

Abstract
Inhibitors of checkpoint kinase 1 (CHK1) are of current interest as potential antitumor agents, but the most advanced inhibitor series reported to date are not orally bioavailable. A novel series of potent and orally bioavailable 3-alkoxyamino-5-(pyridin-2-ylamino)pyrazine-2-carbonitrile CHK1 inhibitors was generated by hybridization of two lead scaffolds derived from fragment-based drug design and optimized for CHK1 potency and high selectivity using a cell-based assay cascade. Efficient in vivo pharmacokinetic assessment was used to identify compounds with prolonged exposure following oral dosing. The optimized compound (CCT244747) was a potent and highly selective CHK1 inhibitor, which modulated the DNA damage response pathway in human tumor xenografts and showed antitumor activity in combination with genotoxic chemotherapies and as a single agent.
Introduction
Checkpoint kinase 1 (CHK1) is an intracellular, serine/threonine kinase that plays a central role in the DNA damage response pathway.1,2 When single or double strand breaks are formed in the DNA in proliferating cells, either by exogenous DNA-damaging events (e.g., exposure to genotoxic chemicals or ionizing radiation) or through faults in the DNA replication process, a signaling cascade is triggered to halt the cell cycle and initiate DNA repair. CHK1 is predominantly, but not exclusively, activated by the upstream kinase, ataxia telangiectasia and rad3 related (ATR), in response to single strand breaks in DNA,3 and in turn CHK1 phosphorylates a number of downstream proteins leading to cell cycle arrest in S-phase or at the G2/M transition.4 As well as establishing S and G2/M cell cycle checkpoints, CHK1 also promotes homologous recombination repair of damaged DNA.5 Cell cycle arrest in response to DNA damage may occur in G1, and the structurally unrelated enzyme checkpoint kinase 2 (CHK2) plays a significant part in the control of the G1 checkpoint.6 The presence of alternative checkpoints and DNA repair mechanisms reduces the sensitivity of normal cells to CHK1 inhibition. However, more than half of solid tumors are deficient for the function of the tumor suppressor p537,8 or contain other defects in cell cycle checkpoints and are more reliant on the late phase cell cycle checkpoints and CHK1-mediated DNA damage response pathways as a result.9
Inhibition of CHK1 is established as a potential therapy for cancer in two distinct contexts: in combination with conventional genotoxic chemotherapy or ionizing radiation, and as a single agent in specific tumors with a genetic background that leads to high levels of intrinsic DNA damage.10 CHK1 inhibition prevents effective repair of lesions in DNA and forces proliferating cells to proceed to mitosis with unrepaired DNA, resulting in aberrant cell division and death. Thus CHK1 inhibition can potentiate the cytotoxicity of genotoxic therapies, as has been extensively demonstrated in preclinical studies with CHK1 RNAi and small molecule CHK1 inhibitors.9,10 CHK1 inhibitors show high potentiation of the efficacy of antimetabolite DNA-damaging agents that act mainly in S-phase (e.g., nucleotide analogues, folate synthesis inhibitors), and selective inhibition of CHK1 over CHK2 has been shown to be beneficial over simultaneous inhibition of CHK1 and CHK2.10 Recent studies have shown that some cancer cells carry a high level of intrinsic DNA damage resulting from the particular genetic defects underlying their transformation and are dependent on CHK1-mediated DNA damage repair for survival. CHK1 inhibition may confer synthetic lethality in these tumors.11,12 For example, pediatric neuroblastomas driven by amplification of the MYCN oncogenic transcription factor have constitutive activation of the DNA damage response pathway and are sensitive to single agent inhibition of CHK1.13
CHK1 inhibitors have been widely studied and a number of compounds have reached early clinical trials.10 Notable among these are the ATP-competitive inhibitors LY260361814 (1), PF0047773615 (2), AZD776216 (3), SCH90077617 (4), and LY260636818 (5) (Figure 1). However, of these agents, only 1 has so far progressed to phase II clinical trials,14 and the clinical benefit of CHK1 inhibition remains to be tested. Most of these compounds have low or no selectivity for inhibition of CHK1 over CHK2, and all are administered intravenously. Thus, there is a need for CHK1 inhibitors with improved selectivity profiles, while orally bioavailable compounds would provide flexibility for dosing in combinations with conventional chemotherapies and would also be advantageous in emerging single agent contexts in oncology where more frequent administration may be required. Oral CHK1 inhibitors have been recently reported but not yet fully described.18
Figure 1.

Structures of the intravenous, clinical candidate checkpoint kinase inhibitors LY2603618 (1), PF00477736 (2), AZD7762 (3), SCH900776 (4), and LY2606368 (5).
We have previously detailed the fragment-based discovery and optimization of a series of 2-aminoisoquinoline CHK1 inhibitors, exemplified by SAR-02010619 (6, Figure 2), that potentiated genotoxic drug efficacy in cellular assays and in human tumor xenografts. Although a potent and selective CHK1 inhibitor, compound 6 lacked oral bioavailability. To address this, we pursued a hybridization strategy, combining the structural elements conferring CHK1 selectivity in 6 with an alternative pyridine scaffold which had shown more promising in vitro ADME properties. This approach generated a novel series of 3-alkoxyamino-5-(pyridin-2-ylamino)pyrazine-2-carbonitriles, which we have optimized for potency and efficacy in cells, and for ADME properties, leading to the highly selective CHK1 inhibitor 26. Compound 26 has good oral bioavailability and demonstrates biomarker modulation and enhancement of genotoxic drug efficacy in multiple xenograft models. Additionally, 26 shows strong single agent activity in a MYCN-driven transgenic mouse model of pediatric neuroblastoma.
Figure 2.

Design of novel 3-alkoxyamino-5-(pyridin-2-ylamino)pyrazine-2-carbonitrile CHK1 inhibitors by hybridization of the 2-aminoisoquinoline 6 and the 2-aminopyridine 7.
Results and Discussion
The isoquinoline 6 was a potent CHK1 inhibitor with high selectivity over inhibition of CHK2 and showed cellular effects characteristic of selective CHK1 inhibition. However, the compound suffered from high metabolism in mouse liver microsomes (MLM) and had minimal oral bioavailability (F = 5%). We therefore sought to modify the chemical scaffold to increase metabolic stability, with the aim of enhancing oral bioavailability. During the evolution of the lead compound 6 by scaffold morphing from a purine fragment hit,20 pyridines represented by compound 7 had been prepared and evaluated.19 Compound 7 lacked the potent cellular activity or cellular selectivity of 6 but showed good microsomal stability and equivalent permeability (Figure 2, Table 1). We therefore investigated hybridization of the two scaffolds represented by 6 and 7 to give novel 3-alkoxyamino-5-(pyridin-2-ylamino)pyrazine-2-carbonitriles that could allow the maintenance of the cellular potency and selectivity of 6 while conferring the improved in vitro ADME properties observed for 7. Compound 6 had shown micromolar inhibition of the hERG ion channel, whereas an apparent reduction in hERG inhibition was observed for 7. An additional aim was therefore to minimize hERG channel inhibition in the novel series of CHK1 inhibitors.
Table 1. CHK1 Inhibition and Cellular Activity of 3-Alkoxyamino-5-(5-carbomethoxypyridin-2-ylamino)pyrazine-2-carbonitriles.

| potentiation
of gemcitabine cytotoxicity (fold)d |
||||||||
|---|---|---|---|---|---|---|---|---|
| no. | R1 | R2 | CHK1 IC50 (nM)a | checkpoint abrogation IC50 (nM)b | cellular selectivity (fold)c | HT29 | SW620 | hERG IC50 (μM)e |
| 6 | – | – | 6.1 (±1.2)f | 55 (±19)f | 8.5 | 3.0 | 12 | 5 |
| 7 | – | – | 21 (15, 27) | 825 (950, 700) | 3.8 | –g | – | 39% @10 μMh |
| 8 | NHMe | A | 7.0 (7.5, 6.4) | 40 (47, 33) | 17 | 6.9 | 11 | 5 |
| 9 | NHMe | B | 5.4 (5.3, 5.5) | 28 (38, 18) | 12 | 3.6 | 7.1 | 16 |
| 10 | NMe2 | B | 8.8 (8.5, 9.0) | 200i | 11 | 4.1 | 7.5 | 30 |
| 11 | OMe | B | 46.5 (47, 46) | 1700i | 8.0 | 1.2 | 1.7 | 44% @11 μMh |
| 12 | SMe | B | 18 (21, 14) | 390i | 6.8 | 1.6 | 2.3 | 25% @10 μMh |
Determined in Caliper microfluidic assay,40 mean of n = 2, individual values in parentheses.
Abrogation of etoposide-induced G2 checkpoint arrest in HT29 human colon cancer cells, mean of n = 2, individual values in parentheses.
Ratio of cytotoxicity GI50 (measured by SRB assay41) to IC50 for CHK1-mediated abrogation of etoposide-induced G2 checkpoint arrest in HT29 human colon cancer cells.
Potentiation by CHK1 inhibitor of gemcitabine cytotoxicity in HT29 or SW620 human colon cancer cells.
Inhibition of hERG ion current in HEK cells overexpressing hERG ion channel (PatchExpress, Millipore Inc.).
Mean (±SD), n ≥ 3.
Not determined.
Percent inhibition at single concentration.
Single determination.
