

Abstract
Selectivity in the catalytic functionalization of complex molecules is a major challenge in chemical synthesis. The problem is magnified when there are several possible stereochemical outcomes and when similar functional groups occur repeatedly within the same molecule. Selective polyene oxidation provides an archetypical example of this challenge. Historically, enzymatic catalysis provided the only precedents. Although nonenzymatic catalysts are now known that meet some of these challenges, a comprehensive solution has remained elusive. Here, we describe low molecular weight, peptide-based catalysts discovered through a combinatorial synthesis and screening protocol that exhibit site- and enantioselective oxidation of certain positions of various isoprenols. This diversity-based approach, which exhibits features remiscent of the directed evolution of enzymes, delivers catalysts that compare favourably to the state-of-the-art for asymmertric oxidation of these compounds. Moreover, the approach culminated in catalysts that exhibit alternate site-selectivity in comparison to previously described oxidation catalysts.
Subject terms: chemistry, catalysis, epoxidation, chemical evolution, biomimetic
Analysis of biosynthetic pathways reveals that functional group selectivity in enzymatic reactions is a persistent issue. For example, olefins may undergo oxidation in favor of C-H bonds, or instead of alcohols. And different enzymes are engaged to reverse the order of events. Likewise, the derivation of strategies for the chemical synthesis of complex structures requires constant consideration of selectivity. The complexity of the situation is amplified when identical functional groups occur repeatedly within the same molecule. Now, for example, catalysts may be called upon to oxidize a singular olefin in the presense of others, and different catalysts may be needed to target alternative sites. Even so, selectivity for one of the multiple sites represents but one aspect of the chemical challenge, since many functional groups may also react in ways that yield new isomers, adding stereochemical diversity to the positional selectivity outcomes. Thus, the selective oxidation of polyenes presents a profound challenge.
For many years, the selective functionalization of similarly reactive olefins within polyenes was primarily observed through the study of enzymes1. Today, small molecule catalysts are known that also meet some of these challenges. Yet, catalyst types that deliver substantially different site-selectivity are not well established, nor are generalizable protocols for their discovery. Venerable catalysts provide solutions to problems that enable allylic olefin epoxidation2,3,4,5, oxidation of subterminal olefins of polyenes6,7, and selective oxidation of conjugated systems8,9. Some strategies provide access to internal epoxides of polyenes using stoichiometric, auxiliary-based, “template-directed” strategies10,11,12. In this article, we describe low molecular weight, peptide-based catalysts13,14, discovered through a combinatorial synthesis and screening protocol, that exhibit site- and enantioselective oxidation of various isoprenols. The diversity-based approach15,16 delivers catalysts that compare favorably to the state-of-the-art for asymmetric oxidation of certain isoprenols, such as geraniol, farnesol, and geranylgeraniol. Moreover, the approach leads to catalysts that offer alternate site-selectivity in ways that are not known with previously described oxidation catalysts.
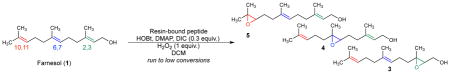
We have previously developed a catalytic cycle employing an aspartatic acid-based carboxylic acid and have applied this methodology to the asymmetric oxidation of allylic urethanes17 and indoles18. During the course of the catalytic cycle, the carboxylic acid side chain of aspartic acid is activated with a carbodiimide, generating a transient peracid upon reaction with hydrogen peroxide (Fig. 1a). Catalytic turnover of this cycle is enhanced through addition of common nucleophilic co-catalysts such as 1-hydroxybenzotriazole (HOBt) and N,N-dimethylaminopyridine (DMAP). Notably, this approach is distinct from enzymatic “epoxidases”19, which often exploit cofactors. When applied to farnesol (1), this catalytic cycle, employing propionic acid (2) as the catalyst, furnishes the same product selectivity as the textbook-level reagent meta-chloroperbenzoic acid (m-CPBA), which is used as a stoichiometric reagent (Fig. 1b).
Figure 1. Precedent and goal of catalytic oxidation of farnesol.

