

Abstract
Background
A common and severe neural tube defect (NTD) phenotype, myelomeningocele (MM), results from the defective closure of the caudal end of the neural tube with herniation of the spinal cord and meninges through the vertebral column. The exact mechanisms for NTDs are unknown, but excessive oxidative stress, particularly in association with maternal diabetes, has been postulated as a mechanism for MM.
Methods
The SNPlex Genotyping (ABI, Foster City, CA) platform was used to investigate single nucleotide polymorphisms (SNPs) across the superoxide dismutase (SOD) 1 and 2 genes to assess their association with MM risk. The study population included 329 trio (affected child and both parents) and 281 duo (affected child and one parent) families. Only cases with documented MM were studied. Seventeen SNPs across the SOD1 and SOD2 genes met the quality control criteria to be considered for statistical analysis. Genetic association was assessed using the family-based Transmission Disequilibrium Test in PLINK.
Results
Four SNPs in the SOD1 gene (rs 202446, rs202447, rs4816405 and rs2070424) and one SNP in the SOD2 gene (rs5746105) appeared to be associated with MM risk in our population. After adjusting for multiple testing, these SNPs remained significant.
Conclusion
The study provides the first genetic evidence to support association of myelomeningocele with superoxide scavenging. The rare alleles of the five specific SNPs within SOD1 and SOD2 appear to confer a protective effect on the susceptibility for MM risk in the MM population tested. Further evaluation of the roles of superoxide scavenging and neural tube development is warranted.
INTRODUCTION
Congenital anomalies are the leading cause of infant death in the United States (Mathews and MacDorman, 2011). Neural tube defects (NTD) are a heterogeneous condition resulting from failure of normal neural tube closure during embryogenesis with a birth incidence of approximately 1 in 1,000 (Greene and others, 2009). A common and severe NTD phenotype, myelomeningocele (MM), results from the defective closure of the caudal end of the neural tube with herniation of the spinal cord and meninges through the vertebral column. The mortality rate for children with MM is increased over the general population and morbidity varies depending on the size and location of the defect (Fletcher and others, 2005).
The exact cause of NTDs is unknown, but the existence of the most common presentations within families as well as an association with known genetic syndromes raises the question of whether these phenotypes are related and due to a common underlying genetic susceptibility (Au and others, 2010; Greene and others, 2009). NTDs exhibit a multifactorial inheritance pattern, with suspected genetic and environmental factors. Identifying the hereditary basis of NTDs and the contributing genetic factors is thus critical for characterizing the interactions between genes and the environment. To date the search for genes responsible for NTDs has focused extensively on folate metabolism.
Numerous environmental factors have demonstrated an association with NTDs (Czeizel and Dudas, 1992; Finnell and others, 2000). An increased risk has been definitively associated with maternal diabetes and maternal obesity, both supporting an involvement with aberrant glucose metabolism (Hendricks and others, 2001; Shaw and others, 1996; Waller and others, 2007) However, the exact mechanism behind the increased risk for NTD is unknown. It has been postulated that increased oxidative stress associated with maternal diabetes influences a change in embryonic gene expression and enzymatic pathways that contribute to key developmental processes, resulting in an increased risk of congenital defects (Chang and others, 2003). The relative immaturity of antioxidant defense mechanisms during early embryonic development also contributes to the influences of reactive oxygen species on the developing embryo(Zaken and others, 2000; Zangen and others, 2002)
The superoxide dismutase (SOD) enzymes, that produce hydrogen peroxide from superoxide radicals, are among the body’s key protective mechanisms against reactive oxygen species. Studies have found that adding SOD or antioxidants to diabetic culture medium reduces the rate of embryonic anomalies (Pippenger, 2003). Alterations of SOD function are therefore a possible mechanism by which increased oxidative stress may contribute to the overall risk of congenital anomalies, including NTDs. Two types of SOD were considered in this study -copper-zinc superoxide dismutase (CuZnSOD or SOD1) located in the cytoplasm and manganese superoxide dismutase (MnSOD or SOD2) found in the mitochondria. The human SOD1 is 9,309 bp in length on chromosome 21q22.11, and SOD2 is 14,205 bp in length on chromosome 6q25.3. A diabetic sensitive animal model demonstrated that a reduced SOD1 activity was associated with an increased rate of NTDs, while fewer embryonic malformations were seen in embryos that over expressed SOD1 (Zangen and others, 2002). Data also demonstrates that a SOD2 deficiency results in alterations in several key signaling pathways, resulting in a lethal phenotype of Sod2−/− mice (Zhang and others, 2010).
Our objective was to explore genetic variants of SOD1 and SOD2 that may predispose an embryo to the development of neural tube defects, specifically MM. We analyzed 17 single nucleotide polymorphisms (SNPs) known among the SOD1 and SOD2 genes and their association with MM. We hypothesized that SNPs may result in a functionally inactive enzyme or altered gene expression or associate with other similar type of SNPs, inhibiting the pro-survival functions and ultimately leading to disconcerted cell death in the developing neural tube subsequently causing the NTD phenotype.
METHODS
Experimental protocols were approved by the Institutional Review Board (IRB) at the University of Texas Health Science Center at Houston.
Patients with MM and their parents were enrolled in the study from 1997 to 2006 (Au et al., 2008). Sites for enrollment of subjects included Spina Bifida and specialty clinics located at the Shriners Hospital for Children at Houston, Texas; the Shriners Hospital for Children at Los Angeles, California; the Shriners Hospital for Children at Lexington, Kentucky; Texas Children’s Hospital, Houston, Texas; and the Hospital for Sick Kids, Toronto, Canada.