Compounds were assessed in an assay cascade designed to identify inhibitors with potent and selective effects on CHK1 activity in cancer cells. As well as measuring inhibition of CHK1 in a biochemical assay, compounds were tested for their ability to abrogate an etoposide-induced G2 checkpoint arrest in HT29 colon cancer cells, a specific CHK1-mediated effect.21 In parallel, compounds were tested in an antiproliferative assay in HT29 cells. Many cancer cell lines, including HT29, are not anticipated to be sensitive to specific CHK1 inhibition. The comparison of the checkpoint abrogation and antiproliferative assays therefore informed on the degree of selectivity for CHK1 inhibition versus off-target effects in HT29 cells. Our target profile was to achieve, as a minimum, CHK1 IC50 < 20 nM with cellular checkpoint abrogation IC50 < 150 nM, and at least 5-fold selectivity for checkpoint abrogation over antiproliferative activity in the HT29 cells. Suitable compounds were further tested for their ability to enhance the cytotoxicity of gemcitabine in HT29 and SW620 colon cancer cells as a measure of efficacy, where at least a 5-fold potentiation was considered desirable.
Hybridization of the scaffolds represented by 6 and 7 led to the pyridine ester 8 and analogues (Table 1). The importance of the cyanopyrazine and aminoalkoxy groups in binding to selectivity determinants in the CHK1 ATP site had previously been shown by the X-ray structure of 6 complexed with CHK1.19 The 4-aminomethyl substituent retained in the hybrid molecule was anticipated to generate a pseudobicycle to replace the isoquinoline of 6 through forming an intramolecular hydrogen bond to the adjacent ester. Compound 8 showed equivalent potency and cellular activities to 6. The microsomal stability of 8 was somewhat improved over 6 (MLM: 49% metabolized @30 min) as anticipated, although not to the level of 7. Previous studies with 6 had shown that the (S)-1-(dimethylamino)propan-2-oxy side chain could be productively replaced by pyrrolidin-3-ols, and CHK1 inhibition and cellular activity were retained in the pyrrolidine 9. Variation of the pyridine 4-substituent was examined on this scaffold (9–12, Table 1), showing that the 4-aminomethyl substituent was more favorable for cellular activity than the dimethylamino, methoxy, or thiomethyl groups. Selectivity for CHK1 inhibition over CHK2 and CDK1 was high for both 6 and 7 (Figure 2), and greater than 100-fold selectivity was maintained in the hybrid compounds 9 and 10 over both CHK2 and CDK1 (data not shown). Encouragingly, hERG inhibition was generally lower for the hybrid pyridine esters than for 6 (Table 1).
We investigated variation of the pyridine 5-substituent in the new series. Although ester 7 had shown microsomal stability, we wished to replace this functionality to avoid possible hydrolysis by plasma or intracellular esterases.22 The vector of the pyridine 5-substituent in these compounds is aligned to a solvent-accessible surface that is part of the canonical ATP binding site pharmacophore for protein kinases.23 In CHK1, this region is tolerant of a range of functional groups. Analysis of the known or predicted binding modes of published CHK1 inhibitors18 suggested that the small halo or methyl substituents of 1, 4, and 6, the larger fluorophenyl group of 3, or the 1-methylpyrazole of 2 could all occupy the relevant space, and these analogues were therefore prepared. In addition to these extremes of substituent size and shape, groups of intermediate size, such as cyclopropyl and substituted alkynyl were also studied (13–20, Table 2).
Table 2. CHK1 Inhibition and Cellular Activity of (R)-3-((1-(Dimethylamino)propan-2-yl)oxy)-5-((5-substituted-4-(methylamino)pyridin-2-yl)amino)pyrazine-2-carbonitriles.

| potentiation of gemcitabine cytotoxicity (fold)d |
|||||||
|---|---|---|---|---|---|---|---|
| no. | R | CHK1 IC50 (nM)a | checkpoint abrogation IC50 (nM)b | cellular selectivity (fold)c | HT29 | SW620 | hERG IC50 (μM)e |
| 13 | H | 97 (93, 102) | 126 (190, 62) | 22 | 3.0 | 3.9 | 71% @10 μMf |
| 14 | Cl | 20 (15, 25) | 27g | 22 | 3.8 | 3.9 | 1 |
| 15 | F3C | 21 (±4.5)h | 28 (21, 34) | 21 | 3.2 | 5.6 | 1 |
| 16 | cyc-C3H5 | 26 (23, 28) | 10 (17, 3.7) | 90 | 9 | 7.3 | 6 |
| 17 | HC≡C | 11 (11, 11) | 45 (32, 58) | 11 | 3.2 | 3.5 | 8 |
| 18 | HO(Me2)CC≡C | 2.9 (3.1, 2.7) | 65 (100, 29) | 6.5 | 2.8 | 6.5 | 59%@10 μMf |
| 19 | 3-fluorophenyl | 36 (41, 31) | 125 (140, 110) | 9.3 | 3.3 | 8.5 | 103% @11 μMf |
| 20 | 1-methyl-pyrazol-4-yl | 3.1 (3.5, 2.7) | 27 (±5)h | 10 | 6.9 | 6.5 | 12 |
Determined in Caliper microfluidic assay,40 mean of n = 2, individual values in parentheses.
Abrogation of etoposide-induced G2 checkpoint arrest in HT29 human colon cancer cells, mean of n = 2, individual values in parentheses.
Ratio of cytotoxicity GI50 (measured by SRB assay41) to IC50 for CHK1-mediated abrogation of etoposide-induced G2 checkpoint arrest in HT29 human colon cancer cells.
Potentiation by CHK1 inhibitor of gemcitabine cytotoxicity in HT29 or SW620 human colon cancer cells.
Inhibition of hERG ion current in HEK cells overexpressing hERG ion channel (PatchExpress, Millipore Inc.).
Percent inhibition at single concentration.
Single determination.
Mean (±SD), n ≥ 3.
The complete removal of the 5-substituent, as in 13, compromised potency and cellular efficacy, while larger groups were better tolerated. Potency increased generally with size, except that the large, lipophilic 3-fluorophenyl analogue 19 showed a drop in biochemical and cellular activity. The potent CHK1 biochemical inhibition by the 2-methylpent-3-yn-2-ol 18 failed to translate to increased checkpoint abrogation in cells. This may reflect the incorporation of an additional hydrogen bond donor in 18, leading to compromised membrane permeability compared to other molecules in this set. Of the compounds studied, the 1-methylpyrazole 20 was the most favorable replacement for the ester, giving a balance of high potency and cellular efficacy with reduced hERG activity relative to 6. The 3-fluorophenyl group 19 and the small lipophilic substituents (14, 15) were associated with increased hERG activity. Compound 20 maintained selectivity for CHK1 over CHK2 (IC50 = 3.2 μM) and CDK1 (IC50 ∼ 10 μM). Importantly, 20 also showed low microsomal turnover and moderate permeability (Table 4), achieving the original aim of combining the in vitro ADME performance of the pyridines (e.g., 7) with the in vitro activity profile of the isoquinolines (e.g., 6).
Table 4. In Vitro ADME and Mouse in Vivo Plasma Concentrations for Selected Potent CHK1 Inhibitors.
| in vitro
stability and permeability |
mouse plasma levels following 10 mg/kg iv or pod |
||||||
|---|---|---|---|---|---|---|---|
| no. | mouse liver microsome metabolism (%)a | CaCo-2 permeability (10–6cm/s)b | efflux ratioc | iv, 1 h (nM) | iv, 6 h (nM) | po, 1 h (nM) | po, 6 h (nM) |
| 9 | 17 (±9) | –e | – | 877 (±215)f | 74 (±19)f | 90 (±64) | 24 (±15) |
| 15 | 43 (±4) | <4.6 | >6 | 1044 (±106) | 33 (±11) | 809 (±114) | 26 (±2) |
| 16 | 42 (±16) | – | – | 417 (±81) | 29 (±2) | 80 (±30) | 8 (±2) |
| 17 | 40 (±5) | – | – | 833 (±58) | 7 (±1) | 439 (±112) | 6 (±2) |
| 20 | 30 (±13) | 8.8 (±0.4)g | 4.8 | 910 (±23)h | 45 (±10)h | 555 (±82)h | 57 (±7)h |
| 23 | 12 (±23) | – | – | 254 (±61) | 53 (±59) | 3 (±1) | 2 (±1) |
| 24 | 17 (±4) | – | – | 320 (±30)i | <20i,j | 193 (±72) | 12 (±9) |
| 25 | 19 (±6) | 147 (±5) | 8 (±1) | 18 (±8) | 4 (±1) | ||
| 26 | 38 (±13) | 29 | 2.4 | 1850 (±217) | 27 (±6) | 1125 (±264) | 52 (±15) |
| 28 | 18 (±13) | – | – | 1184 (±61) | 7 (±3) | 264 (±168) | 6 (±2) |
Percent metabolized after 30 min incubation, mean (±SD), n ≥ 3.
Permeability A→B across CaCo-2 cell monolayer, single determination.
Ratio of permeability A→B/B→A across CaCo-2 cell monolayer.
Plasma levels at 1 and 6 h following 10 mg/kg iv or po of test compound.
Not determined.
Normalized from 5 mg/kg dose.
Mean (±SD), n ≥ 3.
Data abstracted from full PK determination.
Normalized from 1 mg/kg dose.
Below level of detection.
