

Abstract
Safe and effective vaccines are crucial for maintaining public health and reducing the global burden of infectious disease. Here we introduce a new vaccine platform that uses hydrogen peroxide (H2O2) to inactivate viruses for vaccine production. H2O2 rapidly inactivates both RNA and DNA viruses with minimal damage to antigenic structure or immunogenicity and is a highly effective method when compared with conventional vaccine inactivation approaches such as formaldehyde or β-propiolactone. Mice immunized with H2O2-inactivated lymphocytic choriomeningitis virus (LCMV) generated cytolytic, multifunctional virus-specific CD8+ T cells that conferred protection against chronic LCMV infection. Likewise, mice vaccinated with H2O2-inactivated vaccinia virus or H2O2-inactivated West Nile virus showed high virus-specific neutralizing antibody titers and were fully protected against lethal challenge. Together, these studies demonstrate that H2O2-based vaccines are highly immunogenic, provide protection against a range of viral pathogens in mice and represent a promising new approach to future vaccine development.
Routine vaccination represents one of the most effective approaches to safeguard public health against infectious disease. All currently licensed antiviral vaccines fall into one of two broad categories: replicating, live, attenuated vaccines and noninfectious whole or subunit vaccines. Formaldehyde, the most common reagent used for vaccine production, was first identified by serendipity in the 1920s as a chemical means for inactivation of bacterial toxins1. β-propiolactone (BPL, first described in 1955 (ref. 2)), is second only to formaldehyde as the most commonly used inactivation method for vaccine development. Despite the routine use of formaldehyde, vaccinologists have known for decades that it is a cross-linking agent that can damage key antigenic epitopes, leading to reduced immunogenicity or even exacerbated disease under certain circumstances3. One example of vaccine-induced exacerbation of disease is the case of the formaldehyde-inactivated respiratory syncytial virus (RSV) vaccine developed in the 1960s4. Though the vaccine was well tolerated, severe complications arose following exposure to wild-type RSV, leading to 16 hospitalizations and the deaths of two children4. Recent studies indicate that formaldehyde destroys key neutralizing epitopes, resulting in exacerbated disease following wild-type challenge5. Similarly, clinical trials involving formaldehyde-inactivated measles vaccine failed to protect against wild-type infection and instead led to an atypical form of the disease6. This was also associated with an inadequate antiviral antibody response, linked to formaldehyde-induced alteration of the measles hemolysin (F protein)6. Inactivation of viruses with BPL may also trigger adverse immune reactions, including the induction of allergic responses through chemical modifications of vaccine components7,8. It is unclear whether this was a factor in a recent phase 1 clinical trial in which one of 20 subjects developed urticaria shortly after booster vaccination with a 4.8-μg dose of BPL-inactivated yellow fever vaccine9. Bearing these concerns in mind, there is clearly an unmet need for identifying new and improved strategies for preparing inactivated vaccines.
H2O2 is an oxidizing agent that is well established as a potent antimicrobial agent and antiseptic10. The belief that strong oxidizing agents irreversibly damage the basic molecular structure of proteins may be one reason why H2O2 has not previously been tested as a means for producing inactivated viral vaccines11. However, inactivation of microbes with H2O2 (as well as other oxidizing agents such as superoxide and nitric oxide) represents a key element of the innate mammalian immune system and functions in endosomal compartments to inactivate intracellular pathogens12. In addition, H2O2 has also been used to detoxify pertussis toxin13. In these current studies we have used three unrelated virus model systems to show that H2O2-based vaccines can protect against chronic or lethal viral infection, and we believe that this approach represents a new concept in vaccine development.
RESULTS
H2O2 inactivates pathogens while maintaining antigenicity
To determine the feasibility of H2O2-based vaccine development, we first examined the inactivating potential of H2O2 against a spectrum of viral pathogens. A 3% aqueous solution of H2O2 inactivated both RNA and DNA viruses with up to a 6-log10 reduction in titer observed in less than 2 h (Fig. 1a). One mechanism for virus inactivation is through genomic damage caused by hydroxyl radicals that attack carbon double bonds in the nucleosides or abstract hydrogen atoms, both of which are processes that lead to carbon radicals with the potential for further downstream oxidation, and this ultimately results in single- or double-strand breaks that destroy viability14. For the viruses described here, H2O2-based inactivation followed first-order rate kinetics, with half-life decay rates of <4 min. This is in contrast to formaldehyde, with which complete virus inactivation often requires 2–3 weeks of treatment15–17.
