

Abstract
The posttranslational modification of chromatin through acetylation at selected histone lysine residues is governed by histone acetyltransferases (HATs) and histone deacetylases (HDACs). The significance of this subset of the epigenetic code is interrogated and interpreted by an acetyllysine-specific protein–protein interaction with bromodomain reader modules. Selective inhibition of the bromo and extra C-terminal domain (BET) family of bromodomains with a small molecule is feasible, and this may represent an opportunity for disease intervention through the recently disclosed antiproliferative and anti-inflammatory properties of such inhibitors. Herein, we describe the discovery and structure–activity relationship (SAR) of a novel, small-molecule chemical probe for BET family inhibition that was identified through the application of structure-based fragment assessment and optimization techniques. This has yielded a potent, selective compound with cell-based activity (PFI-1) that may further add to the understanding of BET family function within the bromodomains.
Introduction
Bromodomains are protein interaction modules that recognize the ε-N-acetylation state of specific lysine residues found within histone tails and other proteins.1 Histones are lysine-rich proteins that, left unmodified, are highly basic in character. Modification of these hallmark lysine residues through acetylation or iterative methylation causes changes in the structural and physicochemical properties of the histone protein,2,3 affecting the structure of nucleosomes that organize the protein–DNA hybrid arrangement. Alteration of these ε-NH2 sites on lysine represents the protein-held part of the epigenetic code within the chromatin of each cell nucleus. The acetylation level of these initially basic residues is controlled by the action of histone acetyltransferases (HATs) and histone deacetylases (HDACs); however, the significance of these modulations is relayed by the bromodomains and their histone tail recognition function.4 The consequences of this acetylation reading process may trigger further remodeling at the epigenetically modifiable sites within the protein or DNA components of chromatin, ultimately manifesting themselves in transcriptional activity control.5 The number of proteins susceptible to lysine ε-NH2 acetylation state changes reaches into the thousands, and these have been shown to play a diverse range of functions.6 Bromodomains are the only modules that can specifically recognize acetylated linear motifs. They are found within large multidomain nuclear proteins tasked with controlling processes such as methyl transfer, transcription coactivation, and motor protein (helicase) activity. There are 61 bromodomains in the human proteome and these are further classified into eight families, one of which is the bromodomain and extra C-terminal domain (BET) family.7 BET family function has been studied through protein expression quantification or knockdown experiments of individual BET family members. This has highlighted the control of processes that mediate cancer,8 inflammation,9 and viral infection,10 among others. This renders the bromodomains within the BET protein family attractive targets for drug discovery, at least in the context of potential efficacy.1 Chemical probes against bromodomain families, or perhaps in time highly selective inhibitors of each of the 61 family members, will be useful tools in fully establishing the role of these proteins. Chemical probes that can help validate the potential efficacy and, of equal importance, safety of bromodomain inhibition will be of great utility in this emerging target class. The first chemotypes for BET family inhibitors have recently been disclosed by members of this group11 and others.12,13 The structurally related triazolodiazepine compounds (+)-JQ1 1 and I-BET762 2 (Figure 1) are the most potent (nanomolar), BET family-selective, and cell-active BET family inhibitors disclosed thus far. I-BET762 has recently entered clinical trials for NUT midline carcinoma.14
Figure 1.

First-generation BET family inhibitors.
(+)-JQ1 1 showed anti-cancer activity in patient-derived xenografts,11 while I-BET762 exhibited anti-inflammatory effects in mice.12 A small number of orthogonal chemotypes have also been recently disclosed. Conway and co-workers15 (compounds 3 and 4, Figure 2) and Prinjha and co-workers16 (I-BET151, 5, Figure 2) have independently described 3,5-dimethylisoxazole as a viable acetyllysine mimetic in identifying novel BET binders. Bamborough and Chung and co-workers17 have also described fragment-based approaches to identifying novel chemotypes (such as compound 6, Figure 2) through a designed selection of fragments with the potential to be N-acetyllysine mimetics. This yielded novel hits of moderate efficiency in this proof-of-principle study.
Figure 2.

Orthogonal bromodomain chemotype hits with BRD4 activity.
Herein we describe the discovery and optimization of a novel BET family inhibitor chemotype from similar fragmentlike starting points as those of Chung and co-workers.17 The most optimized compounds disclosed meet the criteria we defined at the outset of the collaboration for a chemical probe, that being ∼100–300 nM activity in a biochemical assay; cell-based activity; family selectivity within bromodomains; and broader polypharmacological selectivity against a panel of kinases, G protein-coupled receptors (GPCRs), enzymes, and ion channels.