Patient selection has previously been described (Au and others, 2008; Davidson and others, 2008; Tran and others, 2011). Briefly, patients born between 1955 and 2008 (ages 0.4–53 years) with a diagnosis of an isolated non-syndromic MM at birth were eligible for study inclusion. The study primarily recruited Hispanics of Mexican descent (54.8%) and North American Caucasians of European descent (37.1%). Other ethnicities included were African-Americans (3%), and Asian/Pacific Islanders (0.7%). As previously described (Au 2008, Tran 2010), 92 Hispanic individuals were recruited locally, and 92 Caucasian individuals were selected from the HD100CAU (Corriell Institute, Camden, NJ) to serve as genotyping quality controls.
DNA Collection and Preparation
As described previously by Au et al 2008, Davidson et al 2008, and Tran et al 2011, whole blood samples and/or saliva samples were collected from patients with MM and parents when feasible. Genomic DNA from blood cells was extracted using the Puregene DNA extraction Kit (Gentra Systems, Inc., Minneapolis, MN ) and from saliva using Oragene DNA Collection Kit (DNA Genotek, Inc., Ottawa, Ontario, Canada).
Genetic Marker SNP Selection
We searched the public database (http://www.ncbi.nlm.nih.gov/SNP/ and http://genome.cse.ucsc.edu/) for potentially functionally significant SNPs in our candidate genes. A total of 17 coding SNPs were chosen for evaluation, 7 involving SOD1 (NM_000454) and 10 involving SOD2 (NM_001024465), and are further described in Tables 1 and 2. These SNPs were selected primarily based on putative functional importance and also include tagSNPs. SNPs within the genes are in high linkage disequilibrium (LD) and the LD information is publicly available from HapMap and UCSC Genome Browser LD track (http://hapmap.ncbi.nlm.nih.gov/ AND http://genome.ucsc.edu/) Figure 1 demonstrates the SOD 1 gene, as well as the locus for each selected SNP.
Table 1.
Population frequency of rare allele of selected SOD1 SNPs
| Gene | Chr | SNP | chr loc | Signif. | A1 | A2 | CaucA. C n=290 f(A1) | CauA Pat n=458 F(A1) | MexA C n=184 f(A1) | MexA Pat n=672 f(A1) |
|---|---|---|---|---|---|---|---|---|---|---|
| SOD1 | 21 | rs202446 | 31950512 | Pro/enh | A | C | 0.11 | 0.05 | 0.13 | 0.03 |
| SOD1 | 21 | rs202447 | 31951244 | Pro/enh | T | G | 0.01 | 0.02 | 0.02 | 0.00 |
| SOD1 | 21 | rs11910115 | 31951960 | Pro/enh | C | A | 0.00 | 0.00 | 0.00 | 0.01 |
| SOD1 | 21 | rs4816405 | 31954871 | int1 | G | C | 0.07 | 0.10 | 0.36* | 0.30* |
| SOD1 | 21 | rs4998557 | 31956763 | int1 | A | G | 0.10 | 0.12 | 0.39* | 0.32* |
| SOD1 | 21 | rs2070424 | 31961191 | int3 | G | A | 0.08 | 0.08 | 0.36* | 0.31* |
| SOD1 | 21 | rs1041740 | 31962032 | int4 | T | C | 0.33 | 0.29 | 0.35 | 0.35 |
Notes: Chr – chromosome number, loc – location, signif – significance of SNP by function or location with respect to gene loci, int1-intron 1, A1 – rare allele, A2 – common allele, CauA – Caucasian American, MexA – Mexican American, Pat – patient with MM, C- SNP control population, n=number of chromosomes tested, f(A1) – frequency of rare allele.
significant difference (p<0.05) in the rare allele A1 frequency between Caucasian and Mexican Americans
Table 2.
Population frequency of rare allele of selected SOD2 SNPs
| Gene | Chr | SNP | chr loc | Signif. | A1 | A2 | CaucA. C n=290 f(A1) | CauA Pat. n=458 F(A1) | MexA C n=184 f(A1) | MexA Pat n=672 f(A1) |
|---|---|---|---|---|---|---|---|---|---|---|
| SOD2 | 6 | rs5746136 | 160023074 | int5 | A | G | 0.30 | 0.31 | 0.26 | 0.28 |
| SOD2 | 6 | rs2758331 | 160025060 | int4 | A | C | 0.45 | 0.50 | 0.58 | 0.52 |
| SOD2 | 6 | rs5746129 | 160025933 | p.W156R | T | C | 0.004 | 0.00 | 0.01 | 0.00 |
| SOD2 | 6 | rs2855116 | 160026115 | int3 | G | T | 0.46 | 0.48 | 0.55 | 0.53 |
| SOD2 | 6 | rs2070994 | 160029041 | int3 | A | T | 0.45 | 0.50 | 0.57 | 0.54 |
| SOD2 | 6 | rs2758339 | 160032574 | int2 | C | A | 0.53 | 0.50 | 0.42 | 0.43 |
| SOD2 | 6 | rs5746105 | 160032628 | int2 | C | T | 0.31 | 0.31 | 0.28 | 0.28 |
| SOD2 | 6 | rs4987023 | 160033683 | p.R76G | A | G | 0.00 | 0.00 | 0.02 | 0.00 |
| SOD2 | 6 | rs5746097 | 160033712 | p.V66E | T | A | 0.01 | 0.00 | 0.00 | 0.00 |
| SOD2 | 6 | rs1799725 | 160033862 | p.A16V | T | C | 0.18 | 0.14 | 0.10 | 0.11 |
Notes: Chr – chromosome number, loc – location, signif – significance of SNP by function or location with respect to gene loci, int-intron, A1 – rare allele, A2 – common allele, CauA – Caucasian American, MexA – Mexican American, Pat – patient with MM, C- SNP control population, n=number of chromosomes tested, f(A1) – frequency of rare allele
Figure 1.

