

Abstract
Background & Aims
In polycystic kidney (PKD) and liver (PLD) diseases, the normally non-proliferative hepato-renal epithelia acquire a proliferative, cystic phenotype, which is linked to overexpression of Cdc25A and cell cycle deregulation. We investigated the effects of Cdc25A inhibition in mice and rats, via genetic and pharmacological approaches.
Methods
Cdc25A+/− mice (which have reduced levels of Cdc25A) were cross-bred with Pkhd1del2/del2 mice (which have increased levels of Cdc25A and develop hepatic cysts). Cdc25A expression was analyzed in livers of control and PCK rats, control and Pkd2ws25/− mice, healthy individuals, and patients with PLD. We examined effects of pharmacologic inhibition of Cdc25A with Vitamin K3 (VK3) on the cell cycle, proliferation, and cyst expansion in vitro; hepato-renal cystogenesis in PCK rats and Pkd2ws25/− mice; and expression of Cdc25A and the cell cycle proteins regulated by Cdc25A. We also examined effects of the Cdc25A inhibitor PM-20 on hepato-renal cystogenesis in Pkd2ws25/− mice.
Results
Liver weights and hepatic and fibrotic areas were decreased by 32%–52% in Cdc25A+/−:Pkhd1del2/del2 mice, compared to Pkhd1del2/del2 mice.VK3 altered the cell cycle and reduced proliferation of cultured cholangiocytes by 32%–83% and decreased growth of cultured cysts by 23%–67%. In PCK rats and Pkd2ws25/− mice, VK3 reduced liver and kidney weights and hepato-renal cystic and fibrotic areas by 18%–34%. PM-20 decreased hepato-renal cystogenesis in Pkd2ws25/− mice by 15%.
Conclusions
Cdc25A inhibitors block cell cycle progression and proliferation, reduce liver and kidney weights and cyst growth in animal models of PKD and PLD, and might be developed as therapeutics for these diseases.
Keywords: animal model, phosphatase, therapeutic strategy, cell division
Introduction
Different genetic mutations in polycystic kidney and liver diseases (PKD/PLD) initiate the formation of hepato-renal cysts that continue to grow progressively.1-3 Substantial evidence suggests that a switch from a non-proliferative normal epithelia to a proliferative cystic epithelia is associated, in particular, with accelerated cell proliferation (due to elevated cAMP and decreased [Ca2+]i) and cell cycle deregulation (due to overexpression of the Cdc25A phosphatase).4-10 Indeed, suppression of cAMP and restoration of [Ca2+]i decreases abnormal cell proliferation and inhibits cyst growth.4, 5, 8, 9 Moreover, in PKD/PLD patients, treatment with somatostatin analogs halted progression of hepatic and renal cysts and improved quality of life.11-14
The cell cycle regulator, Cdc25A is of particular interest to us for the following reasons: (i) Cdc25A regulates all phases of the cell cycle;15, 16 (ii) Cdc25A is overexpressed in cancers and its suppression reduces cancer cell growth;17, 18 and (iii) Cdc25A elevation in PCK rats, an animal model of PKD/PLD, is linked to decreased levels of its regulator, microRNA-15a (miR-15A); experimental increase of miR-15A in PCK cholangiocytes reduced Cdc25A expression inhibiting cyst growth.4 Taken together, these observations suggest that Cdc25A might represent a potential therapeutic target in the treatment of PKD/PLD.
Multiple agents including analogues of natural Vitamin K (VK) have been developed as pharmacological inhibitors of Cdc25 phosphatases. VK3 (2-methyl-1, 4-naphtoquinone or menadione), in particular, has anti-proliferative capacity irreversibly inhibiting Cdc25A through covalent modification of its active site.19-21 Another Cdc25A inhibitor, PM-20 [N-(4-biphenyl)-3, 4-bis-(2-hydroxy-ethylsulphanyl)-maleimide], acts as a phosphatase antagonist binding to Cdc25A active site and subsequently inhibiting cancer cell growth in vitro and in vivo.20, 22 While VK3 and PM-20 as anti-cancer agents had been tested in vitro and in vivo and recently in clinical trials,19, 23, 24 their role in benign hyperproliferative conditions in general and in PKD/PLD in particular has not been explored.
In the present study, we used genetic and pharmacological approaches to assess further the pathogenic role of Cdc25A in hepato-cystogenesis and to provide evidence supporting its therapeutic potential in PKD/PLD. First, we crossbred Cdc25A+/−mice (characterized by reduced levels of Cdc25A)25 with our own model of PKD/PLD, Pkhd1del2/del2 mice (characterized by progressive hepatic but not renal cyst growth and elevated Cdc25A levels).26 We hypothesized that if Cdc25A is involved in hepatic cystogenesis, the crossbred mice would have less severe cystic disease due to decreased Cdc25A levels. Second, we examined the effects of pharmacological targeting of Cdc25A by VK3 on: (i) cell cycle and proliferation in vitro; (ii) cyst expansion in 3-D cultures; (iii) hepato-renal cystic areas and fibrosis in vivo in the PCK rat and Pkd2ws25/− mice; and (iv) the expression of Cdc25A and the cell cycle proteins known to be regulated by Cdc25A (i.e., Cdk1, 2, 4/6, cyclin A, B, D and E, Rb and PCNA). We also tested the effect of PM-20 on progression of hepato-renal cystic disease in vivo in Pkd2ws25/− mice. Both the PCK rat and Pkd2ws25/− mice resemble human pathology in PKD/PLD and have been used in preclinical trials.2, 6 The PCK rat (a model of Autosomal Recessive PKD/PLD) develops liver and kidney cysts at birth which gradually expand over time. Pkd2ws25/− mice (a model of Autosomal Dominant PKD/PLD) have progressive hepato-renal cystic disease with onset at 5-6 month of age. We recently described the phenotype of PCK rats and Pkd2ws25/− mice in detail.27, 28
We showed that hepatic cystogenesis is inhibited in Cdc25A+/−:Pkhd1del2/del2 crossbreds compared to their Pkhd1del2/del2 counterparts, providing support for an essential role of Cdc25A in cyst growth. VK3 reduced the proliferation of cystic cholangiocytes, caused cell cycle arrest, suppressed cyst growth in 3-D cultures, inhibited progression of hepato-renal cystic disease in vivo in animal models of PKD/PLD, and affected the expression of cell cycle proteins. Targeting of Cdc25A with PM-20 also attenuated hepato-renal cystogenesis in Pkd2ws25/− mice. These data suggest that Cdc25A represents a potential molecular target in PKD/PLD and its inhibitors might be beneficial in the treatment of these pathological conditions.