Results and Discussion
Despite the protein–protein interaction nature of the histone and its associated bromodomain reader, the pockets for binding acetylated lysine within the bromodomain proteins are sufficiently deep and defined to potentially accommodate inhibitory small molecules with high affinity and efficiency.7 Given this potential, and the knowledge that acetylated lysine is the endogenous binding substrate for the pocket, we (and others)15,17 embarked on screening fragment-sized potential acetyllysine mimetics that may be weak, nonselective pan-bromodomain binders. Much like starting points for Conway and co-workers15 and Chung and co-workers,17 3,4-dihydro-3methyl-2(1H)-quinazolinones seemed like attractive fragments to test this hypothesis. Commercially available 3,4-dihydro-3-methyl-2(1H)-quinazolinone 7 [also identified by Chung as a bromodomain fragment, leading to a BRD2(1) cocrystal deposition in the Protein Data Bank, accession code 4A9E]17 and its 6-brominated equivalent 8 both proved to be active in a peptide displacement biochemical assay (AlphaScreen format) in the 30–90 μM range against BRD4 and cyclic AMP response binding element binding protein (CREBBP).18 They were not universally active in all bromodomains screened in this format, with no activity seen at 200 μM in the BAZ2B and FALZ AlphaScreens.
With sub-30 μM pharmacology found in such small molecules, the consequent ligand efficiencies19 of >0.45 made for a very promising fragment-derived starting point. The 3,4-dihydro-3-methyl-2(1H)-quinazolinone screening lead 3 of Conway and co-workers15 (larger than conventional fragments) had the ambiguity of two potential binding modes as it also contained a 3,5-dimethylisoxazole at the 6-position of the bicyclic core. This question was solved through their first crystal structure in BRD4(1), which also showed the unexpected oxidative incorporation of an ethylene glycol unit under the conditions of crystallization. The compound to actually cocrystallize was found to have the structure 4 (Figure 2). This structure showed the 3,5-dimethylisoxazole making the key interaction at the base of the binding pocket, accepting a hydrogen bond from Asn140. The 3,5-dimethylisoxazole was then optimized further by Conway and co-workers,15 moving away from the dihydroquinazolinone substructure found in the other half of the original screening lead 3.
The smaller fragments 7 and 8 screened in our campaign (featuring only H and Br at the core’s 6-position) contained the cyclic urea as the only potential interaction functionality available to Asn140. Chronologically in our study, a crystal structure was first solved for 8 in CREBBP (not a BET family member). This was followed up by crystallizing 8 in BRD4(1) in a 1.63 Å structure, with no oxidative reactivity observed in the crystallization protocol. Both the CREBBP and BRD4(1) crystal structures for 8 showed the cyclic urea buried in the pocket mimicking the acetyl NH of the endogenous histone lysine. It also overlaid well in comparison to the BRD2(1) structure of 7 (PDB code 4A9E).17 This reconfirms that both binding modes screened for by Conway are indeed possible and that 3,4-dihydro-3-methyl-2(1H)-quinazolinones (6-brominated or not) are viable N-acetyl mimetics. The binding of 8 in BRD4 (1) (Figure 3) is characterized by a lipophilic sandwich of the bicyclic core between residues Val87, Leu92, Leu94, Tyr97 and Phe83, Ile146 in the protein. The cyclic urea meets Asn140 slightly offset with a hydrogen bond accepted by the urea carbonyl from the amino acid carboxamide.
Figure 3.

Two views (A and C) of fragment hit 8 (B) cocrystallized in BRD4(1).