SOD1 gene with SNPs selected for study. Cartoon of gene structure of SOD1 is shown as a line with arrow in the direction of gene from promoter/enhancer region to exons 1 through 5. Untranslated region of exons are shown in black boxes and coding sequences of exons are shown in open boxes. SNPs are shown on top of cartoon with locations in base-pair relative to the transcription start site nucleotide 1 at exon 1 shown beneath the cartoon. SNPs upstream in the promoter enhancer regions are shown with location in bp of negative values.
From the SOD2 gene, there were a 10 SNPs selected for association study. They are further described in Table 2. Figure 2 demonstrates the SOD2 gene, as well as the locus for each selected SNP.
Figure 2.

SOD2 gene with SNPs selected for study. Cartoon of gene structure of SOD2 is shown as a line with arrow in the direction of gene from promoter enhancer region to exons 1 through 6. Untranslated region of exons are shown in black boxes and coding sequences of exons are shown in open boxes. SNPs are shown on top of cartoon with locations in base-pair relative to the transcription start site nucleotide 1 at exon 1 shown underneath the cartoon.
SNPlex Genotyping
As described previously (Davidson and others, 2008), SNP genotyping was performed using the SNPlex Genotyping platform (Applied Biosystems, Foster City, CA). The genotypes initially assigned by the GeneMapper 4.7 software were examined by at least two investigators before exporting and compiling for statistical analyses. Duplicate samples were included, and CEPH samples were selected from the HapMap project to verify genotype call accuracy.
Statistical Analysis
Allele frequencies, Hardy-Weinberg equilibrium, and a triad-based association analyses were performed using a transmission disequilibrium test (TDT) in the PLINK v1.07 (http://pngu.mgh.harvard.edu/purcell/plink/) whole genome association analysis toolset (Purcell and others, 2007). In addition, we also performed reconstruction-combined transmission/disequilibrium test (RC-TDT) to analyze incomplete trios (Knapp 1999). To adjust for multiple testing, we chose to use the PLINK tool package to evaluate the significance levels of our findings from the empirical p-value instead of the overly stringent Bonferroni method (Churchill and Doerge, 1994; Perneger, 1998). Hardy-Weinberg equilibrium tests were done separately for each ethnicity. All SNPs were in Hardy-Weinberg equilibrium.
RESULTS
Overall, there were 329 trios (affected child and both parents) and 279 duos (affected child and one parent) whose DNA samples were available for testing. The patient demographics have been previously described and are available for review (Tran and others, 2011).
The majority of the 17 SNPs in the SOD1 and SOD2 loci examined did not show a significant difference in the frequency of rare alleles between Caucasian and Mexican subjects in the study (Tables 1 & 2). However, a significant difference between Caucasian and Mexican patients was observed for three SNPs (rs4816405, rs4998557 and rs2070424) in SOD1as shown in Table 1.
After performing the PLINK TDT, four SNPs of the SOD1 gene were found to have a statistically significant association with MM (p<0.05) among all patients, and are indicated by bold font as further described in Table 3. SNP rs 202446 and rs 202447 are both located in the promoter/enhancer regions of the SOD1 gene. SNP rs4816405 and rs2070424 are located in intron 1 and intron 3 respectively of the SOD1 gene. Table 4 demonstrates the SNP association significance when stratified by ethnicity for SOD1. After performing the PLINK TDT, one SNP of the SOD2 gene was found to have a statistically significant association with MM (p<0.05) among all patients, and is indicated by bold font as shown in Table 5. SNP rs5746105 is located in an intronic region of the gene. Table 6 demonstrates the SNP association significance when stratified by ethnicity for SOD2. Empirical probability tests by permutation of all SNPs show significant association with MM for the same five SNPs (Tables 3 & 5).
Table 3.
TDT results on SOD1 SNPs association with MM among all patients
| CHR | SNP | A1 | A2 | T | U | OR | CHISQ | P | EMP-P |
|---|---|---|---|---|---|---|---|---|---|
| SOD1 | rs202446 | A | C | 20 | 40 | 0.5000 | 6.6670 | 0.0098 | 0.0133 |
| SOD1 | rs202447 | T | G | 6 | 20 | 0.3000 | 7.5380 | 0.0060 | 0.0154 |
| SOD1 | rs11910115 | C | A | 6 | 7 | 0.8571 | 0.0769 | 0.7815 | 1.0000 |
| SOD1 | rs4816405 | G | C | 85 | 122 | 0.6967 | 6.6140 | 0.0101 | 0.0125 |
| SOD1 | rs4998557 | A | G | 77 | 95 | 0.8105 | 1.8840 | 0.1699 | 0.1629 |
| SOD1 | rs2070424 | G | A | 69 | 104 | 0.6635 | 7.0810 | 0.0078 | 0.0071 |
| SOD1 | rs1041740 | T | C | 142 | 163 | 0.8712 | 1.4460 | 0.2292 | 0.2304 |
Notes: A1 – rare allele, A2 – common allele, T – number of A1 Transmitted, U – number of A1 not transmitted, OR – Odds Ratio, CHISQ – chi square, p-value – PLINK TDT probability, EMP-p – p-value from empirical probability test by permutation. Significant SNPs are bolded.
Table 4.
