

Abstract
Administration of non-myeloablative chemotherapeutic agents or total body irradiation (TBI) prior to adoptive transfer of tumor-specific T cells may reduce or eliminate immunosuppressive populations such as T regulatory cells (Tregs) and myeloid derived suppressor cells (MDSC). Little is known about these populations during immune reconstitution. This study was designed to understand the reconstitution rate and function of these populations post TBI in melanoma tumor bearing mice. Reconstitution rate and suppressive activity of CD4+CD25+Foxp3+ Tregs and CD11b+Gr1+ MDSC following TBI-induced lymphopenia was measured in B16 melanoma tumor-bearing mice. In order to ablate the rapid reconstitution of suppressive populations, we treated mice with docetaxel (DTX), a known chemotherapeutic agent that targets MDSC, in combination with adoptive T cell transfer and DC immunotherapy. Both Treg and MDSC populations exhibited rapid reconstitution after TBI-induced lymphopenia. While reconstituted Tregs were just as suppressive as Tregs from untreated mice, MDSC demonstrated enhanced suppressive activity of CD8+ T cell proliferation compared to endogenous MDSC from tumor bearing mice. TBI-induced lymphopenia followed by DTX treatment improved the efficacy of adoptive T cell transfer and DC immunotherapy in melanoma-bearing mice, inducing a significant reduction in tumor growth and enhancing survival. Tumor regression correlated with increased CTL activity and persistence of adoptively transferred T cells. Overall, these findings suggest that TBI-induced MDSC are highly immunosuppressive and blocking their rapid reconstitution may improve the efficacy of vaccination strategies and adoptive immunotherapy.
Keywords: Myeloid derived suppressor cells, T regulatory cells, Immunosuppression, Immunomodulation, T cells, B16 melanoma, Lymphopenia, homeostatic proliferation, Docetaxel, chemotherapeutic agents
Introduction
Melanoma is a leading cause of cancer mortality in the United States, resulting in one American dying every sixty minutes. Currently, there are few non-surgical options for the treatment of metastatic melanoma (1, 2). Cancer immunotherapy, which focuses on the induction of immunity against tumor cells, is a promising approach for the treatment of melanoma. A major hurdle in the development of effective immunotherapy is tumor-induced suppression, including factors such as regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSC) that can limit the effectiveness of tumor-specific T cells (3, 4). Tregs have been shown to suppress anti-tumor activity and ablation of this population has led to tumor eradication (5). In pre-clinical melanoma models as well as in cancer patients, depletion of Tregs followed by dendritic cell (DC) -based vaccination has led to enhanced vaccine-mediated tumor-specific T cell responses (3, 6, 7). MDSC are implicated in the maintenance of immune tolerance to tumors (4, 8, 9). MDSC are a phenotypically heterogeneous cell population consists of myeloid progenitor cells and immature myeloid cells. In mice, these cells are broadly defined as CD11b+ Gr-1+ cells. Recent reports have shown that MDSC by themselves can induce tumor-specific Tregs (10). Increased levels of circulating MDSC have been correlated with advanced disease stage and extensive metastatic tumor burden in cancer patients (11).
There have been several reports showing that lymphopenia induced by non-myeloablative chemotherapeutic treatments or total body irradiation (TBI) can modulate immune responses and increase anti-tumor immunity (12–14). These treatments induce a severe lymphopenic condition in the host and leads to the subsequent expansion of T cells in the periphery in a process known as homeostatic proliferation. Homeostatic proliferation can lead to the proliferation and activation of T cells specific for self-antigens (15, 16). Studies have shown increased anti-tumor responses following irradiation and adoptive T cell transfer in preclinical murine models (17, 18). Adoptive transfer of autologous tumor-infiltrating lymphocytes (TIL) after lymphodepleting chemotherapy has resulted in objective responses in melanoma patients (12, 19). However, only one fifth of these patients demonstrated complete tumor regressions. While it has been shown that administration of cyclophosphamide and fludarabine or TBI in mice before adoptive transfer reduces or eliminates immunosuppressive populations (13), little is known about the effects of TBI on reconstitution of Tregs and MDSC. Hence, it is important to understand the role of these suppressor populations after the induction of lymphopenia in order to design effective immunotherapeutic treatments to achieve complete tumor regression.
Conventional anti-cancer agents have been explored for their ability to target suppressor populations (20–24). Gemcitabine, a nucleoside analog has been shown to reduce splenic MDSC in mice bearing large tumors without affecting T cells, NK cells, macrophages or B cells (25). Serafini et al showed that sildenafil downregulates arginase activity and nitric oxide synthase expression, thereby reducing the suppressive function of MDSC (26). Anti-microtubule agents, such as DTX and paclitaxel have immune enhancing properties when used alone or in combination with immunotherapy (22, 27, 28). Recent study has shown that paclitaxel promotes differentiation of MDSC into dendritic cells in vitro in a TLR4-independent manner (29). Paclitaxel has also been shown to reduce Tregs and their inhibitory function (28). DTX has been shown to decrease splenic MDSC in mice bearing mammary tumors, leading to an increased anti-tumor response (22).
Although recent reports have shown the effect of docetaxel on MDSC, it has not been used for specific inhibition of MDSC in a setting of lymphopenia for the treatment of melanoma. Hence, in this study, we investigated the reconstitution of suppressor populations (Tregs and MDSC) after lymphodepletion. Further, we investigated if blockade of reconstituting MDSC leads to persistence of adoptively transferred T cells and enhances the efficacy of adoptive T cell transfer and vaccination in a murine model of melanoma.
Materials and methods
Mice
Six to eight week-old C57BL/6 mice were purchased from Harlan Laboratories Indianapolis, IN). OT-I mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were housed at the Animal Research Facility of the H. Lee Moffitt Cancer Center and Research Institute. All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee at the University of South Florida.
Cell lines
B16 melanoma was maintained by serial in vitro passages in complete medium (CM) consisted of RPMI 1640 supplemented with 10% heat-inactivated FCS, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 2 mM fresh L-glutamine, 100 mg/ml streptomycin, 100 U/mL penicillin, 50 mg/mL gentamycin, 0.5 mg/mL fungizone (all from Life Technologies, Rockville, MD), and 0.05 mM 2-ME (Sigma-Aldrich, St. Louis, MO). M05 was generated by transfection of B16 melanoma with pAc-neo-OVA plasmid and was provided by Dr. Kenneth Rock (Dana-Farber Cancer Institute, Boston, MA). M05 cells were maintained by serial in vitro passages in CM supplemented with 0.8mg/ml of G418.
