

Abstract
Astrocytes are the most abundant cell type in the brain and play a critical role in maintaining healthy nervous tissue. In Alzheimer's disease (AD) and most other neurodegenerative disorders, many astrocytes convert to a chronically “activated” phenotype characterized by morphologic and biochemical changes that appear to compromise protective properties and/or promote harmful neuroinflammatory processes. Activated astrocytes emerge early in the course of AD and become increasingly prominent as clinical and pathological symptoms progress, but few studies have tested the potential of astrocyte-targeted therapeutics in an intact animal model of AD. Here, we used adeno-associated virus (AAV) vectors containing the astrocyte-specific Gfa2 promoter to target hippocampal astrocytes in APP/PS1 mice. AAV–Gfa2 vectors drove the expression of VIVIT, a peptide that interferes with the immune/inflammatory calcineurin/NFAT (nuclear factor of activated T-cells) signaling pathway, shown by our laboratory and others to orchestrate biochemical cascades leading to astrocyte activation. After several months of treatment with Gfa2–VIVIT, APP/PS1 mice exhibited improved cognitive and synaptic function, reduced glial activation, and lower amyloid levels. The results confirm a deleterious role for activated astrocytes in AD and lay the groundwork for exploration of other novel astrocyte-based therapies.
Introduction
Astrocyte activation, characterized by hypertrophic somata and processes, is pervasive in most neurodegenerative conditions, including Alzheimer's disease (AD) (Verkhratsky et al., 2010; Vincent et al., 2010; Sidoryk-Wegrzynowicz et al., 2011). Signs of astrocyte activation appear early in the clinical progression of AD (Schipper et al., 2006; Owen et al., 2009; Carter et al., 2012) and are especially conspicuous in later disease stages when amyloid and neurofibrillary tangle pathology are extensive. Despite the clear physical association between activated astrocytes and AD biomarkers, the functional impact of these cells and their therapeutic potential have remained elusive. However, recent advances in cell-type-specific gene delivery techniques have helped identify unique beneficial and detrimental roles of astrocytes in other neurodegenerative disorders (Sofroniew, 2009), suggesting that astrocytic signaling cascades can be selectively exploited for treating AD.
Astrocytes host a complex network of signaling pathways, providing an abundance of potential molecular targets. Many activated astrocytes in AD brain tissue and AD mouse models express high levels of calcineurin (CN) (Norris et al., 2005; Celsi et al., 2007; Abdul et al., 2009), a protein phosphatase widely known for orchestrating immune/inflammatory responses (Im and Rao, 2004). Mounting evidence suggests that CN signaling is increased during AD (Liu et al., 2005; Abdul et al., 2009; Wu et al., 2010; Mohmmad Abdul et al., 2011) and linked to numerous disease biomarkers, including synapse loss/altered plasticity, reduced neuronal viability, and impaired cognition (Shankar et al., 2007; Agostinho et al., 2008; Reese et al., 2008; Dineley et al., 2010; Mohmmad Abdul et al., 2011; Hudry et al., 2012). In astrocytes, CN strongly promotes the activated phenotype through dephosphorylation of NFAT (nuclear factor of activated T-cells) transcription factors involved in cytokine production, phenotype switching, and many other functions (Crabtree and Olson, 2002; Horsley and Pavlath, 2002). CN/NFAT signaling is robustly activated in astrocytes by neurotoxic factors implicated in AD, including amyloid peptides and cytokines (Fernandez et al., 2007; Canellada et al., 2008; Sama et al., 2008; Abdul et al., 2009; Furman et al., 2010). In turn, hyperactivation of astrocytic CN/NFAT induces numerous transcriptional programs associated with aging and early-stage AD (Norris et al., 2005).
Disrupting the physical interaction between CN and NFATs with the synthetic peptide VIVIT prevents NFAT activation (Aramburu et al., 1999) and reduces immune/inflammatory signaling in many model systems. In primary neural cultures, deleterious actions of activated astrocytes on neighboring neurons and glia are minimized by targeting VIVIT directly to astrocytes using recombinant viruses (Sama et al., 2008; Abdul et al., 2009). Here, we used adeno-associated virus (AAV) expressing the astrocyte-specific promoter Gfa2 and the CN/NFAT inhibitor VIVIT to target astrocytes in an intact mouse model of AD. Bilateral administration of Gfa2–VIVIT into the hippocampus of 7- to 8-month-old APP/PS1 mice, before extensive amyloid pathology, was associated with reduced glial activation, lower amyloid levels, improved synaptic plasticity, and greater cognitive function at 16–17 months of age. The results reveal a deleterious role for activated astrocytes in neurologic function and lay the groundwork for exploring similar astrocyte-based strategies in the treatment of AD.
Materials and Methods
Animals.
Male transgenic (Tg) mice expressing a chimeric mouse/human amyloid precursor protein (Mo/HuAPP695swe) and a mutant human presenilin 1 (PSEN1dE9) under the control of a mouse prion protein promoter were purchased from The Jackson Laboratory (stock #005864). Homozygotes from the same colonies lacking these mutations [i.e., wild type (WT)] served as control (Ct) animals. All animals were provided with food and water ad libitum and were maintained on a 12 h light/dark schedule. Mice were treated in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals.
AAV–Gfa2 vectors.
cDNA encoding enhanced green fluorescent protein (EGFP) from the pEGFPn1 vector (Clontech) and VIVIT–EGFP (gift from Dr. Anjana Rao, Harvard University, Boston, MA) was extracted and inserted into modified pAdlink vectors downstream of the human GFAP promoter Gfa2 (gift from Dr. Michael Brenner, University of Alabama, Birmingham, AL) as described previously (Abdul et al., 2009). pGfa2–EGFP and pGfa2–VIVIT–EGFP constructs were then inserted into pENN.AAV2/5 vectors for creation of high-titer (1012 infectious units/ml) AAV2/5 vectors at the University of Pennsylvania Viral Vector Core (Philadelphia, PA). Our previous work on primary cultures shows that Gfa2–VIVIT–EGFP potently inhibits NFAT transcriptional regulation and NFAT-dependent signaling, selectively in astrocytes (Abdul et al., 2009).
Surgeries.
All animals underwent stereotaxic surgery. Mice were anesthetized with isoflurane (2.5%) throughout the duration of surgery. Once immobilized in a stereotaxic frame, AAV vectors (Gfa2–VIVIT or Gfa2–EGFP) or 5% glycerol solution (i.e., vehicle) was delivered bilaterally into the hippocampus (4 μl/hemisphere) at a rate of 0.2 μl/min using a stereotaxic injector (Stoelting). Syringe needles were left in place for 2 min after completion to limit reflux. Coordinates for injection relative to bregma were +2.0 mm anteroposterior, ±1.5 mm mediolateral, and −1.5 mm dorsoventral.
Active avoidance.
Methods for measuring avoidance behavior in mice were similar to those used in our previous work (Thibault et al., 2012). Animals were placed into the dark compartment of a two-compartment light/dark apparatus. After 7 s latency, a 0.8 mA footshock was administered in the dark chamber that lasted 24 s. Mice had free access to the light chamber, in which no shock was given. On training days 1–3, animals underwent four trials, with a 1 min intertrial rest period, in which they learned to avoid footshock by escaping into the light chamber. On day 4, a probe trial was administered, in which animals were placed into the dark chamber but no footshock was given. On the probe trial, escape latency to the light chamber was measured (with 31 s being the maximum time allotted) for each mouse. Because these values necessarily showed a non-normal distribution, performance of each mouse was ranked relative to all other mice (i.e., the lower the escape latency, the lower the ranking) and compared across treatment conditions using a nonparametric test (see below).
Immunohistochemistry.
After electrophysiological recordings (see below), sagittal slices from the rostral to middle region of the hippocampus were fixed overnight in 4% paraformaldehyde, preserved in sucrose buffer, and stored in phosphate buffer. Slices were further cut on a freezing microtome to 50 μm thickness. When necessary, either heat-induced or enzymatic epitope retrieval was performed to enhance antigen binding. Sections were blocked in normal serum and incubated overnight with the following primary antibodies: mouse anti-GFAP (1:50; Cell Signaling Technology), rabbit anti-Iba-1 (1:400; Wako), or mouse anti-β-amyloid (Aβ) (1:50; Vector Laboratories). Iba-1 antibody was tagged with Cy3-conjugated fluorescent secondary antibody (1:500; Wako). GFAP and Aβ antibodies were tagged with biotinylated secondary antibodies (1:100), amplified with the avidin–biotin complex, and visualized with Nova Red or 3,3′-diaminobenzidine tetrahydrochloride. In some cases, sections were counterstained with hematoxylin or DAPI (Invitrogen) to identify neuronal cell layers. Unless stated otherwise, all reagents were from Vector Laboratories. Images were visualized and captured on either an Aperio ScanScope XT digital slide scanner (Aperio) or with an inverted epifluorescence confocal microscope (DMIRE-2; Leica).
Morphometrical analysis of astrocytes.