Figure 1.

H2O2 inactivates viruses without substantial damage to antigenic epitopes. (a) H2O2 was used to inactivate a group of viruses including YFV, WNV, LCMV, VV and monkeypox virus (MPV). After 2 h of incubation with or without H2O2, infectious virus was measured by plaque assay. The limit of detection is indicated by the dashed line. (b,c) Purified YFV was treated with H2O2, β-propiolactone, or formaldehyde (Form.) for 2 h (b) or 16 h (c) at 20–24 °C and compared to untreated, live virus for retained antigenicity. ELISA plates were coated with each purified virus preparation, and screened with convalescent serum from YFV-infected mice (n = 3). ELISA results were normalized to the levels observed when live virus was used as antigen (that is, percentage of live virus signal), and error bars represent s.d.
To broadly assess the consequences of H2O2 inactivation on antigenicity, we compared yellow fever virus (YFV) inactivated by H2O2 to YFV inactivated by formaldehyde or BPL (Fig. 1b,c). Samples of purified virus that underwent these three different forms of inactivation were coated onto ELISA plates and we used YFV-immune serum from mice infected with YFV-17D (220 d after infection), to probe the samples and determine end-point antibody titers. We chose YFV-immune serum to assess the reactivity of antibodies to native YFV that would be generated during live viral infection. After 2 h of treatment (Fig. 1b), virus inactivation by H2O2 had only a minor effect on antigenicity and maintained 87–98% of the maximum antibody binding response observed with live virus. This is in contrast to the effects observed with BPL and formaldehyde, which significantly reduced the antigenicity of YFV at 3.0% and 1.0% concentrations (Fig. 1b). At 0.1%, H2O2 still performed significantly better than formaldehyde (P = 0.005), whereas 0.1% BPL was not significantly different from 0.1% H2O2 (P = 0.30). In practice, however, both BPL18 and formaldehyde15–17 are used for more extended periods of time to provide complete virus inactivation. Therefore, we exposed samples to H2O2, BPL, or formaldehyde at concentrations of 3.0%, 1.0% or 0.10% for 16 h before determining viral antigenicity. Similar to the results observed after 2 h of exposure, H2O2 maintained significantly greater antibody binding compared to formaldehyde or BPL (Fig. 1c). We observed analogous results showing retained antigenicity in preliminary experiments in which we used YFV-immune serum from YFV-17D–infected human subjects or YFV-17D–infected rhesus macaques (data not shown). Although this ELISA approach does not distinguish between neutralizing and non-neutralizing epitopes, these studies indicate that H2O2-based inactivation of a virus is less damaging to native antibody binding sites than other currently used approaches to virus inactivation such as BPL or formaldehyde.
H2O2-LCMV vaccination induces CD8+ T cell–mediated immunity
One concern with inactivated vaccines is that many are suboptimal at inducing protective CD8+ T cell responses19. To determine whether the H2O2-based vaccine approach is capable of generating effective antiviral CD8+ T cell responses, we chose the LCMV model, wherein protective immunity is mediated by CD8+ T cells20. We vaccinated mice with purified H2O2-inactivated LCMV-Armstrong and compared them to mice acutely infected with LCMV-Armstrong (Fig. 2). At 8 d after vaccination, the frequency of LCMV-specific CD8+ T cells reached about 1–2% of the total splenic CD8+ T cell response (Fig. 2a), similar to what is observed following live vaccination with recombinant vaccinia virus (VV) expressing LCMV nucleoprotein (NP)21 or glycoprotein (GP)22. Comparable levels of HLA-A*0201-restricted CD8+ T cell immunity have been observed in HLA-A*0201-transgenic mice vaccinated with H2O2-inactivated West Nile virus (WNV) (M.S. Diamond (Washington University– St. Louis), personal communication), indicating these results are not unique to the H-2Ld–restricted NP118 epitope or to the LCMV model system. At 8 d after LCMV-Armstrong infection, 40–50% of splenic CD8+ T cells were specific for LCMV NP118 and expressed interferon-γ (IFN-γ) after 6 h stimulation with peptide directly ex vivo (Fig. 2a). However, CD8+ T cells generated by live viral infection were significantly less efficient at co-expressing tumor necrosis factor-α (TNF-α) when compared to T cells generated by H2O2-LCMV vaccination (62 ± 9% IFN-γ+TNF-α+CD8+ T cells versus 89 ± 5% IFN-γ+TNF-α+CD8+ T cells, respectively; P = 0.0002). Notably, there was also more than a tenfold difference in the ability to produce interleukin-2 (IL-2) after peptide stimulation, with 51 ± 5% of IFN-γ+CD8+ T cells from H2O2-LCMV–vaccinated mice co-expressing IL-2 compared to only 3 ± 1% of virus-specific T cells from LCMV-Armstrong–infected mice (P = 0.0001, Fig. 2a).
