 and distal Chr 16 sequences as indicated in red. Bacterial artificial chromosomes from the RP23 library that contain genomic sequences flanking the breakpoints are depicted as colored boxes with the colors corresponding to the fluorophores used to label these clones. The letters within the boxes indicate which pairs of probes were used to generate the images below. The IDs of the clones are also noted. B. Hybridization with probes RP23-325G3 and -67A9 showed that marker chromosome contains sequences from the proximal Chr 17 but not Chr 16 probe. C. Hybridization with probes RP23-18M23 and -178A3 revealed that the marker chromosome includes sequences from the distal Chr 16 but not Chr 17 probe.
Results
High throughput sequencing approaches, including targeted sequencing of samples enriched for specific regions of interest and whole genome sequencing, have recently proven useful for identification of chromosomal breakpoints at base pair resolution in genomes harboring chromosomal rearrangements (Talkowski et al. 2011). To determine the optimum approach (whole genome or targeted sequencing), the final coverage requirements were estimated. Based on previous sequence capture data, a minimum read depth requirement of 5 reads was established to ensure sufficient sampling of each haplotype in the region of interest. Therefore, since Ts65Dn mice are trisomic for the regions of interest, a minimum read depth of 15 reads was required for sufficient sampling. Data from our previous targeted re-sequencing efforts indicated that 150X oversampling was sufficient to ensure 15 reads minimum coverage within the targeted region (D’Ascenzo et al. 2009). Taking into account that some reads spanning the 1716 junction might fail to successfully align to the reference genome, an oversampling goal of 200X was established. Considering a target region of ~13 Mb (based on the combined mapping data for the Chr 16 and Chr 17 breakpoints), we estimated that 2.6 Gb (13 Mb × 200) of sequence would be needed to sufficiently cover the targeted regions at a minimum depth of 15 reads. To maximize oversampling of the target region, two 1M feature arrays with overlapping DNA probes (3 bp offset, 60 bp probes) were used to enrich for Chr 16 sequence between Ncam2 and Mrpl39, as well as Chr 17 sequence between D17Mit19 and D17Mit 58.
A total of 37 million, 76 bp, paired end reads were generated on an Illumina GIIAx platform and nearly half of the reads mapped to the desired regions on Chr 16 and Chr 17, resulting in ~2.8 Gb of sequence data representing the target region and an average read depth of 94 reads. Further analysis revealed that 99.9% of the targeted bases were covered by at least one read and 90.5% were covered by at least 15 reads. These coverage statistics indicated that sufficient coverage of the target region was obtained with 37 milllion, 76 bp, paired end reads.
The sequence data were mapped to the C57BL/6 reference genome (NCBI m37/mm9) and analysis of the resulting alignments revealed fourteen paired-end reads flanking the 1716 junction, with one mate mapping properly to Chr 16 and the other mapping to Chr 17 (Figure 2). Furthermore, among the reads flagged as ‘not properly mapped’ were a total of 55 individual 76 bp reads spanning the junction, consisting of both Chr 16 and Chr 17 sequence. De-novo assembly of these reads revealed the precise location of the Chr 16 and Chr 17 breakpoints to be 84,351,351 bp and 9,426,822 bp, respectively. In depth analysis of the aligned reads the 1716 junction revealed that approximately 12.4% (17/137) of properly mapped reads were from the 1716 chromosome. This was significantly lower than expected assuming equal representation (33%) of each allele in the sequencing data; however, since many of the reads from the translocation chromosome failed to properly map to reference, it was not surprising to find bias in the mapped data.
Figure 2.