a, Catalytic cycle of aspartic acid-mediated epoxidations. b, Site-selectivity of m-CPBA and propionic acid. aDetermined by uncalibrated GC integrations (see Supplementary Information for details). b1.0 equiv. m-CPBA, Na2HPO4 (2.0 equiv.), DCM, H2O. c10 mol% acid, HOBt (10 mol%), DMAP (10 mol%), 1.0 equiv. N,N′-diisopropylcarbodiimide (DIC), 2.0 equiv. H2O2. c, Precedents of the Sharpless epoxidation of 1. d, The goal of the present study.
The work began with an assessment of peptide-embedded aspartates to realize site-selective oxidation of farnesol (1). Could aspartate-based catalysts be found that would oxidize farnesol with high levels of site-selectivity and ultimately enantiocontrol? Would catalysts be found that would functionalize the allylic alkene20, or other olefins of 1? Oxidation of 1 with (+) or (−)-DIPT/Ti(Oi-Pr)4 under the conditions of Sharpless has been reported over the years to deliver 3 with excellent selectivity (82–95% ee; Fig 1c)21,22,23,24,25,26. However, we lacked a clear structure-based hypothesis for how peptide-based catalysts might achieve our goals. Therefore, we applied an on-bead screening protocol for discovering peptide oxidation catalysts27 for selective oxidation of 1 (Fig. 1d).
Results and Discussion
Our approach employs the one-bead-one-compound approach to library design29, which capitalizes on split-pool combinatorial synthesis30,31. This approach also allows for an evolutionary approach to catalyst optimization32, in analogy to the strategy employed in the directed evolution of enzymes33,34,35.
For our initial screening, we made a number of strategic decisions. For example, we elected to run reactions to low conversion initially, allowing preliminary assessment of catalyst krel values. We define krel as the relative rate of epoxidation at each olefinic site of 1 (2,3-position to give 3; 6,7-position, 4; 10,11-position, 5). It is well appreciated that reaction conversion (i.e., the extent of overoxidation) can influence product distributions and muddle the extraction of meaningful krel comparisons among catalysts36. Thus, we planned to validate the best “hit” catalysts at a later stage under synthetically meaningful conditions, with sufficient stoichiometry of terminal oxidant to achieve good yields of products (vide infra).
We began with evaluation of a small library of resin-bound peptide catalysts we had synthesized in a parallel fashion, as well as a diverse split-and-pool library for the oxidation of 1. (The data from initial screen is in the Supplementary Information.) Notably, both the parallel synthesis library and many members of the unbiased split-pool library displayed some selectivity, mostly toward the 2,3-position of 1 (Table 1, selected results). These observations were auspicious, given that many catalysts exhibited significant differences from the product distribution observed with catalytic propionic acid (5:4:3 = 2.6:2.2:1.0; Fig. 1b). Catalyst 6 displayed the most significant deviation from the distribution afforded by propionic acid (5:4:3 = 1.0:1.0:3.9; Table 1, entry 1).
Table 1. Results from on-bead screening.
Reactions for 2,3-selective catalysts were run in triplicate from trials run at the same time with the same batch of reagents. Conversions and monoepoxide ratios are estimated by GC. See Supplementary Information for details.
| ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | i | i+1 | i+2 | i+3 | i+4 | i+5 | Catalyst | Approx. conv. | 10,11- (5) | 6,7- (4) | 2,3- (3) | Std. dev. | Library | |
| 1 | Boc-Asp | Pro | D-Thr(OtBu) | Asn(Trt) | Leu | – | 6 | 13% | 1.0 | 1.0 | 3.9 | 0.1 | Initial screening | |
| 2 | Boc-Asp | Pro | Asn(Trt) | D-Phe | Thr(OBn) | – | 7 | 13% | 1.3 | 1.0 | 8.2 | 0.1 | Directed library #1 | |
| 3 | Boc-Asp | Pro | Asn(Trt) | D-Phe | Thr(OBn) | Asn(Trt) | 8a | 15% | 1.2 | 1.0 | 18.0 | 0.5 |
|
Directed library #2 |
| 4 | Boc-Asp | Pro | Asn(Trt) | D-Phe | Pro | Asn(Trt) | 9a | 12% | 1.2 | 1.0 | 14.6 | 0.7 | ||
| 5 | Boc-Asp | Pro | Asn(Trt) | D-Phe | – | – | 10 | 14% | 1.3 | 1.0 | 5.0 | 0.0 | ||
| 6 | Boc-Asp | D-Pro | Thr(OBn) | Leu | – | – | 11 | 14% | 1.5 | 1.9 | 1.0 | 0.0 | Truncated directed library #2 | |
| 7 | Boc-Asp | D-Pro | Thr(OBn) | Asn(Trt) | Tyr(OtBu) | – | 12a | 16% | 1.5 | 2.9 | 1.0 | 0.0 | Directed library #3 | |
Variations of selective peptide catalysts:
To enhance selectivity for 3, a small one-bead-one-compound (OBOC) combinatorial library was synthesized and biased toward peptide 6, with four variable positions adjacent to the catalytic aspartic acid (Fig. 2a). Biasing of the library was accomplished as follows: at each variable position, approximately 50% of the functionalized polystyrene beads were coupled to the parent residue (corresponding to catalyst 6) at each position, while the remaining 50% of the beads were evenly distributed for coupling to either seven different residues (positions i+1 and i+2) or six different residues (i+3 and i+4 positions; Fig. 2). The resulting library possesses a theoretical size of about 3,000 unique peptide sequences (n = 7×7×8×8; see Supplementary Information). Upon completion, the theoretical distribution of peptides in this library follows the histogram shown in Fig. 2b: 6% of beads contain the parent sequence (6), 25% contain a single substitution, 38% contain two substitutions, etc.
Figure 2. Catalyst library design by split and pool synthesis via the one-bead-one-compound library method.

a, Design of first directed library. b, Histogram of theoretical library composition.
Approximately 228 beads from this library were tested, including catalyst 7, which favored epoxide 3 with approximate doubling of selectivity (5:4:3 = 1.3:1.0:8.2, Table 1, entry 2). This result represented a further departure of the performance of a peptide-based catalyst from the control catalyst 2. This data stimulated the evaluation of a second biased library.
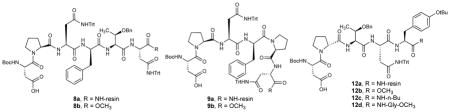
For the new OBOC library, now biased toward 7, we fixed the i+1 residue as Pro and i+2 position as Asn(Trt), hypothesizing that each was important for selectivity. The identities of three adjacent residues (i+3, i+4 and i+5) were then varied. Biased library 2 (Table 1, entries 3 and 4) produced catalysts 8a and 9a, which exhibited even greater site-selectivity, again nearly doubling the preference for 3. Catalyst 8a demonstrated a 5:4:3 ratio of 1.2:1.0:18.0. Catalyst 9a produced a ratio of 1.2:1.0:14.6. In contrast, truncated analog 10 exhibits reduced selectivity of 1.3:1.0:5.0 (entry 5).
We then assessed the performance of the catalysts upon resynthesis and evaluation in solution, and under synthetically meaningful conditions (e.g., 1.0 theoretical equiv oxidant, controlled by stoichiometry of DIC). Two points are of note. First, as noted below, we observe that catalysts provide better results when they are dissolved in solution, as opposed to when they are immobilized on beads. Second, and perhaps of greater significance, was our observation of significant enantioselectivity in these experiments. We had hypothesized that catalyst-dependent regioselectivity might also be coupled to the observation of enantioselectivity for a given monoepoxide isomer. The notion that the same intrinsic transition state organization associated with a specific regioselectivity might also translate into the organization required for enantioselectivity finds analogy in our previous rate-based selections for catalysts that also turned out to be enantioselective29. With these considerations in mind, we resynthesized 8a and 9a as their corresponding C-terminal methyl esters, 8b and 9b. When examined under conditions designed to lead to full conversion to monoepoxide (1.0 theoretical equiv oxidant: 1.0 equiv DIC, 2.0 equiv H2O2), methyl esters 8b and 9b both provide excellent overall selectivity. Catalyst 8b delivers 3 with essentially total regioselectivity, and in 82% ee (Table 2, entry 1a). Catalyst 9b also exhibits high regioselectivity, with 3 showing a slightly improved 86% ee (entry 1b). Strikingly, these peptide-based catalysts exhibit site- and enantioselectivities that compare favorably to the Sharpless catalyst for oxidation of 1 (Equation 1, above).