Reagents
For analysis of immune cell populations, the following anti-mouse antibodies were purchased from BD Biosciences (San Diego, CA): anti-Ly6G and -Ly6C biotinylated antibodies, anti-CD11b PerCP-Cy5.5, anti-CD11b APC, anti- Gr-1 PE, anti-Gr-1 PE Cy7, anti-CD4 pacific blue, anti-CD8 Alexafluor 780 and anti-CD3 FITC. The CD4+CD25+ regulatory T cell isolation kit and streptavidin microbeads were purchased from Miltenyi Biotech (Auburn, CA). Anti-mouse Foxp3 T regulatory staining kit was purchased from eBiosciences, San Diego, CA). Clinical grade DTX (Taxotere; Sanofi- Aventis) was used in this study.
Lymphopenia model
A total of 1×105 B16 or 3×105 M05 tumor cells were injected subcutaneously (s.c.) in the left flank of C57BL/6 or Ly5.2 mice. Three days later, mice received a sublethal dose (600 cGy) of total body irradiation (TBI) administered by a [137Cs] γ radiation source.
Isolation of MDSC and MDSC Suppressor Assay
MDSC were isolated from the spleens of naïve, B16 tumor bearing mice, or B16 tumor bearing mice treated with TBI. Splenocytes were depleted of red blood cells using ACK (Ammonium-Chloride-Potassium chloride) lysis buffer and washed twice with cold MACS buffer (1% BSA in PBS with 2 mmol/L EDTA). Washed cells were resuspended at 2×108 cells in 1 mL of MACS buffer and incubated with 100 mcL of biotinylated anti-Ly6C and Ly6G (Gr-1) antibodies (Miltenyi Biotec) for 20 minutes at 4°C. Labeled splenocytes were then incubated at 4°C with 100 mcl of streptavidin microbeads (Miltenyi Biotec) for 15 minutes. Cells were washed, resuspended in 5 mL of MACS buffer, and applied to a MACS column for positive selection according to the manufacturer’s instructions (Miltenyi Biotec). The purity of cell populations as analyzed by flow cytometery was >95%. For functional assays, splenocytes from OT-I mice were used as responder cells. CD8+ T cells from these mice have a transgenic TCR that recognize the OVA 257-264 peptide. MDSC from naïve, tumor bearing mice with or with out TBI treatment were cultured at different ratios with 2×105 splenocytes from OT-1 mice in the presence of control or specific peptides. Cell proliferation was measured by 3[H] thymidine uptake. All experiments were performed in triplicate.
Functional assays
To test the function of Tregs from TBI-treated or untreated tumor bearing mice, a CD4+CD25+ regulatory T cell isolation kit was used to isolate Tregs on day 21 after B16 injection. For stimulator cells, T cell-depleted splenic cells from C57BL/6 mice were used and cultured at 5×104 cells/well in a 96 well plate coated with 0.25 mcg anti-CD3. Naïve CD4+CD25− T cells were co-cultured at 5×104cells/well. CD4+CD25+ T cells from B16-bearing mice treated with or without TBI were added at 5×104cells/well. Naïve T cells alone, stimulators alone and inhibitory cells alone served as controls. After 72 hours of culture, proliferation was analyzed by the incorporation of 3[H] thymidine during the final six hours of culture. All experiments were performed in triplicate.
ROS production
The oxidation-sensitive dye DCFDA (Molecular Probes/Invitrogen, Eugene, OR) was used for the measurement of reactive oxygen species production by MDSC (30). Cells were incubated at room temperature in serum-free RPMI media in the presence of 3 μmol/L DCFDA with or without 300 nmol/L phorbol 12-myristate 13-acetate for 30 minutes, washed with PBS, and then labeled with anti–CD11b and Gr-1 antibodies. After incubation on ice for 20 minutes, cells were washed with PBS and analyzed using flow cytometry.
Arginase activity
Arginase activity was measured in cell lysates as previously described (31). Briefly, cells were lysed for 30 minutes with 100 mcl of 0.1% Triton X-100. Subsequently, 100 mcl of 25 mM Tris-HCl and 10 mcl of 10 mM MnCl2 were added, and the enzyme was activated by heating for 10 minutes at 56°C. Arginine hydrolysis was conducted by incubating the lysate with 100 mcl of 0.5 M L-arginine (pH 9.7) at 37°C for 2 hours. The reaction was stopped with 900 mcl of H2SO4 (96%)/H3PO4 (85%)/H2O (1/3/7, v/v/v). Urea concentration was measured at 540 nm after addition of 40 mcl of a-isonitrosopropiophenone (dissolved in 100% ethanol), followed by heating at 95°C for 30 minutes.
NO production
Equal volumes of culture supernatants (100 mcl) were mixed with Greiss reagent (1% sulfanilamide in 5% phosphoric acid and 0.1% N-1-naphthylethylenediamine dihydrochloride in double-distilled water). After 10 minutes of incubation at room temperature, the absorbance at 550 nm was measured using a microplate plate reader (Bio-Rad, Hercules, CA). Nitrite concentrations were determined by comparing the absorbance values for the test samples to a standard curve generated by a serial dilution of 0.25 mM sodium nitrite.
Flow cytometry
Spleens were harvested under sterile conditions. Single-cell suspensions were prepared, and red cells were removed using ACK lysing buffer. One million cells (splenocytes or MDSC) were incubated for 30 minutes on ice in staining medium with relevant antibodies for the surface expression analysis according to the standard protocols. After washing, the samples were analyzed using an LSRII (BD Biosciences), and the data were analyzed using FlowJo software (Tree Star).
Generation of bone marrow-derived DCs and antigen pulsing
Erythrocyte-depleted mouse bone marrow cells were cultured in CM supplemented with 20 ng/ml GM-CSF and 10 ng/mL IL-4 (R&D Systems, Minneapolis, MN) as described previously (1). On day 5, cells were harvested by gentle pipetting and layered onto an Optiprep gradient (Axis-Shield, Oslo, Norway). The low-density cell interface was collected and washed twice. Resulting DC were washed one time and resuspended at 1 × 106 cells/ml of CM containing 20 ng/mL GM-CSF and 20 ng/mL IL-4. DC were pulsed overnight with 10 mcg/mL of OVA 257-264 peptide (Invitrogen, Carlsbad, CA)
Immunization schedule
MDSC blockade in TBI treated mice in combination immunotherapy
A total of 3×105 M05 tumor cells were injected s.c. in the left flank of C57BL/6 mice. Three days later, mice received a sublethal dose (600 cGy) of total body irradiation (TBI) administered by a [137Cs] γ radiation source. Mice received 5×106 OT-1 T cells/mouse on day 4 and two injections of DC pulsed with OVA 257-264 peptide (1×106/mouse/vaccination) on days 4 and 10. Four doses of DTX at 16mg/kg were given intraperitoneally, (i.p.), every three days starting on day 4. Control groups received T cells alone, DC alone, DTX alone or PBS. Tumor size was measured and recorded every two days. In another set of experiments, splenocytes were harvested from these mice one week after the final immunization for in vitro assays.