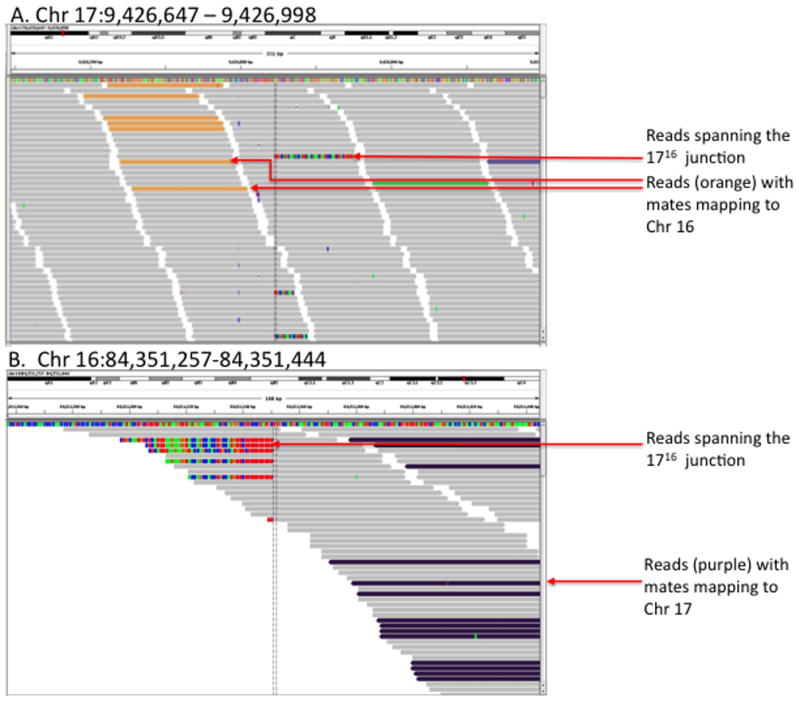
Visualization of reads aligning to the Chr 17 (A) and Chr 16 (B) breakpoint regions using the Integrated Genomics Viewer (IGV). Paired end reads mapping to Chr 17 with mates mapping to Chr 16 are shown in orange (A). Likewise, paired end reads mapping to Chr 16 with mates mapping to Chr 17 are shown in purple (B). Reads that map directly to the 1716 junction map either to Chr 17 or Chr 16, depending on the % of mismatches in the alignment (mismatches within the alignments are indicated in red, blue, green and orange).
To confirm that this breakpoint sequence is present in animals that carry this marker chromosome, more than 300 genomic DNA samples from animals previously genotyped by standard genotyping methods (qPCR and FISH) were subjected to PCR amplification of the junction fragment (Fig 3). In all trisomic animals, the breakpoint sequences were detected. Additionally, fluorescent in situ hybridization using BACs containing genomic DNA from flanking regions within 100 kb of the breakpoints also confirmed the location of the breakpoint (Fig 4).
Figure 3.
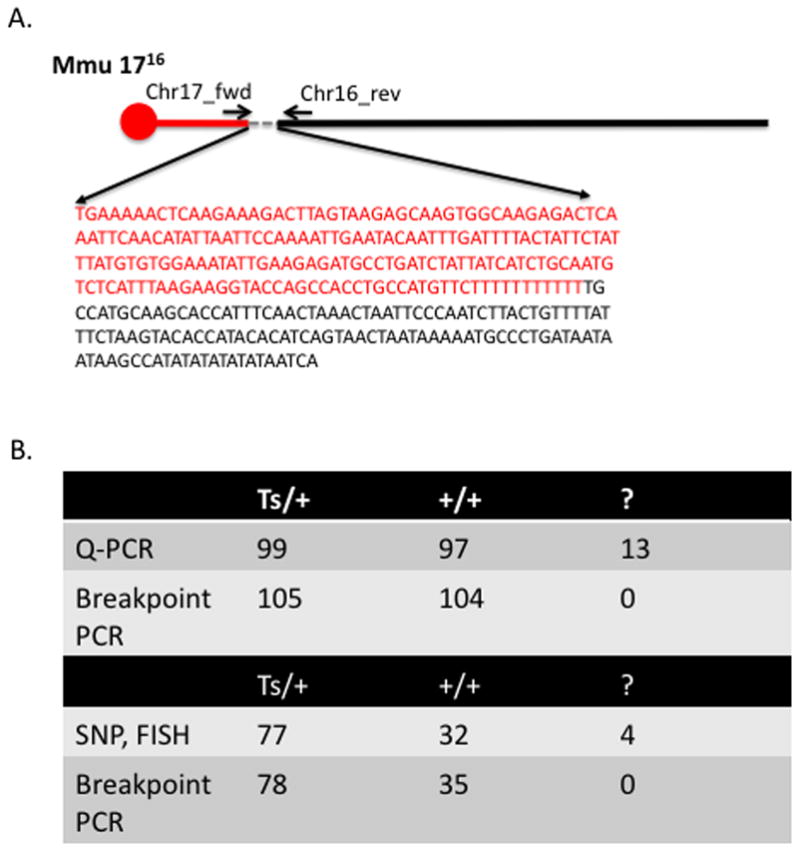
Amplification and sequencing of the 1716 junction. PCR and Sanger sequencing of the junction confirmed the precise sequence of the breakpoint as predicted by de novo assembly of reads spanning the junction (A). PCR amplification of the junction in 209 previously genotyped samples were completely concordant with samples that could be confidently scored as trisomy (Ts/+) or euploid (+/+) by quantitative PCR (Q-PCR) (Liu et al. 2003). Moreover, 13 genotypes (‘?’) that could not be scored due to inconclusive Q-PCR results were successfully scored as either trisomy or euploid based on amplification of the junction (“breakpoint PCR”). Similarly, PCR amplification of the junction in an additional set of 113 previously genotyped samples were also completely concordant with samples that could be confidently scored as trisomy (Ts/+) or euploid (+/+) by SNP prescreen (Lorenzi et al.) and DNA FISH (Moore et al. 1999). In addition, 4 genotypes (‘?’) that could not be confidently genotyped via SNP prescreen or for which SNP prescreen results and DNA FISH were conflicting, could be confidently scored as either trisomy or euploid by PCR amplification of the 1716 junction (B).
Multiple sequence alignment of the junction sequence with homologous regions on Chr 16 and Chr 17 revealed no evidence of gain or loss of sequence from either chromosome. Moreover, there was very little evidence of sequence homology between the breakpoint regions on Chr 16 and Chr 17, with the exception of a short, 7 nt homology region, 15 bp distal to the Chrs 16 and 17 breakpoints (Fig 5). The Chr 16 breakpoint confirms the previously characterized gene dosage data as illustrated in Fig. 1 (Akeson et al. 2001; Kahlem et al. 2004). In addition, the position of the Chr 16 breakpoint also confirms the presence of the microRNA, mir155, on the 1716 chromosome. The position