Materials and Methods
Animals and cell culture
Animals (number of rats and mice used is indicated for each experimental condition) were maintained on a standard diet after Mayo Institutional Animal Care and Use Committee approval. They were anesthetized with pentobarbital (50 mg/kg). Blood was collected from PCK rats by cardiac puncture. Liver and kidneys were fixed and embedded in paraffin for histology. For in vitro study, cholangiocytes were isolated from normal and PCK rats and cultured as described.7 Normal and diseased human liver tissue were obtained from Mayo Clinical Core and National Disease Research Interchange.
Development of Cdc25A+/−:Pkhd1del2/del2 mice
The Cdc25A+/− mice were bred to Pkhd1del2/del2 mice (both of C57BL6 background) and offspring’s (i.e., Cdc25A+/−:Pkhd1+/del2) were backcrossed with Pkhd1del2/del2 mice to produce Cdc25A+/−:Pkhd1del2/del2 animals. Mice were genotyped by PCR. The primers and PCR conditions are described in Supplementary Methods. We analyzed body, liver and kidney weighs and cystic and fibrotic areas in age matched 7-9 month old littermates of Cdc25A+/− (n=3), Pkhd1del2/del2 (n=5) and Cdc25A+/−:Pkhd1del2/del2 (n=3) mice.
Flow cytometry
Normal (n=5) and PCK cholangiocytes (n=6) were treated with 50, 100 and 200 μM of VK3 for 24h, fixed in ethanol and suspended in 50 μg/ml propidium iodide containing 0,1mg/mL RNase. Cell cycle analysis was performed at Mayo Advanced Genomics Technology Center.
Immunofluorescence confocal microscopy
Microscopy was performed with Zeiss LSM-510 microscope using tissue of normal humans (n=5) and patients with ADPKD, ARPKD and CHF (n=5 for each condition); normal (n=5) and PCK (n=6) rats; and normal (n=4) and Pkd2ws25/− (n=5) mice. Liver sections were incubated with antibodies against Cdc25A, Cdk4 and 6, cyclin D1 and D3, p-Rb, (Santa Cruz Biotechnology, Santa Cruz, CA; 1:50); Cdk1 and cyclin B (Cell Signaling, Danvers, MA; 1:50); cyclin A and D2 (Abcam, Cambridge, MA; 1:50); Cdk2 (BD Biosciences, Franklin Lakes, NJ; 1:50); cyclin E (Upstate; Lake Placid, NY; 1:50) and PCNA (Santa Cruz, 1:100). Respective secondary antibodies (Invitrogen, Carlsbad, CA; 1:200) were applied. Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay with the ApopTag Peroxidase In Situ Apoptosis Detection Kit (Chemicon, Billerica, MA) was used to detect apoptosis. For mitotic and apoptotic indices (calculated as a percent of PCNA- or TUNEL-positive cells, respectively), 1000 nuclei in randomly selected fields of liver and kidney sections were counted.
Western Blot
Cholangiocytes isolated from livers of normal humans (n=3) and ADPKD patients (n=3); normal (n=5) and PCK rats [non-treated (n=5) or VK3-treated (n=4)]; normal (n=6), Pkd2ws25/− (n=6), Cdc25A+/− (n=3), Pkhd1del2/de/2 (n=6) mice and Cdc25A+/−:Pkhd1del2/de/2 crossbreeds (n=3); and cultured normal (n=3) and PCK (n=3) cholangiocytes were used. The details are described in Supplementary Methods.
Cell proliferation
Normal (n=5) and PCK (n=5) cholangiocytes (5000 cells/well) were grown in regular DMEM/F12 medium at 37°C (5% CO2 and 100% humidity) for 48 hours before treatment. VK3 (50, 100 and 200 μM) was added daily for a total of 5 days. Cell proliferation were determined every 24 h by CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI) as described.4
3D culture and cyst measurement
Freshly isolated bile ducts of normal (n=5) and PCK (n=7) rats were kept between 2 layers of collagen (1.5 mg/mL, BD Biosciences) plus 10% Matrigel (BD Biosciences).5 VK3 (50, 100 and 200 μM) was added daily. Growth of cystic structures was assessed by light microscopy at days 0 (24 hours after seeding), 1, 3 and 5. The circumferential area of each cyst (n=20 for each condition) was measured using Image J software (National Institutes of Health) as described.29 Data were expressed as a percent change in cystic area compared to day 0.
VK3 and PM-20 treatment protocol
PCK rats (n=22) and Pkd2ws25/− mice (n=11) received 0.15 g/1 L of VK3 (Sigma, St. Louis, MO) dissolved in 1L of drinking water every other day for 4 and 8 weeks; control rats (n=22) and mice (n=10) received water. Pkd2ws25/− mice (n=12) received daily intraperitoneal injections of PM-20 (Sigma; 1 mg/kg for 4 weeks); the control group (n=7) was injected with DMSO (Supplementary Figure 4). Doses of VK3 and PM-20 was chosen based on published studies.20, 22, 30, 31 We assessed: body weight, serum biochemistry, liver and kidney weights, hepatic and renal cystic areas and fibrosis. Since no differences was observed in liver and kidney weights between male and female PCK rats and Pkd2ws25/− mice, for statistical analysis male and female data were combined. Cystic and fibrotic areas were analyzed as described.5 Briefly, livers and kidney sections were stained with H&E or picrosirius red collagen to assess, respectively, cystic or fibrotic areas. Measurements were done by Meta-Morph software (Universal Imaging, West Chester, PA) installed on a Pentium IBM-compatible computer (Dell OptiPlex, Dell Ins, Round Rock, TX) following image acquisition using a light microscope and color digital camera (Nikon DXM 1200). Hepatic and renal cystic and fibrotic areas were expressed as a percent of total hepatic or renal parenchyma, respectively.
3-dimentional reconstruction of the intrahepatic biliary tree
The intrahepatic biliary tree of non-treated (n=4) and VK3-treated for 8 weeks (n=4) PCK rats were reconstructed in three-dimensions. Briefly, anesthetized rats were injected via the common bile duct with a silicone polymer MV-122 (Flow Tech, Carver, MA). Liver lobes were fixed, suspended within a plastic cylinder and scanned using a micro-computed tomography (micro-CT) scanner with 20 μm resolution. Quantitation was performed by Analyze (Biomedical Imaging Resource; Mayo Clinic) as described.27
Results
Cell cycle phase distribution is altered in cystic cholangiocytes and rate of cell proliferation is increased
We assessed the cell cycle distribution in normal and PCK rats. In cystic cholangiocytes, the percent of cells in G0/G1 phase was reduced, in S phase was not altered, and in G2/M phase was increased compared to normal cholangiocytes (Supplementary Figure 1). We also examined the rate of proliferation in humans and rodents by PCNA expression. Most of cystic cholangiocytes in patients with PKD/PLD, in PCK rats and Pkd2ws25/− mice showed PCNA immunoreactivity while in normal cholangiocytes PCNA-positive nuclei were practically undetectable (Supplementary Figure 2A). Western blots further confirmed that PCNA levels are increased in vivo in cholangiocytes isolated from ADPKD patients, PCK rats and Pkd2ws25/− mice and in vitro in PCK-derived cholangiocytes compared to respective controls (Supplementary Figure 2B).