51Cr release cytotoxicity assay
A 51Cr release assay was done as described previously (18). M05 cells were used as targets. Spleens of treated mice were harvested on day 21 and purified T cells were used as effector cells. T cell purity was measured by flow cytometry and cells were 95% positive for CD90 (data not shown). Briefly, target M05 cells were labeled with 100 μCi of 51Cr (Amersham Corp.) at 37°C in a 5% CO2 atmosphere for one and half hours. The labeled tumor cells were washed three times and added to the effector cells in triplicate wells of 96-well round-bottomed microplates at 100:1, 50:1, and 25:1 effector to target ratios. After 5 hours, the percentage of specific 51Cr release was determined by the following equation: [(experimental cpm − spontaneous cpm)/(total cpm incorporated − spontaneous cpm)] × 100. All determinations were done in triplicate, and the SE of all assays was calculated and was typically 5% of the mean or less.
CBA assay
Briefly, splenocytes were plated at 2×106, co-cultured with 2×105 irradiated M05 melanoma tumor cells, and incubated for 48 hours. Culture supernatants were analyzed for IFN-γ production using commercially available cytometry bead array kit (BD Biosciences, San Diego, CA). Briefly, 25 ul of mixed capture beads were incubated with 25 ul of culture supernatant and 25 ul of PE detection reagent for 2 hours at room temperature. The immuno-complexes were then washed and analyzed using a FACS Calibur affixed with a 488-nm laser (BD Biosciences, San Diego, CA), according manufacturer’s protocol.
Statistical analysis
A Mann-Whitney test (unpaired) or a Student’s t-test was used to compare differences between two treatment groups. All statistical evaluations of data were performed using Graph Pad Prism software. Statistical significance was achieved at p<0.05.
Results
Reconstitution of immune subsets after lymphopenia
To evaluate the reconstitution of immune subsets induction of lymphopenia, naive and B16 tumor-bearing mice received 600 rad of TBI. We observed lymphopenia and leukopenia in both naive and B16 tumor bearing mice evidenced by total splenocyte counts post TBI (data not shown). As shown in Figure 1A, CD4+ and CD8+ T cells were detected on day 7 in the wild type mice, with 60% of CD4+ T cells reconstituted (percent of normal) and 40% of CD8+ T cells reconstituted by day 21. In B16 tumor bearing mice, 30% reconstitution of CD4+ T cells and 20% CD8+ T cells was observed (Figure 1B). CD4+CD25+Foxp3+ Tregs were reconstituted to 70% in wild type and 100% in B16 tumor bearing mice by day 10. While CD11b+Gr-1+ MDSC were reconstituted to 100% by day 10 in both wild type and B16 tumor bearing mice. Alternatively, the cell numbers of splenic immune subsets per million of splenocytes were calculated post-TBI are shown as mean ± SD (n=4) in Table-1. These findings demonstrate that in a lymphopenic environment, MDSC and Treg recover from lymphodepletion faster than CD4+ and CD8+ T cells, and this difference is further accentuated in tumor-bearing mice, with proliferation resulting in higher inhibitory cell frequency compared to basline by 21 days.
Figure 1.
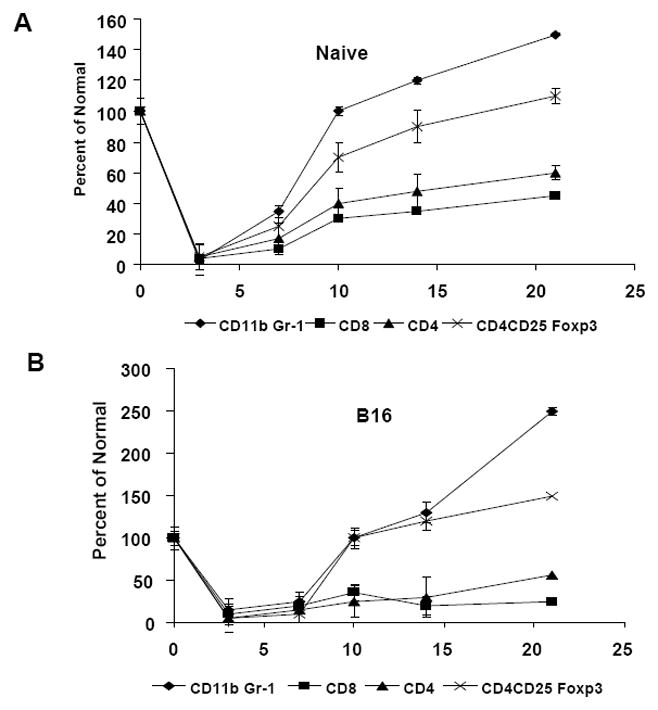
Reconstitution of splenic immune subsets after TBI. Mice receive 1×105 B16 tumor cells on day 0 followed by 600 rad TBI on day 3. Spleen cells from (A) wild type and (B) B16 tumor bearing mice were analyzed for CD4+, CD8+, CD11b+Gr-1+ MDSC and CD4+CD25+foxp3+ Tregs on days 3, 7, 10, 14 and 21. Data represents the percent of normal compared to splenocytes from mice not treated with TBI. Data are representative of three independent experiments (n=4).
Table-1.