of the Chr 17 breakpoint indicates that the 1716 chromosome carries ~10 Mb of Chr 17 and within this region, there are 60 Ensembl and/or NCBI annotations for protein coding genes, pseudogenes and non-coding RNAs genes. The region shares conserved synteny with the human Chr 6 q25.3–q27 and ~70% (42/60) of the genes annotated in the Chr 17 region have a predicted ortholog (Ensembl predictions) in the human regions of conserved synteny (Supplemental Table 1).
Figure 5.

Multiple sequence alignment of breakpoint regions on Chr 17 (red), Chr16 (black) and the junction site sequence on Mmu1716. Multiple sequence alignment of the breakpoint regions revealed a 7 bp homology region (underlined), 15 bp distal to the breakpoint.
Discussion
These data reveal, for the first time, a complete picture of gene dosage in Ts65Dn mice. Of the 50 Ensembl gene annotations in the ~ 9 Mb Chr 17 region, seven have associated phenotype annotations in the Mouse Genome Informatics Database (MGI); however, all of these annotations are associated with mutant alleles. There are currently no published data on phenotypic effects associated with copy number gains for any of these 50 genes. Further work will be needed to determine whether copy number gains for these genes contribute to the phenotypes that are characteristic of Ts65Dn mice.
The Ts65Dn translocation chromosome is the product of a translocation between Chr 16 and Chr 17 that was induced by ionizing radiation (two 600R doses, 24 hours apart from a cesium137 source) of the testes (Davisson et al. 1993). Irradiated males were immediately bred to females of a different inbred strain for just two weeks. This breeding strategy was used to capture chromosomal rearrangements induced in haploid spermatids (Davisson et al. 1993), a stage of spermatogenesis that is particularly sensitive to radiation induced DNA lesions (Russell et al. 1998). In mammalian cells, double strand breaks (DSBs) induced by ionizing radiation are repaired by one of two major DNA repair pathways, homologous recombination (HR) or non-homologous end joining (NHEJ). The pathway by which a DSB is repaired, HR or NHEJ, is influenced by many factors including cell type and cell cycle stage. Since long stretches of homology are not found in the Chr 16 or Chr 17 breakpoint regions and because haploid spermatids are comparable to the G1 stage of the cell cycle, it is likely that the 1716 translocation product arose via NHEJ and not HR. Interestingly, a 7 nucleotide (nt) homology region was found just 15 nts distal to the breakpoint. In mammalian cells that are deficient for the NHEJ repair factors Ku86 and XRCC4, 7 nt homology regions are frequently associated with and are thought to direct end joining (Kabotyanski et al. 1998). In this case, however, the 7 nt homology regions are not found at the breakpoint junction, but a short distance away. Therefore, it is unclear whether this homology region is related to the NHEJ event that generated the 1716 translocation product.