Cdc25A is overexpressed in cystic cholangiocytes
Here we confirm and extended our previous finding4 showing the increase in Cdc25A immunoreactivity in cystic cholangiocytes of patients with PKD/PLD, in PCK rats and Pkd2ws25/− mice compared to normal respective conditions (Figure 1A). In agreement with immunofluorescent data, western blots demonstrate that Cdc25A is increased in vivo in cholangiocytes isolated from ADPKD patients, PCK rats and Pkd2ws25/− mice and in vitro in cultured cholangiocytes derived from PCK rats compared to respective controls (Figure 1B). Interestingly, Cdc25A expression in normal humans and mice appears to be stronger compared to rats which might be a reflection of species differences.
Figure 1.
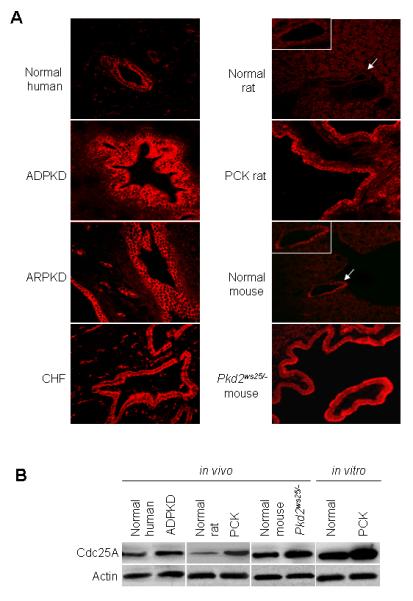
Cdc25A is overexpressed in cystic cholangiocytes. (A) Relatively low expression of Cdc25A (red) is found in normal human (n=5), rat (n=5) and mouse (n=4) cholangiocytes by confocal microscopy. Cdc25A appears to be increased in cystic cholangiocytes of patients with ARPKD, ADPKD, CHF, in PCK rats and Pkd2ws25/− mice (n=5 for each condition). Magnification, x40. White boxes show higher power of bile duct depicted by an arrow. (B) Representative western blots (n=3 for each set of data) demonstrate that Cdc25A levels are elevated in vivo in human patients with ADPKD, in PCK rats and Pkd2ws25/− mice and in vitro in cultured PCK cholangiocytes compared to normal controls, respectively.
Cdc25A plays an important role in hepatic cystogenesis
To assess further the potential pathogenic role of Cdc25A in hepatic cyst growth and provide evidence supporting its role as a therapeutic target, we hypothesized that crossbreeding of Cdc25A+/− mice with our own Pkhd1del2/del2 mice might decrease hepatic cystogenesis. Cdc25A+/− mice have reduced Cdc25A protein by ~50% compared to wild type resulting in impaired proliferative capacity.25 Pkhd1del2/del2 animals develop hepatic cysts by the age of 7-8 months while no renal cysts are present26 and have increased levels of Cdc25A (Supplementary Figure 3). The scheme describing development of Cdc25A+/− :Pkhd1del2/del2 crossbred mice is depicted in Supplementary Figure 3.
Body and kidney weights were not different in age matched 7-9 month old Cdc25A+/−, Pkhd1del2/del2 and Cdc25A+/−:Pkhd1del2/del2 crossbreds (Supplementary Table 1). In contrast, liver weights were higher by 62% in Pkhd1del2/del2 mice compared to Cdc25A+/− mice. A 33% reduction in liver weight was observed in Cdc25A+/−:Pkhd1del2/del2 mice compared to Pkhd1del2/del2 animals (Figure 2A and D, Supplementary Table 1). Multiple cysts occupied a substantial portion of liver in Pkhd1del2/del2 mice; no cysts were present in Cdc25A+/− mice; and hepatic cystogenesis were decreased in Cdc25A+/−:Pkhd1del2/del2 crossbreds (Figure 2A-C). Quantitatively, hepatic cysts represented 40% of total liver parenchyma in Pkhd1del2/del2, 0% in Cdc25A+/− and 18% in Cdc25A+/−:Pkhd1del2/del2 mice (Figure 2E). Hepatic fibrosis was also reduced in Cdc25A+/−:Pkhd1del2/del2 crossbreds compared to Pkhd1del2/del2 mice (Figure 2F).
Figure 2.
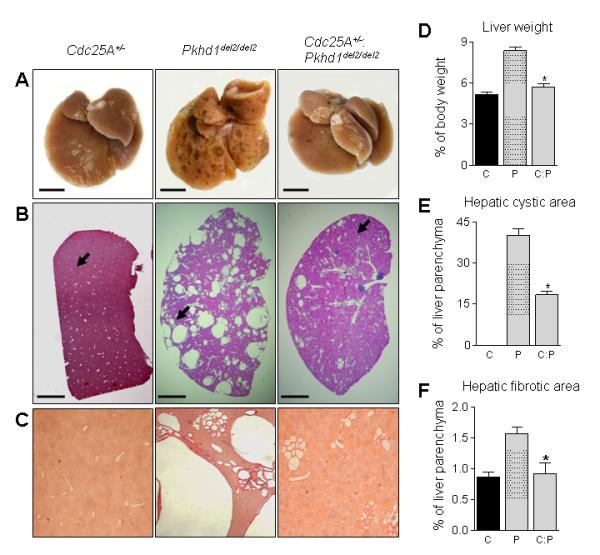
Cdc25A is involved in hepatic cystogenesis. (A) Representative photographs of livers from Cdc25A+/− (n=3), Pkhd1del2/del2 (n=5) and Cdc25A+/− :Pkhd1del2/del2 (n=3) mice. Bar, 10 mm. (B) Representative images (H&E staining) demonstrate that no cysts are present in Cdc25A+/− mice, many hepatic cysts observed in Pkhd1del2/del2 mice, and Cdc25A+/−:Pkhd1del2/del2 crossbreeds have reduced hepatic cystogenesis. Bar, 500 mm. Arrow indicates the liver area shown with the higher magnification in panel C. (C) Representative images (Picrosirius Red staining) of liver areas depicted by arrows in panel B. Magnification, x4. (D) Liver weight, hepatic cystic (E) and fibrotic (F) areas are decreased in Cdc25A+/−:Pkhd1del2/del2 mice compared to Pkhd1del2/del2 counterparts. C - Cdc25A+/−, P - Pkhd1del2/del2 and C:P - Cdc25A+/−:Pkhd1del2/del2. * -P <.05.