To generate a broader range of compounds suitable for in vivo testing, the 5-(1-methylpyrazolyl) substituent of 20 was kept constant and conservative variations of the pyrazine aminoalkoxy group and the 4-substituent of the pyridine were explored (21–29, Table 3). The enantiomer of 20, compound 21, was a significantly less potent CHK1 inhibitor. This mirrored the difference in activity we had observed between 6 and its enantiomer.19 Subnanomolar CHK1 inhibition was seen when the aminoalkoxy group of 20 was replaced with either enantiomer of pyrrolidin-3-ol to give 22 and 23, but neither compound retained potent cell cycle checkpoint abrogation activity. Removal of a hydrogen bond donor functionality by N-methylation of the pyrrolidines gave compounds 24 and 25, which had cellular potency better than that of 22 and 23, despite somewhat reduced CHK1 inhibition in the biochemical assay. It was notable that the increased polarity of 22 and 23 was associated with minimal hERG inhibition.
Table 3. CHK1 Inhibition and Cellular Activity of 3-Aminoalkoxy-5-((5-(1-methyl-1H-pyrazol-4-yl)-pyridin-2-yl)amino)pyrazine-2-carbonitriles.

| potentiation of gemcitabine cytotoxicity (fold)d |
||||||||
|---|---|---|---|---|---|---|---|---|
| no. | R1 | R2 | CHK1 IC50 (nM)a | checkpoint abrogation IC50 (nM)b | cellular selectivity (fold)c | HT29 | SW620 | hERG IC50 (μM)e |
| 20 | NHMe | A | 3.1 (3.5, 2.7) | 27 (±5)f | 10 | 6.9 | 6.5 | 12 |
| 21 | NHMe | C | 169 (±30)f | 2200g | 6.4 | 3.5 | 1.5 | –h |
| 22 | NHMe | B | <1 | 210g | 6.7 | 7.4 | 8.4 | 22% @ 10 μMi |
| 23 | NHMe | D | <1 | 210 (240, 180) | 7.1 | 4.1 | 13 | 37% @ 10 μMi |
| 24 | NHMe | E | 4.4 (4.1, 4.7) | 110 (130, 90) | 11 | 5.6 | 8.6 | 24 |
| 25 | NHMe | F | 3.4 (3.9, 3.0) | 135 (150, 120) | 5.9 | 8.4 | 10 | 19 |
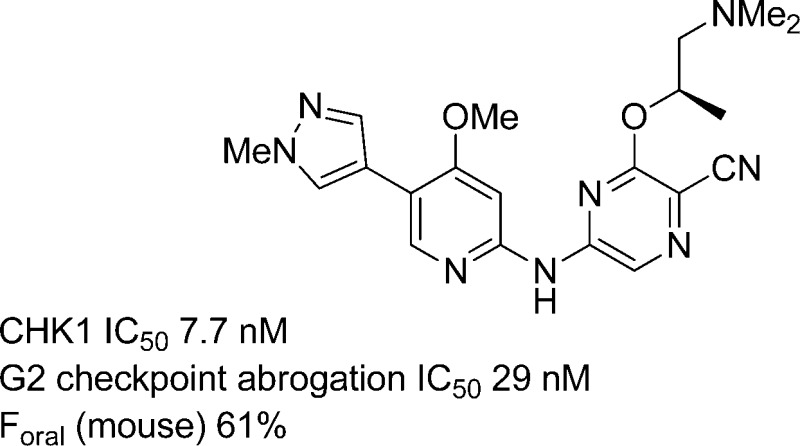
| 26 | OMe | A | 7.7 (±3.1)f | 29 (±8)f | 21 | 8.3 | 11 | 5 |
| 27 | OMe | B | <1 | 29 (38, 19) | 15 | 4.8 | 5.5 | –h |
| 28 | OMe | E | 6.3 (6.1, 6.5) | 56 (54, 57) | 20 | 7.3 | 7.5 | 16 |
| 29 | SMe | A | 27 (28, 26) | 110g | 19 | 9.5 | 4.3 | 8 |
Determined in Caliper microfluidic assay,40 mean of n = 2, individual values in parentheses.
Abrogation of etoposide-induced G2 checkpoint arrest in HT29 human colon cancer cells, mean of n = 2, individual values in parentheses.
Ratio of cytotoxicity GI50 (measured by SRB assay41) to IC50 for CHK1-mediated abrogation of etoposide-induced G2 checkpoint arrest in HT29 human colon cancer cells.
Potentiation by CHK1 inhibitor of gemcitabine cytotoxicity in HT29 or SW620 human colon cancer cells.
Inhibition of hERG ion current in HEK cells overexpressing HERG ion channel (PatchExpress, Millipore Inc.)
Mean (±SD), n ≥ 3.
Single determination.
Not determined.
Percent inhibition at single concentration.
Given the apparent dependence of the translation of biochemical activity to cellular potency on total hydrogen bond donor count, we examined the 4-thiomethyl and 4-methoxy substituents on the pyridine as replacements for the 4-methylamino group. With the optimized 5-(1-methylpyrazolyl) substituent in place of the ester, the change from 4-methylamino to 4-methylthio in 29 was better tolerated than in other examples but still resulted in reduced biochemical and cellular activity. Incorporation of the 4-methoxy group gave excellent potency in the (S)-1-(dimethylamino)propan-2-oxy analogue 26 along with high potentiation of the efficacy of gemcitabine in two cell lines. However, the replacement of the 4-methylamino group by 4-methoxy did not confer any reduction in hERG inhibition, and 26 had in vitro hERG activity similar to the isoquinoline 6. The 4-methoxy-substituted pyrrolidine 27 also showed improved cellular potency compared to the 4-methylamino derivative 22 but with a higher differential between biochemical and cellular activities than that of 26 and reduced potentiation of genotoxic efficacy.
Selected compounds from this series that met the target potencies and showed good cellular effects were assessed for MLM stability (Table 4). The in vitro metabolic stability of these pyridine-based inhibitors was improved over the isoquinolines represented by 6, as had been anticipated. Compounds were screened in vivo for oral exposure to determine if the increased metabolic stability would consistently lead to oral bioavailability. A full pharmacokinetic profile was obtained for 20 (Table 5). Gratifyingly, the compound showed substantial oral bioavailability, despite high in vivo clearance (greater than mouse liver blood flow), and had a high volume of distribution. To carry out a comparative assessment of the in vivo pharmacokinetics of other compounds efficiently, a limited sampling strategy was validated on a series of test compounds, whereby plasma levels from various paired time points were evaluated for their potential to predict plasma clearance. We established that analysis of plasma levels at 1 and 6 h after intravenous and oral dosing of the compounds was useful and applied this to the CHK1 inhibitors (Table 4). Although this approach was not expected to accurately predict the full in vivo pharmacokinetic profiles, certain features could be used for comparison between compounds within the series. In particular, the differences between plasma levels at 1 and 6 h allowed for the identification of compounds most likely to have persistent oral levels, considered desirable because sustained inhibition of CHK1 may be required for optimal potentiation of genotoxic efficacy.18 A comparison of the plasma levels at the 1 h time point following iv and po dosing was used to rank compounds for the likely degree of oral absorption.
Table 5. Mouse in Vivo Pharmacokinetic Data for Compounds 20 and 26.
Determined following 10 mg/kg iv and po dosing.
A range of oral pharmacokinetic behaviors was observed for the compounds tested, despite generally similar in vitro MLM stabilities, emphasizing the value of an efficient in vivo screening approach to complement in vitro ADME assays. The ester 9 showed limited oral exposure compared to the 5-pyrazolyl analogue 20. Substitutions at C-5 by cyclopropyl or alkynyl groups 16 and 17, respectively, were less favorable for sustained oral exposure. The 5-trifluoromethyl analogue 15 maintained the oral bioavailability of 20, although there was a suggestion from the CaCo-2 cell permeability data that this compound could have an increased potential for active efflux. The expected importance of high membrane permeability was demonstrated by the unsubstituted pyrrolidine 23 which, in line with its poor cellular activity, showed negligible oral exposure. N-Methylation to give the pyrrolidine 24, a change which had restored cellular activity, also improved the plasma levels achieved on oral dosing but not to the extent of the parent (S)-1-(dimethylamino)propan-2-oxy analogue 20. Replacement of the 4-methylamino substituent of 20 by 4-methoxy in 26, removing one hydrogen bond donor, gave a 2-fold enhancement in plasma levels at the 1 h time point. A similar change was seen when comparing the N-methylpyrrolidine 28 with the parent unsubstituted pyrrolidine 24. Determination of the full pharmacokinetic profile of 26 (Table 5) confirmed the moderately enhanced oral bioavailability of this compound relative to the analogue 20.
On the basis of their oral pharmacokinetic profiles, in particular the ability to sustain plasma levels at 6 h following a 10 mg/kg dose, and the potency and cellular efficacy of the compounds, 20 and 26 were identified as the most promising compounds and were progressed to more detailed studies. Human plasma protein binding (PPB) was determined as moderate for both compounds (20, PPB = 70%; 26, PPB = 82%), indicating that a substantial fraction of the plasma levels would be available as unbound drug. The thermodynamic solubilities of 20 and 26 in water were 0.1 mg/mL and 0.07 mg/mL, respectively. In line with the presence of a basic amine in the molecules, in pH 6.5 aqueous phosphate buffer their solubilities increased to >8 mg/mL and 6.2 mg/mL. To allow further in vivo studies, the maximum tolerated doses (MTDs) in mice were determined; when administered as single doses in suspension, the individual MTDs were 160 mg/kg po and >300 mg/kg po for 20 and 26, respectively.