GFAP-labeled astrocytes were analyzed in 20× digital images acquired by an Aperio ScanScope. For each slice (up to three per animal), multiple 400 μm2 fields spanning the entire CA1 stratum radiatum region were transferred to the MetaMorph Image Analysis Software Suite (Molecular Devices), in which they were thresholded and converted to binary images for automated and nonbiased morphometric analysis. In this procedure, immunolabeled cells in each field were automatically counted and assigned a pixel value based on size (i.e., larger cells have larger pixel areas). These values were exported to a spreadsheet in which the number of counted cells, along with corresponding pixel areas, were averaged across slices for each animal (n indicates the number of mice). Frequency histograms showing astrocyte size distributions were constructed using SigmaPlot 12 software and subsequently fit with a four-parameter Weibull Function (R2 > 0.9). Weibull function parameters corresponding to distribution amplitude and width were compared across treatment groups using Z tests. In other analyses, binarized astrocytes in each field were sorted into three broad categories (i.e., “small,” “medium,” and “large”) based on pixel area (i.e., 150–500, 500–1000, and >1000 total pixels). The percentage of cells in each size category was calculated relative to the total number of cells within each field and then averaged across fields within each animal for statistical testing.
Quantitative image analysis of amyloid labeling.
Amyloid-labeled hippocampal slices were scanned with the Aperio ScanScope at 20× magnification, and amyloid plaque load analysis was performed as described previously (Bachstetter et al., 2012). Briefly, after setting color and intensity thresholds, the Aperio-positive pixel count algorithm (version 9) was used to distinguish amyloid-specific labeling (i.e., positive pixels) from background (i.e., negative pixels) in the hippocampal CA1 region. Data are presented as the percentage area occupied by immunolabeled Aβ.
Tissue homogenate preparation.
Immediately after the animals were killed, one hippocampus was removed, snap-frozen in liquid nitrogen, and then stored at −80°C until use. Tissue was Polytron homogenized in ice-cold PBS supplemented with protease inhibitor mixture, phosphatase inhibitor mixture, and calpain inhibitor (all from Calbiochem). Samples were centrifuged at 20,800 × g for 30 min at 4°C, and supernatant was collected. The resultant pellet was reextracted by sonication in 2% SDS (containing inhibitors) and centrifuged at 20,800 × g for 30 min at 14°C. Supernatant was collected, and the remaining pellet was again reextracted by sonication in 70% formic acid. Sample was centrifuged at 20,800 × g for 1 h at 4°C, and supernatant was collected. Samples were stored at −80°C until use. Supernatant from the first extraction (i.e., PBS fraction) was used for Western blot analyses. Supernatants from the second and third extractions (i.e., SDS and formic acid, respectively) were used for ELISA analyses of Aβ peptide levels.
Aβ ELISA.
Methods for quantifying soluble and insoluble Aβ(1–42) peptide levels were similar to those used in our previous work (McGowan et al., 2005; Murphy et al., 2007; Abdul et al., 2009). SDS-extracted fractions, representative of soluble Aβ, were diluted in antigen capture (AC) buffer [20 mm Na3PO4, 0.4% Block Ace (AbD Serotec), 0.05% NaN3, 2 mm EDTA, 0.4 m NaCl, 0.2% BSA, and 0.05% CHAPS, pH 7] as needed. Formic acid-extracted fractions, representative of insoluble Aβ, were first neutralized by a 1:20 dilution with TP buffer (1.0 m Tris base and 0.5 m Na2HPO4) and then further diluted in AC buffer as needed. Immulon 4HBX plates were coated with 2.1.3 [end specific for Aβ(1–42)] capture antibody (0.5 μg/well) and blocked with Synblock (AbD Serotec), as per the instructions of the manufacturer. Synthetic human Aβ(1–42) was diluted in AC buffer and used to construct a standard curve. Protein standards and sample extracts were loaded in at least duplicate, and antigen was detected with 4G8 (Aβ17–24; Covance) biotinylated antibody. Reactions were developed with 3,3′,5,5′-tetramethylbenzidine reagent, stopped with 6% o-phosphoric acid, and read at 450 nm using a multiwell plate reader. Aβ levels were calculated relative to the standard curve.
Western blot analysis.
Protein concentrations in PBS homogenates were estimated using the Lowry method. Equal amounts of protein were loaded into individual wells of pre-cast 4–20% gradient gels (Bio-Rad). Proteins were resolved with electrophoresis and transferred to polyvinylidene difluoride membranes for semiquantitative Western blot analysis using the Odyssey Sa Imager System. Membranes were preblocked with Odyssey Blocking Buffer and incubated overnight in blocking buffer plus primary antibodies, including the following: mouse anti-GFAP (1:10,000; Cell Signaling Technology), rabbit anti-Iba-1 (1:1000; Wako), rabbit anti-β-secretase 1 (BACE1) (1:1000; Epitomics), rabbit anti-insulin degrading enzyme (IDE) (1:750; Abcam), mouse anti-neprilysin (1:750; Abcam), and anti-GAPDH (1:10,000; Abcam or Cell Signaling Technology). Primary antibodies were tagged with IRDye-conjugated fluorescent secondary antibodies (1:20,000; Li-Cor), and near-infrared signal was detected on the Odyssey Sa Imager System (Li-Cor). Signal intensity for resultant bands was calculated, and all protein signals were normalized to internal control (i.e., GAPDH) bands.
Hippocampal slice preparation.
All methods for harvesting brain slices for electrophysiological recordings were similar to those described in our previously published work (Norris et al., 1998; Norris and Scheff, 2009; Mathis et al., 2011). Mice were deeply anesthetized with CO2 and decapitated. Brains were removed and stored briefly in Ca2+-free, ice-cold, oxygenated (95% O2, 5% CO2) artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 2 KCl, 1.25 KH2PO4, 2 MgSO4, 26 NaHCO3, and 10 dextrose, pH 7.4. Four- hundred-micrometer-thick sections from one hemisphere, chosen at random, were cut on a vibrating microtome (Leica). Slices were then quickly transferred to netting in a custom Plexiglas holding chamber and maintained in CaCl2-containing (2 mm) ACSF at an interface with warm (32°C), humidified air. Slices were permitted to equilibrate for at least 1.5 h before beginning electrophysiological analysis.
Field EPSP recordings.
Slices were submerged in oxygenated ACSF (32°C) and perfused at a rate of 1–2 ml/min for 15–20 min before the start of each recording session. CA3 Schaffer collaterals were activated with a bipolar platinum–iridium electrode located in stratum radiatum near the CA3 border. Stimulus intensity was controlled by a constant-current stimulus isolation unit (World Precision Instruments), and stimulus timing was controlled by Clampex 9.2 software (Molecular Devices). Field EPSPs were recorded using a glass micropipette (1–6 m), filled with ACSF and containing an Ag–AgCl wire, positioned in stratum radiatum of CA1, ∼1 mm away from the point of stimulation. Field potentials were amplified 100×, Bessel filtered at 1 kHz, and digitized at 10 kHz using a Multiclamp 700B amplifier and a Digidata 1320 digitizer (Molecular Devices).
Synaptic strength and long-term potentiation measures.
For each slice, dual-stimulus pulses (S1 and S2), separated by 50 ms, were delivered at nine different intensity levels (range of 30–500 mA) at a rate of 0.1 Hz to establish a synaptic strength curve. Five field potentials at each stimulus level were averaged and measurements of fiber volley (FV) amplitude (in millivolts) and EPSP slope (millivolts per milliseconds) were performed offline using Clampfit software (Molecular Devices). Averaged EPSP slope measures were plotted against their corresponding FV amplitudes to estimate the strength of CA3–CA1 synaptic contacts. Paired-pulse facilitation (PPF) of the EPSP slope was calculated along the linear portion of the synaptic strength curve by dividing the EPSP slope of S1 by the EPSP slope of S2 and multiplying by 100. Following measurements of synaptic strength, stimulation intensity was readjusted to elicit an EPSP of ∼1 mV, and stimulus pulses were delivered at 0.033 Hz until a stable 20 min baseline was established. High-frequency stimulation (two 100 Hz trains, 1 s each, 10 s intertrain interval) was then delivered at the baseline stimulation intensity to induce long-term potentiation (LTP), followed by an additional 60 min baseline. Within each group, EPSP slope measures from the last 10 min of the post-LTP baseline were averaged across slices within animal and compared with the pre-LTP baseline slope average. For each animal, electrophysiological parameters were averaged across all slices within each animal (one to three slices), and the n used for statistical comparisons reflects the number of animals per genotype and treatment group. All electrophysiological recordings were conducted and analyzed by personnel who were blind to genotype and treatment conditions.
Statistics.
ANOVA was used to detect differences in Western blot protein levels. Student's t test was used to analyze astrocyte size distributions and amyloid levels. Z tests were used to compare Weibull distribution parameters across Tg mice treated with and without AAV–Gfa2–VIVIT. Values greater than |2| were considered statistically significant. Performance across training days on the active avoidance task and changes in synaptic efficacy after high-frequency stimulation were analyzed with repeated-measures ANOVA. Fisher's least significant difference test was used for post hoc comparisons. The Kruskal–Wallis nonparametric test was used to analyze ranked probe trial latency values in the active avoidance task, and the Mann–Whitney U test was used for follow-up pairwise comparisons. Statistical significance for all comparisons was set at p ≤ 0.05.