Targeting of Cdc25A by VK3 in vitro suppresses cholangiocyte proliferation and alters cell cycle phase distribution
Non-treated PCK cholangiocytes proliferate at a much higher rate (Figure 3A) then normal ones (Figure 3B). In response to all VK3 doses (50, 100 and 200 μM), proliferation of both PCK (Figure 3A) and normal cholangiocytes (Figure 3B) was inhibited; however, VK3 decreased the proliferation of cystic cholangiocytes to a higher extent than in normal cholangiocytes (Figure 3C).
Figure 3.
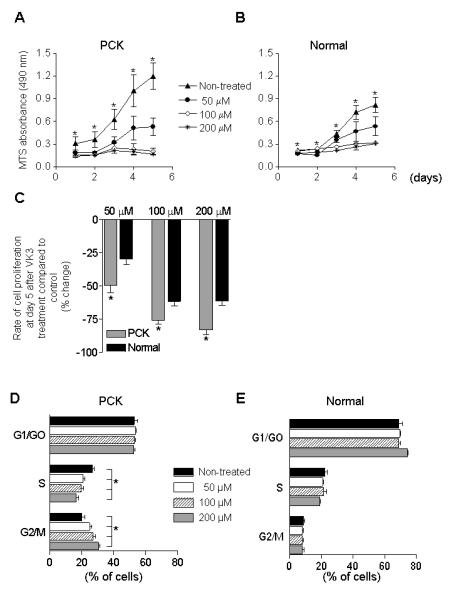
VK3 decreases cholangiocyte proliferation and alters the cell cycle distribution. VK3 inhibits proliferation in both (A) PCK (n=5) and (B) normal (n=5) cholangiocytes. (C) Changes in proliferation of PCK cholangiocytes at day 5 after treatment (compared to non-treated condition) were greater than in normal cholangiocytes. (D) VK3 decreases a percent of PCK cholangiocytes (n=3) during S phase and leads to cell accumulation in G2/M phase. (E) VK3 did not affect the cell cycle in normal cholangiocytes (n=3). * - P<.05.
Non-treated PCK cholangiocytes were distributed during the cell cycle as followed: 53.04% (G0/G1 phase), 26.88% (S phase), and 20.08% (G2/M phase). VK3 in all doses caused cell cycle arrest of PCK cholangiocytes in G2/M phase, did not affect G0/G1 phase and decreased the cell proportion in S phase (Figure 3D). In contrast, VK3 did not change the distribution of normal cholangiocytes (Figure 3E).
VK3 inhibits growth of cystic structures in 3-D culture
PCK cystic structures grown in 3-dimentions expanded more rapidly then normal ones (Figure 4). VK3 suppressed PCK cyst growth as shown by light microscopy and quantitative assessment of their circumferential areas compared to non-treated counterparts (Figure 4A). VK3 also inhibited growth of normal cysts (Figure 4B). However, VK3 suppressed expansion of normal cystic structures to a lesser degree. By day 5, in response to 50, 100 and 200 μM of VK3, the growth of PCK cysts was decreased 1.9, 2.8 and 3.7-fold, respectively; under the same conditions development of normal cystic structures was inhibited 1.2, 1.5 and 1.7-fold.
Figure 4.
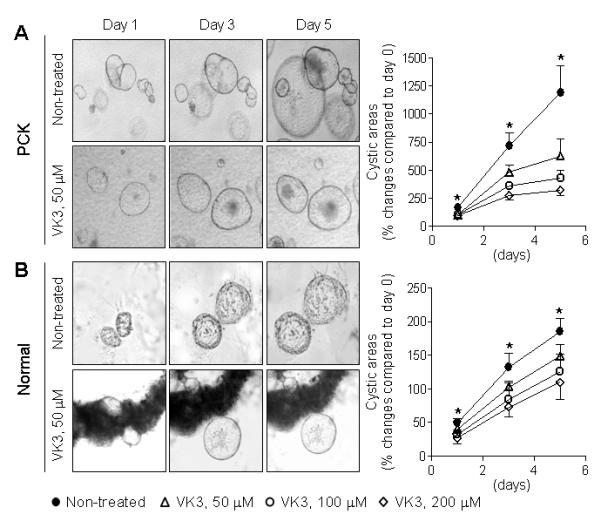
VK3 suppresses growth of hepatic cysts in 3-D cultures. Representative images of cystic structures derived from (A) PCK (n=7) and (B) normal rats (n=5). They were treated with VK3 (50 μM) daily for total of 5 days. Magnification, x4 (A) and x10 (B). Graphs show the effects of different doses of VK3 on cyst enlargement assessed by changes in their circumferential areas. * - P<.05.
VK3 decreases total volume of the intrahepatic biliary tree and hepato-renal cystic and fibrotic areas in vivo
We next tested the effects of Cdc25A targeting by two inhibitors, VK3 and PM-20, in vivo. The detailed treatment protocol is described in Supplementary Figure 4.
No visible defects (such as weight loss) were observed in VK3 and PM-20 treated animals. Serum markers of the PCK rats (which are within the range of normal rats) also showed no differences in treated and non-treated animals (Supplementary Table 2).
We first analyzed the VK3 effects on hepatic cystogenesis in PCK rats by assessing the total volume of the intrahepatic biliary tree. Representative images demonstrate that VK3 improved biliary tree architecture reducing its total volume by 26% (Figure 5A).
Figure 5.
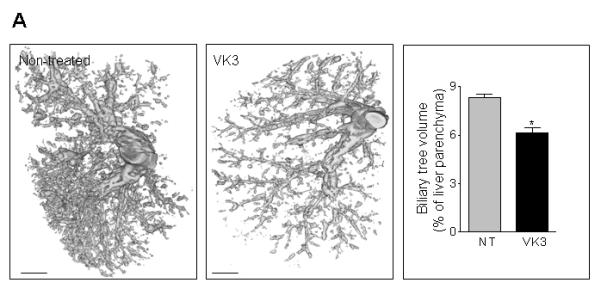
VK3 attenuates hepato-renal cystic disease in PCK rats. (A) Representative images and bar graph demonstrate that VK3 significantly reduced the biliary tree volume in VK3 treated (n=4) for 8 weeks PCK rats compared to non-treated rats (n=4). Bar, 5 mm. (B) Representative images of picrosirius red-stained liver and (C) kidney sections and bar graphs demonstrate that VK3 decreases hepatic and renal cystic and fibrotic areas of PCK rats compared to non-treated counterparts. Magnification, x4. Bar, 500 μm. NT – non-treated. * - P<.05.