The ability of 20 and 26 to inhibit DNA damaging agent-induced CHK1 signaling in human tumors xenografts in athymic mice after oral dosing was assessed (Figure 3). The compounds were given as suspensions at their respective MTDs to athymic mice bearing SW620 human colon cancer xenografts, followed by dosing of gemcitabine (60 mg/kg iv) after 1 h. Plasma and tumor samples were collected at 6 and 12 h after dosing the genotoxic agent. Tumor lysates were analyzed by Western blot for total CHK1 protein, phospho-S317 CHK1 as a marker of activation of CHK1 by the upstream kinase ATR, and phospho-S296 CHK1 autophosphorylation to demonstrate inhibition of CHK1 kinase function.21 Activation of the DNA damage response resulting from gemcitabine treatment was shown by the increase in phospho-S317 CHK1 and was sustained over 12 h. CHK1 autophosphorylation on S296 was also seen at both time points in response to gemcitabine treatment. Both 20 and 26 at their MTDs strongly inhibited CHK1 S296 autophosphorylation at 6 h following gemcitabine treatment. Compound 26 continued to show robust inhibition of CHK1 at the 12 h time point, while a less powerful effect was seen for 20 (Figure 3A). Analysis of the drug levels in plasma and tumors showed micromolar plasma concentrations and high distribution to tumor for both compounds (Figure 3B). The 2-fold higher dose of 26 was associated with an increased exposure compared to 20, as expected. However, a significantly greater fall in drug concentration between 6 and 12 h was seen for 20 (ca. 20-fold decrease) than with 26 (ca. 2–3-fold decrease). The more sustained levels of 26 in plasma and tumor correlated with stronger inhibition of CHK1 autophosphorylation at 12 h. While 26 had shown a slightly lower clearance than 20 at low dose (Table 5), it is possible that the difference in behavior of 26 and 20 in the pharmacodynamic experiment could indicate saturation of metabolism at the high dose of 26. Further pharmacodynamic studies showed that single doses of 100–300 mg/kg po 26 could maintain inhibition of gemcitabine-induced pS296 CHK1 for up to 24 h in HT29 colon tumor xenografts.24
Figure 3.

(A) Effect of single doses of 20 (160 mg/mg po) and 26 (300 mg/kg po) on gemcitabine (60 mg/kg, iv)-induced CHK1 phosphorylation in SW620 human cancer colon xenografts in nude mice, at 6 and 12 h after dosing the genotoxic. (B) Plasma and tumor levels of 20 and 26 at 6 and 12 h after genotoxic drug administration.
The kinase selectivity of 26 was studied in detail. In common with most of the compounds tested in this novel series (data not shown), 26 exhibited excellent selectivity for inhibition of CHK1 versus CHK2 (IC50 >10 μM) and CDK1 (IC50 >10 μM). To expand on this, the compound was profiled against a panel of 140 representative kinases using a radiometric assay format.25 At a test concentration of 1 μM, greater than 50% inhibition was seen for only 13 enzymes, including CHK1, of which just 7 were strongly inhibited (>80%; CHK1, RSK1, RSK2, AMPK, BRSK1, IRAK1, TrkA). The high selectivity of 26 was confirmed by a similar screen against 121 kinases at an increased test concentration of 10 μM. This indicated that the selectivity of 26 for CHK1 was >1000-fold against 89 out of the 121 enzymes.
It was notable that for the new compounds prepared in this study, many of those with reduced in vitro hERG activity relative to the original isoquinoline 6 did not fulfill the target criteria for CHK1-mediated potentiation of genotoxic cellular efficacy or had suboptimal exposure on oral dosing. Thus, 26 showed micromolar inhibition of the hERG ion channel (Table 3). A wider assessment of the compound’s effect on seven other human cardiac ion channels was carried out.26 Pleasingly, 26 showed minimal inhibition of all the other ion channels tested (hNav1.5, hKv4.3/hKChIP2, hCav1.2, hKv1.5, hKCNQ1/hminK, hHCN4; hKir2.1 <25% inhibition @ 10 μM). The potential significance of the in vitro hERG inhibition by 26 for further development would require more detailed assessment in advanced models of cardiac arrhythmic effects.27
Compound 26 showed good oral bioavailability and robust inhibition of CHK1 signaling over 12–24 h in human tumor xenografts following a single dose and was therefore studied for oral antitumor efficacy in combination with genotoxic chemotherapies in vivo.24 It has been suggested that prolonged inhibition of the DNA-damage response following acute treatment with a genotoxic agent is required to maximize the additional cell death from the combination treatment.18,28 Some previously reported CHK1 inhibitors have been studied preclinically using schedules involving delayed administration of the CHK1 inhibitor until up to 24 h after the genotoxic drug dose.15,28,29 The phase I clinical trial of 1 involved sequential treatment with pemetrexed and 1 separated by a 24 h interval.14 We investigated the scheduling of 6 and 26 in combination with gemcitabine in SW620 cells in vitro.24 These studies showed that continuous exposure of the cells to the CHK1 inhibitors during the period 24 – 48 h after gemcitabine addition was required to obtain the maximum in vitro potentiation of gemcitabine cytotoxicity. Provided this late period of exposure was maintained, the degree of potentiation was similar following either simultaneous exposure of the genotoxic and 26 or with a 24 h delay before addition of 26.
On the basis of the above data, we used a treatment schedule in vivo that involved giving 26 for two daily doses, beginning 24 h after each genotoxic treatment. Three cycles of treatment were administered with each cycle beginning at seven- or four-day intervals. For multiple doses over an extended period, the MTD of 26 in combination with gemcitabine was determined as 75 mg/kg po. Pleasingly, and in agreement with the in vitro data, strong potentiation of the efficacy of gemcitabine by 26 (75 mg/kg po) was observed in HT29 and SW620 human colon cancers, and in Calu6 human non-small cell lung cancers, grown as xenografts in athymic mice (Table 6).24 Additionally, potentiation of the efficacy of the topoisomerase I inhibitor irinotecan was also observed in the HT29 xenograft model. Importantly, there were minimal effects on tumor growth of the CHK1 inhibitor alone, reflecting the weak single-agent cytotoxicity observed in the cellular studies. The treatments were well tolerated and neither the single agent nor combination-treated cohorts showed any body weight loss over the period of the experiment. Because single agent activity of CHK1 inhibitors has been proposed in tumors overexpressing the MYC oncogene,11−13 we also investigated the oral efficacy of 26 as a single agent in a hemizygotic mouse transgenic model of neuroblastoma (TH-MYC30) which overexpresses MYCN. Pleasingly, tumors arising in these animals underwent significant regression in response to treatment with 26 (100 mg/kg po; qd7d) with the 26-treated cohort showing a statistically significant reduction in tumor burden compared with solvent (vehicle)-treated controls (T/C = 13%; p < 0.001).24
Table 6. In Vivo Potentiation of Cytotoxic Drug Efficacy by 26 in Human Tumor Xenografts.
| tumor growth
delay (days)c |
||||||
|---|---|---|---|---|---|---|
| tumor | schedulea | control tumor growth (days)b | gemcitabine, 100 mg/kg iv | irinotecan, 12.5 mg/kg i.p. | 26, 75 mg/kg po | combination |
| HT29d | q7d × 3 | 8.8 (±4.8) | 2.3 (±3.3) | –e | –1.8 (±2.1) | 11.4 (±3.3)*** |
| SW620f | q4d × 3 | 5.3 (±1.3) | 1.6 (±2.2) | –e | 2.8 (±2.2) | 6.0 (±2.4)* |
| Calu6g | q7d × 3 | 6.9 (±1.6) | 7.2 (±6.2) | –e | 0.10 (±1.2)* | 15.9 (±2.5)** |
| HT29 | q7d × 3 | 6.9 (±1.5) | –e | 6.4 (±4.6) | 0.30 (±1.4)*** | 13.5 (±2.4)*** |
Each cycle of treatment consisted of the genotoxic agent (dosed at T = 0 h) followed by two doses of 26 (at T = 24 h and T = 48 h).
Time to reach 300% of starting tumor volume, mean (±SD), for four to six mice per treatment group.
Delay in time to reach 300% of starting tumor volume relative to vehicle-treated controls, mean (±SD), for four to six mice per treatment group. Statistical significance of growth delay was determined using a one-way ANOVA with Tukey’s multiple comparison test: *, P < 0.05; **, P < 0.01; ***, P < 0.001, significantly different from cytotoxic treatment alone.
HT29 human colon cancer xenograft.
Not applicable.
SW620 human colon cancer xenograft.
Calu6 human non-small cell lung cancer xenograft.
Conclusions
A novel series of orally active 3-alkoxyamino-5-(pyridin-2-ylamino)pyrazine-2-carbonitrile CHK1 inhibitors was designed through the hybridization of potent and selective 2-aminoisoquinolines with a more metabolically stable 2-aminopyridine scaffold. Such a lead generation strategy is most effective when the desirable properties of the parent moieties translate independently into the merged structure. In this case, the high potency and selectivity of the 2-aminoisoquinolines such as 6 were known to depend to a high degree on the binding of the 5-amino-3-(alkoxyamino)pyrazine-2-carbonitrile group to specific residues in the ATP site of CHK1,19,31 and the potential for these interactions was retained in the merged structures. The in vitro microsomal stability and good membrane permeability of the prototype 2-aminopyridine 7 was observed to be retained in the merged structures, leading to an orally bioavailable hybrid series.