Results
Targeting astrocytes in APP/PS1 mice with AAV–Gfa2 vectors
AAV seems nearly ideal for obtaining long-lasting and widespread transgene expression in the CNS. Here, the fluorescent marker EGFP, with or without the CN/NFAT inhibitor VIVIT (VIVIT–EGFP), was inserted into AAV2/5 vectors downstream of the astrocyte-specific promoter Gfa2 (Lee et al., 2008). As shown in Figure 1, a single injection of AAV–Gfa2 vectors directly into the hippocampus of an adult mouse results in uniform EGFP expression across the entire longitudinal (Fig. 1A) and sagittal (Fig. 1B) axes of the hippocampus. Moreover, transgene expression is very long lasting (>9 months) and limited almost exclusively to astrocytes, with little to no EGFP found in other major cell types, including microglia (Fig. 1C) and neurons (Fig. 1D). Although it is possible that very small amounts of VIVIT–EGFP could be released by astrocytes and taken up in other cell types (for instance, in dendritic spines of closely apposed neurons), such cross-cell contamination was too low for detection using fluorescent microscopy, even at very high magnifications (data not shown).
Figure 1.

AAV–Gfa2 vectors drive long-lasting and astrocyte-specific transgene expression. A–D, Representative confocal fluorescent photomicrographs showing EGFP expression in brain sections (A, longitudinal; B–D, coronal) prepared from mice that received a bilateral injection of AAV–Gfa2 vectors into the CA1 region of the hippocampal formation. At 2 months after injection (A), the longitudinal axis of the hippocampus showed abundant EGFP expression, although the neocortex, which is enclosed by the white dashed line, mostly excluded EGFP expression. At 9 months after injection (B–D), the hippocampal molecular layers, but not the dentate granule and CA1 pyramidal neuron layers (counterlabeled blue with DAPI in B and D), showed high levels of EGFP expression. Microglial cells, positively labeled for the presence of Iba-1 (red), were similarly devoid of EGFP expression (C). In contrast to neurons and microglia, numerous GFAP-positive astrocytes (red) colocalized with EGFP (green) (D), confirming that hippocampal astrocytes exclusively expressed the transgene (note EGFP/GFAP colabel appears orange/yellow). E illustrates the treatment paradigm and endpoint measures investigated in this study. WT and Tg mice received injections of either vehicle, or AAV–Gfa2 vectors containing EGFP control or EGFP coupled to the NFAT inhibitor VIVIT. We treated mice at ∼7–8 months of age, at the early stages of amyloid pathology, and then aged them to ∼16 months, at which time they underwent behavioral characterization. After the animals were killed, we assessed several AD-like biomarkers, including neuroinflammation, amyloid pathology, and hippocampal synaptic dysfunction.
APPswe/PSEN1dE9 (Tg) mice received bilateral hippocampal injections of AAV–Gfa2 vectors (EGFP, n = 8; VIVIT, n = 7) or vehicle (n = 3) at ∼7–8 months of age, a time when amyloid pathology is relatively mild and astrocytic CN expression is just starting to appear in this and similar animal models (Jankowsky et al., 2004; Norris et al., 2005). Hippocampus was targeted because this structure is affected early in the progression of AD (Daulatzai, 2010) and exhibits an AD-related increase in CN/NFAT signaling (Norris et al., 2005; Abdul et al., 2009). After injection, mice were aged for an additional 9 months to permit the extensive presentation of multiple AD biomarkers, including glial activation, amyloid deposition, synaptic dysfunction, and cognitive impairment (Fig. 1E) (Jankowsky et al., 2004). Gfa2–EGFP and vehicle-treated mice were quantitatively similar on every biomarker measure and were therefore combined into a single control group (n = 11) for statistical comparisons. As a genotype control, age-matched WT mice were also investigated in parallel (Ct, n = 9; VIVIT, n = 7).
Astrocytic CN/NFAT inhibition blunts glial activation
Astrocyte activation is indicated by the appearance of hypertrophic somata and processes and is often accompanied by an increase in the expression of GFAP, a major intermediate filament protein specific for astrocytes (Fuller et al., 2009; Rodriguez et al., 2009; Sofroniew and Vinters, 2010). Consistent with previous results on other AD mouse models (Oakley et al., 2006; Yeh et al., 2011), Western blot analyses of hippocampal tissue homogenates (Fig. 2A) revealed a significant, nearly 300% increase in GFAP protein levels in Tg Ct mice relative to age-matched WT Ct mice. However, mice from both genotype groups exhibited lower (>35%) GFAP levels when treated with Gfa2–VIVIT. For the Tg group, the difference between Ct and Gfa2–VIVIT-treated mice closely approached statistical significance (p = 0.06 for Tg Ct vs Tg VIVIT).
Figure 2.

Gfa2–VIVIT reduces astrocyte activation in Tg mice. A, Representative Western blots and mean ± SEM protein levels for the astrocyte marker GFAP from hippocampal homogenates of WT and Tg mice treated with vehicle and Gfa2–EGFP (Ct) or Gfa2–VIVIT (VIV). In the bar graph, GFAP levels are normalized to GAPDH internal controls and expressed as a percentage of the WT Ct group (+p < 0.001 Tg Ct vs WT Ct and WT VIV; #p < 0.01 Tg VIV vs WT VIV; *p < 0.05 Tg VIV vs WT Ct). B–E, Low-magnification (B1–E1) and high-magnification (B2–E2) representative images of astrocytes immunohistochemically labeled in hippocampal CA1 for the presence of GFAP. CA1 s.p., CA1 pyramidal cell layer. Scale bar, 500 μm. F, Representative region showing GFAP immunoreactivity before (raw) and after conversion to a binary image (cells in blue) for quantification of possible changes in astrocyte size. G, Histograms of binary images showing the size of individual astrocytes (total pixel area per cell) counted per unit area (mean ± SEM) in CA1 of Tg Ct and Tg VIVIT mice. The last column in each histogram indicates the number of cells counted with a total pixel area in excess of 1000. H, Weibull distributions for histograms shown in G. Curve parameters were compared using Z tests (see Results). I, Representative binary images of astrocytes sorted into small-, medium-, and large-sized categories based on total pixel area. J, Total number of astrocytes (mean ± SEM per square millimeter) counted in CA1 for Tg Ct and Tg VIVIT mice. K, Mean ± SEM astrocyte size (pixel area) for astrocytes counted in G. L–N, The proportion of small, medium, and large astrocytes (mean ± SEM, expressed as percentage of total cells) in area CA1 for Tg Ct and Tg VIV groups. For all panels, +p < 0.001, #p < 0.01, *p < 0.05.
Gfa2–VIVIT treatment also markedly affected the physical appearance of hippocampal astrocytes, especially those in the Tg group. Representative images showing immunolabeled GFAP in area CA1 are provided in Figure 2B–E. At low magnification (Fig. 2B1–E1), GFAP labeling appeared similar across treatment conditions. However, at higher magnification, many GFAP-positive astrocytes in the Tg Ct group appeared larger and more ramified compared with the Tg VIVIT group (Fig. 2D2 vs E2). To determine whether Gfa2–VIVIT treatment caused a shift in the size distribution of astrocytes, 20× images spanning the entire CA1 region were acquired from Tg Ct and Tg VIVIT mice and converted to binary images for automated morphometric quantification using MetaMorph software (Fig. 2F,I; binarized cells shown in blue). Total area of each astrocyte was recorded and used to construct average histograms (Fig. 2G), which were fit with a Weibull function (Fig. 2H). These comparisons revealed no effect of Gfa2–VIVIT on the total number of GFAP-labeled cells (Fig. 2J). However, relative to Tg Ct mice, the size histogram for the Tg VIVIT group exhibited a higher peak (Z = −5.28) and a narrower distribution (Z = 3.66), reflecting a shift toward smaller astrocytes (Fig. 2G,H). Consistent with this shift, average astrocyte size exhibited a significant reduction in Gfa2–VIVIT mice (p < 0.05; Fig. 2K).
To expand on this observation, we sorted astrocytes into three broad groups based on cell size (Fig. 2I). Small astrocytes (pixel area between 150 and 500), with relatively small somas and few major processes, were most abundant in both treatment groups but were observed in proportionally greater numbers (p < 0.001) in Gfa2–VIVIT-treated mice (Fig. 2L). In contrast, medium astrocytes (pixel area between 500 and 1000), characterized by thicker cell bodies and processes (Fig. 2M), were significantly reduced (p < 0.05) in the Gfa2–VIVIT group. Moreover, large, highly ramified (i.e., hypertrophied) astrocytes (pixel area >1000) (Fig. 2N) were rarely seen after treatment with Gfa2–VIVIT (p < 0.001). Together, these results are consistent with reduced astrocyte activation in Gfa2–VIVIT mice.
Activated astrocytes are widely believed to coordinate with microglial cells to drive neuroinflammatory signaling in AD and most other neurodegenerative conditions (Akiyama et al., 2000; Skaper, 2007). Similar to astrocytes, microglia are strongly activated during the progression of AD and closely associated with disease pathology (Tuppo and Arias, 2005; Van Eldik et al., 2007; Lee and Landreth, 2010). As shown in Figure 3, Tg mice exhibited significantly elevated hippocampal protein levels (p < 0.001; Fig. 3A) and displayed more intense immunohistochemical labeling (Fig. 3B–E) of the microglial marker Iba-1, indicative of pronounced microglial activation. Interestingly, the effects of Gfa2–VIVIT on Iba-1 expression showed a strong interaction with genotype. In Tg mice, Iba-1 levels were reduced by >35% (p < 0.05) after VIVIT treatment, similar to astrocyte measures shown in Figure 2. However, WT mice exhibited an increase in Iba-1 levels after Gfa2–VIVIT treatment (p < 0.05). This observation suggests that astrocytes and microglia may interact very differently depending on disease state. Nonetheless, results from Tg animals demonstrate that key indicators of glial activation and neuroinflammation associated with AD can be significantly ameliorated through selective targeting of astrocytic signaling pathways.