TDT results on SOD1 SNP associations stratified by ethnicity
| CHR | SNP | BP | A1 | A2 | T | U | OR | CHISQ | P |
|---|---|---|---|---|---|---|---|---|---|
| Caucasian Families | |||||||||
|
| |||||||||
| SOD1 | rs202446 | 31950512 | A | C | 12 | 22 | 0.5455 | 2.9410 | 0.0864 |
| SOD1 | rs202447 | 31951244 | T | G | 5 | 8 | 0.6250 | 0.6923 | 0.4054 |
| SOD1 | rs11910115 | 31951960 | C | A | 0 | 5 | 0.0000 | 5.0000 | 0.0254 |
| SOD1 | rs4816405 | 31954871 | G | C | 16 | 26 | 0.6154 | 2.3810 | 0.1228 |
| SOD1 | rs4998557 | 31956763 | A | G | 19 | 20 | 0.9500 | 0.0256 | 0.8728 |
| SOD1 | rs2070424 | 31961191 | G | A | 10 | 20 | 0.5000 | 3.3330 | 0.0679 |
| SOD1 | rs1041740 | 31962032 | T | C | 53 | 86 | 0.6163 | 7.8350 | 0.0051 |
|
| |||||||||
| Mexican Families | |||||||||
|
| |||||||||
| SOD1 | rs202446 | 31950512 | A | C | 7 | 16 | 0.4375 | 3.5220 | 0.0606 |
| SOD1 | rs202447 | 31951244 | T | G | 1 | 11 | 0.0909 | 8.3330 | 0.0039 |
| SOD1 | rs11910115 | 31951960 | C | A | 4 | 0 | NA | 4.0000 | 0.0455 |
| SOD1 | rs4816405 | 31954871 | G | C | 67 | 89 | 0.7528 | 3.1030 | 0.0788 |
| SOD1 | rs4998557 | 31956763 | A | G | 55 | 71 | 0.7746 | 2.0320 | 0.1540 |
| SOD1 | rs2070424 | 31961191 | G | A | 59 | 79 | 0.7468 | 2.8990 | 0.0887 |
| SOD1 | rs1041740 | 31962032 | T | C | 81 | 69 | 1.1740 | 0.9600 | 0.3272 |
Notes: A1 – rare allele, A2 – common allele, T – number of A1 Transmitted, U – number of A1 not transmitted, OR – Odds Ratio, CHISQ – chi square, p-value – PLINK TDT probability, p – p-value from empirical probability test by permutation.
Table 5.
TDT results on SOD2 SNPs association with MM among all patients
| Gene | SNP | A1 | A2 | T | U | OR | CHISQ | P | EMP-P |
|---|---|---|---|---|---|---|---|---|---|
| SOD2 | rs5746136 | A | G | 104 | 122 | 0.8525 | 1.4340 | 0.2312 | 0.2311 |
| SOD2 | rs2758331 | A | C | 119 | 106 | 1.1230 | 0.7511 | 0.3861 | 0.4147 |
| SOD2 | rs5746129 | T | C | 0 | 2 | 0.0000 | 2.0000 | 0.1573 | 0.5018 |
| SOD2 | rs2855116 | T | G | 108 | 121 | 0.8926 | 0.7380 | 0.3903 | 0.4289 |
| SOD2 | rs2070994 | T | A | 137 | 152 | 0.9013 | 0.7785 | 0.3776 | 0.3915 |
| SOD2 | rs2758339 | A | C | 144 | 152 | 0.9474 | 0.2162 | 0.6419 | 0.6812 |
| SOD2 | rs5746105 | C | T | 92 | 124 | 0.7419 | 4.7410 | 0.0295 | 0.0330 |
| SOD2 | rs4987023 | A | G | 0 | 1 | 0.0000 | 1.0000 | 0.3173 | 1.0000 |
| SOD2 | rs5746097 | T | A | 0 | 3 | 0.0000 | 3.0000 | 0.0833 | 0.2515 |
| SOD2 | rs1799725 | T | C | 20 | 33 | 0.6061 | 3.1890 | 0.0742 | 0.0925 |
Notes: A1 – rare allele, A2 – common allele, T – number of A1 Transmitted, U – number of A1 not transmitted, OR – Odds Ratio, CHISQ – chi square, p-value – PLINK TDT probability, EMP-p – p-value from empirical probability test by permutation. Significant SNP is bolded.
Table 6.
TDT Results on SOD2 SNP associations stratified by ethnicity
| CHR | SNP | BP | A1 | A2 | T | U | OR | CHISQ | P |
|---|---|---|---|---|---|---|---|---|---|
| Caucasian Families | |||||||||
|
| |||||||||
| SOD2 | rs5746136 | 160023074 | A | G | 51 | 51 | 1.0000 | 0.0000 | 1.0000 |
| SOD2 | rs2758331 | 160025060 | C | A | 50 | 53 | 0.9434 | 0.0874 | 0.7675 |
| SOD2 | rs5746129 | 160025933 | T | C | 0 | 1 | 0.0000 | 1.0000 | 0.3173 |
| SOD2 | rs2855116 | 160026115 | T | G | 56 | 53 | 1.0570 | 0.0826 | 0.7738 |
| SOD2 | rs2070994 | 160029041 | T | A | 68 | 66 | 1.0300 | 0.0299 | 0.8628 |
| SOD2 | rs2758339 | 160032574 | A | C | 68 | 67 | 1.0150 | 0.0074 | 0.9314 |
| SOD2 | rs5746105 | 160032628 | C | T | 47 | 62 | 0.7581 | 2.0640 | 0.1508 |
| SOD2 | rs4987023 | 160033683 | A | G | 0 | 0 | NA | NA | NA |
| SOD2 | rs5746097 | 160033712 | T | A | 0 | 2 | 0.0000 | 2.0000 | 0.1573 |
| SOD2 | rs1799725 | 160033862 | T | C | 10 | 13 | 0.7692 | 0.3913 | 0.5316 |
|
| |||||||||
| Mexican Families | |||||||||
|
| |||||||||
| SOD2 | rs5746136 | 160023074 | A | G | 47 | 59 | 0.7966 | 1.3580 | 0.2438 |
| SOD2 | rs2758331 | 160025060 | C | A | 54 | 61 | 0.8852 | 0.4261 | 0.5139 |
| SOD2 | rs5746129 | 160025933 | T | C | 0 | 0 | NA | NA | NA |
| SOD2 | rs2855116 | 160026115 | T | G | 49 | 57 | 0.8596 | 0.6038 | 0.4371 |
| SOD2 | rs2070994 | 160029041 | T | A | 65 | 76 | 0.8553 | 0.8582 | 0.3543 |
| SOD2 | rs2758339 | 160032574 | A | C | 71 | 73 | 0.9726 | 0.0278 | 0.8676 |
| SOD2 | rs5746105 | 160032628 | C | T | 40 | 53 | 0.7547 | 1.8170 | 0.1776 |
| SOD2 | rs4987023 | 160033683 | A | G | 0 | 1 | 0.0000 | 1.0000 | 0.3173 |
| SOD2 | rs5746097 | 160033712 | T | A | 0 | 0 | NA | NA | NA |
| SOD2 | rs1799725 | 160033862 | T | C | 10 | 14 | 0.7143 | 0.6667 | 0.4142 |
Notes: A1 – rare allele, A2 – common allele, T – number of A1 Transmitted, U – number of A1 not transmitted, OR – Odds Ratio, CHISQ – chi square, p-value – PLINK TDT probability, p – p-value from empirical probability test by permutation.
