

Abstract
A sensitive cAMP response element (CRE) reporter system is essential for studying the cAMP/protein kinase A/cAMP response element binding protein signal pathway. Here we have tested a few CRE promoters and found one with high sensitivity to external stimuli. By using this optimal CRE promoter and the enhanced green fluorescent protein as the reporter, we have established a CRE reporter cell line. This cell line can be used to study the signal pathway by fluorescent microscope, fluorescence-activated cell analysis and luciferase assay. This cell line’s sensitivity to forskolin, by using the technique of fluorescence-activated cell sorting, was increased to approximately 7 times that of its parental HEK 293 cell line, which is currently the most commonly used cell line in the field for the signal pathway study. Therefore, this newly created cell line is potentially useful for studying the signal pathway’s modulators, which generally have weaker effect than its mediators. Our research has also established a general procedure for optimizing transcription-based reporter cell lines, which might be useful in performing the same task when studying many other transcription-based signal pathways.
Keywords: cAMP, cAMP response element, cell signaling pathway, reporter cell line, memory formation
1. Introduction
Cell function is mediated by various signal pathways. Most signal pathways mediate cell function by altering the transcription activity of certain genes. For example, in the cAMP/protein kinase A/cAMP response element binding protein (cAMP/PKA/CREB) signal pathway, the adenylyl cyclase catalyzes the formation of cAMP upon a Gs protein-coupled receptor activation or calcium entry. The cAMP then activates the PKA, which in turn initiates the downstream kinase cascade. One of the end steps of this cascade is the phosphorylation of the CREB. The phosphorylated CREB, together with other transcription factors, can bind promoter regions containing the 5’-TGACGTCA-3’ sequence, i.e. the cAMP response element (CRE), and in consequence induces the downstream gene transcription. CREs exist in the promoter region of many genes that play important roles in metabolism, cell cycle, development, neurotransmission, memory formation, etc. (Mayr and Montminy 2001; Wang and Storm 2003). This signal pathway has therefore been intensively studied.
A signal pathway’s activity is usually measured by testing its outcome, the transcription products. The transcription products can be endogenous proteins, but, more often they are the products of externally-introduced reporter genes. A reporter gene usually contains both a specific promoter and a reporter gene cDNA. The promoter can be either obtained from one of the endogenous target genes of the specific signal pathway, or made artificially from a few consensus sequences, in this case CREs, together with a minimal promoter. The commonly used reporters are the enzymes luciferase and β-galactosidase (LacZ), the activities of which can be measured by testing their catalysis of their respective substrates. However, the application of these two reporter systems is quite limited. For example, they cannot be used for real-time imaging. To overcome this limitation, we have developed a new CRE-enhanced green fluorescent protein (EGFP) reporter system, which could be used for monitoring real-time transcription under a fluorescent microscope and for testing signal pathway activity by fluorescence-activated cell sorting (FACS).
These reporter systems can be used to measure how a small molecule or a specific gene product regulates the CRE signal pathway. This is usually done by applying the small molecules directly or by transfecting the target genes to the reporter cells, and measuring the change in the level of reporter expression. This approach works well if the change is robust, but it would be difficult to distinguish the signal from the noise when the change is slight. This is especially crucial when studying the effect of modulators, rather than mediators, on the CRE signal pathway, as the former usually have less effect than the latter. Therefore, a more sensitive CRE reporter system is needed. To achieve this, the new CRE-EGFP reporter system was further optimized with a significantly enhanced sensitivity to external stimuli.
2. Materials and methods
2.1. Plasmid construction
The luciferase and EGFP reporter plasmids were constructed as shown in Fig. 1.
Fig. 1.
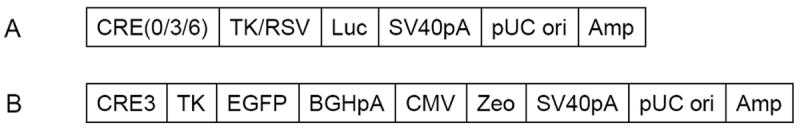
Construction of CRE luciferase (A) and EGFP (B) reporter plasmids.
2.2. Cell culture and transfection
The HEK 293 (ATCC) and HT22 cells (Morimoto and Koshland 1990) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The NIH3T3 cells (ATCC) were maintained in DMEM supplemented with 10% calf serum. Cells were transfected with lipofectamine 2000 (Invitrogen) according to the manufacturer’s guide.