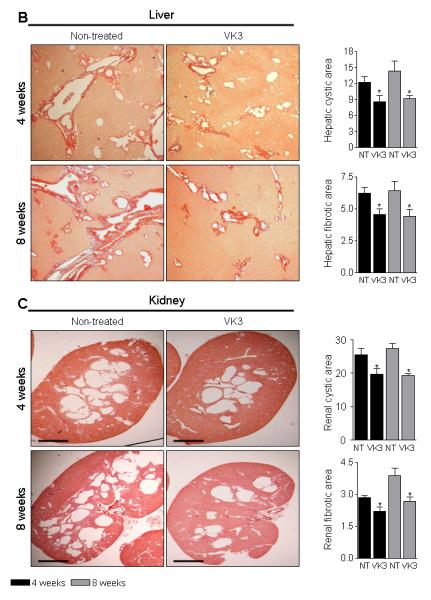
VK3 treatment reduced liver and kidney weights up to 18%, hepatic and renal cyst areas up to 36%, hepatic and renal fibrosis up to 31%, and the mitotic and apoptotic indices up to 34% in both PCK rats (Supplementary Table 3 and Figure 5B and C) and Pkd2ws25/− mice (Supplementary Table 4 and Figure 6A-B). Moreover, longer treatment (i.e., for 8 weeks) led to more significant inhibition of all parameters studied compared to respective controls. PM-20 had similar effects on hepato-renal cystogenesis in Pkd2ws25/− mice: liver and kidney weights, hepatic and renal cyst areas, hepatic and renal fibrosis, and mitotic and apoptotic indices were decreased by 10-15% compared to non-treated mice (Supplementary Table 5 and Figure 6C-E).
Figure 6.
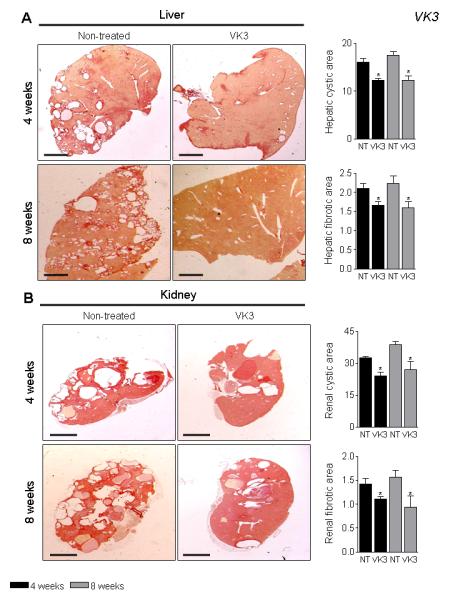
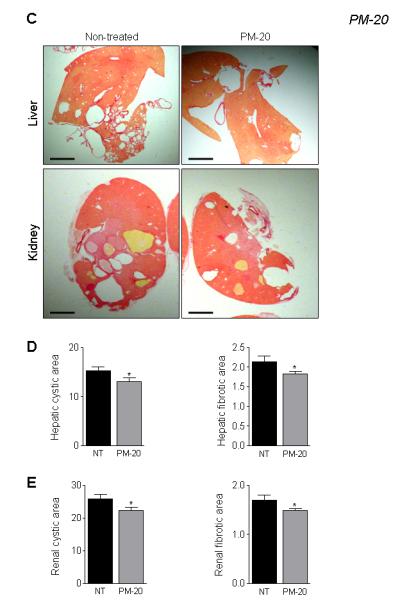
VK3 and PM-20 decreases hepato-renal cystic and fibrotic areas in Pkd2ws25/− mice. Representative images and bar graphs demonstrate that hepatic and renal cystic and fibrotic areas were decreased in VK3-treated (A and B) and PM-20 (C-E) mice compared to non-treated animals. Liver and kidney were stained with picrosirius red. Bar, 2500 μm. * - P<.05.
VK3 affects the expression of cell cycle machinery
Cdc25 phosphatases are recognized as only dephosphorylation molecules of Cdks which in turn are important regulators of the cell cycle transitions.32 Cdk/Cyclin complexes play an important role during cell cycle: G1 phase is regulated by Cdk4/Cyclin D and Cdk6/Cyclin D; G1/S phase - by Cdk2/Cyclin E; S phase - by Cdk2/Cyclin A; G2 phase – by Cdk1/Cyclin A, and M phase - by Cdk1/CyclinB (Supplementary Figure 5).33 Thus, we examined the expression of Cdc25A and its downstream targets in response to VK3 (Figure 7). VK3 inhibited the expression of Cdc25A and the cell cycle proteins known to be regulated by Cdc25A (i.e., Cdk1, 2, 4/6, cyclin A, B, D and E, Rb and PCNA), increased the phosphorylated status of Cdk1 and Cdk2 and decreased Erk1/2 phosphorylation.
Figure 7.
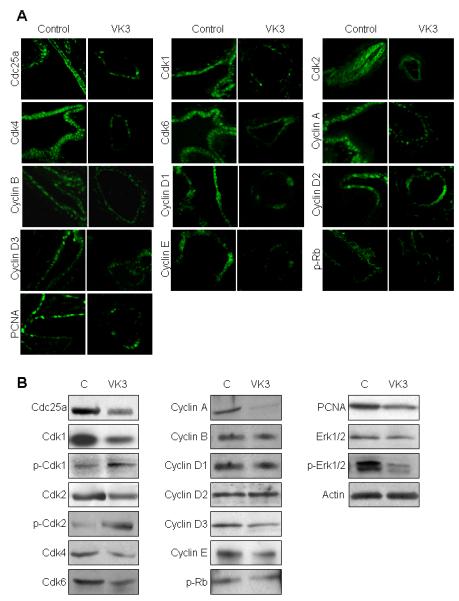
VK3 affects the expression of the cell cycle proteins. (A) Confocal microscopy and western blots (B) demonstrates that VK3 treatment changes the expression of the cell cycle proteins stained in green. Magnifications: x100.
Discussion
We described here that: (a) cystic cholangiocytes are hyper-proliferative and have an abnormal cell cycle; (b) Cdc25A is overexpressed in cystic cholangiocytes of humans and rodents with PKD/PLD; (c) in Cdc25A+/− :Pkhd1del2/del2 crossbred mice, liver weights and cystic and fibrotic areas are decreased compared to Pkhd1del2/del2 counterparts supporting the importance of Cdc25A in hepatic cystogenesis; (d) VK3 decreases proliferation and causes cell cycle arrest of PCK cholangiocytes; (e) VK3 reduces cyst growth in 3-D cultures; (f) the intrahepatic biliary tree volume is significantly less after VK3; (g) in vivo in PCK rats and Pkd2ws25/− mice, liver and kidney weights, hepato-renal cystic and fibrotic areas, and mitotic and apoptotic indices are all decreased after VK3 treatment; (h) PM-20 attenuates hepato-renal cystogenesis in Pkd2ws25/− mice; and (i) VK3 affects the expression of multiple cell cycle proteins. These findings provide new insights into the pathogenesis of PKD/PLD and present a rationale for the use of Cdc25A inhibitors to halt cyst progression.