The binding mode implication of the cyclic urea interaction with Asn140 consequently presents the core’s brominated 6-position in the general direction of solvent. The fragment hits 7 and 8, along with elucidation of the binding mode through X-ray crystallography, efficiently directed design toward replacing the bromine. The possibility for further productive interactions before reaching solvent through extending a larger substituent from the 6-position vector was the basis for the next round of molecular design. This was also expected to improve selectivity for the BET family targets, as greater variation in bromodomain protein structures is found away from the N-acetyl binding residues. Although the bromine atom represents a useful synthetic handle for many bond formations in its own right, the unoccupied regions of the BRD protein in the crystal structure suggested that inducing a pronounced kink in the 6-substituent directly off the template by design may be productive. This was undertaken with access to a vacant lipohilic shelf (the so-called WPF shelf) in mind at the opening of the pocket. Sulfonamides were selected as a linker to explore this conformational plan. The availability of the 6-brominated variant of the original 3,4-dihydro-3-methyl-2(1H)-quinazolinone suggests that reactivity of the unsubstituted parent is directed through the 6-position during electrophilic aromatic substitution reactions. To explore this proposed reactivity, two templates were synthesized with sulfonamide hypotheses in mind. Commercially available 7 was reacted with chlorosulfonic acid to produce a 6-sulfonyl chloride template 9, and the known 6-nitro-3,4-dihydro-3-methyl-2(1H)-quinazolinone 10 was resynthesized and then reduced to the corresponding aniline 11 by hydrogenation. The 6-sulfonyl chloride 9 and the 6-amino 11 synthesized as templates are novel compounds and make for attractive monomers in their own right. In this program they were used as templates in relatively small library designs where diversity was drawn from the corresponding partner amine or sulfonyl chloride monomer sets, held within the Pfizer company collection.
The sulfonamides derived from the 6-sulfonylchloride template were profiled first. Pyrrolidine 12 and ethylamine 13 sulfonamides were 5–20-fold more active than the parent bromide. The pyrrolidine sulfonamide 12 had a respectable ligand efficiency of 0.38. The aniline-derived sulfonamide 14 was of similar activity, suggesting that this sulfonamide regioisomer was not efficiently delivering the lipophilicity into the open WPF shelf. Co-crystal structures of this series show alkylamine sulfonamides (12 and 13) adopting highly comparable conformations on binding, suggesting