Table 2.
Selectivity of peptide 9b with different linear terpenoid alcohols.

| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | Catalyst | Site-selectivity | Yield | ee | Producta | ||
| 10,11- | 6,7- | 2,3- | ||||||
| 1a |
|
8b | 1.0 | 1.0 | >100 | n.d | 82% |
|
| 1b | 9b | 1.0 | 1.0 | >100 | 81% | 86% | ||
| 2 |
|
9b | 1.0 | >100 | 80% | 87% |
|
|
| 3 |
|
9b | 1.0 | >100 | 79% | 93% |
|
|
| 4 |
|
9b | 75%b | 92% |
|
|||
See supplementary information for details of absolute configuration assignment.
GC yield.
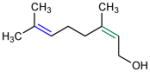
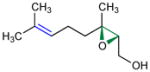
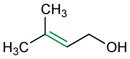
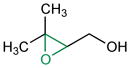
Given the analogy, we also evaluated catalyst 9b for asymmetric epoxidation of other terpenes to assess its generality toward allylic alcohols. Peptide 9b maintains excellent site-selectivity for geraniol (Table 2, entry 2; >100:1 site-selectivity, 87% ee). Nerol shows somewhat better enantioselectivity (entry 3, 93% ee) while retaining site-selectivity for the 2,3-cis-allylic position. Prenol also gives a favorable result, with the corresponding epoxide formed with 92% ee (entry 4). Catalyst 9b shows no selectivity for the 2,3-olefin of farnesyl methyl ether, supporting a hydroxyl directed mechanism.
Initial evaluation of the libraries also revealed catalyst 11, which exhibited a surprising, albeit modest, selectivity for the 6,7-position of 1, to favor 4 (Table 1, entry 6; 5:4:3 = 1.5:1.9:1.0). Catalyst 11, with D-Pro in the i+1 position, was unique in that we had not observed even this modest level of alternate regioselectivity previously. We therefore prepared a new library, biased toward the structure of 11, wherein the i+1 D-Pro was fixed and the adjacent three residues were varied. Therein, we found catalyst 12a, which displayed improved selectivity for 4 (Table 1, entry 7; 5:4:3 = 1.5:2.9:1.0). When resynthesized as the C-terminal methyl ester 12b, and evaluated in CHCl3, this catalyst yielded a product distribution of up to 1.0:4.1:1.3 (5:4:3). We then evaluated a second-generation library deliberately targeting 6,7-selective catalysts. Intriguingly, this study did not reveal catalysts that exhibited dramatically improved selectivity for 4, although several comparable catalysts were present (see Supplementary Table S1). The structures of the peptides unveiled by this library prompted us to prepare analogs of catalyst 12 in a hypothesis-driven manner. We projected that a C-terminal amide might better resemble the catalysts in their on-bead screening form (wherein peptides are linked to the resin as the amide). Catalysts 12c (with C-terminal n-Bu amide) and 12d (with C-terminal Gly-OMe) were examined. While both catalysts favored production of epoxide 4, catalyst 12d exhibited greater selectivity (5:4:3 = 1.2:8.0:1.0) under optimized conditions (Fig. 3a). Furthermore, this product ratio simplifies an otherwise difficult separation of products by chromatography, allowing isolation of 4 in 43% yield as a >94:6 mixture of 4:5 isomers.
Figure 3. Results and raw data for the site-selective oxidation of farnesol and geranylgeraniol with m-CPBA, 2,3-selective catalyst 9b and 6,7-selective catalyst 12d.