Cdc25A over-expression in cystic cholangiocytes is likely linked to disturbances in several signaling pathways. First, elevated Cdc25A is associated with reduced expression of its regulator, miR-15a.4 Second, Cdk2 (i.e., a down-stream effector of Cdc25A) phosphorylates Cdc25A in a positive feedback loop increasing its activity.34 We observed (unpublished data) that Ckd2 is up-regulated in cystic cholangiocytes; therefore its overexpression may add to Cdc25A elevation. Importantly, Cdk2 is also a miR-15a target. Third, the levels of many RTK’ (such as EGFR, VGFR, IGFR, HGFR) and several components of the RTK pathway (i.e., Raf, MEK and ERK) are increased in hepato-renal cysts.2, 7 Raf kinase forms a functional complex with Cdc25A while ERK is known to directly phosphorylate Cdk2 further suggesting a connection between mitogenic signaling and the cell cycle machinery.35 Additional studies are underway in our laboratory to directly assess all these possibilities. Be that as it may, accumulating data suggest a major role for Cdc25A in hepatic cystogenesis.
Based on our published evidence that hepatic cystogenesis is linked to elevated Cdc25A in cholangiocytes,4 we hypothesized that its reduction might decrease cyst growth. To this end, we cross-breed the Pkhd1del2/del2 mice (they develop hepatic cysts and have increased Cdc25A expression) with Cdc25A+/− mice that have reduced Cdc25A levels. We anticipated that in crossbred (i.e., Pkhd1del2/del2:Cdc25A+/− mice), cystogenesis should be less due to Cdc25A diminution. As expected, in Cdc25A+/−:Pkhd1del2/del2 mice, decreased Cdc25A expression was associated with reduced liver weights and less evident cystogenesis. The mechanism/s by which Cdc25A depletion affects hepatic cystogenesis is not yet understood. However, our indirect in vitro and in vivo data suggest that cell cycle progression and cell proliferation might be involved since suppression of Cdc25A by VK3 affected the distribution of the cell cycle phases, inhibited rate of cell proliferation and cyst growth.
Various pharmacological inhibitors of Cdc25 phosphatases have been developed.20, 21, 36, 37 Studies testing the effects of VK family members on cancer cell growth have shown that VK3 has the strongest antiproliferative capacity directly affecting Cdc25A.38 Mechanisms of VK3 action might relate to: (i) an oxidative effect produced by redox cycling of VK3; (ii) modulation of transcriptional factors; (iii) perturbation of cell proliferation; and (iv) cell cycle arrest with consequent inhibition of proliferation due to direct binding of inhibitor to Cdc25A.21, 23, 38-42 Consistent with these possibilities, we observed inhibited proliferation of PCK cholangiocytes which was coupled with cell cycle arrest after treatment. In contrast, VK3 has no effect on the cell cycle in normal cholangiocytes suggesting that suppression of their proliferation might involve other mechanisms including those described above. VK3 also slows down the growth of cystic structures in 3-D cultures. Finally, in vivo in two animal models of PKD/PLD, VK3 halted hepato-renal cystogenesis by decreasing mitotic indices and reducing PCNA expression. Another inhibitor used in this study, PM-20, also attenuated hepato-renal cystogenesis in vivo in Pkd2ws25/− mice. PM-20 is known to bind and inhibit the Cdc25A activity subsequently affecting cell proliferation and tumor growth.20, 22 Indeed, mitotic indices were decreased in PM-20 treated mice.
To begin to elucidate the mechanisms by which VK3 affects hepatic cyst growth, we analyzed the expression of the cell cycle proteins. We found that treatment of PCK rats with VK3 decreased levels of Cdc25A and proteins known to be regulated by Cdc25A; reduced the phosphorylated status of Rb, and increased the phosphorylated status of Cdk1 and Cdk2. These observations are in agreement with previous reports.36, 41, 42 Increased phosphorylation of Cdks suggest that VK3 might affect cell proliferation and cell cycle via direct inactivation of Cdc25A which is usually correlated with protein levels.35, 43, 44 Consistent with this view, treatment of PCK rats with VK3 for 8 weeks decreased Cdc25A expression. Finally, we evaluated the phosphorylated status of Erk1/2. We have recently shown the significance of Erk1/2 signaling in hepatic cystogenesis.7 In particular, expansion of PCK-derived hepatic cysts in 3-D cultures as well as enhanced proliferation of cystic cholangiocytes was Mek/Erk1/2-dependent.7 In the present study, VK3 affected Mek/Erk1/2 pathway by decreasing the level of Erk1/2 activation. The mechanism of this inhibition is currently unclear; however, suppressed Erk1/2 phosphorylation was observed in vivo in jck mice (the animal model of PKD) after 5 weeks of treatment with Cdk inhibitor, roscovitine.45
Taken together, our study provides the first experimental evidence that Cdc25A targeting with VK3 and PM-20 decreases proliferation of cystic cholangiocytes halting the expansion of hepatic cysts. Moreover, VK3 and PM-20 also has positive effects on renal cytogenesis.
In conclusion, we believe that Cdc25A represents a potential therapeutic target and this study provides a rationale for examining the role of VK3, PM-20 or other Cdc25 inhibitors that are now at developmental/testing stage37 in the treatment of PKD/PLD.
Supplementary Material
Acknowledgments
This work was supported by grants DK24031, EB000305 and CA112281 from the NIH, by the PKD Foundation, by the National Disease Research Interchange (NDRI), by the Mayo Clinic, by the Clinical Core and Optical Microscopy Core of the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567) and by the Mayo Translational Polycystic Kidney Disease Center (NIDDK P30DK090728).
Abbreviations
- PKD
polycystic kidney disease
- PLD
polycystic liver disease
- ADPKD
autosomal dominant PKD
- ARPKD
autosomal recessive PKD
- CHF
congenital hepatic fibrosis
- Pkhd1
polycystic kidney and hepatic disease 1
- Pkd2
polycystic kidney 2
- PCK
polycystic kidney
- Cdc25
cell division cycle 25
- VK3
Vitamin K3
- cAMP
cyclic adenosine monophosphate
- miR-15A
microRNA 15A
- C57BL6
c57 black 6
- Cdk
cyclin dependent kinase
- p-Rb
retinoblastoma protein
- PCNA
proliferating cell nuclear antigen
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick-end labeling
- DMEM/F12
Dulbecco Modified Eagle Medium: Nutrient Mixture F-12
- H&E
hematoxylin and eosin
- MV-122
low-viscosity radiopaque polymer Microfil MV-122
- micro-CT
micro-computed tomography
- G0/G1 phase
gap 0/1
- S phase
synthetic phase
- G2/M phase
gap2/mitosis
- 3-D
three dimensional
- RTK
receptor tyrosine kinases
- EGFR
epidermal growth factor receptor
- VGFR
vascular endothelial growth factor receptor
- IGFR
insulin-like growth factor receptor
- HGFR
hepatocyte growth factor receptor
- ERK
extracellular signal-regulated protein kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors have declared that no conflict of interest exists.