2.3. Luciferase assay
Cells were transfected with the CRE-luciferase reporters and EF1-LacZ plasmids. The EF1-LacZ plasmid contains the β-galactosidase cDNA preceded by the constitutively active EF1 promoter. It works as an internal control to normalize luciferase activity. One day after transfection, the cells were treated with either 10 μM forskolin, 100 μM 5,6-Dichlorobenzimidazole riboside (DRB, Sigma), 10 μM forskolin (Sigma) and 100 μM DRB or DMSO, as a control, for 6 hours. The cells were harvested, washed with ice-cold phosphate buffered saline (PBS) twice and lysed in 100 μl of 0.1% Triton X-100/1 mM dithiothreitol/1.5 mM MgSO4/4 mM ATP/100 mM Na2HPO4, pH 7.8. Luciferase and β-galactosidase assays were performed as described previously (Orellana and McKnight 1992; Matthews et al. 1994). The luciferase activity was divided by the β-galactosidase activity to normalize for differences in cell number and transfection efficiency. The assays for each CRE reporter were performed three times and the final data were averaged.
2.4. Fluorescence-activated cell analysis and sorting
Cells were harvested by trypsin treatment and centrifuged at 500 g for 5 minutes. The pellet was resuspended in ice-cold PBS and centrifuged again. The pellet was then resuspended at a concentration of 4 × 106 cells/ml in the sorting buffer, which is PBS containing 100 Kunitz DNase I/ml, 10 μg/ml propidium iodide (Sigma) and 2% FBS. The sorting solution was also supplemented with either 10 μM forskolin, 100 μM 5,6-Dichlorobenzimidazole riboside (DRB, Sigma), 10 μM forskolin (Sigma) and 100 μM DRB or DMSO, as a control. The cells were then sent through a 40 μm filter to remove large clumps and loaded into either a FACScan Flow Cytometer (BD Bioscience) for cell analysis or a FACSVantage SE cell sorter (BD Bioscience) for cell sorting. The cells with positive propidium iodide staining, i.e. the dead cells, were first eliminated from the analysis or sorting pool. For cell sorting, the desired population, either the EGFP brightest or the least bright ones according to the purpose of experiments (see Results), was sorted into either 15ml conical tubes or 96-well plates, which both contained complete DMEM culture media.
2.5. Genotyping
Genotyping was performed by following a protocol modified from the one described in Shan et al. 2008. Briefly, cells were grown in 6-well plates and then harvested by trypsin treatment. The trypsin treatment was terminated by adding 2ml DMEM. The cells in DMEM were centrifuged at 500 g for 5 minutes. The supernatant was discarded and the cell pellet was washed in 0.5 ml ice-cold PBS and centrifuged at 500 g for 5 minutes. The pellet was digested in 0.25 ml of a buffer containing 5 mM EDTA, 200 mM NaCl, 100 mM Tris (pH 8.0), 0.2% (w/v) SDS and l0 mg/ml Proteinase K (Sigma) at 50 degrees for 12-18 hours. The genomic DNA was extracted by phenol/chloroform purification and isopropanol precipitation. The genomic DNA was then used as the template for a PCR, which used two oligos as primers. The primers were designed to complement sequences 5’ to CRE3 and 3’ to BGHpA (Fig. 1B), respectively. Therefore, the essential elements for the inducible EGFP construct, including the sequences of CRE3, TK, EGFP and BGHpA, were amplified. The sequences of the oligos were CTGCGGCCGCAATAAAATATC and CAGAAGCCATAGAGCCCAC.