Cell number (×104)/million of splenocytes
| CD4 | CD8 | CD11bGr-1 | CD4CD25Foxp3 | |||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Naïve | B16 | Naïve | B16 | Naïve | B16 | Naïve | B16 | |
|
| ||||||||
| Pre- TBI | 25 ± 2.3 | 16.3±1.52 | 2.5± 0.31 | 1.2±0.03 | ||||
| Post- TBI | ||||||||
| Day3 | 0.5± 0.01 | 0.2±0.04 | 0.8± 0.01 | 0.6± 0.02 | 0.1±0.02 | 0.2±0.01 | 0.1±0.01 | 0.3±0.01 |
| Day 7 | 2.2±0.03 | 2.3± 0.12 | 1.9±0.02 | 3.5±0.01 | 0.9± 0.01 | 0.9±0.05 | 0.4±0.02 | 0.4±0.02 |
| Day 10 | 9.4 ±0.80 | 7.8±0.61 | 5.8± 0.05 | 6.5±0.21 | 2.5± 0.31 | 2.6±0.26 | 0.9±0.1 | 1.3±0.11 |
| Day 14 | 10.1±1.21 | 11.3±1.10 | 6.7± 0.11 | 4.5±0.80 | 2.9±0.21 | 3.5±0.97 | 1.1±0.32 | 1.5±0.21 |
| Day 21 | 16.5±0.34 | 12.5±0.20 | 7.8±0.21 | 4.9±0.12 | 3.8± 1.14 | 6.3±0.03 | 1.4±0.66 | 1.8±0.05 |
1. n= 4 per group
2. Data compiled from three independent experiments.
Repopulating Tregs are suppressive after TBI
Because Tregs and MDSC undergo rapid reconstitution after TBI, we sought to determine their suppressive function on T cell proliferation. Tregs were purified from the spleens of B16 tumor bearing mice with or without TBI on day 21 post tumor injection. Inhibitor alone, stimulator alone or responder alone served as controls. As shown in Figure 2A, CD4+CD25+ Tregs from the spleens of TBI treated, B16 tumor-bearing mice were as suppressive as the CD4+CD25+ Tregs from tumor-bearing mice with no TBI treatment while CD4+CD25− cells in the absence of CD4+ CD25+ Tregs had significant proliferation (p<0.01). This result shows that reconstituting Tregs post-TBI are suppressive and behave similarly to endogenous Tregs from tumor bearing mice.
Figure 2.

The suppressive activity of reconstituted Tregs/MDSC (A) CD4+CD25+ cells were purified on day 20 from B16 tumor bearing mice treated with or without TBI. T cell depleted splenic cells from C57BL/6 mice were used as stimulators were cultured at 5×104 cells/well in a 96 well plate coated with 0.25ug anti-CD3. Naïve CD4+CD25− T cells were co-cultured at 5×104 cells/well. CD4+CD25+ T cells were added at 5×104 cells/well. After 72 hours of culture, proliferation was analyzed by the addition of 3[H] thymidine during the final six hours of culture. All experiments were performed in triplicate (B) CD11b+Gr-1+ MDSCs were purified using a MACs column and were cultured at various ratios with 2×105 splenocytes from OT-I mice in the presence of OVA257-264 peptide. After 72 hours of culture, proliferation was analyzed by the addition of 3[H] thymidine during the final six hours of culture (C) IFN-gamma production was measured in the supernatants after 48 hours of co-culture with a CBA assay as described in Materials and Methods * indicates p<0.01.
Repopulating MDSC are highly immunosuppressive after TBI
Next, we evaluated the function of TBI-induced MDSC. We purified Gr-1+ cells from the spleens of naive and tumor bearing mice that received TBI on day 21 post tumor injection. B16 tumor bearing mice with no TBI treatment were used as a control. MDSC were co-cultured with OT-I splenocytes at various ratios in the presence of OVA257-264 peptide and T cell proliferation was evaluated. OT-1 cells alone and MDSC alone served as controls. We observed that TBI-induced MDSC from tumor bearing mice were significantly more suppressive compared to the endogenous MDSC from tumor bearing mice that did not receive TBI treatment (Figure 2B). Even at the lowest ratio of 1 MDSC: 12 T cells, there was significant (p<0.01) suppressive activity, while the endogenous MDSC from tumor bearing mice were not able to inhibit T cell proliferation. MDSC derived from TBI treated naive mice (not bearing tumor) did not show any suppressive activity compared to control OT-I cells pulsed with peptide alone. To further analyze suppressive activity, MDSC were added at various ratios to OT-I splenocytes in the presence of OVA257-264 peptide, and IFN-γ production was evaluated. We observed significantly suppressed IFN-γ production by OT-1 cells when co-cultured with MDSC from tumor bearing mice with or without TBI at 1:3 ratios (Figure 2C, p<0.01). However, at both 1:6 and 1:12 ratios, no suppressive activity was observed in MDSC from non-TBI-treated tumor bearing mice, while a significant reduction of IFN-γ production was observed in OT-I cells when co-cultured with MDSC from TBI-treated tumor bearing mice. Collectively, these data show that repopulating MDSC post- TBI has enhanced immunosuppressive activity of MDSC on a per cell basis.
Next, we sought to understand the mechanism for the increased suppressive activity. Several factors have been implicated in MDSC-mediated immune suppression, including the production of arginase, nitric oxide (NO), and reactive oxygen species (ROS) (30–32). We compared the production of these factors in MDSC from B16 tumor-bearing mice treated with or without TBI. Gr-1+ cells were purified from the spleens of TBI-treated or untreated tumor bearing mice on day 21 post-tumor injection. OT-1 splenocytes were cultured at different ratios of MDSC in the presence of OVA257-264 peptide for 48h. OT-1 splenocytes alone and MDSC alone served as controls. OT-I splenocytes cultured with different ratios of MDSC in the presence of OVA257-264 peptide had significant increase in the NO production compared to the endogenous MDSC from the tumor bearing mice (Figure 3A, p<0.05). The oxidation-sensitive dye DCFDA was used for the measurement of reactive oxygen species production by MDSC and analyzed by flow cytometry. As shown in Figure 3B, there was an increased ROS production in TBI-induced MDSC compared to endogenous MDSC from tumor bearing mice. Next, arginase activity in cell lysates was also measured. We observed a similar trend as that seen in the NO and ROS levels, with increased arginase activity in the TBI- induced MDSC compared to the endogenous MDSC (p<0.01, Fig. 3C).
Figure 3.

TBI-induced MDSC express high levels of NO, ROS, and arginase. (A) Nitric oxide (NO) production (data represents the mean ± SD of triplicates * indicates p<0.05). OT-1 cells (1×105 cells per well) were stimulated in triplicates for 48h with OVA257-264 peptide in the presence of different ratios of MDSC and NO levels were measured, B) The levels of ROS production in MDSC from untreated or TBI-treated mice bearing B16 tumor was measured using DCFDA staining and flow cytometry. * indicates p<0.05. Data represents one of the three independent experiments C) Arginase activity was measured in untreated and TBI-treated mice bearing B16 tumors. Mean ± SD of two independent experiments is shown. * indicates p<0.01.