a tolerated sulfonamide replacement of the 6-bromide from fragment hit 8 (Figure 4). The inefficiency of the anilinic sulfonamide 14 is explained by the noticeable change in the SO2 group conformation as a consequence of the phenyl group accommodating itself on the shelf. The electron density on solving the crystal structure also showed multiple conformations of similar occupancy for compound 14, suggesting that there was no single ideal binding mode for the aryl group.
Figure 4.

First-generation sulfonamide cocrystal structures in BRD4(1). (A) Fragment hit 8; (B) ethylsulfonamide 12; (C) anilinesulfonamide 14 with change in NHSO2 conformation; (D) pyrrolidinesulfonamide 13.
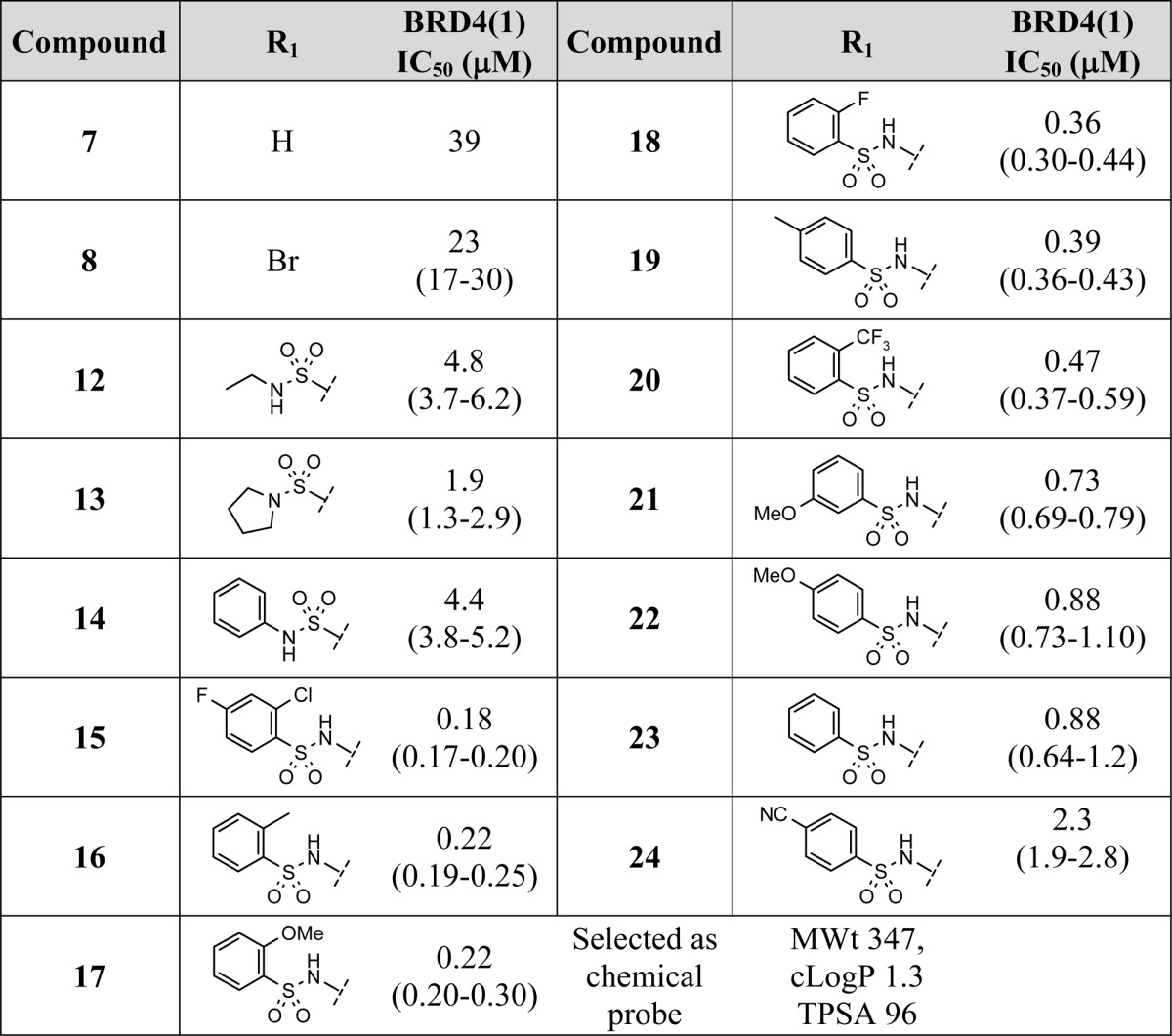
Reversing the sulfonamide by use of the 6-amino template 11 produced a series of compounds with significantly improved BRD4 activity. The abundance of readily available arylsulfonyl chlorides weighted design in favor of aryl substituents. These compounds showed a structure–activity relationship (SAR) implying that they were consistently accessing the lipophilic shelf as targeted in the design hypothesis. A complementary synthesis of this type of compound was also possible from the bromide fragment hit 8 through palladium-catalyzed coupling of a primary arylsulfonamide (Scheme 1). The 10 examples (15–24) in Table 1 show BRD4(1) activity (180 nM–2.3 μM) across a selection of regioisomers of both electron-withdrawing and -donating substituents on a phenyl ring.
Scheme 1. Synthesis of Sulfonamide Variants.

(i) ClSO3H, DCM, 0 °C. (ii) R1R2NH, DCM, RT. (iii) H2SO4, KNO3, 0 °C. (iv) Cat. Raney Ni, H2 (1 atm), AcOH, RT. (v) R3SO2Cl, pyridine, DCM, RT. (vi) Cs2CO3, cat. [(Pd2(dba)3)/X-Phos], R3SO2NH2, dioxane, 100 °C.
Table 1. Structure–Activity Relationship for BRD4 Activity within the Seriesa.
IC50 values are expressed with a 95% confidence interval derived from a minimum of two determinations.
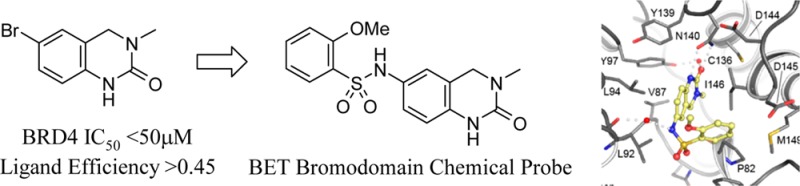
Four out of five of the most active compounds are derived from ortho-substituted arylsulfonyl chlorides, although the entire structure–activity relationship in Table 1 falls within 10-fold of each other for compounds 15–24, regardless of the aryl group used. Given the position of this group, now occupying the solvent-exposed WPF shelf, the key improvement in activity has been attained primarily through the use of the reversed sulfonamide linker. The binding of the now optimally orientated aryl group is less sensitive to substitution patterns around the ring system. Compound 17 (PFI-1) was selected as the nominated chemical probe from the series for further profiling, as it struck the best compromise between lipophilicity and pharmacology (Figure 5).