a, Optimized oxidation conditions with 12d. Comparison of product ratios and GC spectra from crude reaction mixtures with m-CPBA, catalyst 9b, and catalyst 12d. Relevant area of GC spectra with, b, farnesol and, c, geranylgeraniol. aPeak has a shoulder that is integrated, overestimating the area. Conditions for m-CPBA reactions found in Fig. 1b; for reactions with catalyst 9b, in Table 2; for reactions with catalyst 12d, in a.
The appreciable, catalyst-dependent selectivity for the 6,7-position of farnesol is quite unusual. As in the case of the 2,3-selective catalysts, experiments with farnesyl methyl ether exhibited little to no selectivity (data not shown). Thus, the observed 6,7-selectivity may be largely the result of a hydroxyl-directed mechanism, despite the distance of the hydroxyl, six bonds away from the reacting olefin. Yet, unlike the results obtained with the best 2,3-selective catalysts, the production of 4 through the action of catalyst 12d occurs with only modest, but measurable 10% ee. The observation that both high site-selectivity and enantioselectivity can track together qualitatively with 2,3-selective catalysts, but do not appear as tightly coupled with 6,7-selective catalysts is intriguing. This may be due to the higher number of rotatable bonds between the directing hydroxyl group and the preferred site of oxidation. We have observed, in our screening studies (in correspondingly unoptimized experiments), catalysts that give somewhat lower site-selectivity (Supplementary Table S2, 5:4:3, ~1:3–4:1), but slightly higher enantioselectivity (~20–30% ee). Yet, our data sets are not large, and this concept requires further study for a full assessment. Even so, the observed 6,7-site-selectivity exhibited by catalyst 12d greatly contrasts with the product distribution induced by m-CPBA, and any other reported catalyst, to our knowledge (Fig. 3b).
That catalyst 12d shows a preference for the 6,7-position of 1 stimulated its assessment for oxidation of a more complex polyene, geranylgernaniol (13), which presents four competing olefins for functionalization. Fig. 3 shows the raw data for the oxidation of 1 (Fig. 3b) and 13 (Fig. 3c) with m-CPBA and with the catalysts described above. With geranylgernaniol (13; Fig. 3c), the results are quite striking. With m-CPBA, the four monoxides are formed essentially without control. Catalyst 9b, as with the oxidation of 1, exhibits high regioselectivity to give the 2,3-monooxide of geranylgeraniol (14). Catalyst 12d again exhibits striking site-selectivity for an internal alkene, which we have assigned as the 6,7-monooxide 15. The monoepoxide 14, as well as the other monoepoxides (i.e., the 10,11- and 14,15-epoxides; see Supplementary Information) are also produced in the reaction, but as minor products. The catalytic, site-selective epoxidation of geranylgeraniol (or farnesol), at a position other than the 2,3-olefin, has not previously been reported to our knowledge. The challenge of the task is manifest in the nearly equivalent reactivity of the 14,15-, 10,11- and 6,7-olefins. Thus, the observations shown in Figure 3 may represent an important step forward, enabled by the iterative peptide-based catalyst screening approach.
Conclusions
In summary, we have discovered different peptide catalysts capable of furnishing different isomers of epoxyfarnesol and related compounds that are minor products of generic peracid-based olefin epoxidation. Peptide 9b appears to operate via a hydroxyl-directing mechanism, analogous to Sharpless’ asymmetric epoxidation and with comparable selectivity. Peptide 12d, provides unprecedented selectivity for a catalyst for the internal olefin of farnesol, with substantial site-selectivity, but modest enantioselectivity. Discovery of catalysts for complex selectivity situations, wherein myriad possible products may be formed as a function of a catalytic mechanism of bond formation, is a state-of-the-art challenge for asymmetric catalysis endeavors. The application of diversity-based approaches, such as that described above, may prove fruitful, and may also offer some analogy to the directed evolution strategies employed by natural and engineered enzymatic systems.