DTX reduces reconstitution rate of MDSC post TBI
Because MDSC from TBI-treated tumor bearing mice demonstrated enhanced suppressive activity, we wanted to evaluate whether we could abrogate their reconstitution with DTX treatment. To evaluate the effect of DTX on reconstituting MDSC post-TBI, C57BL/6 mice were injected s.c. with B16 cells on day 0 and received 600 rad of TBI on day 3. Mice received four doses of DTX injected i.p, at 3 day or weekly intervals. Spleens were harvested on days 3, 7, 10, 14 and 21 post tumor injection and immune subsets were analyzed by flow cytometry. As shown in Figure 4A, B16 tumor bearing mice that received DTX had a significant reduction in TBI-induced MDSC, regardless of whether they received the treatment on a weekly basis or every three days (p<0.05). By day 10, we observed a reduction in TBI-induced MDSC reconstitution in DTX treated mice compared to the TBI- treated tumor bearing mice that did not receive DTX. By day 21, mice that received DTX every 3 days had a significant reduction in their MDSC reconstitution, up to 50% (p< 0.002) compared to the untreated mice (250%) while we observed 100% reconstitution in mice receiving the treatment on a weekly interval (p<0.01).
Figure 4.
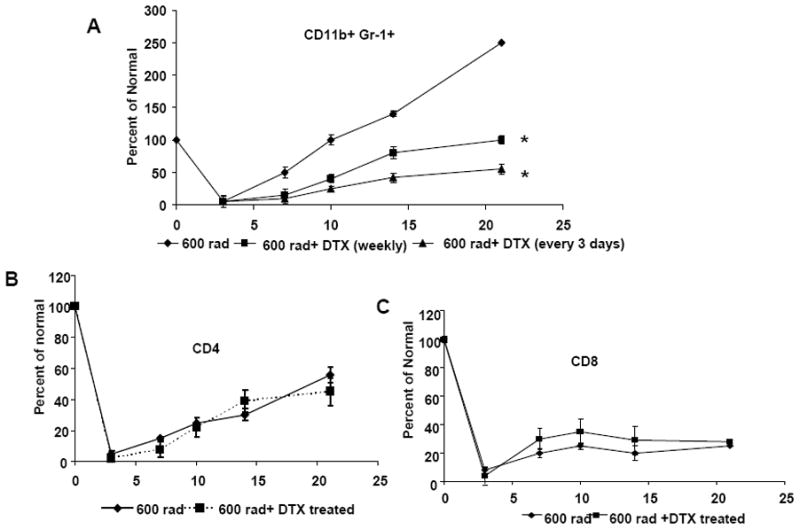
DTX treatment in combination with TBI reduces MDSC in tumor-bearing mice. Mice were injected with B16 tumor cells on day 0 followed by TBI on day 3. Mice received DTX treatment on a weekly basis or every 3 days starting on day 4. (A) Spleen cells from mice that received 600 rad alone or 600 rad +DTX were analyzed for CD11b+Gr-1+MDSC, (B) CD4+ T cells and (C) CD8+ T cells on days 3, 7, 10, 14 and 21 post tumor injection. Graphs represents the percent of normal of CD11b+Gr-1+ MDSC, CD4+ and CD8+ T cells (n=4 per group) * indicates p<0.01.
Previous studies have shown that DTX treatment can increase the number of CD4+ and CD8+ T cells (22, 27). To determine if DTX, given every 3 days, had an effect on reconstituting T cells, we analyzed the CD4+ and CD8+ T cell reconstitution in TBI-treated tumor bearing mice after DTX treatment. As shown in Figures 4B & C, we did not see any significant difference reconstitution of CD4+ and CD8+ on days 7, 10, 14 and 21 in mice that received DTX. This data shows that DTX given at 3 day intervals significantly reduced the MDSC reconstitution post-TBI without modulating T cell populations.
Blockade of TBI-induced MDSC with DTX improves the anti-tumor efficacy of adoptive T cell transfer and DC immunotherapy
Next, we evaluated whether blocking TBI-induced MDSC with DTX would improve the efficacy of adoptive T cell transfer and DC immunotherapy. C57BL/6 mice were injected s.c. with M05 cells on day 0 and received 600 rad of TBI on day 3. Mice received 5×106 OT-I T cells on day 4, and were vaccinated with DC pulsed with OVA 257-264 peptide (DCOVA) on days 4 and 10. DTX was given every three days starting on day 4 for a total of four doses. As shown in Figure 5A, after four doses of DTX treatment, a significant reduction in tumor growth was observed in mice that received OT-I T cells and DCOVA immunotherapy compared to mice that received only T cells and DCOVA vaccination (p<0.001). However, mice that received DTX treatment with DCOVA or T cells alone had a significant reduction in tumor growth compared to the untreated groups (Figure 5B, p<0.001). These data suggest that blockade of TBI-induced MDSC can improve the efficacy of adoptive T cell transfer and DC vaccination.
Figure 5.

DTX treatment in combination with TBI enhances the immunotherapeutic efficacy of adoptive T cell transfer and DC vaccination. Mice received 3×105 M05 tumor cells on day 0 followed by TBI on day 3. On day 4, 5×106 OT-I T cells were adoptively transferred followed by 2 doses of DC pulsed with OVA 257-264 peptide on days 4 and 10. Four doses of DTX treatment were given every 3 days starting from day 4. (A) Tumor growth. (B) Survival curve * indicates p< 0.001
Blockade of TBI-induced MDSC enhances the persistence of adoptively transferred T cells and improves CTL function
We next examined whether adoptively transferred T cells persisted for a longer duration in mice treated with DTX. Ly5.2 mice were injected with 3×105 M05 cells followed by TBI on day 3. Mice received 5×106 OT-I (Ly5.1 mice) T cells on day 4. DCOVA vaccinations were given on days 4 and 10. Four doses of DTX were given every 2–3 days beginning on day 4. Splenocytes were collected at various time points and the percentage of OT-I T cells were measured by flow cytometry. Mice that received therapy with OT-I T cells and DCOVA in combination with DTX had a higher percentage of OVA tetramer specific T cells on days 8, 15 and 20. As shown in Figure 6A, a higher percentage of CD8+ OVA specific T cells were observed in mice that received DTX treatment compared to the mice that received immunotherapy alone. We next evaluated IFN-γ production as a measure of T cell function. As shown in Figure 6B, re-stimulation with OVA peptide for 48 hours resulted in higher IFN-γ production by splenocytes from mice that received DTX treatment in combination with OT-I T cells and DCOVA vaccination (980 ± 35.4 pg/ml) compared to splenocytes from mice that received OT-I T cells and DCOVA vaccination alone (597± 30.4 pg/ml, p<0.01). These data suggest that blockade of TBI-induced MDSC with DTX enhanced the persistence and activity of adoptively transferred T cells.