Figure 3.

Gfa2–VIVIT reduces microglial activation in Tg mice. A, Representative Western blots and mean ± SEM protein levels for the microglial marker Iba-1 from hippocampal homogenates of WT and Tg mice treated with vehicle and Gfa2–EGFP (Ct) or Gfa2–VIVIT (VIV). In the bar graph, Iba-1 levels are normalized to GAPDH internal controls and expressed as a percentage of the WT Ct group. Iba-1 levels were significantly reduced in VIVIT-treated Tg mice relative to Tg controls. Interestingly, the opposite effect was seen in WT animals (#p < 0.01, *p < 0.05). Fluorescent labeling of Iba-1 in CA1 stratum radiatum of WT Ct (B), WT VIV (C), Tg Ct (D), and Tg VIV (E) mice corroborated these treatment effects. Scale bars, 250 μm.
Astrocytic CN/NFAT inhibition ameliorates amyloid pathology
Neurotoxic Aβ(1–42) peptides are the primary constituent of extracellular amyloid deposits in AD and serve as the molecular target for several anti-AD therapeutics currently in clinical trials (Morgan, 2011). The role of neuroglia in amyloid pathology is controversial, with several studies showing that activated astrocytes and microglia can either exacerbate or reduce brain amyloid levels (Sastre et al., 2006; Shaftel et al., 2007). Recent studies have shown that blockade of systemic CN activity using commercially available inhibitors results in lower brain amyloid levels in Tg mice (Hong et al., 2010). Whether or not these beneficial effects were attributable to specific actions on glial signaling was not investigated. To determine the extent to which astrocytic CN/NFAT activity influences amyloid pathology, we immunolabeled hippocampal sections from Tg Ct and Tg VIVIT mice with a monoclonal antibody that recognizes the human Aβ peptide (Fig. 4A,B). Absolute Aβ(1–42) peptide levels were also estimated in whole hippocampal homogenates using ELISA (Fig. 4D–F). As shown in Figure 4, A and B, Gfa2–VIVIT treatment was generally associated with fewer and smaller amyloid deposits in hippocampal CA1, corresponding to a significant (p < 0.01) reduction (>30%) in the percentage area occupied by immunoreactive Aβ (Fig. 4C). Similar to immunohistochemical measures, Gfa2–VIVIT also caused a significant, >20% reduction (p < 0.05) in total Aβ(1–42) peptide levels (Fig. 4F). This effect was observed for both soluble (i.e., SDS-extractable; Fig. 4D) and insoluble (i.e., formic acid-extractable; Fig. 4E) Aβ(1–42) fractions, although differences for insoluble Aβ(1–42) were variable and did not reach statistical significance. These results demonstrate that astrocytic CN/NFAT signaling plays a regulatory role in amyloid pathology.
Figure 4.

Gfa2–VIVIT reduces Aβ pathology in Tg mice. A, B, Representative photomicrographs (scale bar, 100 μm) and accompanying plaque load analysis (C, mean ± SEM) illustrating differences in the immunohistochemical labeling of Aβ deposits (brown) in Tg Ct and Tg VIV mice. Hematoxylin (blue) labels the pyramidal neuron layer in CA1. D–F show ELISA measures (mean ± SEM) of soluble, insoluble, and total (soluble + insoluble) Aβ(1–42) peptide levels in hippocampal homogenates of Tg mice. The Gfa2–VIVIT-treated group showed significantly lower (*p < 0.05) total peptide levels (F), attributable mostly to a significant reduction in the toxic, soluble Aβ fraction (D). G–J, Representative Western blots (G) and mean ± SEM protein levels (H–J) for Aβ metabolic enzymes measured from hippocampal homogenates of WT Ct, WT VIV, Tg Ct, and Tg VIV mice. Values are normalized to GAPDH internal controls and expressed as a percentage of the WT Ct group. H, BACE1, the rate-limiting enzyme in Aβ production, was differentially affected by VIVIT treatment across genotypes (#p < 0.01 Tg Ct vs Tg VIV; *p < 0.05 WT Ct vs WT VIV). I, J, IDE and neprilysin, participants in amyloid clearance, were not affected by VIVIT treatment, although neprilysin did show a genotype difference (+p < 0.001 Tg Ct and Tg VIV vs WT Ct).
Elevations in Aβ peptide levels can arise from changes in several enzyme pathways involved in Aβ production and Aβ clearance. The rate-limiting enzyme in Aβ production, BACE1, is found at elevated levels in human AD subjects (Ahmed et al., 2010) and also in AD mouse models (Zhao et al., 2007), in which it closely correlates to the severity of amyloid pathology. Mounting evidence also suggests that significant amounts of BACE1 are produced in activated astrocytes during AD (Zhao et al., 2011). Interestingly, the CN/NFAT pathway was recently shown to upregulate BACE1 in cell cultures and Tg mice (Cho et al., 2008), although a selective role for astrocytic CN/NFAT activity was not investigated.
Similar to previous studies, we also observed a significant increase in hippocampal BACE1 protein levels in Tg Ct relative to WT Ct mice (p < 0.05; Fig. 4G,H). Furthermore, we found that astrocytic CN/NFAT activity plays a significant role in BACE1 expression. However, the effects of Gfa2–VIVIT were very different for WT and Tg mice. In the Tg group, Gfa2–VIVIT prevented BACE1 elevations, consistent with a positive regulatory role as demonstrated in previous studies (Cho et al., 2008). Conversely, BACE1 levels were potentiated in WT mice treated with Gfa2–VIVIT (Fig. 4G,H). These results suggest that interactions between BACE1 expression and astrocytic CN/NFAT signaling depend critically on the presence of existing amyloid pathology.
In addition to BACE1, we also measured protein levels of enzymes consistently linked to Aβ clearance, including IDE and neprilysin (Fig. 4G,I,J). Of the two, neprilysin (Fig. 4J) showed differences between WT and Tg mice (i.e., neprilysin was elevated in the Tg group, p < 0.001), yet neither enzyme was significantly altered by Gfa2–VIVIT treatment. The results suggest that astrocytic CN/NFAT activity does not regulate amyloid clearance in Tg mice, at least not through IDE and neprilysin-mediated pathways.
Astrocytic CN/NFAT inhibition improves cognition
Impairment on hippocampal-dependent cognitive tasks is perhaps the earliest and most well-recognized clinical feature of AD and is also characteristic of several AD mouse models (Selkoe, 2001; Jankowsky et al., 2004; Mattson, 2004; Oakley et al., 2006). Cognitive deficits in Tg mice were confirmed at ∼16 months of age using a standard hippocampal-dependent active avoidance behavioral task, in which mice are given 7 s to escape from a dark compartment to an illuminated compartment before a 24 s electric shock is delivered through the dark compartment flooring. Successful avoidances, defined by escape before footshock, were recorded across four training trials given each day for 3 d. On day 4 of the task, escape latency to the light compartment was measured for each mouse on a single probe trial in which no shock was delivered.
As shown in Figure 5, Gfa2–VIVIT had little effect on avoidance behavior in WT mice during training (Fig. 5A) or during the probe trial (Fig. 5C). However, Tg mice treated with Gfa2–VIVIT generally outperformed their Ct counterparts by the final training day (Fig. 5B). Similarly, Tg Ct mice were significantly impaired on the day 4 probe trial relative to all other genotype/treatment groups (p < 0.05), whereas Tg mice treated with Gfa2–VIVIT were nearly indistinguishable from the WT groups (Fig. 5C). These results demonstrate that cognitive performance in Tg mice can be protected by selective inhibition of astrocytic CN/NFAT signaling.
Figure 5.