TM & NL supervised project & wrote manuscript. JB, BR, AM, SG, NC, AS, GB, JB, HK & CW performed experiments and contributed to discussion. ER reviewed three-dimensional reconstruction studies.
References
- 1.Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2009;60:321–37. doi: 10.1146/annurev.med.60.101707.125712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Masyuk T, Masyuk A, LaRusso N. Cholangiociliopathies: genetics, molecular mechanisms and potential therapies. Curr Opin Gastroenterol. 2009;25:265–71. doi: 10.1097/MOG.0b013e328328f4ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Onori P, Franchitto A, Mancinelli R, Carpino G, Alvaro D, Francis H, Alpini G, Gaudio E. Polycystic liver diseases. Dig Liver Dis. 2010;42:261–71. doi: 10.1016/j.dld.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee SO, Masyuk T, Splinter P, Banales JM, Masyuk A, Stroope A, Larusso N. MicroRNA15a modulates expression of the cell-cycle regulator Cdc25A and affects hepatic cystogenesis in a rat model of polycystic kidney disease. J Clin Invest. 2008;118:3714–24. doi: 10.1172/JCI34922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3′,5′-cyclic monophosphate. Gastroenterology. 2007;132:1104–16. doi: 10.1053/j.gastro.2006.12.039. [DOI] [PubMed] [Google Scholar]
- 6.Torres VE, Harris PC. Polycystic kidney disease: genes, proteins, animal models, disease mechanisms and therapeutic opportunities. J Intern Med. 2007;261:17–31. doi: 10.1111/j.1365-2796.2006.01743.x. [DOI] [PubMed] [Google Scholar]
- 7.Banales JM, Masyuk TV, Gradilone SA, Masyuk AI, Medina JF, LaRusso NF. The cAMP effectors Epac and protein kinase a (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD) Hepatology. 2009;49:160–74. doi: 10.1002/hep.22636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamaguchi T, Hempson SJ, Reif GA, Hedge AM, Wallace DP. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol. 2006;17:178–87. doi: 10.1681/ASN.2005060645. [DOI] [PubMed] [Google Scholar]
- 9.Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem. 2004;279:40419–30. doi: 10.1074/jbc.M405079200. [DOI] [PubMed] [Google Scholar]
- 10.Gradilone SA, Masyuk TV, Huang BQ, Banales JM, Lehmann GL, Radtke BN, Stroope A, Masyuk AI, Splinter PL, LaRusso NF. Activation of Trpv4 reduces the hyperproliferative phenotype of cystic cholangiocytes from an animal model of ARPKD. Gastroenterology. 2010;139:304–14. doi: 10.1053/j.gastro.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hogan MC, Masyuk TV, Page LJ, Kubly VJ, Bergstralh EJ, Li X, Kim B, King BF, Glockner J, Holmes DR, 3rd, Rossetti S, Harris PC, LaRusso NF, Torres VE. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol. 2010;21:1052–61. doi: 10.1681/ASN.2009121291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Keimpema L, de Man RA, Drenth JP. Somatostatin analogues reduce liver volume in polycystic liver disease. Gut. 2008;57:1338–9. doi: 10.1136/gut.2008.155721. [DOI] [PubMed] [Google Scholar]
- 13.Ruggenenti P, Remuzzi A, Ondei P, Fasolini G, Antiga L, Ene-Iordache B, Remuzzi G, Epstein FH. Safety and efficacy of long-acting somatostatin treatment in autosomal-dominant polycystic kidney disease. Kidney Int. 2005;68:206–16. doi: 10.1111/j.1523-1755.2005.00395.x. [DOI] [PubMed] [Google Scholar]
- 14.van Keimpema L, Nevens F, Vanslembrouck R, van Oijen MG, Hoffmann AL, Dekker HM, de Man RA, Drenth JP. Lanreotide reduces the volume of polycystic liver: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2009;137:1661–8. doi: 10.1053/j.gastro.2009.07.052. [DOI] [PubMed] [Google Scholar]
- 15.Ducruet AP, Vogt A, Wipf P, Lazo JS. Dual specificity protein phosphatases: therapeutic targets for cancer and Alzheimer’s disease. Annu Rev Pharmacol Toxicol. 2005;45:725–50. doi: 10.1146/annurev.pharmtox.45.120403.100040. [DOI] [PubMed] [Google Scholar]
- 16.Boutros R, Dozier C, Ducommun B. The when and wheres of CDC25 phosphatases. Curr Opin Cell Biol. 2006;18:185–91. doi: 10.1016/j.ceb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 17.Aressy B, Ducommun B. Cell cycle control by the CDC25 phosphatases. Anticancer Agents Med Chem. 2008;8:818–24. doi: 10.2174/187152008786847756. [DOI] [PubMed] [Google Scholar]
- 18.Kristjansdottir K, Rudolph J. Cdc25 phosphatases and cancer. Chem Biol. 2004;11:1043–51. doi: 10.1016/j.chembiol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 19.Wu FY, Sun TP. Vitamin K3 induces cell cycle arrest and cell death by inhibiting Cdc25 phosphatase. Eur J Cancer. 1999;35:1388–93. doi: 10.1016/s0959-8049(99)00156-2. [DOI] [PubMed] [Google Scholar]
- 20.Kar S, Wang M, Yao W, Michejda CJ, Carr BI. PM-20, a novel inhibitor of Cdc25A, induces extracellular signal-regulated kinase 1/2 phosphorylation and inhibits hepatocellular carcinoma growth in vitro and in vivo. Mol Cancer Ther. 2006;5:1511–9. doi: 10.1158/1535-7163.MCT-05-0485. [DOI] [PubMed] [Google Scholar]
- 21.Lazo JS, Wipf P. Is Cdc25 a druggable target? Anticancer Agents Med Chem. 2008;8:837–42. doi: 10.2174/187152008786847738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naderi A, Liu J. Inhibition of androgen receptor and Cdc25A phosphatase as a combination targeted therapy in molecular apocrine breast cancer. Cancer Lett. 2010;298:74–87. doi: 10.1016/j.canlet.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 23.Osada S, Tomita H, Tanaka Y, Tokuyama Y, Tanaka H, Sakashita F, Takahashi T. The utility of vitamin K3 (menadione) against pancreatic cancer. Anticancer Res. 2008;28:45–50. [PubMed] [Google Scholar]