The targeted sequencing approach allowed for robust oversampling of the 1716 junction in Ts65Dn. Oversampling allowed for enrichment of the most informative reads: paired end reads with mate pairs mapping to both Chr 17 and Chr 16 (reads that flank the breakpoints) and individual reads that span the 1716. As the per base cost of high throughput sequencing continues to plummet and access to high throughput sequencing services expands, it is likely that whole genome sequencing will prove cost effective for routine discovery of translocation breakpoints. Unfortunately, the development of tools for data management and data analysis has not kept pace with the rapidly declining cost of whole genome sequencing, therefore for many laboratories, targeted sequencing approaches are a useful alternative, especially where higher coverage is desirable. Here, the successful use of targeted, high throughput sequencing for the discovery of the translocation breakpoints in Ts65Dn provides the basis for cheaper and more reliable genotyping assays, and, perhaps more importantly, allows for complete characterization of gene dosage in this important disease model. Moreover, the high throughput sequencing analysis used for this study provides the basis for the development of analytical tools that can be applied not only to the discovery of translocation breakpoints, but potentially to the discovery of transgene insertion sites, which in most transgenic disease models, remain uncharacterized.
Supplementary Material
Supplementary Figure 1. Ensemble and NCBI gene annotations and Ensembl predicted human orthologs for Mmu17, 0 bp to 9,426,822 bp. Ensembl prediction of orthology is based on maximum likelihood phylogenetic gene trees (Vilella et al. 2009). Orthology information is not available (N/A) from Ensembl for non-coding RNA genes and for genes that are predicted by NCBI (RefSeq) only.
Acknowledgments
We are grateful to the High Throughput Sequencing and Genome Sciences cores at The Jackson Laboratory for their excellent services and advice. The Transgenic Genotyping core at The Jackson Laboratory provided DNA samples and genotyping data that were critical for validation. This work was supported by funding from NICHD CYTO-01, a Cancer Center Core Grant at The Jackson Laboratory, CA34196, and NIH DE021034 (RJR)
References
- Akeson EC, Davisson MT. Mitotic chromosome preparations from mouse cells for karyotyping. Curr Protoc Hum Genet. 2001;Chapter 4(Unit 4.10) doi: 10.1002/0471142905.hg0410s25. [DOI] [PubMed] [Google Scholar]
- Akeson EC, Lambert JP, Narayanswami S, Gardiner K, Bechtel LJ, Davisson MT. Ts65Dn --localization of the translocation breakpoint and trisomic gene content in a mouse model for Down syndrome. Cytogenet Cell Genet. 2001;93:270–276. doi: 10.1159/000056997. [DOI] [PubMed] [Google Scholar]
- Costa AC, Stasko MR, Schmidt C, Davisson MT. Behavioral validation of the Ts65Dn mouse model for Down syndrome of a genetic background free of the retinal degeneration mutation Pde6b(rd1) Behav Brain Res. 2010;206:52–62. doi: 10.1016/j.bbr.2009.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ascenzo M, Meacham C, Kitzman J, Middle C, Knight J, Winer R, Kukricar M, Richmond T, Albert TJ, Czechanski A, Donahue LR, Affourtit J, Jeddeloh JA, Reinholdt L. Mutation discovery in the mouse using genetically guided array capture and resequencing. Mamm Genome. 2009;20:424–436. doi: 10.1007/s00335-009-9200-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davisson M, Akeson E, Schmidt C, Harris B, Farley J, Handel MA. Impact of trisomy on fertility and meiosis in male mice. Human reproduction (Oxford, England) 2007;22:468–476. doi: 10.1093/humrep/del397. [DOI] [PubMed] [Google Scholar]