We further investigated the functional significance associated with the alleles of the five SNPs we identified. We explored the correlation of the genotypes of SOD1 (NM_000454) and SOD2 (NM_000636) in CEPH cell lines with the expression profile of the genes from the Gene Expression Omnibus (GEO) dataset GDS1048 (Monks and others, 2004). Our laboratory previously genotyped the CEPH cell lines. We then used our data to generate the genotypes of the SNPs with demonstrated association with MM on the four SNPs (rs202446, rs202447, rs4816405, and rs2070424) in SOD1 in 69 CEPH cell lines, and one SNP of the SOD2 gene in 61 CEPH cell lines for evaluation. Analysis of rare alleles of individual SNPs to SOD1 expression levels did not show significance for three SNPs and a suggestive significance for rs202446. The TDT results suggest a protective effect for all of the rare alleles of the four significant SNPs for SOD1, so we compared the SOD1 expression levels between the CEU samples that have no rare alleles versus the group that has one or multiple rare alleles to increase the power of the analysis. Forty-seven CEPH samples with no rare allele for the four significant SNPs have an average SOD1 mRNA log2 expression value of −0.0038 compared to the −0.0417 for the 22 samples with at least one rare allele (p=0.0462 by Student t-test) (Figure 3). Thirty-one samples without the rare allele C of rs5746105 have an average SOD2 mRNA log2 expression value of 0.0752 compared to 0.0125 for the 30 samples have rare allele C (p=0.003) (Figure 4). The difference in sample numbers (69 for SOD1 and 61 for SOD2) is due to different genotyping success rates and overlapping between the 69 and 61 samples.
Figure 3.

Correlation of allele genotype of significant SNPs with SOD1 mRNA expression profiles. Expression profiles of SOD1 mRNA for CEPH cell lines were downloaded from Gene Expression Omnibus (GEO) dataset GDS1048 (Monks and others, 2004). The genotypes of the selected SNPs of SOD1 in the study for CEPH DNA we obtained were generated by SNPlex platform and 69 CEPH samples were matched to the GDS1048 dataset for analysis. We are comparing a group of 22 samples present with one or more rare alleles of the four significant SNPs (rs202446, rs202447, rs4816405, and rs2070424) to another group of 47 samples without rare allele in their genotypes. Expression level of SOD1 (NM_000454) mRNA was normalized using the expression level of GAPDH mRNA of individual sample before analysis. We perform Student t-test to compare expression of the two groups and a p-value <0.05 is considered significant.
Figure 4.
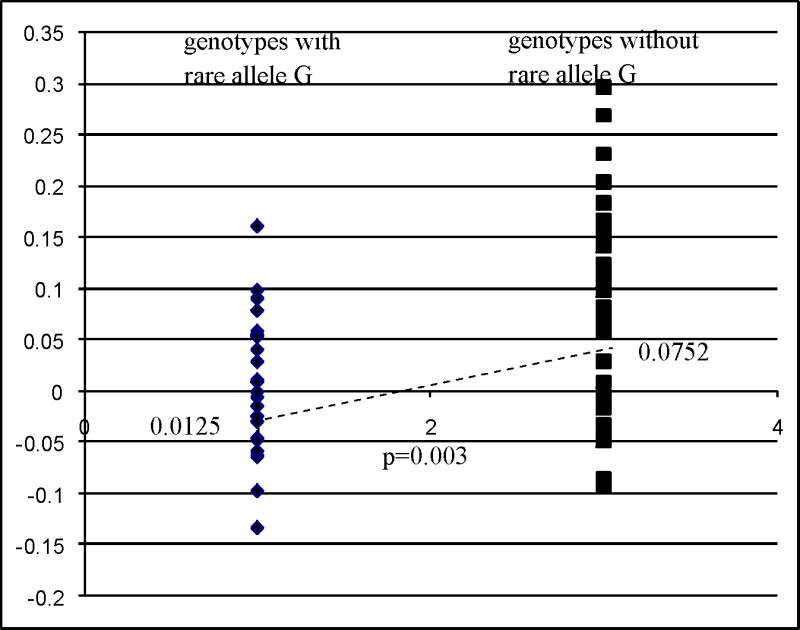
Correlation of allele genotype of significant SNPs with SOD2 mRNA expression profiles. Expression profiles of SOD2 mRNA for CEPH cell lines were downloaded from Gene Expression Omnibus (GEO) dataset GDS1048 (Monks and others, 2004). The genotypes of the selected SNPs of SOD2 in the study for CEPH DNA we obtained were generated by SNPlex platform and 61 CEPH samples were matched to the GDS1048 dataset for analysis. We are comparing a group of 30 samples present with one or more rare alleles of SNP rs5746105 to another group of 31 samples without rare allele in their genotypes. Expression level of SOD2 (NM_000636) mRNA was normalized using the expression level of GAPDH mRNA of individual sample before analysis. We perform Student t-test to compare expression of the two groups and a p-value <0.05 is considered significant.