Methods
General procedure for epoxidation of allylic alcohols with peptide 9b
To a test tube with stir bar and Teflon-lined screw cap was added peptide 9b (0.1 equiv); allylic alcohol (1.0 equiv.); a freshly prepared/sonicated solution containing HOBt•H2O (0.1 equiv.) and DMAP (0.1 equiv.) in DCM (to a concentration of 0.2 M of substrate); and 30% aqueous H2O2 (2.0 equiv.). The test tube was placed in ice (if running reaction below room temperature) and allowed to chill before adding DIC (1.0 equiv.). The reaction tube was then sealed with a screw-cap under ambient conditions (without exclusion of air) and brought to a cold room (4 °C) where the reaction was stirred vigorously. After 7 h following addition of DIC, the reaction was quenched with a saturated aqueous solution of Na2SO3 and stirred for a few moments, sitting in ice, before allowing to warm to room temperature. Saturated aqueous NaHCO3 and hexane or EtOAc were added, the mixture was vortexed, allowed to settle, and then the organic layer was removed, followed by two to three additional hexane or EtOAc extracts, peformed similarly. If site-selectivity was to be determined, an aliquot of the combined organics was removed, diluted with additional hexanes and analyzed by GC. The GC sample was returned to the organics, which were then concentrated in vacuo or under a stream of N2, and purified by flash silica gel column chromatography.
General procedure for epoxidation of 6,7-olefin in polyisoprenols with peptide 12d
To a test tube with stir bar and Teflon-lined screw cap was added peptide 12d (0.1 equiv); substrate (1.0 equiv.); about two thirds of a freshly prepared/sonicated solution containing HOBt•H2O (0.1 equiv.) and DMAP (0.1 equiv.) in CHCl3 (the CHCl3 was passed through basic alumina prior to mixing with DMAP/HOBt); and 30% aqueous H2O2 (2.0 equiv.). The test tube was placed in ice and allowed to chill before adding DIC (1.0 equiv.) followed by the remaining CHCl3 solution (to a concentration of 0.2 M of substrate), rinsing down the sides of the reaction tube. The reaction tube was then sealed with a screw-cap under ambient conditions without exclusion of air and placed in an iPrOH bath maintained at −12 °C to −18 °C by cryostat. The reaction was then stirred vigorously. 25–48 h following addition of DIC, the reaction was quenched with a saturated aqueous solution of Na2SO3 and stirred for a few moments, still at low temperature, before allowing to warm to room temperature. Saturated aqueous NaHCO3 and EtOAc were then added and the mixture was vortexed, allowed to settle, and then the organic layer was removed, along with three additional EtOAc extracts, peformed similarly. An aliquot of the combined organics was removed, diluted with additional EtOAc, and analyzed by GC. The organics were concentrated in vacuo and then purified by flash column chromatography.
Additional experimental procedures, descriptions of compounds, and analytical methods may be found in the Supplementary Information.
Supplementary Material
Acknowledgments
This work is supported by National Institutes of Health (R01-GM096403) to SJM. P.A.L. was partially supported by NIH CBI-TG-GM-067543. P.A.L. thanks Brandon Fowler for collaborating to build the library resulting in catalyst 2 and thanks Seth Alexander and the Schepartz laboratory for assistance.