The in vitro optimization of the 3-alkoxyamino-5-(pyridin-2-ylamino)pyrazine-2-carbonitriles demonstrates the usefulness of a cell-based mechanistic assay cascade to guide the achievement of high selectivity in a series of ATP-competitive kinase inhibitors, a task often considered challenging.32 Seeking well-defined functional selectivity in cells may provide a potential alternative to iterative large-scale kinome profiling during lead optimization.17 Ultimately, the optimized molecule in this study, 26, indeed showed high biochemical kinase selectivity as well as selectivity for CHK1-dependent mechanism-of-action in human cancer cells. For CHK1, specificity of binding in the ATP site is aided by the presence of structural features, such as protein-bound water molecules and polar residues deep in the ATP pocket, that are unique to CHK1 and are effectively targeted by the 5-amino-3-(alkoxyamino)pyrazine-2-carbonitrile group.19,20,31,33,42
Some inhibition of the hERG ion channel was observed with several of the inhibitors in this series. Increased polarity generally led to decreased hERG inhibition34 but was often accompanied by reduced cellular potency or efficacy, particularly where the polarity arose from an increase in the number of hydrogen bond donors. The micromolar hERG inhibition observed for 26 requires further studies to determine the therapeutic index relevant to potential clinical development, but it is notable that other cardiac ion channels were not inhibited by 26.
Significantly improved in vitro microsomal stability, compared to that of the parent isoquinoline 6, was observed in the hybrid compounds. This led to oral bioavailability provided that high membrane permeability was maintained. However, the in vivo behavior of the compounds was not uniform, with varying levels of oral exposure and in vivo clearance observed even where similar metabolic stability was observed in vitro. The limited sampling strategy for in vivo pharmacokinetics presented here was useful in optimizing compound oral pharmacokinetics and distinguishing between compounds with similar in vitro profiles, without the need for determining multiple full PK profiles.
We focused on achieving sustained high concentrations of the compounds in plasma and xenograft tumors over at least a 6 h period, because sustained inhibition of CHK1 signaling is postulated to be necessary for efficacy.10,18,28 This approach successfully translated into sustained inhibition of CHK1 signaling in vivo, as shown by the inhibition of genotoxic agent-induced biomarkers of the DNA damage response by compounds 20 and 26. Importantly, the pharmacodynamic effects of 26 in xenograft tumors translated to efficacy in a range of tumor xenograft models. As anticipated from the cellular activities, the selective oral CHK1 inhibitor 26 potentiated the antiproliferative effect of the DNA-damaging agents gemcitabine and irinotecan in p53-defective solid tumors. Additionally, oral compound 26 was effective as a single agent in a genetically engineered model of MYCN-driven neuroblastoma, confirming with a distinct chemical class of CHK1 inhibitor the results reported for the intravenous inhibitor 2 in a tumor xenograft neuroblastoma model.13
In summary, we have identified a novel series of potent and highly selective, orally bioavailable CHK1 inhibitors with oral antitumor activity in vivo. This represents one of the first series of orally bioavailable and efficacious CHK1 inhibitors to be described.18 The profile of the highlighted compound 26 (CCT244747) shows this molecule to be an excellent CHK1-selective research tool for in vitro and in vivo pharmacological studies35 and to possess many of the properties desirable in a clinical development compound.
Experimental Section
Synthetic Chemistry
Modular syntheses using sequential chemo- or regioselective SNAr reactions and palladium-catalyzed couplings were developed to prepare the novel 3-alkoxyamino-5-(pyridin-2-ylamino)pyrazine-2-carbonitriles. 5-Amino-3-(alkoxy)pyrazine-2-carbonitriles 30–35 were synthesized from 5-amino-3-chloropyrazine-2-carbonitrile19 by SNAr displacement with the appropriate alcohol under basic conditions (Scheme 1). The enantiomerically pure 1-(dimethylamino)propan-2-ols were prepared from the appropriate enantiomer of glycidol as previously described.19
Scheme 1. Synthesis of 5-Amino-3-alkoxypyrazine-2-carbonitrile Intermediates.

Reagents and conditions: (i) R–OH, NaH, dioxane, 90 °C (16–69%).
The 5-carboxymethyl-substituted compounds 8–12 were made from methyl 4,6-dichloronicotinate 36 by displacement of the more reactive 4-chloro substituent with methylamine, dimethylamine, methanol, or methanethiol, followed by Buchwald coupling to the appropriate 5-amino-3-(alkoxy)pyrazine-2-carbonitrile 30 or 32 and N-deprotection where required (Scheme 2).
Scheme 2. Synthesis of Compounds 8–12.

Reagents and conditions: (i) MeNH2 or Me2NH, MeCN, 0 °C to rt, (71–80%); (ii) MeONa, THF, rt (56%); (iii) MeSNa, THF–H2O, rt (56%); (iv) 30, Pd2dba3, xantphos, Cs2CO3, toluene, 130 °C, microwave (92%); (v) (a) 32, Pd2dba3, xantphos, Cs2CO3, toluene or dioxane, 130 °C, microwave, and then (b) CF3CO2H, CH2Cl2, rt (16–31%).
To prepare the 5-unsubstituted pyridine analogue 13, commercially available 2-chloro-N-methylpyridin-4-amine 41 was coupled with the aminopyrazine 30 (Scheme 3). The 5-chloropyridine 14 was also prepared starting from 41, which was chlorinated with N-chlorosuccinimide to give a mixture of 2,3-dichloro-, 2,5-dichloro-, and 2,3,5-trichloropyridine products, from which 2,5-dichloro-N-methyl-pyridin-4-amine 42 was isolated by column chromatography. Subsequent Buchwald reaction of 42 proceeded selectively at the more reactive 2-chloro substituent. The 5-trifluoromethyl analogue 15 was prepared starting with the selective SNAr displacement of the 4-iodo substituent of 2-chloro-4-iodo-5-trifluoromethylpyridine 43 with methylamine to give 2-chloro-N-methyl-5-(trifluoromethyl)pyridin-4-amine 44, followed by Buchwald coupling to 30.
Scheme 3. Synthesis of Compounds 15–25.

Reagents and conditions: (i) 30, 31, 34, or 35, Pd2dba3, xantphos, Cs2CO3, toluene or dioxane, 130 °C, microwave (6–66%); (ii) NCS, KOAc, AcOH, 80 °C (13%); (iii) MeNH2, MeOH, 130 °C, microwave (31%); (iv) ICl, KOAc, AcOH, 80 °C (43%); (v) (H2CO)n, NaB(OAc)3H, AcOH, 40 °C (78%); (vi) cyc-(C3H5)B(OH)2, Pd(PPh3)4, Na2CO3, MeCN, 100 °C, microwave (49%); (vii) R-CC-H, PdCl2(PPh3)2, CuI, Et3N, DMF, 120 °C, microwave (50–75%); (viii) 3-F-(C6H4)B(OH)2, Pd(PPh3)4, Na2CO3, MeCN, 100 °C, microwave (79%); (ix) 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole, Pd(PPh3)4, Na2CO3, MeCN–H2O, 100 °C, microwave (96%); (x) (a) 32 or 33, Pd2dba3, xantphos, Cs2CO3, toluene or dioxane, 130 °C, microwave, and then (b) CF3CO2H, CH2Cl2, rt (31–47%).
To vary the 5-substituent on the pyridine ring more widely, 2-chloro-5-iodo-N-methylpyridin-4-amine 46 was prepared as a key intermediate (Scheme 3). Iodination of 2-chloropyridin-4-amine with iodine monochloride on a multigram scale gave a mixture of 3-iodo-, 5-iodo-, and 3,5-diiodopyridine products, from which 45 was isolated in moderate yield (43%) by column chromatography.36 Monomethylation of 45 was achieved by reductive amination using paraformaldehyde and sodium triacetoxyborohydride. Suzuki and Sonogashira coupling reactions of 46 were found to occur selectively at the 5-iodo substituent, and subsequent Buchwald coupling to the remaining 2-chloro group gave compounds 16–19. For compound 20 and analogues 21–25, the 5-(1-methylpyrazol-3-yl) substituent was introduced first by Suzuki coupling, followed by Buchwald coupling to a variety of 5-amino-3-(alkoxy)pyrazine-2-carbonitriles.
To prepare the 5-(1-methylpyrazol-3-yl)-4-methoxy-substituted pyridines 26–28, commercially available 2-chloro-4-methoxypyridine 48 was first iodinated with N-iodosuccinimide in sulfuric acid to give 2-chloro-5-iodo-4-methoxypyridine, which was then subject to a chemoselective Suzuki coupling at the iodo substituent to give the 5-substituted intermediate 49 (Scheme 4). For small-scale reactions, microwave heating and homogeneous tetrakis(triphenylphosphine)palladium(0) catalyst provided suitable conditions for these couplings. For larger scale preparations of 26, the use of a microencapsulated palladium source (Pd EnCat TPP30; palladium(II) acetate and triphenylphosphine microencapsulated in polyurea matrix)37,38 as the catalyst and a standard heat source simplified the preparation and purification. Buchwald couplings to the remaining chloro group of 49 using microwave heating gave 26 and related target compounds on a small scale. For large scale preparations of 26, the Buchwald coupling could be conveniently carried out using several microwave reactions in parallel, which were combined for purification, or alternatively by nonmicrowave conditions. To introduce a 4-thiomethyl substituent to the pyridine, 2-chloro-5-iodopyridine 50 was selectively lithiated at the 4-position39 and quenched with dimethyl disulfide to give 2-chloro-5-iodo-4-(methylthio)pyridine 51, which was coupled with 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole to yield 52. Subsequent Buchwald coupling to 52 gave the 4-thiomethyl analogue 29.
Scheme 4. Synthesis of Compounds 26–29.