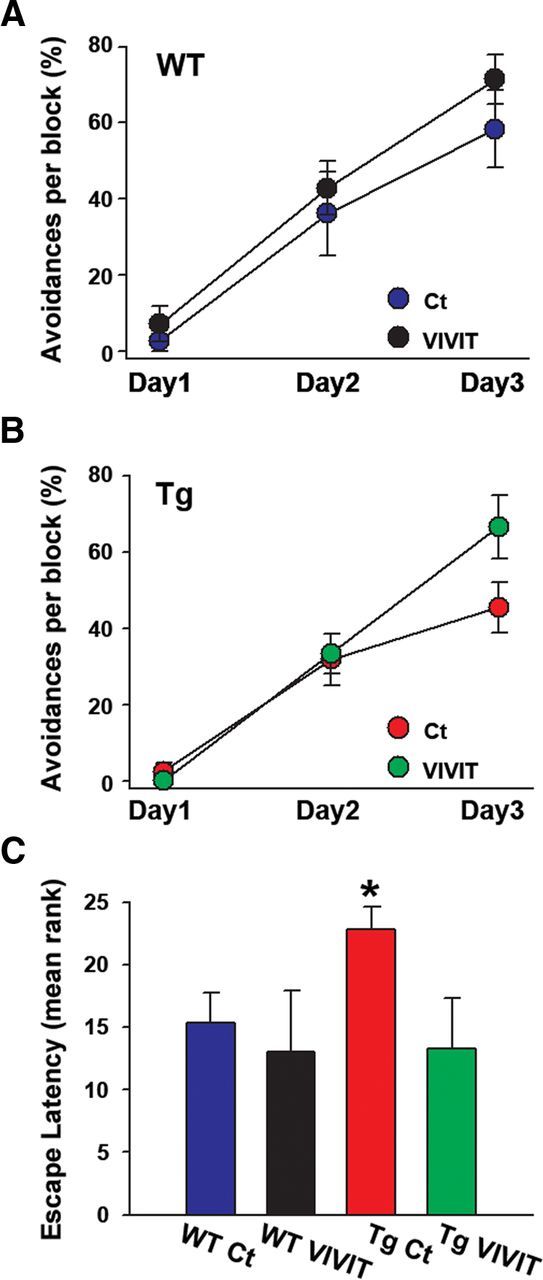
Gfa2–VIVIT improves cognitive performance in Tg mice. Percentage avoidances (mean ± SEM) by WT (A) and Tg (B) mice exhibited on three training days (Day 1, Day 2, Day 3) in a one-way active avoidance task. Gfa2–VIVIT had little effect on avoidance behavior in WT mice (A) but tended to improve performance in the Tg group by the end of training (B). On day 4 of the task, mice completed a single probe trial (C) in the absence of footshock and received rankings (mean ± SEM ranking) according to their escape times (lower ranks correspond to quicker escape latencies). Mean rankings for the WTs and Tg VIVIT groups were very similar on the probe trial and significantly lower than the Tg Ct group (* p > 0.05 Tg Ct vs all other groups).
Astrocytic CN/NFAT inhibition ameliorates synaptic dysfunction and plasticity
Because synaptic dysfunction is among the most reliable biomarkers of impaired cognition, we also investigated the effects of Gfa2–VIVIT on several synaptic transmission properties using electrophysiological methods in acutely prepared sagittal brain slices. For each brain slice, basal synaptic strength curves were constructed in CA1 stratum radiatum by plotting EPSP slope amplitudes against presynaptic FV amplitudes (Fig. 6A–C). Possible genotype/treatment effects on PPF (Fig. 6D) and population spike threshold (Fig. 6E) across synaptic activation levels were also investigated.
Figure 6.

Gfa2–VIVIT improves synaptic function in Tg mice. Representative electrophysiological waveforms recorded in CA1 stratum radiatum of brain slices from WT (A1) and Tg (B1) mice in response to electrical stimulation of CA3 Schaffer collaterals. Calibration: 0.5 mV, 2.5 ms. Waveforms in each treatment group were matched to similar FV amplitudes to illustrate differences in the amplitude of the corresponding postsynaptic response. A2 and B2 show synaptic strength curves for WT (A2) and Tg (B2) mice in which mean EPSP slope (millivolts per milliseconds) amplitudes (SEM, vertical error bars) are plotted against FV (millivolts) amplitudes (SEM, horizontal error bars) across nine stimulus intensity levels. C, Mean ± SEM EPSP/FV ratios calculated from the upper two stimulus intensity levels shown in A2 and B2. The Tg Ct group exhibited a significantly reduced EPSP/FV ratio relative to both the WT Ct and Tg VIVIT groups. D, In contrast to synaptic strength, levels of PPF (mean ± SEM) did not differ across treatment group. E, The mean ± SEM EPSP slope amplitude at which a population spike appeared (i.e., population spike threshold) in the ascending phase of the field potential (see A1 and B1). The WT VIVIT and Tg Ct groups showed a reduced population spike threshold compared with WT Ct and Tg VIVIT mice. This difference reached significance for the Tg Ct group (p < 0.01 vs WT Ct).
In WT animals, Gfa2–VIVIT caused a slight depression (rightward shift) of the synaptic strength curve (Fig. 6A2) and a modest, although insignificant (p = 0.19), reduction in the EPSP/FV ratio (Fig. 6A2,C). Gfa2–VIVIT was also associated with a slight reduction (p = 0.06) in the population spike threshold (Fig. 6E), suggesting that CA1 neurons in Gfa2–VIVIT mice may be more excitable, although synaptic effectiveness is weakened somewhat. In contrast to WT mice, Gfa2–VIVIT pushed the synaptic strength curve in Tg animals to the left (Fig. 6B2) and significantly increased (p < 0.05) the EPSP/FV ratio closer to WT Ct levels (Tg VIVIT vs WT Ct, p = 0.13; Fig. 6C). The population spike threshold was also modestly but insignificantly elevated (p = 0.07) in Tg VIVIT mice relative to Tg Ct mice (Fig. 6E). There were no transgene and/or treatment effects on PPF (Fig. 6D).
After synaptic strength curves were established, stimulus intensity was reset to evoke an ∼1 mV EPSP at a rate of 0.033 Hz. After a stable baseline period (≥20 min), each slice received two 1-s-duration 100 Hz stimulus trains (10 s intertrain interval) to induce LTP, a form of synaptic plasticity widely believed to underlie learning and memory processes (Malenka, 2003). Many animal models of aging and AD show impaired LTP coincident with cognitive decline (Foster and Norris, 1997; Foster, 2002; Marchetti and Marie, 2011; Spires-Jones and Knafo, 2012). As shown in Figure 7A, Gfa2–VIVIT had little-to-no effect on LTP levels in WT animals, whereas LTP in Tg mice was significantly enhanced (p < 0.001; Fig. 7B) by Gfa2–VIVIT. Thus, of all treatment groups investigated, LTP deficits were only observed in the Tg Ct group (Fig. 7C). Together, the results demonstrate that targeting CN/NFAT inhibitors selectively to astrocytes can improve synaptic function and plasticity in AD model mice.
Figure 7.
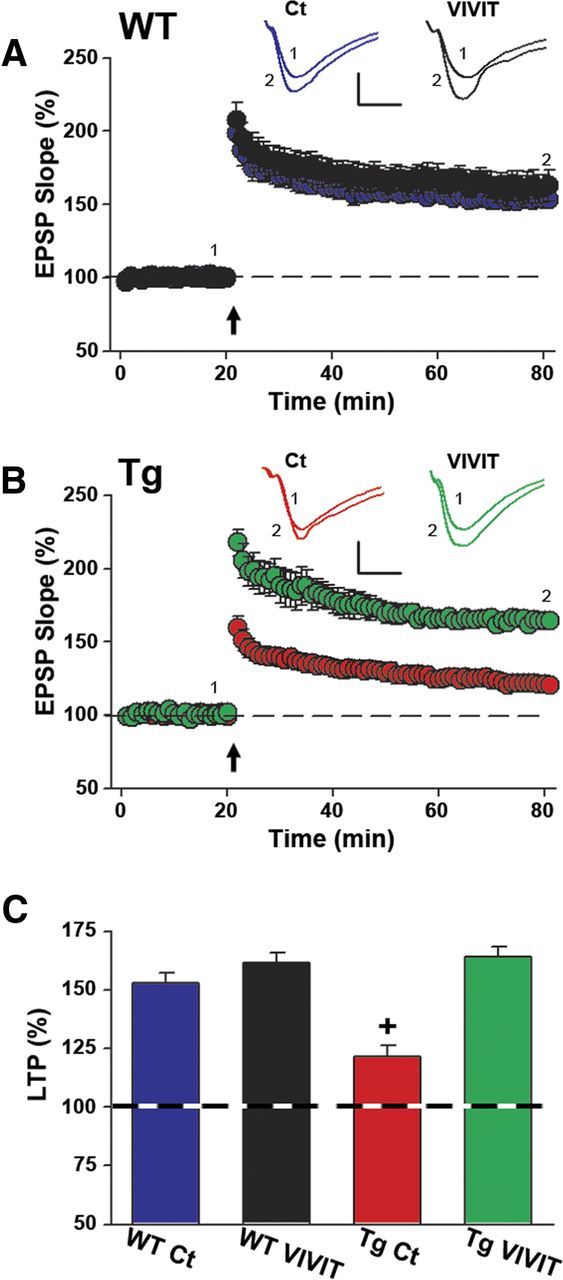
Gfa2–VIVIT improves LTP in Tg mice. Time plots of mean ± SEM EPSP slope values in CA1 stratum radiatum from WT (A) and Tg (B) mice. Insets show representative EPSP waveforms averaged in individual slices immediately before (1) and 60 min after (2) the delivery of two 1 s trains of 100 Hz stimulation (arrow). Calibration: 0.5 mV, 5 ms. LTP levels did not differ with Gfa2 treatment in WT mice (A); however, Tg Ct mice showed a substantial LTP deficit relative to the Tg VIVIT group (B). The bar graph in C shows LTP levels (mean ± SEM, percentage of baseline) at 60 min after 100 Hz stimulation and illustrates the LTP deficit in Tg Ct mice (+p < 0.001 Tg Ct vs all other groups).