Footnotes
Author contributions P.A.L. designed and performed experiments. S.J.M. oversaw the project. Both authors analysed data and wrote manuscript.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
References
- 1.van Tamelen EE, Heys JR. Enzymic epoxidation of squalene variants. J Am Chem Soc. 1974;97:1252–1253. doi: 10.1021/ja00838a054. [DOI] [PubMed] [Google Scholar]
- 2.Katsuki T, Martin VS. Asymmetric epoxidation of allylic alcohols: The Katsuki-Sharpless epoxidation reaction. Org React. 1996;48:1–299. [Google Scholar]
- 3.Zhang W, Basak A, Kosugi Y, Hoshino Y, Yamamoto H. Enantioselective epoxidation of allylic alcohols by a chiral complex of vanadium: an effective controller system and a rational mechanistic model. Angew Chem Int Ed. 2005;44:4389–4391. doi: 10.1002/anie.200500938. [DOI] [PubMed] [Google Scholar]
- 4.Malkov AV, Czemerys L, Malyshev DA. Vanadium-catalyzed asymmetric epoxidation of allylic alcohols in water. J Org Chem. 2009;74:3350–3355. doi: 10.1021/jo900294h. [DOI] [PubMed] [Google Scholar]
- 5.Egami H, Oguma T, Katsuki T. Oxidation catalysis of Nb(salan) complexes: asymmetric epoxidation of allylic alcohols using aqueous hydrogen peroxide. J Am Chem Soc. 2010;132:5886–5895. doi: 10.1021/ja100795k. [DOI] [PubMed] [Google Scholar]
- 6.Barlan AU, Basak A, Yamamoto H. Enantioselective oxidation of olefins catalyzed by a chiral bishydroxamic acid complex of molybdenum. Angew Chem Int Ed. 2006;45:5849–5852. doi: 10.1002/anie.200601742. [DOI] [PubMed] [Google Scholar]
- 7.Corey EJ, Zhang J. Highly effective transition structure designed catalyst for the enantio- and position-selective dihydroxylation of polyisoprenoids. Org Lett. 2001;3:3211–3214. doi: 10.1021/ol016577i. [DOI] [PubMed] [Google Scholar]
- 8.Chang S, Lee NH, Jacobsen EN. Regio- and enantioselective catalytic epoxidation of conjugated polyenes. Formal synthesis of LTA4 methyl ester. J Org Chem. 1993;58:6939–6941. [Google Scholar]
- 9.Burke CP, Shi Y. Regio- and enantioselective epoxidation of dienes by a chiral dioxirane: synthesis of optically active vinyl cis-epoxides. Angew Chem Int Ed. 2006;45:4475–44478. doi: 10.1002/anie.200600840. [DOI] [PubMed] [Google Scholar]
- 10.Breslow R, Maresca LM. Template-directed epoxidation of farnesol and geranylgeraniol as conformational probes. Tetrahedron Lett. 1978;10:887–890. [Google Scholar]
- 11.Saito I, Mano T, Nagata R, Matsuura T. Inter- and intramolecular epoxidation utilizing silyl-protected peroxy esters and copper salt. Tetrahedron Lett. 1987;28:1909–1912. [Google Scholar]
- 12.Gnanadesikan V, Corey EJ. A strategy for position-selective epoxidation of polyprenols. J Am Chem Soc. 2008;130:8089–8093. doi: 10.1021/ja801899v. [DOI] [PubMed] [Google Scholar]
- 13.Colby Davie EA, Mennen SM, Xu Y, Miller SJ. Asymmetric catalysis mediated by synthetic peptides. Chem Rev. 2007;107:5759–5812. doi: 10.1021/cr068377w. [DOI] [PubMed] [Google Scholar]
- 14.Wennemers H. Asymmetric catalysis with peptides. Chem Comm. 2011;47:12036–12041. doi: 10.1039/c1cc15237h. [DOI] [PubMed] [Google Scholar]
- 15.Francis MB, Jamison TF, Jacobsen EN. Combinatorial libraries of transition-metal complexes, catalysts and materials. Curr Opin Chem Biol. 1998;2:422–428. doi: 10.1016/s1367-5931(98)80019-7. [DOI] [PubMed] [Google Scholar]
- 16.Kuntz KW, Snapper ML, Hoveyda AH. Combinatorial catalyst discovery. Curr Opin Chem Biol. 1999;3:313–319. doi: 10.1016/S1367-5931(99)80048-9. [DOI] [PubMed] [Google Scholar]