3. Results
The quality of a reporter system is determined by two factors, i.e. cell type and reporter gene construction. In order to make an optimal CRE-reporter cell line, we tested 3 cell lines, HEK 293, NIH3T3 and HT22. The HEK 293 and NIH3T3 cell lines are widely used in reporter assays because of their easy maintenance. The HT22 cell line is an immortalized hippocampal cell line (Morimoto and Koshland 1990) and has the advantage of possessing some neuronal characteristics. Since our lab is working in the learning and memory field, any result from the HT22 cell line might be more readily extrapolated into neurons in vivo. Besides the cell type, the reporter gene construction is also essential for the quality of the reporter system. Generally, in order to boost assay outcome, multiple consensus sequences followed by a mini-promoter are used to construct the promoter region. However, having too few consensus elements causes a low overall expression level, while having too many induces a high basal level. Therefore, it is necessary to identify the optimal number of consensus elements, in this case CREs, to achieve the best signal to noise ratio. Here we chose to test reporter genes containing three and six CREs (CRE3 and CRE6). In addition, mini-promoters of different origin may have different transcription strengths and therefore differently affect the basal expression level of a reporter gene. We chose to test the mini-promoters from the thymidine kinase gene of human herpesvirus (TK) and the terminal repeat of Rous sarcoma virus (RSV). Overall we constructed six promoters, i.e. TK, CRE3-TK, CRE6-TK, RSV, CRE3-RSV and CRE6-RSV, among which the TK and RSV served as controls. We fused them to the cDNA of luciferase (Fig. 1A) and luciferase assays were subsequently performed. In the luciferase assay, we treated cells with 10 μM forskolin for 6 hours to induce CRE activity, because forskolin of this concentration usually induces a half-saturating response and 6 hours is usually the time point when the luciferase expression reaches saturation (unpublished data).
As shown in Fig. 2A, in all cases when the CRE was present in the promoter region, there was some level of basal luciferase expression. This is not surprising since a basal CRE signal activity is essential for cell viability (Mayr and Montminy 2001). Moreover, as expected, the luciferase activity is enhanced following forskolin stimulation. In each cell line, a difference in luciferase activities resulted when either the number of CREs was altered or the mini promoter changed. The CRE6 and the RSV seemed to have higher transcription strength than the CRE3 and the TK, respectively. This applied to both the DMSO and forskolin-treated groups among the three cell lines tested. However, although a high level of absolute luciferase activity is essential (Fig. 2A), what is more important in determining system sensitivity is the relative luciferase activity, i.e. the ratio of luciferase activity with forskolin stimulation to luciferase activity with DMSO treatment (Fig. 2B). Unlike in the case of absolute luciferase activity, CRE3 demonstrated a slightly higher relative luciferase activity than CRE6, while there was no significant difference between the TK and RSV mini-promoters (Fig. 2B). A more dramatic difference in relative luciferase activity was seen between the three cell lines we tested, i.e. the HEK 293 showed the highest relative luciferase activity (9.2-11.6 fold change), in contrast to NIT3T3 and HT22, which only had 1.9-3.4 and 2.4-4.0 fold changes, respectively (Fig. 2B).
Fig. 2.
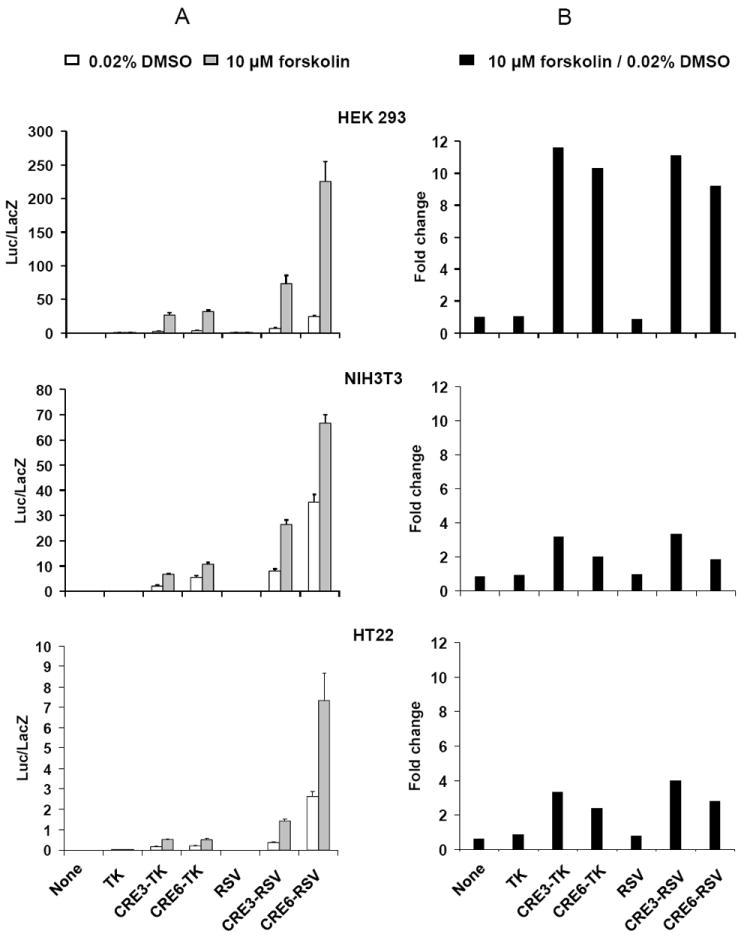
Luciferase assay of various promoters in three cell lines (HEK 293, NIH3T3, and HT22). The normalized luciferase activities for the transfected cells treated with either DMSO or 10 μM forskolin for 6 hours are shown in the left panels (A). The fold changes, i.e. the value of the forskolin group divided by the value of the DMSO group, are shown in the right panels (B).