Figure 6.

Addition of DTX treatment in combination with adoptive T cell transfer and DC vaccination increases the persistence OT-I T cells in mice. Ly5.2 mice were injected with 3×105 M05 cells followed by TBI on day 3. Mice received 5×106 OT-I (Ly5.1 mice) T cells on day 4. DCOVA vaccinations were given on days 4 and 10. Four doses of DTX were given every 2–3 days beginning on day 4. A) The percentage of CD8+ and OVA tetramer positive cells was measured by flow cytometry. B) IFN-γ production was measured in the splenocytes of treated mice. Splenocytes were plated at 2×106, co-cultured with 2×105 irradiated M05 cells, and incubated for 48 hours. Culture supernatants were analyzed for IFN-γ production using commercially available cytometry bead array kit. Data shows the mean ± SD of triplicates * indicates p<0.01. C) A five hour 51Cr release assay was performed using M05 tumor cells as targets. Spleens of treated mice were harvested on day 21 and purified T cells were used as effector cells. Data shows the mean ± SE of triplicates from a chromium release assay * indicates p<0.05
Next we examined whether reduction of TBI-induced MDSC by DTX enhances the function of CD8+ T cells. Splenic T cells from mice receiving T cells and DCOVA, treated with or without DTX, were purified on day 21 and co-cultured with 51Cr-labeled M05 melanoma tumor cells at effector to target ratios of 100:1, 50:1, or 25:1. T cells from mice that received DCOVA and OT-I T cells in combination with DTX treatment exhibited higher cytotoxicity against M05 melanoma tumor cells at a 100:1 ratio compared to mice that did not receive DTX treatment (p<0.05, Figure 6C). However, no significant differences were measured at lower ratios. This data suggests that blockade of TBI-induced MDSC with DTX improves the CTL function in vaccinated mice against M05 melanoma tumor cells.
Discussion
Impaired immune function is often associated with negative regulatory factors such as regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSC) (3, 9, 33). These populations can inhibit T cell activation and may limit the efficacy of immunotherapies (34). Induction of lymphopenia by TBI or chemotherapy can eliminate these immunosuppressive populations. Our laboratory and others have shown that adoptive T cell therapy and DC vaccination after the induction of lymphopenia enhances T cell expansion and activation and leads to improved anti-tumor immune responses against melanoma (13, 17, 18). Adoptive transfer of T cells following the induction of lymphopenia has led to enhanced anti-tumor immunity in animal models and significant clinical responses in patients with melanoma (14, 17, 19, 35). However, there are few reports on the reconstitution and function of suppressor populations of suppressive populations after the induction of lymphopenia.
In this study, we demonstrate that in a setting of lymphopenia, MDSC and Tregs reconstitute faster than CD4+ and CD8+ T cells. TBI-induced Tregs were as suppressive as the endogenous Tregs. Depletion of TBI-induced Tregs with anti-CD25 antibodies enhanced the anti-tumor efficacy of adoptive T cell transfer and DC immunotherapy in melanoma bearing mice (data not shown). Our findings suggest that depletion of TBI-induced Tregs improves anti-tumor efficacy of adoptive T cell transfer and DC immunotherapy. This data is supported by a recent report showing an increase in the frequency of Tregs in B16 tumor bearing mice after temozliamide-induced lymphopenia. Systemic Treg depletion during lymphopenia resulted in enhanced anti-tumor immunity. (36). It has been shown that lymphopenia induces expansion of Tregs during immune reconstitution in individuals with cancer (37, 38). A study has shown that Denileukin diftitox (DAB/IL2; Ontak) administration to stage IV melanoma patients depletes peripheral blood Tregs and causes the regression of metastatic tumors in a subset of patients (39). CD25 depletion in combination with lymphopenia has been shown to result in a potent tumor rejection in a B16 melanoma model and may enhance immunity in lymphopenic patients with metastatic melanoma (40, 41).
Our findings demonstrate that TBI-induced MDSC are more immunosuppressive on a per cell basis compared to endogenous MDSC isolated from B16 tumor bearing mice. This is a novel finding that repopulating MDSC after the induction of lymphopenia are highly immunosuppressive Based on these initial findings, this study focused on evaluating the immunosuppressive function of TBI-induced MDSC. We tested NO, arginase and ROS production as possible mechanisms for the observed increase in suppressive function and found increased production of all three suppressive factors in the reconstituted MDSC following lymphodepletion. It has been reported that granulocytic subsets of MDSC express a high level of ROS and very little NO, while the monocytic subsets have very little ROS but high levels of NO (31). These suppressive factors have been shown to have direct effect in the inhibition of T cell function (42–45). Although, it has been shown in several studies that MDSC uses multiple factors such as NO, ROS and arginase for their suppressive function on T cells, this is the first study to show that MDSC that regenerate after the induction of lymphopenia has increased production of these factors for their immunosuppressive function compared to the endogenous MDSC from non-TBI treated tumor bearing mice. It has been shown that various tumor derived factors define the expansion of these MDSC subsets. However, in our study, we observed elevated levels of all of these suppressive factors. It is possible that both monocytic and granulocytic MDSC are being reconstituted following lymphodepletion and that each population contributes to increased immunosuppressive function. Studies to define the factors responsible for the rapid expansion of these MDSC subsets in a lymphopenic environment are ongoing.
MDSC frequency is increased in melanoma patients and is associated with disease progression (46, 47). Strategies to target MDSC include inhibition of expansion or promotion of differentiation into mature cells that no longer possess suppressive activity (21, 48). All-trans-retinoic acid (ATRA) has been shown to induce MDSC differentiation that leads to neutralization of ROS production in both mice and in cancer patients (23, 49). Several studies have also shown that MDSC can be directly eliminated using chemotherapeutic drugs such as Gemcitabine, DTX, suntinib, or flurouracil (20, 22, 23, 26, 28). Treatment of mice bearing large tumors with chemotherapeutic drugs has been shown to result in dramatic reductions in the number of splenic MDSC and a marked improvement in immunotherapeutic efficacy (24, 25). In order to block rapidly reconstituting MDSC in our model, we used DTX, a chemotherapeutic drug previously shown to block MDSC expansion (22). In melanoma-bearing lymphopenic mice, DTX treatment decreased MDSC reconstitution and increased the persistence of adoptively transferred T cells, resulting in delayed tumor growth and enhanced survival. These are the key findings that can potentially improve the design of clinical trials in patients with melanoma.