- 17.Peris G, Jakobsche CE, Miller SJ. Aspartate-catalyzed asymmetric epoxidation reactions. J Am Chem Soc. 2007;129:8710–8711. doi: 10.1021/ja073055a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolundzic F, Noshi MN, Tjandra M, Movassaghi M, Miller SJ. Chemoselective and enantioselective oxidation of indoles employing aspartyl peptide catalysts. J Am Chem Soc. 2011;133:9104–9111. doi: 10.1021/ja202706g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thibodeaux CJ, Chang WC, Liu HW. Enzymatic chemistry of Cyclopropane, Epoxide and Aziridine Biosynthesis. Chem Rev. 2012;112:1681–1709. doi: 10.1021/cr200073d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharpless KB. Searching for new reactivity (Nobel Lecture) Angew Chem Int Ed. 2002;41:2024–2032. [PubMed] [Google Scholar]
- 21.Kotaki T, Shinada T, Kaihara K. Structure determination of a new juvenile hormone from a Heteropteran insect. Org Lett. 2009;11:5234–5237. doi: 10.1021/ol902161x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koohang A, et al. Enantioselective inhibition of squalene synthase by aziridine analogues of presqualene diphosphate. J Org Chem. 2010;75:4769–4777. doi: 10.1021/jo100718z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanuwidjaja J, Ng SS, Jamison TF. Total synthesis of ent-dioxepandehydrothyrsiferol via a bromonium-initiated epoxide-opening cascade. J Am Chem Soc. 2009;131:12084–12085. doi: 10.1021/ja9052366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uyanik M, Ishibashi H, Ishihara K, Yamamoto H. Biomimetic synthesis of acid-sensitive (−)-caparrapi oxide and (+)-8-epicaparrapi oxide induced by artificial cyclases. Org Lett. 2005;7:1601–1604. doi: 10.1021/ol050295r. [DOI] [PubMed] [Google Scholar]
- 25.Marshall JA, Hann RK. A cascade cyclization route to adjacent bistetrahydrofurans from chiral triepoxyfarnesyl bromides. J Org Chem. 2008;73:6753–6757. doi: 10.1021/jo801188w. [DOI] [PubMed] [Google Scholar]
- 26.Dittmer DC, et al. A tellurium transposition route to allylic alcohols: overcoming some limitations of the Sharpless-Katsuki asymmetric epoxidation. J Org Chem. 1993;58:718–731. [Google Scholar]
- 27.Lichtor PA, Miller SJ. One-bead-one-catalyst approach to aspartic acid-based oxidation catalyst discovery. ACS Comb Sci. 2011;13:321–326. doi: 10.1021/co200010v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lam KS, Lebl M, Krchnák V. The “one-bead-one-compound” combinatorial library method. Chem Rev. 1997;97:411–448. doi: 10.1021/cr9600114. [DOI] [PubMed] [Google Scholar]
- 30.Furka A, Sebestyen F, Asgedom M, Dibo G. General method for rapid synthesis of multicomponent peptide mixtures. Int J Pept Protein Res. 1991;37:487–493. doi: 10.1111/j.1399-3011.1991.tb00765.x. [DOI] [PubMed] [Google Scholar]
- 31.Lam KS, et al. A new type of synthetic peptide library for identifying ligand-binding activity. Nature. 1991;354:82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 32.Singh J, et al. Application of genetic algorithms to combinatorial synthesis: a computational approach to lead identification and lead optimization. J Am Chem Soc. 1996;118:1669–1676. [Google Scholar]
- 33.Reetz MT. Laboratory evolution of stereoselective enzymes: a prolific source of catalysts for asymmetric reactions. Angew Chem Int Ed. 2011;50:138–174. doi: 10.1002/anie.201000826. [DOI] [PubMed] [Google Scholar]
- 34.Brustad EM, Arnold FH. Optimizing non-natural protein function with directed evolution. Curr Opin Chem Biol. 2011;15:201–210. doi: 10.1016/j.cbpa.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Copeland GT, Miller SJ. Selection of enantioselective acyl transfer catalysts from a pooled peptide library through a fluorescence-based activity assay: an approach to kinetic resolution of secondary alcohols of broad substrate scope. J Am Chem Soc. 2001;123:6496–6502. doi: 10.1021/ja0108584. [DOI] [PubMed] [Google Scholar]
- 36.Schreiber SL, Schreiber TS, Smith DB. Reactions that proceed with a combination of enantiotopic group and diastereotopic face selectivity can deliver products with very high enantiomeric excess: experimental support of a mathematical model. J Am Chem Soc. 1987;109:1525–1529. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.