Reagents and conditions: (i) NIS, H2SO4, 55 °C (22%); (ii) 1-methyl-4-(4,4,5,5-tetramethyl1,3,2-dioxaborolan-2-yl)-1H-pyrazole, PdEnCat TPP30, Na2CO3, MeCN–H2O, 105 °C (59%); (iii) 30, Pd2dba3, xantphos, Cs2CO3, dioxane, 100 °C (71%); (iv) 32, Pd2dba3, xantphos, Cs2CO3, toluene, 130 °C, microwave, and then (b) CF3CO2H, CH2Cl2, rt (27%); (v) LDA, MeSSMe, THF, −78 °C – rt (11%); (vi) 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole, Pd(PPh3)4, Na2CO3, MeCN, 100 °C, microwave (72%); (vii)) 30, Pd2dba3, xantphos, Cs2CO3, dioxane, 130 °C, microwave (5%).
General Synthetic Chemistry
Reactions were carried out under N2. Organic solutions were dried over MgSO4 or Na2SO4. Starting materials and solvents were purchased from commercial suppliers and were used without further purification. Microwave reactions were carried out using a Biotage Initiator microwave reactor. Flash silica column chromatography was performed using Merck silica gel 60 (0.025–0.04 mm). Gradient silica column chromatography was performed with a Biotage SP1 medium pressure chromatography system, using prepacked silica gel cartridges. Ion exchange chromatography was performed using Isolute Flash SCX-II (acidic) or Flash NH2 (basic) resin cartridges. 1H NMR spectra were recorded on a Bruker AMX500 instrument at 500 MHz or on a Bruker Avance instrument at 400 MHz using internal deuterium locks. Chemical shifts (δ) are reported relative to TMS (δ = 0) and/or referenced to the solvent in which they were measured. Combined HPLC-MS analyses were recorded using a Waters Alliance 2795 separations module and Waters/Micromass LCT mass detector with HPLC performed using Supelco DISCOVERY C18, 50 mm × 4.6 mm or 30 mm × 4.6 mm i.d. columns, or using an Agilent 6210 TOF HPLC-MS with a Phenomenex Gemini 3 μm C18 (3 cm × 4.6 mm i.d.) column. Both HPLC systems were run at a temperature of 22 °C with gradient elution of 10–90% MeOH/0.1% aqueous formic acid at a flow rate of 1 mL/min and a run time of 3.5, 4, or 6 min as indicated. UV detection was at 254 nm, and ionization was by positive or negative ion electrospray as indicated. The molecular weight scan range was 50–1000 amu. All tested compounds gave >95% purity as determined by these methods. All purified synthetic intermediates gave >95% purity as determined by these methods except where indicated in the text.
Synthesis of Compound 20
2-Chloro-5-iodopyridin-4-amine (45)
ICl (10.0 g, 61.6 mmol) was added to a solution of 2-chloropyridin-4-amine (6.60 g, 51.3 mmol) and KOAc (10.1 g, 103 mmol) in AcOH (325 mL). The reaction was heated to 80 °C and stirred for 3 h. AcOH was removed by evaporation. Toluene was added, and the mixture was evaporated to remove residual AcOH (×2). The crude products were dissolved in CH2Cl2/MeOH and absorbed on to silica gel. The silica gel was dried under high vacuum for 18 h. Gradient silica column chromatography, eluting with 5–20% EtOAc in cyclohexane and collecting the second major component (Rf = 0.5 in 1:1 EtOAc/cyclohexane), gave 45 (5.66 g, 22.2 mmol, 43%) as an off white powder. 1H NMR (500 MHz, CD3OD) δ 8.18 (s, 1H), 6.68 (s, 1H), 4.81 (s, 2H); LC-MS (3.5 min) tR = 1.66 min; m/z (ESI) 255 (M + H); HRMS m/z calcd for C5H5ClIN2 (M + H) 254.9180, found 254.9189.
2-Chloro-5-iodo-N-methylpyridin-4-amine (46)
45 (2.00 g, 7.86 mmol) and paraformaldehyde (0.472 g, 15.7 mmol) were dissolved in AcOH (56.1 mL) and stirred for 2.5 h at 40 °C. NaB(OAc)3H (3.66 g, 17.3 mmol) was added, and the mixture was stirred at 40 °C for 1.5 h. Further NaB(OAc)3H (3.66 g, 17.3 mmol) was added, and the mixture was stirred for 19 h. The reaction mixture was reduced in volume by half by evaporation. Water was added to the mixture, followed by basification with NaHCO3. The mixture was extracted with EtOAc (3 × 70 mL), the combined organic layers were dried and absorbed onto silica gel, and solvent was removed by evaporation. Gradient silica column chromatography, eluting with 5–10% EtOAc in cyclohexane, gave 46 (1.65 g, 6.14 mmol, 78%) as a white powder. 1H NMR (500 MHz, CDCl3) δ 8.27 (s, 1H), 6.41 (s, 1H), 4.85 (brs, 1H), 2.93 (d, J = 5.0, 3H); LC-MS (3.5 min) tR = 1.90 min; m/z (ESI) 268 (M + H); HRMS m/z calcd for C6H7ClIN2 (M + H) 268.9337, found 268.9339.
2-Chloro-N-methyl-5-(1-methyl-1H-pyrazol-4-yl)pyridin-4-amine (47)
A mixture of 46 (0.083 g, 0.309 mmol), 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.068 g, 0.309 mmol), Pd(PPh3)4 (0.018 g, 0.015 mmol), and aq Na2CO3 (0.5 M; 0.927 mL) in MeCN (2.1 mL) was heated at 100 °C in a microwave reactor for 30 min. The cooled reaction mixture was absorbed onto silica gel, and solvent was removed by evaporation. Gradient silica column chromatography, eluting with 1–10% MeOH in CH2Cl2, gave 47 (0.066 g, 0.296 mmol, 96%) as a colorless solid. 1H NMR (500 MHz, CDCl3) δ 7.83 (s, 1H), 7.56 (s, 1H), 7.45 (s, 1H), 6.48 (s, 1H), 4.68 (brs, 1H), 3.96 (s, 3H), 2.84 (d, J = 5.1 Hz, 3H). LC-MS (3.5 min) tR = 1.09 min; m/z (ESI) 223 (M + H). HRMS m/z calcd for C10H12ClN4 (M + H) 223.0745, found 223.0744.
(R)-3-((1-(Dimethylamino)propan-2-yl)oxy)-5-((5-(1-methyl-1H-pyrazol-4-yl)-4-(methylamino)pyridin-2-yl)amino)pyrazine-2-carbonitrile (20)
A mixture of 47 (0.066 g, 0.296 mmol), 30(19) (0.066 g, 0.296 mmol), xantphos (0.035 g, 0.059 mmol), Pd2dba3 (0.027 g, 0.030 mmol), and Cs2CO3 (0.193 g, 0.593 mmol) in toluene (2.12 mL) was heated at 130 °C in a microwave reactor for 45 min. The cooled reaction mixture was diluted with MeOH (10 mL) and absorbed onto an SCX-2 acidic ion exchange column (2 g). The column was eluted with MeOH (4 × 20 mL), then NH3 in MeOH (2 M; 4 × 20 mL). The basic eluent fractions were combined and absorbed onto silica gel. Solvent was removed by evaporation. Gradient silica column chromatography, eluting with 1–15% MeOH in CH2Cl2, gave 20 (0.040 g, 0.076 mmol, 26%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.23 (brs, 1H), 7.85 (s, 1H), 7.58 (s, 1H), 7.47 (s, 1H), 7.03 (s, 1H), 5.48–5.37 (m, 1H), 4.72 (q, J = 5 Hz, 1H), 3.98 (s, 3H), 2.90 (d, J = 5 Hz, 3H), 2.74 (dd, J = 13.4, 7.2 Hz, 1H), 2.51 (dd, J = 13.4, 4.4 Hz, 1H), 2.31 (s, 6H), 1.41 (d, J = 6.3 Hz, 3H). LC-MS (3.5 min) tR = 1.09 min; m/z (ESI) 408 (M + H). HRMS m/z calcd for C20H26N9O (M + H) 408.2255, found 408.2255.
Synthesis of Compound 26
2-Chloro-4-methoxy-5-(1-methyl-1H-pyrazol-4-yl)pyridine (49)
NIS (25.3 g, 107 mmol) was added portion wise at rt to a solution of 2-chloro-4-methoxypyridine 48 (15.35 g, 107 mmol) in H2SO4 (76 mL). The mixture was heated and stirred at 55 °C for 2 h. The reaction mixture was poured onto ice–water (400 mL), and aq NaOH (8 M; 500 mL) was added slowly to basify the mixture (pH ∼ 14), after which the dark brown solution became pale yellow. The mixture was extracted with CH2Cl2 (2 × 600 mL). The combined organic layers were washed with brine (300 mL) and absorbed onto silica gel. Solvent was removed by evaporation. Dry flash silica column chromatography, eluting with 50% EtOAc in cyclohexane and collecting the first major product (Rf = 0.66 in 50% EtOAc/cyclohexane), followed by flash silica column chromatography of mixed fractions, gave 2-chloro-5-iodo-4-methoxypyridine as a white solid (6.20 g, 23.0 mmol, 22%). 1H NMR (500 MHz, DMSO) δ 8.52 (s, 1H), 7.19 (s, 1H), 3.96 (s, 3H); LC-MS (4 min) tR = 2.56 min; m/z (ESI) 270 (M + H). HRMS m/z calcd for C6H5ClIN2O2 (M + H) 269.9177, found 299.9181. Aqueous Na2CO3 (0.5M; 24.5 mL, 12.25 mmol) was added to a mixture of 2-chloro-5-iodo-4-methoxypyridine (2.20 g, 8.16 mmol), 1-methyl-4-(4,4,5,5-tetramethyl1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1.70 g, 8.16 mmol), and Pd EnCat TPP30 (Sigma-Aldrich Co.; 2.04 g, 0.816 mmol) in MeCN (48.6 mL) and EtOH (12.4 mL). N2 was bubbled through the mixture for 15 min. The reaction mixture was heated at reflux (105 °C) for 3 h. The mixture was cooled and filtered to recover the microencapsulated palladium catalyst. The filtrate was absorbed onto silica gel, and solvent was removed by evaporation. Gradient silica column chromatography, eluting with 1–10% EtOH in CH2Cl2, gave the partially purified product, which was subjected to ion exchange chromatography on acidic SCX-II resin, eluting with MeOH and then with 2 M NH3 in MeOH. The basic fractions were combined and concentrated to give 49 (1.08 g, 4.84 mmol, 59%) as an off white solid. 1H NMR (500 MHz, CDCl3) δ 8.39 (s, 1H), 7.84 (s, 1H), 7.78 (s, 1H), 6.87 (s, 1H), 3.97 (s, 3H), 3.95 (s, 3H). LC-MS (3.5 min) tR = 2.38 min; m/z (ESI) 224 (M + H). HRMS m/z calcd for C10H11ClN3O (M + H) 224.0585, found 224.0583.