Figure 5.

Chemical probe selected for profiling.
A 1.52 Å cocrystal structure was obtained for 17 in BRD4(1). Details of the crystal structure have been deposited in the PDB by members of this collaboration.20 The view shown in Figure 6 reveals a right-angled turn in the molecule induced by the sulfonamide. The key interaction with Asn140 is maintained and the urea carbonyl also makes a water-bridged hydrogen-bond interaction with the phenolic OH of Tyr97.
Figure 6.

Two views (A and B) of 17 (PFI-1) cocrystallized to 1.52 Å in BRD4(1).
The most potent example 15 (2-Cl, 4-F) is >200-fold more active than the fragment hit. The ligand efficiency of the initial hit has been largely preserved in the series, and this indicates that improvements in potency have been attained through the incorporation of appropriately balanced lipophilicity. Beyond the expectation for improved potency through functionalizing the 6-position of the template, there was a BET family selectivity requirement for the novel series. The fragment hits 7 and 8 were unselective starting points for bromodomain selectivity with respect to CREBBP. This is attributed to the fact that, due to their small size, the fragments only occupy the region close to the conserved asparagine residue common to all the bromodomains and are unable to exploit binding site differences elsewhere within some of the more closely related bromodomain proteins. As one of those most closely related non-BET family bromodomains,7 CREBBP activity was assessed on the more elaborated compound 17 from the best sulfonamide series as a surrogate for broader bromdomain selectivity. Surface plasmon resonance (SPR) determined a KD of 49 μM for compound 17 in CREBBP. A Tm shift assay in CREBBP also showed a significantly lower melting temperature for 17 of 1.7 °C at 10 μM. The BET family protein members showed Tm shifts of 2.1–6.5 °C for compound 17. Tm shifts for BAZ2B, PB1(5), and PCAF were less than 1 °C as representative members of bromodomains more distantly related to the BET family.21 Compound 17 is a selective chemical probe for the BET bromodomains.
Having studied the activity of 17 as a representative member of this BET family inhibitor series in biochemical and biophysical assays, we further studied the compound’s credentials as a chemical probe for broader pharmacological selectivity, a cell-based inflammatory end-point assay, and rodent pharmacokinetics. The compound showed <50% inhibition against a panel of 15 targets made up of GPCRs, ion channels, and enzymes at 10 μM at Cerep and <20% inhibition against 50 kinases at 1 μM at Invitrogen. Cell-based activity was tested in an LPS challenge assay in PBMCs. Compound 17 had an EC50 of 1.89 μM (n = 6) for the inhibition of IL6 production from human blood mononuclear cells stimulated by LPS. Rodent pharmacokinetics for 17 were studied in both rat (1 mg/kg iv and 2 mg/kg po) and mouse (2 mg/kg sc). After iv administration in the rat, the volume of distribution was 1 L/kg and the plasma clearance was 18/mL·min–1·kg–1, giving a 1 h half-life. The volume of distribution is consistent with the physicochemical properties of the compound, and the clearance is in line with estimates from in vitro rat liver microsome (RLM) data (RLM t1/2 >120 min).22 After oral dosing in the rat, the oral bioavailability was low at 32%. This is inconsistent with the clearance and is believed to be due to suboptimal compound solubility in the gut. This was further supported by the oral exposure profile in rat, where the compound was present at a steady yet low concentration (within 2-fold of the 1 h Cmax concentration of 120 ng/mL), out to the last time point of the study (62 ng/mL at 7 h). This implies a depot of insoluble material in the gut after oral dosing, gradually delivering low concentrations to the systemic circulation over an extended time course. The compound was 69% plasma protein-bound. The subcutaneous mouse pharmacokinetics indicated exposure out to 4 h based on the 2 mg/kg dose, giving a Cmax of 58 ng/mL with a Tmax of 1 h and a half-life of approximately 2 h.
In conclusion, 17 is a novel chemical probe for the BET family of bromodomain epigenetic reader proteins. With a molecular weight of 347, a cLogP of 1.3, and TPSA of 96, 17 has been optimized such that it has a low probability of polypharmacology and conforms to most of the guidelines on the design of druglike molecules.23 It is a small, nonlipophilic, cell-penetrant compound with selectivity for the BET family within the bromodomains. It has rodent oral bioavailability and pharmacokinetics in line with its neutral template and associated solubility profile. Compound 17 is structurally orthogonal to the triazolodiazepines and isoxazoles first discovered as selective BET family inhibitors and so offers a complementary molecular approach for the use of chemical probes in this field. It was discovered in two design cycles of less than 250 compounds in total, based on an efficient fragment-based starting point.