Although lymphodepletion strategies are an attractive tool for adoptive immunotherapy protocols, our results demonstrate that MDSC and Tregs are rapidly reconstituted and are highly immunosuppressive after the induction of lymphopenia. It is possible that the rapid reconstitution and suppressive functions of TBI-induced MDSC and Tregs may limit the effectiveness of immunotherapy to induce anti-tumor T cell responses which in turn, contribute to failure at inducing complete tumor regressions. Blocking the reconstitution of TBI-induced MDSC or Tregs enhanced anti-tumor immunity in a murine model and may improve current adoptive therapy strategies for the treatment of metastatic melanoma.
Acknowledgments
This study was supported by the National Institute of Health (R00 CA128825-01) to Dr. Shari Pilon-Thomas.
We thank Dr. Dmitry Gabrilovich for his valuable comments during the preparation of this manuscript. We also thank the Flow Cytometry Core Facility at the H. Lee Moffitt Cancer Center and Research Institute for its contribution to this work.
ABBREVIATIONS
- MDSC
myeloid derived suppressor cells
- DTX
Docetaxel
- Tregs
T regulatory cells
- TBI
total body irradiation
- ROS
reactive oxygen species
- ATRA
All-trans-retinoic acid
- DC
dendritic cells
Footnotes
There are no conflicts of interest to disclose
References
- 1.Jemal A, Devesa SS, Hartge P, Tucker MA. Recent trends in cutaneous melanoma incidence among whites in the United States. J Natl Cancer Inst. 2001;93:678–683. doi: 10.1093/jnci/93.9.678. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 3.Viguier M, Lemaitre F, Verola O, Cho MS, Gorochov G, Dubertret L, Bachelez H, Kourilsky P, Ferradini L. Foxp3 expressing CD4+CD25(high) regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J Immunol. 2004;173:1444–1453. doi: 10.4049/jimmunol.173.2.1444. [DOI] [PubMed] [Google Scholar]
- 4.Sinha P, V, Clements K, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol. 2005;174:636–645. doi: 10.4049/jimmunol.174.2.636. [DOI] [PubMed] [Google Scholar]
- 5.Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, Yancey D, Zhang A, Dahm P, Chao N, Gilboa E, Vieweg J. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. The Journal of clinical investigation. 2005;115:3623–3633. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacobs JF, Punt CJ, Lesterhuis WJ, Sutmuller RP, Brouwer HM, Scharenborg NM, Klasen IS, Hilbrands LB, Figdor CG, de Vries IJ, Adema GJ. Dendritic cell vaccination in combination with anti-CD25 monoclonal antibody treatment: a phase I/II study in metastatic melanoma patients. Clin Cancer Res. 16:5067–5078. doi: 10.1158/1078-0432.CCR-10-1757. [DOI] [PubMed] [Google Scholar]
- 7.Powell DJ, Jr, Attia P, Ghetie V, Schindler J, Vitetta ES, Rosenberg SA. Partial reduction of human FOXP3+ CD4 T cells in vivo after CD25-directed recombinant immunotoxin administration. J Immunother. 2008;31:189–198. doi: 10.1097/CJI.0b013e31815dc0e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kusmartsev S, Gabrilovich DI. Immature myeloid cells and cancer-associated immune suppression. Cancer Immunol Immunother. 2002;51:293–298. doi: 10.1007/s00262-002-0280-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother. 2006;55:237–245. doi: 10.1007/s00262-005-0048-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66:1123–1131. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 11.Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 12.Dudley ME, Wunderlich JR, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry RM, Marincola FM, Leitman SF, Seipp CA, Rogers-Freezer L, Morton KE, Nahvi A, Mavroukakis SA, White DE, Rosenberg SA. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother. 2002;25:243–251. doi: 10.1097/01.CJI.0000016820.36510.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, Hwang LN, Yu Z, Wrzesinski C, Heimann DM, Surh CD, Rosenberg SA, Restifo NP. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. The Journal of experimental medicine. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rapoport AP, Stadtmauer EA, Aqui N, Badros A, Cotte J, Chrisley L, Veloso E, Zheng Z, Westphal S, Mair R, Chi N, Ratterree B, Pochran MF, Natt S, Hinkle J, Sickles C, Sohal A, Ruehle K, Lynch C, Zhang L, Porter DL, Luger S, Guo C, Fang HB, Blackwelder W, Hankey K, Mann D, Edelman R, Frasch C, Levine BL, Cross A, June CH. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nat Med. 2005;11:1230–1237. doi: 10.1038/nm1310. [DOI] [PubMed] [Google Scholar]
- 15.Cho BK, V, Rao P, Ge Q, Eisen HN, Chen J. Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. The Journal of experimental medicine. 2000;192:549–556. doi: 10.1084/jem.192.4.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu HM, Poehlein CH, Urba WJ, Fox BA. Development of antitumor immune responses in reconstituted lymphopenic hosts. Cancer Res. 2002;62:3914–3919. [PubMed] [Google Scholar]
- 17.Koike N, Pilon-Thomas S, Mule JJ. Nonmyeloablative chemotherapy followed by T-cell adoptive transfer and dendritic cell-based vaccination results in rejection of established melanoma. J Immunother. 2008;31:402–412. doi: 10.1097/CJI.0b013e31816cabbb. [DOI] [PubMed] [Google Scholar]
- 18.Pilon-Thomas S, Mackay A, Vohra N, Mule JJ. Blockade of programmed death ligand 1 enhances the therapeutic efficacy of combination immunotherapy against melanoma. J Immunol. 184:3442–3449. doi: 10.4049/jimmunol.0904114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, Rogers LJ, Gracia GJ, Jones SA, Mangiameli DP, Pelletier MM, Gea-Banacloche J, Robinson MR, Berman DM, Filie AC, Abati A, Rosenberg SA. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, Wood L, Elson P, Garcia J, Dreicer R, Bukowski R. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008;14:6674–6682. doi: 10.1158/1078-0432.CCR-07-5212. [DOI] [PubMed] [Google Scholar]
- 21.Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 11:215–233. doi: 10.1038/nrd3626. [DOI] [PubMed] [Google Scholar]