- 24.Lim D, Morgan RJ, Jr., Akman S, Margolin K, Carr BI, Leong L, Odujinrin O, Doroshow JH. Phase I trial of menadiol diphosphate (vitamin K3) in advanced malignancy. Invest New Drugs. 2005;23:235–9. doi: 10.1007/s10637-005-6731-2. [DOI] [PubMed] [Google Scholar]
- 25.Ray D, Terao Y, Nimbalkar D, Hirai H, Osmundson EC, Zou X, Franks R, Christov K, Kiyokawa H. Hemizygous disruption of Cdc25A inhibits cellular transformation and mammary tumorigenesis in mice. Cancer Res. 2007;67:6605–11. doi: 10.1158/0008-5472.CAN-06-4815. [DOI] [PubMed] [Google Scholar]
- 26.Woollard JR, Punyashtiti R, Richardson S, Masyuk TV, Whelan S, Huang BQ, Lager DJ, vanDeursen J, Torres VE, Gattone VH, LaRusso NF, Harris PC, Ward CJ. A mouse model of autosomal recessive polycystic kidney disease with biliary duct and proximal tubule dilatation. Kidney Int. 2007;72:328–36. doi: 10.1038/sj.ki.5002294. [DOI] [PubMed] [Google Scholar]
- 27.Masyuk TV, Huang BQ, Masyuk AI, Ritman EL, Torres VE, Wang X, Harris PC, Larusso NF. Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease. Am J Pathol. 2004;165:1719–30. doi: 10.1016/S0002-9440(10)63427-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stroope A, Radtke B, Huang B, Masyuk T, Torres V, Ritman E, LaRusso N. Hepato-renal pathology in pkd2ws25/-mice, an animal model of autosomal dominant polycystic kidney disease. Am J Pathol. 2010;176:1282–91. doi: 10.2353/ajpath.2010.090658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banales JM, Masyuk TV, Bogert PS, Huang BQ, Gradilone SA, Lee SO, Stroope AJ, Masyuk AI, Medina JF, LaRusso NF. Hepatic cystogenesis is associated with abnormal expression and location of ion transporters and water channels in an animal model of autosomal recessive polycystic kidney disease. Am J Pathol. 2008;173:1637–46. doi: 10.2353/ajpath.2008.080125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taper HS, Jamison JM, Gilloteaux J, Summers JL, Calderon PB. Inhibition of the development of metastases by dietary vitamin C:K3 combination. Life Sci. 2004;75:955–67. doi: 10.1016/j.lfs.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 31.Jamison JM, Gilloteaux J, Taper HS, Calderon PB, Summers JL. Autoschizis: a novel cell death. Biochem Pharmacol. 2002;63:1773–83. doi: 10.1016/s0006-2952(02)00904-8. [DOI] [PubMed] [Google Scholar]
- 32.Boutros R, Lobjois V, Ducommun B. CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer. 2007;7:495–507. doi: 10.1038/nrc2169. [DOI] [PubMed] [Google Scholar]
- 33.Markovits J, Wang Z, Carr BI, Sun TP, Mintz P, Le Bret M, Wu CW, Wu FY. Differential effects of two growth inhibitory K vitamin analogs on cell cycle regulating proteins in human hepatoma cells. Life Sci. 2003;72:2769–84. doi: 10.1016/s0024-3205(03)00188-7. [DOI] [PubMed] [Google Scholar]
- 34.Ducruet AP, Lazo JS. Regulation of Cdc25A half-life in interphase by cyclin-dependent kinase 2 activity. J Biol Chem. 2003;278:31838–42. doi: 10.1074/jbc.M303604200. [DOI] [PubMed] [Google Scholar]
- 35.Nemoto K, Vogt A, Oguri T, Lazo JS. Activation of the Raf-1/MEK/Erk kinase pathway by a novel Cdc25 inhibitor in human prostate cancer cells. Prostate. 2004;58:95–102. doi: 10.1002/pros.10292. [DOI] [PubMed] [Google Scholar]
- 36.Kar S, Wang M, Ham SW, Carr BI. H32, a non-quinone sulfone analog of vitamin K3, inhibits human hepatoma cell growth by inhibiting Cdc25 and activating ERK. Cancer Biol Ther. 2006;5:1340–7. doi: 10.4161/cbt.5.10.3223. [DOI] [PubMed] [Google Scholar]
- 37.Lavecchia A, Di Giovanni C, Novellino E. Inhibitors of Cdc25 phosphatases as anticancer agents: a patent review. Expert Opin Ther Pat. 2010;20:405–25. doi: 10.1517/13543771003623232. [DOI] [PubMed] [Google Scholar]
- 38.Hitomi M, Yokoyama F, Kita Y, Nonomura T, Masaki T, Yoshiji H, Inoue H, Kinekawa F, Kurokohchi K, Uchida N, Watanabe S, Kuriyama S. Antitumor effects of vitamins K1, K2 and K3 on hepatocellular carcinoma in vitro and in vivo. Int J Oncol. 2005;26:713–20. [PubMed] [Google Scholar]
- 39.Acharya BR, Choudhury D, Das A, Chakrabarti G. Vitamin K3 disrupts the microtubule networks by binding to tubulin: a novel mechanism of its antiproliferative activity. Biochemistry. 2009;48:6963–74. doi: 10.1021/bi900152k. [DOI] [PubMed] [Google Scholar]
- 40.Lamson DW, Plaza SM. The anticancer effects of vitamin K. Altern Med Rev. 2003;8:303–18. [PubMed] [Google Scholar]
- 41.Ma WW, Adjei AA. Novel agents on the horizon for cancer therapy. CA Cancer J Clin. 2009;59:111–37. doi: 10.3322/caac.20003. [DOI] [PubMed] [Google Scholar]
- 42.Matzno S, Yamaguchi Y, Akiyoshi T, Nakabayashi T, Matsuyama K. An attempt to evaluate the effect of vitamin K3 using as an enhancer of anticancer agents. Biol Pharm Bull. 2008;31:1270–3. doi: 10.1248/bpb.31.1270. [DOI] [PubMed] [Google Scholar]
- 43.Fernandez-Vidal A, Mazars A, Manenti S. CDC25A: a rebel within the CDC25 phosphatases family? Anticancer Agents Med Chem. 2008;8:825–31. doi: 10.2174/187152008786847684. [DOI] [PubMed] [Google Scholar]
- 44.Brisson M, Foster C, Wipf P, Joo B, Tomko RJ, Jr., Nguyen T, Lazo JS. Independent mechanistic inhibition of cdc25 phosphatases by a natural product caulibugulone. Mol Pharmacol. 2007;71:184–92. doi: 10.1124/mol.106.028589. [DOI] [PubMed] [Google Scholar]
- 45.Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O. Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature. 2006;444:949–52. doi: 10.1038/nature05348. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.