Discussion
In this study, we targeted astrocytic CN/NFAT signaling in a mouse model of AD using novel AAV–Gfa2 vectors expressing the CN/NFAT inhibitor VIVIT. AAV treatment administered to Tg mice during early stages of disease progression reduced the appearance of glial activation, amyloid pathology, cognitive deficits, and synaptic dysfunction assayed at mid-age. Our findings suggest that activated astrocytes and/or astrocytic CN/NFAT signaling play integral roles in driving or maintaining multiple pathological and clinical symptoms of AD and lay the groundwork for future investigation of astrocyte-specific molecular targeting as a therapeutic strategy.
Astrocytic CN/NFAT and glial activation
Advances in cell-specific manipulations and assays have greatly increased our understanding of the role of activated astrocytes in neurodegeneration. In recent years, several molecular pathways involved in orchestrating glial activation such, as STAT3, NFκB, and MAP kinases, have been investigated in injury/disease models, and their selective knockdown in astrocytes produces variable degrees of benefit or detriment on neuroinflammation, lesion repair, and functional recovery (Sofroniew, 2009; Munoz and Ammit, 2010). In addition to these critical intracellular constituents, work from our laboratory and others has shown that the CN/NFAT pathway also regulates several components of the activated astrocyte phenotype, including cellular hypertrophy, cytokine expression, and glutamate dysregulation (Norris et al., 2005; Fernandez et al., 2007; Canellada et al., 2008; Sama et al., 2008; Abdul et al., 2009). The VIVIT peptide provides a powerful tool to disrupt this CN/NFAT signaling cascade. By mimicking the endogenous CN/NFAT docking site (i.e., PxIxIT sequence), VIVIT prevents CN-dependent dephosphorylation and activation of NFATs (Aramburu et al., 1999), making it far more specific than other commercially available CN inhibitors, which inhibit all CN signaling pathways and have off-target effects on immunophilins. However, it deserves noting that PxIxIT, or similar sequences, have more recently been identified in other CN substrates (Li et al., 2011), allowing the possibility that some VIVIT-mediated effects occur independently of NFATs. Nevertheless, our results show that targeting astrocytic signaling with VIVIT or other molecular reagents could prove very useful in pinpointing the role of activated astrocytes in a variety of neurodegenerative conditions.
The increased colocalization of CN to astrocytes occurs early in the clinical progression of AD (Norris et al., 2005; Abdul et al., 2009) when signs of glial activation and synapse dysfunction begin to emerge (Carter et al., 2012; Mufson et al., 2012). The present study suggests that early alterations in astrocytic CN/NFAT signaling drives or exacerbates pathologic changes associated with later disease stages. It seems likely that the deleterious effects of astrocytic CN/NFATs are rooted, at least partly, in chronic neuroinflammation. Indeed, astrocyte activation is generally regarded as a primary marker for neuroinflammation associated with most neurodegenerative conditions (Verkhratsky et al., 2010; Vincent et al., 2010; Sidoryk-Wegrzynowicz et al., 2011). Moreover, the effects of Gfa2–VIVIT vectors in Tg mice, as shown here, are similar in many ways to those obtained previously with NSAIDs and other anti-inflammatory drugs (Rowan et al., 2007; Kotilinek et al., 2008; Yirmiya and Goshen, 2011). Previous work has shown that the transition of astrocytes from an acute to a chronically activated “proinflammatory” state can directly arise from the dysregulation of CN/NFAT signaling (Fernandez et al., 2007; Sama et al., 2008). Positive feedback interactions between CN/NFATs and numerous other cytokines, membrane receptors, and Ca2+ sources (Norris et al., 2005; Fernandez et al., 2007; Canellada et al., 2008; Sama et al., 2008; Norris et al., 2010) suggest that the astrocytic CN/NFAT pathway is ideally suited to perpetuate harmful immune/inflammatory signaling cascades. Consistent with this idea, delivery of Gfa2–VIVIT to Tg mice in the present study not only suppressed morphologic features of astrocyte activation (Fig. 2) but also ameliorated signs of microglial activation (Fig. 3). In primary cultures, hyperactive CN/NFAT activity can spread from one astrocyte population to another (Sama et al., 2008). Recent evidence implicating the CN/NFAT pathway as a major regulator of the proinflammatory microglial phenotype (Nagamoto-Combs and Combs, 2010) suggests that CN/NFAT dysregulation in astrocytes may also spread to microglia and vice versa. It would therefore be interesting to determine whether or not inhibition of microglial CN/NFAT signaling in AD mice produces beneficial effects comparable with Gfa2–VIVIT.
Astrocytic CN/NFAT and amyloid pathology
Several clinical and pathological features of AD appear to involve a strong bidirectional interaction between Aβ peptides and CN/NFAT signaling (Agostinho et al., 2008; Reese et al., 2008; Abdul et al., 2009; Hong et al., 2010; Wu et al., 2010; Mohmmad Abdul et al., 2011; Wu et al., 2012). In the present study, Aβ peptide levels and amyloid plaque load were reduced in Tg mice treated with Gfa2–VIVIT (Fig. 4), suggesting that astrocytes are a critical site for CN/NFAT–amyloid interactions. CN/NFATs would seem to be more closely linked to Aβ production than to clearance, because Gfa2–VIVIT significantly reduced BACE1 levels but did not affect the expression of either IDE or neprilysin (Fig. 4) in Tg mice. Although Aβ pathology in these mice is attributable primarily to the presence of human APP and PSEN1 gene mutations, an increase in BACE1 levels, as reported in other AD mouse models (Zhao et al., 2007), would be predicted to exacerbate Aβ production. BACE1 is commonly found in neurons but is also regulated by numerous glial-derived factors that may be affected by Gfa2–VIVIT. For instance, BACE1 levels are increased in neural cultures treated with cytokines (Sastre et al., 2003), several of which are produced in astrocytes and sensitive to CN/NFATs (Sama et al., 2008). Gfa2–VIVIT may also directly influence astrocytic BACE1 expression. Recent evidence suggests that BACE1 transcription depends critically on NFAT1 (Cho et al., 2008, 2009), the NFAT isoform preferentially activated in hippocampal astrocytes during early stages of AD (Abdul et al., 2009). Although BACE1 levels are generally far lower in glial cells relative to neurons (Vassar et al., 1999; Laird et al. 2005), the sheer abundance of astrocytes in brain may still provide a significant source for the generation of Aβ peptides (Zhao et al., 2011). There is also some evidence to suggest that BACE1 is significantly elevated in reactive astrocytes surrounding amyloid deposits (Rossner et al., 2001). Clearly, future studies will be required to sort out the mechanistic interactions between astrocytes, CN/NFAT signaling, and Aβ pathology.
Effects of Gfa2–VIVIT on WT mice
Although important biomarkers of neurologic function (e.g., active avoidance learning and LTP) were not appreciably altered in WT mice treated with Gfa2–VIVIT, other biomarkers (e.g., Iba-1, BACE1, and synaptic strength) changed in ways that may be detrimental, suggesting that astrocyte activation could serve different roles during normal versus pathologic aging. Astrocytic CN/NFAT signaling, in particular, may coordinate both the maintenance and the resolution of neuroinflammatory signaling through its interactions with different transcription factors, including Foxo3 and NFκB (Fernandez et al., 2007, 2012). One possibility is that astrocyte activation (or astrocytic CN/NFAT signaling) provides a vital compensatory function that helps to stem harmful neuroinflammatory responses during middle age but then transitions to a primary mechanism for driving neuroinflammation and subsequent damage, during late aging and AD. In this case, blockade of astrocytic CN/NFATs in healthy, mid-aged subjects, similar to the ones modeled in this study, may be predicted to exacerbate microglial activation and proinflammatory cytokine production leading to increased BACE1 expression (Sastre et al., 2003), synaptic dysfunction (Di Filippo et al., 2008), or other problems. Future studies will need to investigate additional biomeasures and/or use more sensitive functional assays to identify other possible Gfa2–VIVIT-induced changes during the aging process. Such work could be critical for guiding potential therapeutic strategies, especially in regard to older individuals who are more likely to be seen in the clinic for memory problems and/or other symptoms of AD.
Potential of AAV vectors in the treatment of AD
Our results, combined with recent work from another group (Hudry et al., 2012), suggest that the targeted delivery of CN or NFAT inhibitors to select brain regions and/or cell types with AAV can help reduce AD-related biomarkers but also minimizing the possibility of detrimental off-target treatment effects. Because of its specificity, lack of toxicity, and capacity for widespread and long-lasting transgene expression, AAV appears to be an ideal vehicle for directing therapeutics to astrocytes and other cell types. In recent years, AAV has emerged as a promising treatment option for muscle disease (DiPrimio et al., 2010), retinal disorders (Stieger et al., 2011), and Parkinson's disease (PD). Intracranial delivery of AAV vectors to subcortical regions is presently under investigation in Phase I and II clinical trials for the treatment of PD, in which it has been shown to be safe and well tolerated (Kaplitt et al., 2007; Bartus et al., 2011). Although it will first be crucial to understand the response profile of healthy, aged individuals, we suggest that a similar AAV-based strategy targeting astrocytic CN/NFAT signaling, or other astrocytic cascades, could lead to viable new treatment options for AD and other neurodegenerative disorders.
Conclusions
This study is among the relatively few to directly confirm a deleterious role of astrocytes in the progression of multiple AD biomarkers. The results also add to a rapidly growing body of evidence implicating CN/NFAT signaling as an important, and possibly driving, force in the pathophysiology of AD. We suggest that astrocytes in general, and astrocytic CN/NFAT pathways in particular, provide useful molecular targets for the development of new anti-AD therapeutics.