Based on the overall result, we chose to use the CRE3-TK promoter and the HEK 293 cell line to construct the stable CRE reporter cell line.
We next subcloned the CRE3-TK DNA followed by the EGFP cDNA into the modified pcDNA3.1zeo+ vector (Invitrogen) (Fig. 1B). However, one day after this construct transfected the HEK 293 cells, a strong green fluorescence appeared without any forskolin stimulation. In addition, its brightness could not be distinguished from the case after forskolin stimulation (data not shown). Our interpretation is that the EGFP has a very stable structure and it can accumulate and saturate within cells. In consequence, the forskolin stimulation could not further the expression or the fluorescence level of the EGFP. One strategy to circumvent this problem is to make the EGFP less stable, i.e. more amenable to degradation. We achieved this goal by fusing a PEST motif from mouse ornithine decarboxylase to the C-terminal of EGFP. The PEST motif imparts instability to its host proteins (Li et al. 1998; Loetscher et al. 1991). This CRE3-TK-EGFP construct was then used to stably transfect the HEK 293 cells.
This stable cell line could be selected by a traditional one-time zeocin treatment, however, we instead employed an alternative method combining the zeocin selection and FACS in order to obtain a cell clone which is more sensitive to external stimuli. The procedure is described as follows.
The HEK 293 cells were transfected with the CRE3-TK-EGFP construct. One day after transfection, the cells were 1:10 split and treated with 100 μg/ml zeocin (Invitrogen) until they were 80% confluent. The cells were then treated with 10 μM forskolin for 6 hours and subjected to FACS. To isolate the most forskolin-responsive cells, the brightest 15% were sorted out and allowed to continue to grow in the culture medium containing zeocin until they were 80% confluent again. This population, however, might also have consisted of the cells with high constitutive, rather than forskolin-responsive, EGFP expression. They were unwanted since, as discussed above, the quality of the reported cell line is determined by the ratio of the reporter activity of the activated group to that of the control group. To eliminate this unwanted constitutively bright cell population, we starved the cells cultured from the first FACS with serum-free DMEM for 6 hours and sorted out the least bright 15% of cells through FACS. The purpose of the serum starvation was to eliminate any serum-induced CRE activity. The sorted cells were again grown in the zeocin-containing DMEM until they were 80% confluent. The cells were treated with 10 μM forskolin for 6 hours and the brightest 192 cells were sorted individually into 96-well plates coated with poly-D-lysine. The cells were maintained in zeocin-containing DMEM and finally about 20% of them survived.
Each single cell clone was propagated and subjected to fluorescence-activated cell analysis. One of the resulting cell clones indicated a dramatic increase in fluorescence intensity after 10 μM forskolin stimulation. As shown in Fig. 3A, about 70% of the cell population in the forskolin-treatment group of this clone demonstrated fluorescence intensity above 4 × 103, in contrast to the control DMSO treatment group in which less than 5% of cells fell into the same range. To confirm that forskolin induced EGFP expression was through CRE transcription rather than cAMP regulated translation, a transcription inhibitor DRB was used together with forskolin. As shown in Fig. 3A, when 10 μM forskolin was applied in the presence of 100 μM DRB, only about 10% of cells demonstrated fluorescence intensity above 4 × 103. This is much lower than in the forskolin-only treatment group, but just slightly higher than in the control DRB-only treatment group, in which about 5% of cells fell into the same range. We therefore concluded that this new CRE-reporter cell line mostly reported the CRE-transcription activity.