Selected Experimental Procedures: Chemistry Methods and Compound Characterization
Proton (1H NMR) and carbon (13C NMR) magnetic resonance spectra where obtained in DMSO-d6 at 400 and 100 MHz, respectively unless otherwise noted. The following abbreviations were utilized to describe peak patterns when appropriate: br = broad, s = singlet, d = doublet, and m = multiplet. Accurate mass spectrometry (MS) analyses were conducted on an Agilent 6220 TOF mass spectrometer in positive or negative electrospray mode. The samples were separated by UHPLC on an Agilent 1200 system prior to mass spectrometric analysis. The mass accuracy was calculated for all observed isotopes against the theoretical mass ions derived from the chemical formula by use of MassHunter software. All air- and moisture-sensitive reactions were carried out under an atmosphere of dry nitrogen with heat-dried glassware and standard syringe techniques. Tetrahydrofuran (THF) and acetonitrile were purchased from EMD anhydrous and were used without further drying. Flash chromatography was performed on an Analogix Intelliflash 280 with Sepra Si 50 silica gel with ethyl acetate/heptane mixtures as solvent unless otherwise indicated. Certain compounds from the examples described were purified on reversed-phase via automated preparative high-performance liquid chromatography (HPLC) on a Gilson GX281, Shimadzu CL-2010C, or Agilent 1200 system on an Agella Venusil ASB C18 column. Detection was achieved by use of a Shimadzu SPD-20AV or Waters 2998 PDA wavelength absorbance detector set at 220 or 200 nm, followed in series by a Shimadzu MS2010EV or Waters 3100 mass spectrometer. Quality control (QC) for purity analysis was performed by HPLC-MS on a Shimadzu with XB-C18, X-Bridge, Gemini NX C18, or Gemini NX C18 columns. Detection was achieved by use of a Shimadzu 10A detector set at 220 or 260 nm, followed in series by a Shimadzu MS2010EV or Applied Biosystems API 2000 mass spectrometer in parallel. Compound purity was >95% other than intermediate 9 (91%) which, as a reactive sulfonyl chloride, was used satisfactorily with no further chromatographic purification.
6-Amino-3-methyl-3,4-dihydroquinazolin-2(1H)-one (11)
To a solution of 3-methyl-6-nitro-3,4-dihydroquinazolin-2(1H)-one (5.00g, 24.13 mmol, 1.0 equiv) in acetic acid (50 mL) was added Raney nickel (500 mg), and the reaction mixture was stirred at room temperature under H2 balloon (1 atm) for 16 h. After completion of the reaction [via thin-layer chromatography (TLC) with 10% MeOH in dichloromethane (DCM)], the reaction mixture was filtered through a Celite pad and evaporated to provided the crude product. The crude material was purified by titration with ethyl acetate to provide the desired material as a brown solid, which was an acetic acid salt (4.1 g, 72%). 1H NMR (400 MHz, DMSO-d6) δ 2.83 (s, 3H), 4.23 (s, 2H), 6.29 (s, 1H), 6.35 (d, J = 8.36 Hz, 1H), 6.47 (d, J = 8.36 Hz, 1H), 8.73 (s, 1H); 13C NMR (DMSO-d6) δ 33.7, 50.1, 110.7, 113.6, 113.9, 118.2, 127.6, 142.8, 153.9; HRMS [M + H] for C9H11N3O, calcd 178.0975, found 178.0977; LCMS [M + H] = 178, 99% (t = 1.33 min).