Footnotes
This work was supported by National Institutes of Health Grants AG027297 (C.M.N.) and AG005119 (L.V.E. and M.P.M.), a PhRMA Foundation Predoctoral Fellowship (J.L.F.), a Kleberg Foundation Award (C.M.N.), and a gift from Jeffrey and Patti Tautenhan. We thank Drs. Eric Blalock and Stephen Scheff for helpful comments and suggestions.
The authors declare no competing financial interests.
References
- Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM, Patel ES, Baig I, Murphy MP, LeVine H, 3rd, Kraner SD, Norris CM. Cognitive decline in Alzheimer's disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci. 2009;29:12957–12969. doi: 10.1523/JNEUROSCI.1064-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agostinho P, Lopes JP, Velez Z, Oliveira CR. Overactivation of calcineurin induced by amyloid-beta and prion proteins. Neurochem Int. 2008;52:1226–1233. doi: 10.1016/j.neuint.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Ahmed RR, Holler CJ, Webb RL, Li F, Beckett TL, Murphy MP. BACE1 and BACE2 enzymatic activities in Alzheimer's disease. J Neurochem. 2010;112:1045–1053. doi: 10.1111/j.1471-4159.2009.06528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramburu J, Yaffe MB, López-Rodríguez C, Cantley LC, Hogan PG, Rao A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science. 1999;285:2129–2133. doi: 10.1126/science.285.5436.2129. [DOI] [PubMed] [Google Scholar]
- Bachstetter AD, Norris CM, Sompol P, Wilcock DM, Goulding D, Neltner JH, St Clair D, Watterson DM, Van Eldik LJ. Early stage drug treatment that normalizes proinflammatory cytokine production attenuates synaptic dysfunction in a mouse model that exhibits age-dependent progression of Alzheimer's disease-related pathology. J Neurosci. 2012;32:10201–10210. doi: 10.1523/JNEUROSCI.1496-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartus RT, Herzog CD, Chu Y, Wilson A, Brown L, Siffert J, Johnson EM, Jr, Olanow CW, Mufson EJ, Kordower JH. Bioactivity of AAV2-neurturin gene therapy (CERE-120): differences between Parkinson's disease and nonhuman primate brains. Mov Disord. 2011;26:27–36. doi: 10.1002/mds.23442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canellada A, Ramirez BG, Minami T, Redondo JM, Cano E. Calcium/calcineurin signaling in primary cortical astrocyte cultures: Rcan1–4 and cyclooxygenase-2 as NFAT target genes. Glia. 2008;56:709–722. doi: 10.1002/glia.20647. [DOI] [PubMed] [Google Scholar]
- Carter SF, Schöll M, Almkvist O, Wall A, Engler H, Långström B, Nordberg A. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-l-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J Nucl Med. 2012;53:37–46. doi: 10.2967/jnumed.110.087031. [DOI] [PubMed] [Google Scholar]
- Celsi F, Svedberg M, Unger C, Cotman CW, Carrì MT, Ottersen OP, Nordberg A, Torp R. Beta-amyloid causes downregulation of calcineurin in neurons through induction of oxidative stress. Neurobiol Dis. 2007;26:342–352. doi: 10.1016/j.nbd.2006.12.022. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Jin SM, Youn HD, Huh K, Mook-Jung I. Disrupted intracellular calcium regulates BACE1 gene expression via nuclear factor of activated T cells 1 (NFAT 1) signaling. Aging Cell. 2008;7:137–147. doi: 10.1111/j.1474-9726.2007.00360.x. [DOI] [PubMed] [Google Scholar]
- Cho HJ, Son SM, Jin SM, Hong HS, Shin DH, Kim SJ, Huh K, Mook-Jung I. RAGE regulates BACE1 and Abeta generation via NFAT1 activation in Alzheimer's disease animal model. FASEB J. 2009;23:2639–2649. doi: 10.1096/fj.08-126383. [DOI] [PubMed] [Google Scholar]
- Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell [Suppl] 2002;109:S67–S79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- Daulatzai MA. Early stages of pathogenesis in memory impairment during normal senescence and Alzheimer's disease. J Alzheimers Dis. 2010;20:355–367. doi: 10.3233/JAD-2010-1374. [DOI] [PubMed] [Google Scholar]
- Di Filippo M, Sarchielli P, Picconi B, Calabresi P. Neuroinflammation and synaptic plasticity: theoretical basis for a novel, immune-centred, therapeutic approach to neurological disorders. Trends Pharmacol Sci. 2008;29:402–412. doi: 10.1016/j.tips.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Kayed R, Neugebauer V, Fu Y, Zhang W, Reese LC, Taglialatela G. Amyloid-beta oligomers impair fear conditioned memory in a calcineurin-dependent fashion in mice. J Neurosci Res. 2010;88:2923–2932. doi: 10.1002/jnr.22445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPrimio N, McPhee SW, Samulski RJ. Adeno-associated virus for the treatment of muscle diseases: toward clinical trials. Curr Opin Mol Ther. 2010;12:553–560. [PubMed] [Google Scholar]
- Fernandez AM, Fernandez S, Carrero P, Garcia-Garcia M, Torres-Aleman I. Calcineurin in reactive astrocytes plays a key role in the interplay between proinflammatory and anti-inflammatory signals. J Neurosci. 2007;27:8745–8756. doi: 10.1523/JNEUROSCI.1002-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez AM, Jimenez S, Mecha M, Dávila D, Guaza C, Vitorica J, Torres-Aleman I. Regulation of the phosphatase calcineurin by insulin-like growth factor I unveils a key role of astrocytes in Alzheimer's pathology. Mol Psychiatry. 2012;17:705–718. doi: 10.1038/mp.2011.128. [DOI] [PubMed] [Google Scholar]
- Foster TC. Regulation of synaptic plasticity in memory and memory decline with aging. Prog Brain Res. 2002;138:283–303. doi: 10.1016/S0079-6123(02)38083-X. [DOI] [PubMed] [Google Scholar]
- Foster TC, Norris CM. Age-associated changes in Ca2+-dependent processes: relation to hippocampal synaptic plasticity. Hippocampus. 1997;7:602–612. doi: 10.1002/(SICI)1098-1063(1997)7:6<602::AID-HIPO3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Fuller S, Münch G, Steele M. Activated astrocytes: a therapeutic target in Alzheimer's disease? Expert Rev Neurother. 2009;9:1585–1594. doi: 10.1586/ern.09.111. [DOI] [PubMed] [Google Scholar]
- Furman JL, Artiushin IA, Norris CM. Disparate effects of serum on basal and evoked NFAT activity in primary astrocyte cultures. Neurosci Lett. 2010;469:365–369. doi: 10.1016/j.neulet.2009.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong HS, Hwang JY, Son SM, Kim YH, Moon M, Inhee MJ. FK506 reduces amyloid plaque burden and induces MMP-9 in AbetaPP/PS1 double transgenic mice. J Alzheimers Dis. 2010;22:97–105. doi: 10.3233/JAD-2010-100261. [DOI] [PubMed] [Google Scholar]
- Horsley V, Pavlath GK. NFAT: ubiquitous regulator of cell differentiation and adaptation. J Cell Biol. 2002;156:771–774. doi: 10.1083/jcb.200111073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudry E, Wu HY, Arbel-Ornath M, Hashimoto T, Matsouaka R, Fan Z, Spires-Jones TL, Betensky RA, Bacskai BJ, Hyman BT. Inhibition of the NFAT pathway alleviates amyloid-β neurotoxicity in a mouse model of Alzheimer's disease. J Neurosci. 2012;32:3176–3192. doi: 10.1523/JNEUROSCI.6439-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SH, Rao A. Activation and deactivation of gene expression by Ca2+/calcineurin-NFAT-mediated signaling. Mol Cells. 2004;18:1–9. [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA, Bland RJ, Young D, Strybing K, Eidelberg D, During MJ. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson's disease: an open label, phase I trial. Lancet. 2007;369:2097–2105. doi: 10.1016/S0140-6736(07)60982-9. [DOI] [PubMed] [Google Scholar]
- Kotilinek LA, Westerman MA, Wang Q, Panizzon K, Lim GP, Simonyi A, Lesne S, Falinska A, Younkin LH, Younkin SG, Rowan M, Cleary J, Wallis RA, Sun GY, Cole G, Frautschy S, Anwyl R, Ashe KH. Cyclooxygenase-2 inhibition improves amyloid-beta-mediated suppression of memory and synaptic plasticity. Brain. 2008;131:651–664. doi: 10.1093/brain/awn008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC. BACE1, a major determinant of selective vulnerability of the brain to amyloid-β amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci. 2005;25:11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CY, Landreth GE. The role of microglia in amyloid clearance from the AD brain. J Neural Transm. 2010;117:949–960. doi: 10.1007/s00702-010-0433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Messing A, Su M, Brenner M. GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia. 2008;56:481–493. doi: 10.1002/glia.20622. [DOI] [PubMed] [Google Scholar]
- Li H, Rao A, Hogan PG. Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol. 2011;21:91–103. doi: 10.1016/j.tcb.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Oda Y, Tomizawa K, Gong CX. Truncation and activation of calcineurin A by calpain I in Alzheimer disease brain. J Biol Chem. 2005;280:37755–37762. doi: 10.1074/jbc.M507475200. [DOI] [PubMed] [Google Scholar]
- Malenka RC. The long-term potential of LTP. Nat Rev Neurosci. 2003;4:923–926. doi: 10.1038/nrn1258. [DOI] [PubMed] [Google Scholar]