DISCUSSION
Of the 17 genetic variants selected from the SOD1 and SOD2 genes, five SNPs met our threshold for significance for an association with MM among all patients. We additionally investigated the functional significance of these allelic variants, and noted that the rare alleles were under-transmitted to the affected patients. Our data suggests that the rare alleles confer a protective benefit against the development of MM. Rare alleles of the four SNPs (rs202446, rs202447, rs4816405 and rs2070424) are suggested to associate with a higher expression of SOD1 mRNA in the CEPH cell lines examined consistent with the protective benefit we observed. However, it is not known how the lower expression associated rare allele C of rs5746105 protects against MM development. Further functional studies are required to elucidate the correlation of SNP genotype with SOD activity.
The SNPs that demonstrated an association with MM in this study are located within the proximity of transcription regulatory sites implicated by the Encyclopedia of DNA Elements (ENCODE) integrated regulation studies (i.e. ENCODE Transcription, Enhancer/Promoter associated histone marks, DNase hypersensitivity cluster, and transcription factor ChIP-Seq that are available at the University of California Santa Cruz Genome Browser [UCSC Genome Browser] http://genome.ucsc.edu/cgi-bin/hgGateway). For example, rs202446 is approximately 2.5Kbp upstream of the SOD1 exon 1 transcription start site together with rs202447 and flanks a 733bp region that is hypersensitive to DNase I and shown to be target of several transcription factors that regulate cell growth and development (e.g. JunD and PAX5). Similarly, rs4816405 is located in a c-Myc transcription regulatory target site and rs2070424 locates in a DNase hypersensitivity site and these SNPs are not in LD. In addition, the reference allele of these five SNPs appears to be highly conserved among primates and mammals as shown by the UCSC Genome Browser 44 vertebrate alignment tracks. There is no evidence to suggest the four SNPs in SOD1 are in LD. Further transcription regulation assays are necessary to demonstrate the role of these SNPs and/or the proximal regions of expression with the SOD genes. We must also acknowledge the possibility that the associations noted may be due to LD between those SNPs we selected for study and for SNPs not genotyped (or not yet known).
Our report is the first to identify superoxide scavenging genes associated with MM. While previous investigators have identified a role for glutathione peroxidase as well as catalase in the development of NTDs (Sivan and others, 1997), our study did not find an association with these enzymes (unpublished data). As discussed previously, copper and zinc are necessary for normal SOD1 function. In a mouse model, Hawk et al. (2003) demonstrated that copper deficiency was associated with higher frequency malformed embryos, with considerably reduced SOD activity and increased reactive oxygen species concentrations. Their findings support the contribution of dietary deficiencies to increased oxidative stress leading to congenital anomalies (Hawk and others, 2003). Human studies have also demonstrated a role for SOD enzyme cofactors deficiencies in association with NTDs. In 2004, a study of the effect of pollution in Mexico demonstrated that although high levels of lead and cadmium were common among newborns with NTDs, after multivariate logistic regression analyses, only low zinc was found to be associated with NTDs (OR 5.0, 95% CI=1.07–23.0, p<0.04) (Carrillo-Ponce Mde and others, 2004). A 2008 case control study evaluating copper and zinc levels in umbilical cord blood of newborn babies, also demonstrated that low zinc levels and high copper levels, were associated with NTDs (Zeyrek and others, 2009). More recently, Huang and colleagues (2011) demonstrated that copper levels in soil (along with other elements), were associated with NTDs among babies born in the Shanxi province region of China (Huang and others, 2011). The finding of SOD1 associated with MM is consistent with the effect of Cu/Zn and NTDs in both animal and human experience.
SOD2 requires the elements manganese and iron as co-factors to function normally. Previous studies have demonstrated iron deficiency to be associated with NTDs. In a 2004 Dutch case control study, investigators demonstrated that among a population of women with children known to have non-syndromic MM, there was a significant association between low dietary iron intake and development of MM (Groenen and others, 2004). After adjusting for dietary folic acid intake, there remained an association between low dietary iron and the development of MM (Groenen and others, 2004). In a 2010 mouse model, investigators demonstrated iron’s critical role in neural tube development (Mao and others, 2010). Among mice with mutant iron exporter Fpn1 genes, there was a gradient of progressively more severe NTDs observed in embryos directly dependent on the residual activity of the iron transport function (Mao and others, 2010).
Our findings are also consistent with existing animal models that the SOD genes are associated with the development of MM. In 2002, it was demonstrated that impaired expression of free radical scavenging enzymes may be genetically determined (Zangen and others, 2002). Reduced SOD activity was observed in malformed embryos of diabetic mothers. The role of antioxidant defense mechanism in diabetes induced anomalies was studied in the Cohen rat model – a genetic model of nutritionally induced type 2 diabetes mellitus. Diabetes-sensitive (CDs) and diabetes-resistant (CDr) rats were fed a regular diet (RD) or a diabetogenic high-sucrose diet (HSD) and monitored for congenital anomalies. The CDr embryos were normal when fed RD or HSD. In contrast, 7% CDs embryos exhibited NTDs when fed RD, increasing to 27% on HSD (Zangen and others, 2002).