2-Methoxy-N-(3-methyl-2-oxo-1,2,3,4-tetrahydroquinazolin-6-yl)benzenesulfonamide (17)24
To a stirred suspension of 6-amino-3-methyl-3,4-dihydroquinazolin-2(1H)-one (11) (100 mg, 0.421 mmol, 1.0 equiv) in dichloromethane (10 mL) was added pyridine (0.20 mL, 2.48 mmol, 5.9 equiv), followed by 2-methoxybenzene-1-sulfonyl chloride (130 mg, 0.631 mmol, 1.5 equiv). After 2 h, the solvent was evaporated and the residue was partitioned between ethyl acetate and aqueous 2 M HCl. The organic layer was collected, washed with water and brine, dried over magnesium sulfate, filtered, and concentrated to a residue. The residue was dissolved in dimethyl sulfoxide (DMSO, ∼1 mL) and purified by automated HPLC to provided the desired material (50 mg, 34%). 1H NMR (400 MHz, DMSO-d6) δ 2.78 (s, 3H), 3.91 (s, 3H), 4.25 (s, 2H), 6.59 (d, J = 9.16 Hz, 1H), 6.79–6.81 (m, 2H), 6.97 (t, J = 7.5 Hz, 1H), 7.14 (d, J = 8.2 Hz, 1H), 7.52–7.56 (m, 1H), 7.68 (dd, J = 1.4, 7.8 Hz, 1H), 9.09 (s, 1H), 9.64 (s, 1H); HRMS [M + H] for C16H18N3O4S, calcd 348.1013, found 348.1019; LCMS [M + H] = 384.1, >99% (t = 1.23 min).
Acknowledgments
The Structural Genomics Consortium is a registered charity (1097737) that receives funds from the Canadian Institutes for Health Research, the Canada Foundation for Innovation, Genome Canada, Pfizer, GlaxoSmithKline, Eli Lilly, the Novartis Research Foundation, Abbott, the Ontario Ministry of Research and Innovation, and the Wellcome Trust.
Glossary
Abbreviations
- BET
bromodomain and extra C-terminal domain
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- GPCR
G protein-coupled receptor
- cLogP
calculated LogP
- CREB
cyclic AMP response binding element
- CREBBP
cyclic AMP response binding element binding protein
- BAZ2B
bromodomain adjacent to zinc finger domain 2B
- FALZ
fetal Alzheimer-50 clone 1 protein
- SPR
surface plasmon resonance
- DCM
dichloromethane
- RT
room temperature
- cat.
catalytic
- dba
dibenzylidene acetone
- IL6
interleukin 6
- LPS
lipopolysaccharide
- PBMC
peripheral blood mononuclear cell
- sc
subcutaneous
- po
per os (orally)
- iv
intravenous
- TPSA
topological polar surface area
Supporting Information Available
Additional text describing biological methods and chemistry experimental procedures; five figures showing rodent pharmacokinetic profiles and cellular activity; and two tables listing bromodomain selectivity and X-ray statistics. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Present Address
⊥ University College London School of Pharmacy, 29−39 Brunswick Square, London WC1N 1AX, U.K.
Author Present Address
# Pfizer Worldwide Medicinal Chemistry, Pfizer Worldwide R&D, 200 Cambridgepark Dr., Cambridge, MA 02140.
Accession Codes
Coordinates for five compounds have been deposited with the Protein Data Bank under the following accession codes: 8 (4HBV), 12 (4HBW), 13 (4HBX), 14 (4HBY), and 17 (4E96).
Supplementary Material
References
- Chung C.-w.; Tough D. F. Bromodomains: a new target class for small molecule drug discovery. Drug Discovery Today: Ther. Strategies 2012, 10.1016/j.ddstr.2011.12.002. [DOI] [Google Scholar]
- Jenuwein T.; Allis C. D. Translating the histone code. Science 2001, 293, 1074–1080. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation?. EMBO J. 2000, 19, 1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S. D.; Li H.; Ruthenburg A. J. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 2007, 14, 1025–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C.; Kumar C.; Gnad F.; Nielsen M. L.; Rehman M.; Walther T. C.; Olsen J. V.; Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos; Picaud S.; Mangos M.; Keates T.; Lambert J.-P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Müller S.; Pawson T.; Gingras A.-C.; Arrowsmith C. H.; Knapp S. Histone recognition and large scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J.; Li Q.; Wu C.; Kim J.; Ottinger M.; Howley P. M. Regulation of aurora B expression by the bromodomain protein Brd4. Mol. Cell. Biol. 2009, 29, 5094–5103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B.; Yang X.-D.; Zhou M.-M.; Ozato K.; Chen L.