(R)-3-((1-(Dimethylamino)propan-2-yl)oxy)-5-((4-methoxy-5-(1-methyl-1H-pyrazol-4-yl)pyridin-2-yl)amino)pyrazine-2-carbonitrile (26)
N2 was passed through a mixture of 49 (0.532 g, 2.38 mmol), 30(19) (0.526 g, 2.38 mmol), xantphos (0.275 g, 0.476 mmol), Pd2dba3 (0.218 g, 0.238 mmol), and Cs2CO3 (1.55 g, 0.593 mmol) in dioxane (24 mL) for 10 min. The mixture was heated at 100 °C for 17 h. The cooled mixture was absorbed onto SCX-II acidic ion-exchange resin (20 g) and eluted with MeOH/CH2Cl2 and then with 2 M NH3 in MeOH and finally with 7 M NH3 in MeOH. The basic eluent fractions containing the crude product were absorbed onto silica gel, and solvents were removed by evaporation. Gradient silica column chromatography, eluting with 1–14% MeOH in CH2Cl2, gave 26 (0.693 g, 1.68 mmol, 71%) as a yellow solid. 1H NMR (500 MHz, CDCl3/CD3OD) δ 9.14 (s, 1H), 8.35 (s, 1H), 7.83 (s, 1H), 7.76 (s, 1H), 7.06 (s, 1H), 5.87–5.75 (m, 1H), 4.00 (s, 3H), 3.94 (s, 3H), 3.35 (dd, J = 13.7, 6.7 Hz, 1H), 3.21 (d, J = 13.7 Hz, 1H), 2.87 (s, 6H), 1.53 (d, J = 6.4 Hz, 3H). LC-MS (3.5 min) tR = 2.11 min; m/z (ESI) 409 (M + H). HRMS m/z calcd for C20H25N8O2 (M + H) 409.2095, found 409.2103.
Acknowledgments
We thank Dr. A. Mirza, M. Richards, and Dr. M. Liu for assistance in the spectroscopic characterization of test compounds, and Dr. N. Proisy for early studies on the synthesis of 45. We thank Dr. L. Chesler for studies in transgenic models of MYCN-neuroblastoma. This work was supported by Cancer Research UK [CUK] grant numbers C309/A8274 and C309/A11566, and by The Institute of Cancer Research. We acknowledge NHS funding to the NIHR Biomedical Research Centre.
Glossary
Abbreviations Used
- ATP
adenosine triphosphate
- ATR
ataxia telangiectasia and rad3 related
- CDK1
cyclin dependent kinase 1
- CHK1
checkpoint kinase 1
- CHK2
checkpoint kinase 2
- DELFIA
dissociation-enhanced lanthanide fluorescent immunoassay
- ELISA
enzyme-linked immunosorbent assay
- hERG
human ether-a-go-go related gene product
- MLM
mouse liver microsomes
- MPM2
M-phase phosphoprotein 2
- MYCN
V-myc myelocytomatosis viral related oncogene, neuroblastoma derived
- RNAi
RNA interference
- SRB
sulforhodamine B
Supporting Information Available
Experimental methods for the synthesis and characterization of compounds 8–19, 21–25, 27–35, 37–40, 42, 44, 51, and 52; experimental methods for the determination of CHK1 and CHK2 inhibition; experimental methods for the determination of checkpoint abrogation, antiproliferative activity, and potentiation of genotoxic drug efficacy in cancer cell lines; experimental method for the determination of compound concentrations in vivo at selected time points following iv and oral dosing; kinase inhibition profile for 26 at 1 μM and 10 μM test concentrations. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): M. Lainchbury, T. P. Matthews, T. McHardy, K. J. Boxall, M. I. Walton, P. D. Eve, A. Hayes, M. R. Valenti, A. K. de Haven Brandon, G. Box, G. W. Aherne, F. I. Raynaud, S. A. Eccles, M. D. Garrett, I. Collins are or have been employees of The Institute of Cancer Research which has a commercial interest in the development of CHK1 inhibitors. Authors who are, or have been, employed by The Institute of Cancer Research are subject to a “Rewards to Inventors Scheme” which may reward contributors to a program that is subsequently licensed. The authors have or have had direct or indirect commercial interactions with some or all of AstraZeneca plc, Vernalis Ltd., Genentech Inc, Cancer Research Technology Ltd.
Supplementary Material
References
- Kastan M. B.; Bartek J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [DOI] [PubMed] [Google Scholar]
- Dai Y.; Grant S. New insights into checkpoint kinase 1 in the DNA damage response signalling network. Clin. Cancer Res. 2010, 16, 376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.; Tho L. M.; Xu N.; Gillespie D. A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [DOI] [PubMed] [Google Scholar]
- Xiao Z.; Chen Z.; Gunasekera A. H.; Sowin T. J.; Rosenberg S. H.; Fesik S.; Zhang H. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J. Biol. Chem. 2003, 278, 21767–21773. [DOI] [PubMed] [Google Scholar]
- Sorensen C. S.; Hansen L. T.; Dziegielewski J.; Syljuasen R. G.; Lundin C.; Bartek J.; Helleday T. The cell-cycle checkpoint kinase chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005, 7, 195–201. [DOI] [PubMed] [Google Scholar]
- Antoni L.; Sodha N.; Collins I.; Garrett M. D. CHK2 kinase: cancer susceptibility and cancer therapy – two sides of the same coin?. Nat. Rev. Cancer 2007, 7, 925–936. [DOI] [PubMed] [Google Scholar]
- Hollstein M.; Sidransky D.; Vogelstein B.; Harris C. C. P53 mutations in human cancer. Science 1991, 253, 49–53. [DOI] [PubMed] [Google Scholar]
- Giono L. E.; Manfredi J. J. The p53 tumor suppressor participates in multiple cell cycle checkpoints. J. Cell Physiol. 2006, 209, 13–20. [DOI] [PubMed] [Google Scholar]
- Ma C. X.; Janetka J. W.; Piwnica-Worms H. Death by releasing the breaks: Chk1 inhibitors as cancer therapeutics. Trends Mol. Med. 2011, 17, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett M. D.; Collins I. Anticancer therapy with checkpoint kinase inhibitors: what, when and where?. Trends Pharmacol. Sci. 2011, 32, 308–316. [DOI] [PubMed] [Google Scholar]
- Murga M.; Campaner S.; Lopez-Contreras A. J.; Toledo L. I.; Soria R.; Montaña M. F.; D’Artista L.; Schleker T.; Guerra C.; Garcia E.; Barbacid M.; Hidalgo M.; Amati B.; Fernandez-Capetillo O. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höglund A.; Nilsson L. M.; Muralidharan S. V.; Hasvold L. A.; Merta P.; Rudelius M.; Nikolova V.; Keller U.; Nilsson J. A. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clin. Cancer Res. 2011, 17, 7067–7079. [DOI] [PubMed] [Google Scholar]
- Cole K. A.; Huggins J.; Laquaglia M.; Hulderman C. E.; Russell M. R.; Bosse K.; Diskin S. J.; Attiyeh E. F.; Sennett R.; Norris G.; Laudenslager M.; Wood A. C.; Mayes P. A.; Jagannathan J.; Winter C.; Mosse Y. P.; Maris J. M. RNAi screen of the protein kinome identifies checkpoint kinase 1 (chk1) as a therapeutic target in neuroblastoma. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 3336–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss G. J.; Donehower R. C.; Iyengar T.; Ramanathan R. K.; Lewandowski K.; Westin E.; Hurt K.; Hynes S. M.; Anthony S. P.; McKane S.. Phase I dose-escalation study to examine the safety and tolerability of LY2603618, a checkpoint 1 kinase inhibitor, administered 1 day after pemetrexed 500 mg/m(2) every 21 days in patients with cancer. Invest. New Drugs [Online early access]. DOI: 10.1007/s10637-012-9815-9. Published Online: Apr 10, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasina A.; Hallin J.; Chen E.; Arango M. E.; Kraynov E.; Register J.; Grant S.; Ninkovic S.; Chen P.; Nichols T.; O’Connor P.; Anderes K. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol. Cancer Ther. 2008, 7, 2394–404. [DOI] [PubMed] [Google Scholar]