Interestingly, in 2008, it was demonstrated that the spatio-temporal pattern of the neuronal cells of the brain and spinal cord have high SOD1 activity during mouse embryogenesis. Their findings indicated that SOD1 has an important role as a free radical scavenger in the normal development of CNS (Yon and others, 2008). More recently, Zhang and others (2010) investigated changes of the mitochondrial ROS balance and its impact on cellular signaling. Homozygous Sod2 knockout mice were found to be a lethal phenotype in the neonatal period, demonstrating a critical role of Sod2 in early development and survival. The loss of Sod2 dramatically altered several markers involved in cell proliferation and growth (Zhang and others, 2010). The most significant strength of our study is the large population size, and a significant representation of Hispanic individuals of Mexican descent. In the United States, Hispanics of Mexican descent is at the highest risk of all ethnic groups for the development of NTDs (Canfield et al 1996). An additional strength of our study is the use of TDT as an analytic tool for familial association, and eliminating complications of population structure in case versus control analyses.
Though only available for approximately half of the study cohort (262 mothers), we also collected environmental and nutritional data on the patient’s mothers. Of those with available data, greater than 50% were overweight and approximately 25% were obese as defined in the 1998 evidence report from the National Heart Lung and Blood Institute and National Institute of Diabetes and Digestive and Kidney Disease. More patients’ mothers recruited in the Houston area were overweight or obese. Overall, daily food consumption of affected patients’ mothers were higher than the average adult female diet (United States. Dept. of Agriculture. Human Nutrition Information Service. Dietary Guidelines Advisory Committee. and United States. Agricultural Research Service., 2010) and this was observed more frequently for patients’ mothers from the Houston area.
In summary, our study is the first investigation of high density SNP screen evaluating SOD1 and SOD2 in a large population including approximately 55% Hispanics of Mexican descent. After adjusting for multiple comparisons, five SNPs across the SOD1 and SOD2 genes were found to be associated with a protective effect for vulnerability to MM risk in our population. Our study results suggest the needs for replication studies using these five SNPs by other human NTDs research groups in their study cohorts. We next plan to sequence SOD1, SOD2 and SOD3 exons of our study subjects to identify novel variants that may contribute to non-syndromic myelomeningocele.
Acknowledgments
We thank the patients and their families for their participation in this study and C Tsai, and P.X. Tran for technical assistance.
This research was supported in part by grant P01 HD35946-06A2, Spina Bifida: Cognitive and Neurobiological Variability, from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NICHD or the National Institutes of Health.
This work was also supported by Grants from Shriners Hospital for Children (Grant # 8580) to HN and KSA.
Footnotes
The authors have no conflict of interest to declare.
References
- Au KS, Ashley-Koch A, Northrup H. Epidemiologic and genetic aspects of spina bifida and other neural tube defects. Dev Disabil Res Rev. 2010;16(1):6–15. doi: 10.1002/ddrr.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au KS, Tran PX, Tsai CC, O’Byrne MR, Lin JI, Morrison AC, Hampson AW, Cirino P, Fletcher JM, Ostermaier KK, Tyerman GH, Doebel S, Northrup H. Characteristics of a spina bifida population including North American Caucasian and Hispanic individuals. Birth Defects Res A Clin Mol Teratol. 2008;82(10):692–700. doi: 10.1002/bdra.20499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Ponce Mde L, Martinez-Ordaz VA, Velasco-Rodriguez VM, Hernandez-Garcia A, Hernandez-Serrano MC, Sanmiguel F. Serum lead, cadmium, and zinc levels in newborns with neural tube defects from a polluted zone in Mexico. Reprod Toxicol. 2004;19(2):149–154. doi: 10.1016/j.reprotox.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Chang TI, Horal M, Jain SK, Wang F, Patel R, Loeken MR. Oxidant regulation of gene expression and neural tube development: Insights gained from diabetic pregnancy on molecular causes of neural tube defects. Diabetologia. 2003;46(4):538–545. doi: 10.1007/s00125-003-1063-2. [DOI] [PubMed] [Google Scholar]
- Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138(3):963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czeizel AE, Dudas I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med. 1992;327(26):1832–1835. doi: 10.1056/NEJM199212243272602. [DOI] [PubMed] [Google Scholar]
- Davidson CM, Northrup H, King TM, Fletcher JM, Townsend I, Tyerman GH, Au KS. Genes in glucose metabolism and association with spina bifida. Reprod Sci. 2008;15(1):51–58. doi: 10.1177/1933719107309590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnell RH, Gelineau-van Waes J, Bennett GD, Barber RC, Wlodarczyk B, Shaw GM, Lammer EJ, Piedrahita JA, Eberwine JH. Genetic basis of susceptibility to environmentally induced neural tube defects. Ann N Y Acad Sci. 2000;919:261–277. doi: 10.1111/j.1749-6632.2000.tb06886.x. [DOI] [PubMed] [Google Scholar]
- Fletcher JM, Copeland K, Frederick JA, Blaser SE, Kramer LA, Northrup H, Hannay HJ, Brandt ME, Francis DJ, Villarreal G, Drake JM, Laurent JP, Townsend I, Inwood S, Boudousquie A, Dennis M. Spinal lesion level in spina bifida: a source of neural and cognitive heterogeneity. J Neurosurg. 2005;102(3 Suppl):268–279. doi: 10.3171/ped.2005.102.3.0268. [DOI] [PubMed] [Google Scholar]