Fig. 3.
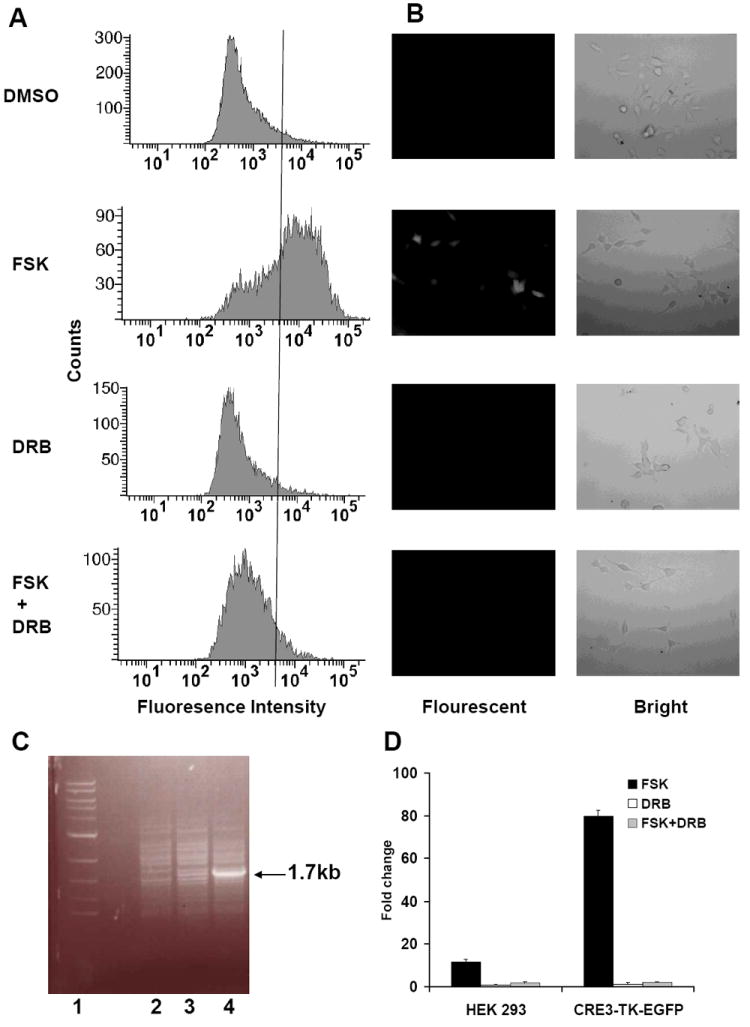
Characterization of the CRE3-TK-EGFP stable cell line. The cells were treated with either DMSO, 10 μM forskolin (FSK), 100 μM DRB or 10 μM forskolin and 100 μM DRB (FSK + DRB) for 6 hours and subjected to fluorescence-activated cell analysis (A) or observed under an inverted fluorescent microscope (B). In (A), a line is drawn crossing the 4 × 103 tick of the x-axis. In (B), the left photos are from the fluorescent field while the right ones are from the bright field. The reporter DNA integration was verified by a PCR on the genomic DNA (C). Lane 1 is the DNA marker; lanes 2, 3 and 4 are the PCR products from the HEK 293 cells, one of the resulting stable cell clones not responsive to forskolin stimulation and the CRE3-TK-EGFP cell line, respectively. An extra 1.7kb band in lane 4 represents the reporter gene. Luciferase assays were also performed on the CRE3-TK-EGFP cells as well as the HEK 293 cells and the fold changes after 10 μM forskolin, 100 μM DRB or 10 μM forskolin and 100 μM DRB stimulation are shown (D).
On the other hand, under a fluorescent microscope, this cell clone barely showed any fluorescence in the control DMSO treatment group while it became fluorescent after 6 hours of 10 μM forskolin treatment (Fig. 3B). Again, 10 μM forskolin in the presence of 100 μM DRB did not induce any detectable fluorescence, which confirmed that this cell line mainly reported CRE-transcription activity. In addition, the stable integration was verified by a PCR amplification of the genomic DNA using a pair of primers flanking the CRE3-TK-EGFP-BGHpA reporter gene (Fig. 3C).