- Marchetti C, Marie H. Hippocampal synaptic plasticity in Alzheimer's disease: what have we learned so far from transgenic models? Rev Neurosci. 2011;22:373–402. doi: 10.1515/RNS.2011.035. [DOI] [PubMed] [Google Scholar]
- Mathis DM, Furman JL, Norris CM. Preparation of acute hippocampal slices from rats and transgenic mice for the study of synaptic alterations during aging and amyloid pathology. J Vis Exp. 2011;pii:2330. doi: 10.3791/2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohmmad Abdul H, Baig I, Levine H, 3rd, Guttmann RP, Norris CM. Proteolysis of calcineurin is increased in human hippocampus during mild cognitive impairment and is stimulated by oligomeric Abeta in primary cell culture. Aging Cell. 2011;10:103–113. doi: 10.1111/j.1474-9726.2010.00645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D. Immunotherapy for Alzheimer's disease. J Int Med. 2011;269:54–63. doi: 10.1111/j.1365-2796.2010.02315.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson EJ, Binder L, Counts SE, DeKosky ST, de Toledo-Morrell L, Ginsberg SD, Ikonomovic MD, Perez SE, Scheff SW. Mild cognitive impairment: pathology and mechanisms. Acta Neuropathologica. 2012;123:13–30. doi: 10.1007/s00401-011-0884-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz L, Ammit AJ. Targeting p38 MAPK pathway for the treatment of Alzheimer's disease. Neuropharmacology. 2010;58:561–568. doi: 10.1016/j.neuropharm.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Beckett TL, Ding Q, Patel E, Markesbery WR, St Clair DK, LeVine H, 3rd, Keller JN. Abeta solubility and deposition during AD progression and in APPxPS-1 knock-in mice. Neurobiol Dis. 2007;27:301–311. doi: 10.1016/j.nbd.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Nagamoto-Combs K, Combs CK. Microglial phenotype is regulated by activity of the transcription factor, NFAT (nuclear factor of activated T cells) J Neurosci. 2010;30:9641–9646. doi: 10.1523/JNEUROSCI.0828-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Scheff SW. Recovery of afferent function and synaptic strength in hippocampal CA1 following traumatic brain injury. J Neurotrauma. 2009;26:2269–2278. doi: 10.1089/neu.2009.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Halpain S, Foster TC. Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2+ channels. J Neurosci. 1998;18:3171–3179. doi: 10.1523/JNEUROSCI.18-09-03171.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Kadish I, Blalock EM, Chen KC, Thibault V, Porter NM, Landfield PW, Kraner SD. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer's models. J Neurosci. 2005;25:4649–4658. doi: 10.1523/JNEUROSCI.0365-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Blalock EM, Chen KC, Porter NM, Thibault O, Kraner SD, Landfield PW. Hippocampal “zipper” slice studies reveal a necessary role for calcineurin in the increased activity of L-type Ca2+ channels with aging. Neurobiol Aging. 2010;31:328–338. doi: 10.1016/j.neurobiolaging.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen JB, Di Domenico F, Sultana R, Perluigi M, Cini C, Pierce WM, Butterfield DA. Proteomics-determined differences in the concanavalin-A-fractionated proteome of hippocampus and inferior parietal lobule in subjects with Alzheimer's disease and mild cognitive impairment: implications for progression of AD. J Proteome Res. 2009;8:471–482. doi: 10.1021/pr800667a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese LC, Zhang W, Dineley KT, Kayed R, Taglialatela G. Selective induction of calcineurin activity and signaling by oligomeric amyloid beta. Aging Cell. 2008;7:824–835. doi: 10.1111/j.1474-9726.2008.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez JJ, Olabarria M, Chvatal A, Verkhratsky A. Astroglia in dementia and Alzheimer's disease. Cell Death Differ. 2009;16:378–385. doi: 10.1038/cdd.2008.172. [DOI] [PubMed] [Google Scholar]
- Rossner S, Apelt J, Schliebs R, Perez-Polo JR, Bigl V. Neuronal and glial beta-secretase (BACE) protein expression in transgenic Tg2576 mice with amyloid plaque pathology. J Neurosci Res. 2001;64:437–446. doi: 10.1002/jnr.1095. [DOI] [PubMed] [Google Scholar]
- Rowan MJ, Klyubin I, Wang Q, Hu NW, Anwyl R. Synaptic memory mechanisms: Alzheimer's disease amyloid beta-peptide-induced dysfunction. Biochem Soc Trans. 2007;35:1219–1223. doi: 10.1042/BST0351219. [DOI] [PubMed] [Google Scholar]
- Sama MA, Mathis DM, Furman JL, Abdul HM, Artiushin IA, Kraner SD, Norris CM. Interleukin-1beta-dependent signaling between astrocytes and neurons depends critically on astrocytic calcineurin/NFAT activity. J Biol Chem. 2008;283:21953–21964. doi: 10.1074/jbc.M800148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, Heneka MT. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-γ agonists modulate immunostimulated processing of amyloid precursor protein through regulation of β-secretase. J Neurosci. 2003;23:9796–9804. doi: 10.1523/JNEUROSCI.23-30-09796.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre M, Klockgether T, Heneka MT. Contribution of inflammatory processes to Alzheimer's disease: molecular mechanisms. Int J Dev Neurosci. 2006;24:167–176. doi: 10.1016/j.ijdevneu.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Schipper HM, Bennett DA, Liberman A, Bienias JL, Schneider JA, Kelly J, Arvanitakis Z. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol Aging. 2006;27:252–261. doi: 10.1016/j.neurobiolaging.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O'Banion MK. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest. 2007;117:1595–1604. doi: 10.1172/JCI31450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoryk-Wegrzynowicz M, Wegrzynowicz M, Lee E, Bowman AB, Aschner M. Role of astrocytes in brain function and disease. Toxicol Pathol. 2011;39:115–123. doi: 10.1177/0192623310385254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper SD. The brain as a target for inflammatory processes and neuroprotective strategies. Ann N Y Acad Sci. 2007;1122:23–34. doi: 10.1196/annals.1403.002. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathologica. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones T, Knafo S. Spines, plasticity, and cognition in Alzheimer's model mice. Neural Plast. 2012;2012:319836. doi: 10.1155/2012/319836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieger K, Cronin T, Bennett J, Rolling F. Adeno-associated virus mediated gene therapy for retinal degenerative diseases. Methods Mol Biol. 2011;807:179–218. doi: 10.1007/978-1-61779-370-7_8. [DOI] [PubMed] [Google Scholar]
- Thibault O, Pancani T, Landfield PW, Norris CM. Reduction in neuronal L-type calcium channel activity in a double knock-in mouse model of Alzheimer's disease. Biochim Biophys Acta. 2012;1822:546–549. doi: 10.1016/j.bbadis.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuppo EE, Arias HR. The role of inflammation in Alzheimer's disease. Int J Biochem Cell Biol. 2005;37:289–305. doi: 10.1016/j.biocel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Van Eldik LJ, Thompson WL, Ralay Ranaivo H, Behanna HA, Martin Watterson D. Glia proinflammatory cytokine upregulation as a therapeutic target for neurodegenerative diseases: function-based and target-based discovery approaches. Int Rev Neurobiol. 2007;82:277–296. doi: 10.1016/S0074-7742(07)82015-0. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Olabarria M, Noristani HN, Yeh CY, Rodriguez JJ. Astrocytes in Alzheimer's disease. Neurotherapeutics. 2010;7:399–412. doi: 10.1016/j.nurt.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AJ, Gasperini R, Foa L, Small DH. Astrocytes in Alzheimer's disease: emerging roles in calcium dysregulation and synaptic plasticity. J Alzheimers Dis. 2010;22:699–714. doi: 10.3233/JAD-2010-101089. [DOI] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, Bacskai BJ, Hyman BT. Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30:2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Hudry E, Hashimoto T, Uemura K, Fan ZY, Berezovska O, Grosskreutz CL, Bacskai BJ, Hyman BT. Distinct dendritic spine and nuclear phases of calcineurin activation after exposure to amyloid-β revealed by a novel fluorescence resonance energy transfer assay. J Neurosci. 2012;32:5298–5309. doi: 10.1523/JNEUROSCI.0227-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh CY, Vadhwana B, Verkhratsky A, Rodríguez JJ. Early astrocytic atrophy in the entorhinal cortex of a triple transgenic animal model of Alzheimer's disease. ASN Neuro. 2011;3:271–279. doi: 10.1042/AN20110025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O'Connor T, Logan S, Maus E, Citron M, Berry R, Binder L, Vassar R. β-Site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer's disease pathogenesis. J Neurosci. 2007;27:3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, O'Connor T, Vassar R. The contribution of activated astrocytes to Abeta production: implications for Alzheimer's disease pathogenesis. J Neuroinflammation. 2011;8:150. doi: 10.1186/1742-2094-8-150. [DOI] [PMC free article] [PubMed] [Google Scholar]