- 22.Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, Djeu JY. A novel chemoimmunomodulating property of docetaxel: suppression of myeloid-derived suppressor cells in tumor bearers. Clin Cancer Res. 16:4583–4594. doi: 10.1158/1078-0432.CCR-10-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, Lush RM, Antonia S, Gabrilovich DI. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006;66:9299–9307. doi: 10.1158/0008-5472.CAN-06-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, Schwartz M, Divino CM, Pan PY, Chen SH. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514–2522. doi: 10.1158/0008-5472.CAN-08-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713–6721. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 26.Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, Dolcetti L, Bronte V, Borrello I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. The Journal of experimental medicine. 2006;203:2691–2702. doi: 10.1084/jem.20061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garnett CT, Schlom J, Hodge JW. Combination of docetaxel and recombinant vaccine enhances T-cell responses and antitumor activity: effects of docetaxel on immune enhancement. Clin Cancer Res. 2008;14:3536–3544. doi: 10.1158/1078-0432.CCR-07-4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vicari AP, Luu R, Zhang N, Patel S, Makinen SR, Hanson DC, Weeratna RD, Krieg AM. Paclitaxel reduces regulatory T cell numbers and inhibitory function and enhances the anti-tumor effects of the TLR9 agonist PF-3512676 in the mouse. Cancer Immunol Immunother. 2009;58:615–628. doi: 10.1007/s00262-008-0586-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michels T, Shurin GV, Naiditch H, Sevko A, Umansky V, Shurin MR. Paclitaxel promotes differentiation of myeloid-derived suppressor cells into dendritic cells in vitro in a TLR4-independent manner. J Immunotoxicol. doi: 10.3109/1547691X.2011.642418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagaraj S, Youn JI, Weber H, Iclozan C, Lu L, Cotter MJ, Meyer C, Becerra CR, Fishman M, Antonia S, Sporn MB, Liby KT, Rawal B, Lee JH, Gabrilovich DI. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res. 16:1812–1823. doi: 10.1158/1078-0432.CCR-09-3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–5802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Corraliza IM, Campo ML, Soler G, Modolell M. Determination of arginase activity in macrophages: a micromethod. Journal of immunological methods. 1994;174:231–235. doi: 10.1016/0022-1759(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 33.Bronte V, Serafini P, Apolloni E, Zanovello P. Tumor-induced immune dysfunctions caused by myeloid suppressor cells. J Immunother. 2001;24:431–446. doi: 10.1097/00002371-200111000-00001. [DOI] [PubMed] [Google Scholar]
- 34.Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R. Immature immunosuppressive CD14+HLA-DR-/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res. 70:4335–4345. doi: 10.1158/0008-5472.CAN-09-3767. [DOI] [PubMed] [Google Scholar]
- 35.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, Wunderlich J, Restifo NP, Thomasian A, Downey SG, Smith FO, Klapper J, Morton K, Laurencot C, White DE, Rosenberg SA. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitchell DA, Cui X, Schmittling RJ, Sanchez-Perez L, Snyder DJ, Congdon KL, Archer GE, Desjardins A, Friedman AH, Friedman HS, Herndon JE. Monoclonal antibody blockade of IL-2R{alpha} during lymphopenia selectively depletes regulatory T cells in mice and humans. In: McLendon RE, Reardon DA, Vredenburgh JJ, Bigner DD, Sampson JH, editors. Blood. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitchell DA, Cui X, Schmittling RJ, Sanchez-Perez L, Snyder DJ, Congdon KL, Archer GE, Desjardins A, Friedman AH, Friedman HS, Herndon JE. Monoclonal antibody blockade of IL-2 receptor alpha during lymphopenia selectively depletes regulatory T cells in mice and humans. In: McLendon RE, Reardon DA, Vredenburgh JJ, Bigner DD, Sampson JH, editors. Blood. 2. Vol. 118. pp. 3003–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang H, Chua KS, Guimond M, Kapoor V, Brown MV, Fleisher TA, Long LM, Bernstein D, Hill BJ, Douek DC, Berzofsky JA, Carter CS, Read EJ, Helman LJ, Mackall CL. Lymphopenia and interleukin-2 therapy alter homeostasis of CD4+CD25+ regulatory T cells. Nat Med. 2005;11:1238–1243. doi: 10.1038/nm1312. [DOI] [PubMed] [Google Scholar]
- 39.Rasku MA, Clem AL, Telang S, Taft B, Gettings K, Gragg H, Cramer D, Lear SC, McMasters KM, Miller DM, Chesney J. Transient T cell depletion causes regression of melanoma metastases. J Transl Med. 2008;6:12. doi: 10.1186/1479-5876-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poehlein CH, Haley DP, Walker EB, Fox BA. Depletion of tumor-induced Treg prior to reconstitution rescues enhanced priming of tumor-specific, therapeutic effector T cells in lymphopenic hosts. Eur J Immunol. 2009;39:3121–3133. doi: 10.1002/eji.200939453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kline J, I, Brown E, Zha YY, Blank C, Strickler J, Wouters H, Zhang L, Gajewski TF. Homeostatic proliferation plus regulatory T-cell depletion promotes potent rejection of B16 melanoma. Clin Cancer Res. 2008;14:3156–3167. doi: 10.1158/1078-0432.CCR-07-4696. [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunological reviews. 2008;222:180–191. doi: 10.1111/j.1600-065X.2008.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172:989–999. doi: 10.4049/jimmunol.172.2.989. [DOI] [PubMed] [Google Scholar]
- 44.Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nature reviews. 2005;5:641–654. doi: 10.1038/nri1668. [DOI] [PubMed] [Google Scholar]
- 45.Bingisser RM, Tilbrook PA, Holt PG, Kees UR. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol. 1998;160:5729–5734. [PubMed] [Google Scholar]
- 46.Daud AI, Mirza N, Lenox B, Andrews S, Urbas P, Gao GX, Lee JH, Sondak VK, Riker AI, Deconti RC, Gabrilovich D. Phenotypic and functional analysis of dendritic cells and clinical outcome in patients with high-risk melanoma treated with adjuvant granulocyte macrophage colony-stimulating factor. J Clin Oncol. 2008;26:3235–3241. doi: 10.1200/JCO.2007.13.9048. [DOI] [PubMed] [Google Scholar]
- 47.Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58:49–59. doi: 10.1007/s00262-008-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nature reviews. 12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007;67:11021–11028. doi: 10.1158/0008-5472.CAN-07-2593. [DOI] [PubMed] [Google Scholar]