-F. Brd4 coactivates transcriptional activation of NF-κB via specific binding to acetylated RelA. Mol. Cell. Biol. 2009, 29, 1375–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.-Y.; Lee A.-Y.; Hou S. Y.; Kemper J. K.; Erdjument-Bromage H.; Tempst P.; Chiang C.-M. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006, 20, 2383–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue. N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E.; Jeffrey K. L.; Schaefer U.; Beinke S.; Dewell S.; Chung C.-w.; Chandwani R.; Marazzi I.; Wilson P.; Coste H.; White J.; Kirilovsky J.; Rice C. M.; Lora J. M.; Prinjha R. K.; Lee K.; Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C.-w.; Coste H.; White J. H.; Mirguet O.; Wilde J.; Gosmini R. L.; Delves C.; Magny S. M.; Woodward R.; Hughes S. A.; Boursier E. V.; Flynn H.; Bouillot A. M.; Bamborough P.; Brusq J.-M. G.; Gellibert F. J.; Jones E. J.; Riou A. M.; Homes P.; Martin S. L.; Uings I. J.; Toum J.; Clement C. A.; Boullay A.-B.; Grimley R. L.; Blandel F. M.; Prinjha R. K.; Lee K.; Kirilovsky J.; Nicodeme E. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J. Med. Chem. 2011, 54, 3827–3838. [DOI] [PubMed] [Google Scholar]
- a French C. A. Nut midline carcinoma. Cancer Genet. Cytogenet. 2010, 203, 16–20. [DOI] [PMC free article] [PubMed] [Google Scholar]; b A study to investigate the safety, pharmacokinetics, pharmacodynamics, and clinical activity of GSK525762 in subjects with NUT midline carcinoma (NMC). ClinicalTrials.gov identifier NCT01587703.
- Hewings D. S.; Wang M.; Philpott M.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud S.; Vuppusetty C.; Marsden B.; Knapp S.; Conway S. J; Heightman T. D. 3,5-Dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. J. Med. Chem. 2011, 54, 6761–6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M. A.; Prinjha R. K.; Dittman A.; Giotopoulos G.; Bantscheff M.; Chan W.-I.; Robson S. C.; Chung C.-w.; Hopf C.; Savitski M. M.; Huthmacher C.; Gudgin E.; Lugo D.; Beinke S.; Chapman T. D.; Roberts E. J.; Soden P. E.; Auger K. R.; Mirguet O.; Doehner K.; Delwel R.; Burnett A. K.; Jeffrey P.; Drewes G.; Lee K.; Huntly B. J. P.; Kouzarides T. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chung C.-w.; Dean A. W.; Woolven J. M.; Bamborough P. Fragment-based discovery of bromodomain inhibitors, part 1: Inhibitor binding modes and implications for lead discovery. J. Med. Chem. 2012, 55, 576–586. [DOI] [PubMed] [Google Scholar]; b Bamborough P.; Diallo H.; Goodacre J. D.; Gordon L.; Lewis A.; Seal J. T.; Wilson D. M.; Woodrow M. D.; Chung C.-w. Fragment-based discovery of bromodomain inhibitors, part 2: Optimization of phenylisoxazole sulfonamides. J. Med. Chem. 2012, 55, 587–596. [DOI] [PubMed] [Google Scholar]
- Philpott M.; Yang J.; Tumber T.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud P.; Keates T.; Felletar I.; Ciulli A.; Knapp S.; Heightman T. D. Bromodomain-peptide displacement assays for interactome mapping and inhibitor discovery. Mol. BioSyst. 2011, 7, 2899–2908. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Felletar I.; Fedorov O.; von Delft F.; Arrowsmith C. H.; Edwards A. M.; Weigelt J.; Fish P.; Bunnage M.; Owen D.; Knapp S.; Cook A.. Crystal structure of the first bromodomain of human BRD4 in complex with the inhibitor PFI-1. RCSB PDB 4E96, 2012, DOI: 10.2210/pdb4e96/pdb [DOI]
- http://www.thesgc.org/scientists/chemical_probes/PFI-1/.
- Poulin P.; Theil F.-P. Prediction of pharmacokinetics prior to in vivo studies. II. Generic physiologically based pharmacokinetic models of drug disposition. J. Pharm. Sci. 2002, 91, 1358–1370. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. [DOI] [PubMed] [Google Scholar]
- Prolonged solublized storage of 17 (for example, in DMSO) should be avoided, as it is prone to oxidative insertion of nucleophilic solvents such as alcohols or water at C-4. Cell-based experimentation and in vivo work were reliably and reproducibly performed from fresh solid sample. (17, PFI-1, Pfizer number PF-6405761, will be made commercially available from Sigma Aldrich, Tocris Bioscience, and Toronto Research Chemical.)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.