- Greene ND, Stanier P, Copp AJ. Genetics of human neural tube defects. Hum Mol Genet. 2009;18(R2):R113–129. doi: 10.1093/hmg/ddp347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenen PM, van Rooij IA, Peer PG, Ocke MC, Zielhuis GA, Steegers-Theunissen RP. Low maternal dietary intakes of iron, magnesium, and niacin are associated with spina bifida in the offspring. J Nutr. 2004;134(6):1516–1522. doi: 10.1093/jn/134.6.1516. [DOI] [PubMed] [Google Scholar]
- Hawk SN, Lanoue L, Keen CL, Kwik-Uribe CL, Rucker RB, Uriu-Adams JY. Copper-deficient rat embryos are characterized by low superoxide dismutase activity and elevated superoxide anions. Biol Reprod. 2003;68(3):896–903. doi: 10.1095/biolreprod.102.009167. [DOI] [PubMed] [Google Scholar]
- Hendricks KA, Nuno OM, Suarez L, Larsen R. Effects of hyperinsulinemia and obesity on risk of neural tube defects among Mexican Americans. Epidemiology. 2001;12(6):630–635. doi: 10.1097/00001648-200111000-00009. [DOI] [PubMed] [Google Scholar]
- Huang J, Wu J, Li T, Song X, Zhang B, Zhang P, Zheng X. Effect of exposure to trace elements in the soil on the prevalence of neural tube defects in a high-risk area of China. Biomed Environ Sci. 2011;24(2):94–101. doi: 10.3967/0895-3988.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Knapp M. The transmission/disequilibrium test and parental-genotype reconstruction: the reconstruction-combined transmission/disequilibrium test. American Journal of Human Genetics. 1999;64:861–870. doi: 10.1086/302285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, McKean DM, Warrier S, Corbin JG, Niswander L, Zohn IE. The iron exporter ferroportin 1 is essential for development of the mouse embryo, forebrain patterning and neural tube closure. Development. 2010;137(18):3079–3088. doi: 10.1242/dev.048744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews TJ, MacDorman MF. Infant mortality statistics from the 2007 period linked birth/infant death data set. Natl Vital Stat Rep. 2011;59(6):1–30. [PubMed] [Google Scholar]
- Monks SA, Leonardson A, Zhu H, Cundiff P, Pietrusiak P, Edwards S, Phillips JW, Sachs A, Schadt EE. Genetic inheritance of gene expression in human cell lines. Am J Hum Genet. 2004;75(6):1094–1105. doi: 10.1086/426461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Heart Lung and Blood Institute., National Institute of Diabetes and Digestive and Kidney Diseases (U.S.) Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults: the evidence report. xxx. Bethesda, Md.?: National Institutes of Health, National Heart, Lung, and Blood Institute; 1998. p. 228. http://www.nhlbi.nih.gov/guidelines/obesity/ob_gdlns.pdf. [Google Scholar]
- Perneger TV. What’s wrong with Bonferroni adjustments. BMJ. 1998;316(7139):1236–1238. doi: 10.1136/bmj.316.7139.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pippenger CE. Pharmacology of neural tube defects. Epilepsia. 2003;44(Suppl 3):24–32. doi: 10.1046/j.1528-1157.44.s3.3.x. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw GM, Velie EM, Schaffer D. Risk of neural tube defect-affected pregnancies among obese women. JAMA. 1996;275(14):1093–1096. doi: 10.1001/jama.1996.03530380035028. [DOI] [PubMed] [Google Scholar]
- Sivan E, Lee YC, Wu YK, Reece EA. Free radical scavenging enzymes in fetal dysmorphogenesis among offspring of diabetic rats. Teratology. 1997;56(6):343–349. doi: 10.1002/(SICI)1096-9926(199712)56:6<343::AID-TERA1>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Tran PX, Au KS, Morrison AC, Fletcher JM, Ostermaier KK, Tyerman GH, Northrup H. Association of retinoic acid receptor genes with meningomyelocele. Birth Defects Res A Clin Mol Teratol. 2011;91(1):39–43. doi: 10.1002/bdra.20744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- United States. Dept. of Agriculture. Human Nutrition Information Service. Dietary Guidelines Advisory Committee., United States. Agricultural Research Service. Report of the Dietary Guidelines Advisory Committee on the dietary guidelines for Americans, 2010: to the Secretary of Agriculture and the Secretary of Health and Human Services. vi. Washington, D.C: United States Dept. of Agriculture: United States Dept. of Health and Human Services; 2010. p. 445. [Google Scholar]
- Waller DK, Shaw GM, Rasmussen SA, Hobbs CA, Canfield MA, Siega-Riz AM, Gallaway MS, Correa A. Prepregnancy obesity as a risk factor for structural birth defects. Arch Pediatr Adolesc Med. 2007;161(8):745–750. doi: 10.1001/archpedi.161.8.745. [DOI] [PubMed] [Google Scholar]
- Yon JM, Baek IJ, Lee SR, Jin Y, Kim MR, Nahm SS, Kim JS, Ahn B, Lee BJ, Yun YW, Nam SY. The spatio-temporal expression pattern of cytoplasmic Cu/Zn superoxide dismutase (SOD1) mRNA during mouse embryogenesis. J Mol Histol. 2008;39(1):95–103. doi: 10.1007/s10735-007-9134-1. [DOI] [PubMed] [Google Scholar]
- Zaken V, Kohen R, Ornoy A. The development of antioxidant defense mechanism in young rat embryos in vivo and in vitro. Early Pregnancy. 2000;4(2):110–123. [PubMed] [Google Scholar]
- Zangen SW, Yaffe P, Shechtman S, Zangen DH, Ornoy A. The role of reactive oxygen species in diabetes-induced anomalies in embryos of Cohen diabetic rats. Int J Exp Diabetes Res. 2002;3(4):247–255. doi: 10.1080/15604280214933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeyrek D, Soran M, Cakmak A, Kocyigit A, Iscan A. Serum copper and zinc levels in mothers and cord blood of their newborn infants with neural tube defects: a case-control study. Indian Pediatr. 2009;46(8):675–680. [PubMed] [Google Scholar]
- Zhang Y, Zhang HM, Shi Y, Lustgarten M, Li Y, Qi W, Zhang BX, Van Remmen H. Loss of manganese superoxide dismutase leads to abnormal growth and signal transduction in mouse embryonic fibroblasts. Free Radic Biol Med. 2010;49(8):1255–1262. doi: 10.1016/j.freeradbiomed.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]