- Davisson MT, Schmidt C, Reeves RH, Irving NG, Akeson EC, Harris BS, Bronson RT. Segmental trisomy as a mouse model for Down syndrome. Progress in clinical and biological research. 1993;384:117–133. [PubMed] [Google Scholar]
- Hodges E, Rooks M, Xuan Z, Bhattacharjee A, Benjamin Gordon D, Brizuela L, Richard McCombie W, Hannon GJ. Hybrid selection of discrete genomic intervals on custom-designed microarrays for massively parallel sequencing. Nat Protoc. 2009;4:960–974. doi: 10.1038/nprot.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabotyanski EB, Gomelsky L, Han JO, Stamato TD, Roth DB. Double-strand break repair in Ku86-and XRCC4-deficient cells. Nucleic Acids Res. 1998;26:5333–5342. doi: 10.1093/nar/26.23.5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlem P, Sultan M, Herwig R, Steinfath M, Balzereit D, Eppens B, Saran NG, Pletcher MT, South ST, Stetten G, Lehrach H, Reeves RH, Yaspo ML. Transcript level alterations reflect gene dosage effects across multiple tissues in a mouse model of down syndrome. Genome research. 2004;14:1258–1267. doi: 10.1101/gr.1951304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Yu T, Morishima M, Pao A, LaDuca J, Conroy J, Nowak N, Matsui S, Shiraishi I, Yu YE. Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum Mol Genet. 2007;16:1359–1366. doi: 10.1093/hmg/ddm086. [DOI] [PubMed] [Google Scholar]
- Liu DP, Schmidt C, Billings T, Davisson MT. Quantitative PCR genotyping assay for the Ts65Dn mouse model of Down syndrome. Bio Techniques. 2003;35:1170–1174. doi: 10.2144/03356st02. [DOI] [PubMed] [Google Scholar]
- Lorenzi H, Duvall N, Cherry SM, Reeves RH, Roper RJ. PCR prescreen for genotyping the Ts65Dn mouse model of Down syndrome. Bio Techniques. 48:35–38. doi: 10.2144/000113342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CS, Lee JS, Birren B, Stetten G, Baxter LL, Reeves RH. Integration of cytogenetic with recombinational and physical maps of mouse chromosome 16. Genomics. 1999;59:1–5. doi: 10.1006/geno.1999.5812. [DOI] [PubMed] [Google Scholar]
- Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, Davisson MT. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nature genetics. 1995;11:177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- Reinholdt LG, Czechanski A, Kamdar S, King BL, Sun F, Handel MA. Meiotic behavior of aneuploid chromatin in mouse models of Down syndrome. Chromosoma. 2009;118:723–736. doi: 10.1007/s00412-009-0230-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigby PW, Dieckmann M, Rhodes C, Berg P. Labeling deoxyribonucleic acid to high specific activity in vitro by nick translation with DNA polymerase I. J Mol Biol. 1977;113:237–251. doi: 10.1016/0022-2836(77)90052-3. [DOI] [PubMed] [Google Scholar]
- Russell WL, Bangham JW, Russell LB. Differential response of mouse male germ-cell stages to radiation-induced specific-locus and dominant mutations. Genetics. 1998;148:1567–1578. doi: 10.1093/genetics/148.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Ernst C, Heilbut A, Chiang C, Hanscom C, Lindgren A, Kirby A, Liu S, Muddukrishna B, Ohsumi TK, Shen Y, Borowsky M, Daly MJ, Morton CC, Gusella JF. Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. American journal of human genetics. 2011;88:469–481. doi: 10.1016/j.ajhg.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome research. 2009;19:327–335. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Ensemble and NCBI gene annotations and Ensembl predicted human orthologs for Mmu17, 0 bp to 9,426,822 bp. Ensembl prediction of orthology is based on maximum likelihood phylogenetic gene trees (Vilella et al. 2009). Orthology information is not available (N/A) from Ensembl for non-coding RNA genes and for genes that are predicted by NCBI (RefSeq) only.