More interestingly, when the CRE3-TK-EGFP stable cell line was transfected with the CRE3-TK-luciferase reporter plasmid, the luciferase activity was enhanced 80 times after 10 μM forskolin stimulation (Fig. 3D). This stable cell line has thus shown much improvement in sensitivity towards forskolin stimulation compared to its parental HEK 293 cell line, which was only enhanced 11.6 times. This improvement was apparently achieved by the procedure of selecting the most forskolin-sensitive cell clone through FACS. In addition, when either the CRE3-TK-EGFP or the HEK 293 cell lines was treated by 10 μM forskolin in the presence of 100 μM DRB, the induced luciferase activity was minimal. This is consistent with the results from the FACS and the microscope studies, confirming that this CRE3-TK-EGFP cell line mainly reported the CRE-transcription activity.
4. Discussion
Here we have tested a few CRE promoters, which, besides directing the construction of a stable CRE reporter cell line, might also provide useful knowledge for in vivo study. For example, we previously made a transgenic mouse line expressing CRE6-RSV-LacZ, which, although useful, tends to have an unwanted high basal LacZ expression in neurons (Impey et al. 1996; Impey et al. 1998). This high basal activity of the CRE6-RSV was coincidently reflected in the luciferase assay we performed (Fig. 2A). However, compared with the CRE6-RSV, the CRE3-TK seemed to have much lower basal activity while slightly higher relative activity (Fig. 2A and B) and therefore promises to be a better CRE reporter promoter for in vivo study.
We have also constructed and optimized a stable CRE reporter cell line. This cell line can be used to study the signal pathway by fluorescent microscope, fluorescence-activated cell analysis and luciferase assay. This cell line can be used to test whether a given protein, or small molecule, regulates the cAMP/PKA/CREB signal pathway. This is especially useful for those that only mildly modulate the signal pathway activity, as this cell line is much more sensitive to external stimuli than any other widely used cell line, such as the HEK 293, NIH3T3 or HT22 cell lines.
Our experiment has also established a general procedure for optimizing transcription-based reporter cell lines, which might be useful in performing the same task when studying many other transcription-based signal pathways.
Acknowledgments
This project was supported by NIH grants NS20498 and MH073601.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Impey S, Mark M, Villacres EC, Poser S, Chavkin C, Storm DR. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–82. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR. Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci. 1998;1:595–601. doi: 10.1038/2830. [DOI] [PubMed] [Google Scholar]
- Li X, Zhao X, Fang Y, Jiang X, Duong T, Fan C, Huang CC, Kain SR. Generation of destabilized green fluorescent protein as a transcription reporter. J Biol Chem. 1998;273:34970–5. doi: 10.1074/jbc.273.52.34970. [DOI] [PubMed] [Google Scholar]
- Loetscher P, Pratt G, Rechsteiner M. The C terminus of mouse ornithine decarboxylase confers rapid degradation on dihydrofolate reductase. Support for the pest hypothesis. J Biol Chem. 1991;266:11213–20. [PubMed] [Google Scholar]
- Matthews RP, Guthrie CR, Wailes LM, Zhao X, Means AR, McKnight GS. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol Cell Biol. 1994;14:6107–16. doi: 10.1128/mcb.14.9.6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- Morimoto BH, Koshland DE., Jr Induction and expression of long- and short-term neurosecretory potentiation in a neural cell line. Neuron. 1990;5:875–80. doi: 10.1016/0896-6273(90)90347-i. [DOI] [PubMed] [Google Scholar]
- Orellana SA, McKnight GS. Mutations in the catalytic subunit of cAMP-dependent protein kinase result in unregulated biological activity. Proc Natl Acad Sci U S A. 1992;89:4726–30. doi: 10.1073/pnas.89.10.4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Q, Chan GC, Storm DR. Type 1 adenylyl cyclase is essential for maintenance of remote contextual fear memory. J Neurosci. 2008;28:12864–7. doi: 10.1523/JNEUROSCI.2413-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Storm DR. Calmodulin-regulated adenylyl cyclases: cross-talk and plasticity in the central nervous system. Mol Pharmacol. 2003;63:463–8. doi: 10.1124/mol.63.3.463. [DOI] [PubMed] [Google Scholar]