- Oza V.; Ashwell S.; Almeida L.; Brassil P.; Breed J.; Deng C.; Gero T.; Grondine M.; Horn C.; Ioannidis S.; Liu D.; Lyne P.; Newcombe N.; Pass M.; Read J.; Ready S.; Rowsell S.; Su M.; Toader; Vasbinder M.; Yu D.; Yu Y.; Xue Y.; Zabludoff S.; Janetka J. Discovery of checkpoint kinase inhibitor (S)-5-(3-fluorophenyl)-N-(piperidin-3-yl)-3-ureidothiophene-2-carboxamide (AZD7762) by structure-based design and optimization of thiophenecarboxamide ureas. J. Med. Chem. 2012, 55, 5130–5142. [DOI] [PubMed] [Google Scholar]
- Guzi T. J.; Paruch K.; Dwyer M. P.; Labroli M.; Shanahan F.; Davis N.; Taricani L.; Wiswell D.; Seghezzi W.; Penaflor E.; Bhagwat B.; Wang W.; Gu D.; Hsieh Y.; Lee S.; Liu M.; Parry D. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol. Cancer Ther. 2011, 10, 591–602. [DOI] [PubMed] [Google Scholar]
- Lainchbury M.; Collins I. Checkpoint kinase inhibitors: a patent review (2009 - 2010). Expert Opin. Ther. Pat. 2011, 21, 1191–210. [DOI] [PubMed] [Google Scholar]
- Reader J. C.; Matthews T. P.; Klair S.; Cheung K. M.; Scanlon J.; Proisy N.; Addison G.; Ellard J.; Piton N.; Taylor S.; Cherry M.; Fisher M.; Boxall K.; Burns S.; Walton M, I.; Westwood I. M.; Hayes A.; Eve P.; Valenti M.; de Haven Brandon A.; Box G.; van Montfort R. L.; Williams D. H.; Aherne G. W.; Raynaud F. I.; Eccles S. A.; Garrett M. D.; Collins I. Structure-guided evolution of potent and selective CHK1 inhibitors through scaffold morphing. J. Med. Chem. 2011, 54, 8328–8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews T. P.; Klair S.; Burns S.; Boxall K.; Cherry M.; Fisher M.; Westwood I. M.; Walton M. I.; McHardy T.; Cheung K. M.; van Montfort R.; Williams D.; Aherne G. W.; Garrett M. D.; Reader J.; Collins I. Identification of inhibitors of checkpoint kinase 1 through template screening. J. Med. Chem. 2009, 52, 4810–4819. [DOI] [PubMed] [Google Scholar]
- Walton M. I.; Eve P. D.; Hayes A.; Valenti M.; De Haven Brandon A.; Box G.; Boxall K. J.; Aherne G. W.; Eccles S. A.; Raynaud F. I.; Williams D. H.; Reader J. C.; Collins I.; Garrett M. D. The preclinical pharmacology and therapeutic activity of the novel CHK1 inhibitor SAR-020106. Mol. Cancer Ther. 2010, 9, 89–100. [DOI] [PubMed] [Google Scholar]
- Williams F. M. Clinical significance of esterases in man. Clin. Pharmacokinet. 1985, 10, 392–403. [DOI] [PubMed] [Google Scholar]
- Traxler P.; Furet P.; Mett H.; Buchdunger E.; Meyer T.; Lydon N. Design and synthesis of novel tyrosine kinase inhibitors using a pharmacophore model of the ATP-binding site of the EGF-R. J. Pharm. Belg. 1997, 52, 88–96. [PubMed] [Google Scholar]
- Walton M. I.; Eve P. D.; Hayes A.; Valenti M. R.; De Haven Brandon A. K.; Box G.; Hallsworth A.; Smith E. L.; Boxall K. J.; Lainchbury M.; Matthews T. P.; Jamin Y.; Robinson S. P.; Aherne G. W.; Reader J. C.; Chesler L.; Raynaud F. I.; Eccles S. A.; Collins I.; Garrett M. D. CCT244747 is a novel, potent and selective CHK1 inhibitor with oral efficacy alone and in combination with genotoxic anticancer drugs. Clin. Cancer Res. 2012, 18, 5650–5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Centre for Kinase Profiling, MRC Protein Phosphorylation Unit, University of Dundee, UK; http://www.kinase-screen.mrc.ac.uk [accessed July 12, 2012]. [Google Scholar]
- Ion Channel CardiacProfiler Panel, Millipore Corporation, St. Charles, MO; http://www.millipore.com/ionchannels [accessed July 12, 2012]. [Google Scholar]
- Roche M.; Renauleaud C.; Ballet. V.; Doubovetzky M.; Guillon J. M. The isolated rabbit heart and Purkinje fibers as models for identifying proarrhythmic liability. J. Pharmacol. Toxicol. Methods 2010, 61, 238–250. [DOI] [PubMed] [Google Scholar]
- Humphries M.Targeting checkpoint kinase 1: a study in the application of preclinical data to inform clinical strategy. Presentation at the 2nd Annual Cancer Targets & Therapeutics Conference; October 21, 2010. www.arraybiopharma.com/_documents/Publication/PubAttachment410.pdf [accessed July 11, 2012].
- Tse A. N.; Rendahl K. G.; Sheikh T.; Cheema H.; Aardalen K.; Embry M.; Ma S.; Moler E. J.; Ni Z. J.; Lopes de Menenzes D. E.; Hibner B.; Gesner T. G.; Schwartz G. K. CHIR-124, a novel potent inhibitor of Chk1, potentiates the cytotoxicity of topoisomerase I poisons in vitro and in vivo. Clin. Cancer Res. 2007, 13, 591–602. [DOI] [PubMed] [Google Scholar]
- Weiss W. A.; Aldape K.; Mohapatra G.; Feuerstein B. G.; Bishop J. M. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997, 16, 2985–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Z.; Wang L.; Stewart K. D.; Chen Z.; Gu W.; Bui M.; Merta P.; Zhang H.; Kovar P.; Johnson E.; Park C.; Judge R.; Rosenberg S.; Sowin T.; Lin N. Structure-based design, synthesis, and biological evaluation of potent and selective macrocyclic checkpoint kinase 1 inhibitors. J. Med. Chem. 2007, 50, 1514–1527. [DOI] [PubMed] [Google Scholar]
- Sawa M. Strategies for the design of selective protein kinase inhibitors. Mini-Rev. Med. Chem. 2008, 8, 1291–1297. [DOI] [PubMed] [Google Scholar]
- Foloppe N.; Fisher L. M.; Francis G.; Howes R.; Kierstan P.; Potter A. Identification of a buried pocket for potent and selective inhibition of Chk1: Prediction and verification. Bioorg. Med. Chem. 2006, 14, 1792–1804. [DOI] [PubMed] [Google Scholar]
- Jamieson C.; Moir E. M.; Rankovic Z.; Wishart G. Medicinal chemistry of hERG optimizations: Highlights and hang-ups. J. Med. Chem. 2006, 49, 5029–5046. [DOI] [PubMed] [Google Scholar]
- Workman P.; Collins I. Probing the probes: fitness factors for small molecule tools. Chem. Biol. 2010, 17, 561–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H.; Kolesnikov A.; Riggs J. R.; Wesson K. E.; Stephens R.; Leahy E. M.; Shrader W. D.; Sprengeler P. A.; Green M. J.; Sanford E.; Nguyen M.; Gjerstad E.; Cabuslay R.; Young W. B. Potent 4-amino-5-azaindole factor VIIa inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 4567–4570. [DOI] [PubMed] [Google Scholar]
- Encapsulation of palladium in polyurea microcapsules.Ramarao C.; Ley S. V.; Smith S. C.; Shirley I. M.; DeAlmeida N. Chem. Commun. 2002, 10, 1132–1133. [DOI] [PubMed] [Google Scholar]
- Pd-EnCat TPP30 as a catalyst for the generation of highly functionalized aryl- and alkenyl-substituted acetylenes via microwave-assisted Sonogashira type reactions. Sedelmeier J.; Ley S. V.; Lange H.; Baxendale I. R. Eur. J. Org. Chem. 2009, 4412–4420. [Google Scholar]
- Godel T.; Hoffmann T.; Schnider P.; Stadler H.. Preparation of 4-phenylpyridines as neurokinin-1 receptor antagonists. Patent Appl. WO 2002016324, 2002;; Chem. Abstr. 2002, 136, 216651. [Google Scholar]
- Dunne J.; Reardon H.; Trinh V.; Li E.; Farinas J. Comparison of on-chip and off-chip microfluidic kinase assay formats. Assay Drug Dev. Technol. 2004, 2, 121–129. [DOI] [PubMed] [Google Scholar]
- Skehan P.; Storeng R.; Scudiero D.; Monks A.; McMahon J.; Vistica D.; Warren J. T.; Bokesch H.; Kenney S.; Boyd M. R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [DOI] [PubMed] [Google Scholar]
- Tao Z. F.; Li G.; Tong Y.; Stewart K. D.; Chen Z.; Bui M. H.; Merta P.; Park C.; Kovar P.; Zhang H.; Sham H. L.; Rosenberg S. H.; Sowin T. J.; Lin N. H. Discovery of 4′-(1,4-dihydro-indeno[1,2-c]pyrazol-3-yl)-benzonitriles and 4′-(1,4-dihydro-indeno[1,2-c]pyrazol-3-yl)-pyridine-2′-carbonitriles as potent checkpoint kinase 1 (Chk1) inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 5944–5951. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.