Figure 2.

Vaccination with H2O2-LCMV induces multifunctional, cytolytic CD8+ T cells that protect against chronic virus infection. (a) BALB/c mice were infected with LCMV-Armstrong or vaccinated with H2O2-LCMV (n = 6 mice per group from three experiments). At 8 d after infection or vaccination, CD8+ T cells were stimulated with NP118 peptide and analyzed by flow cytometry for IFN-γ, TNF-α, and IL-2 production. Numbers in the top quadrants of the dot plots show the percentage of T cells producing the indicated cytokines after background subtraction, and the numbers in parentheses represent the percentage of T cells that are IFN-γ+TNF-α+ or IFN-γ+IL-2+. (b) The percentage of LCMV-specific IFN-γ+CD8+ T cell responses (top right quadrant), as measured at 28 d after primary infection or H2O2-LCMV vaccination (top) or 4 d after challenge (bottom) with LCMV-Armstrong (n = 3 or 4 mice per group). (c) Granzyme B expression assessed directly ex vivo in NP118-tetramer+CD8+ T cells from representative LCMV-Armstrong infected mice (live) or H2O2-LCMV–vaccinated mice (H2O2) at 28 d after infection or vaccination or at 4 d after challenge with LCMV-Armstrong (day 32). (d) In vivo lysis of peptide-coated targets at 8 d after H2O2-LCMV vaccination or LCMV-Armstrong infection, compared to naïve controls (n = 4 mice per group). Error bars represent s.d. (e) At 28 d after LCMV-Armstrong infection or at 7 d or 28 d after H2O2-LCMV vaccination, mice were challenged with LCMV clone 13, and viremia was monitored by plaque assay (n = 4 mice per group).
To examine the proliferative capacity of H2O2-LCMV vaccine-induced CD8+ T cells in vivo, we challenged mice with 2 × 105 plaque-forming units (PFU) of LCMV-Armstrong at 28 d after vaccination and determined virus-specific CD8+ T cell frequencies before challenge (that is, day 28) and 4 d after challenge (that is, day 32). H2O2-LCMV–vaccinated mice showed a >40-fold increase in LCMV NP118–specific T cells within 4 d after challenge, reaching frequencies of 22 ± 6% of the CD8+ T cell compartment (Fig. 2b). This expansion eclipsed the CD8+ T cell responses observed at this early time point in naïve mice during primary infection (0.11 ± 0.04%, P = 0.0002) and was higher than that observed in LCMV-immune mice that were challenged in parallel (10 ± 2%, P = 0.005). Although LCMV-specific memory T cells expressed little or no granzyme B at 28 d after infection or vaccination, they rapidly upregulated granzyme B expression within 96 h after acute viral challenge (Fig. 2c). Of note, H2O2-LCMV vaccine–induced CD8+ T cells expressed higher amounts of granzyme B after secondary challenge than did LCMV-specific T cells elicited by prior acute viral infection (Fig. 2c). These results indicate that vaccine-induced CD8+ T cells rapidly upregulate cytolytic proteins, but it was unclear from these studies whether they have direct cytolytic activity in the absence of infection. To address this question, we used an in vivo cytotoxic T lymphocyte (CTL) assay to measure antiviral CTL activity at 8 d after H2O2-LCMV vaccination or infection. We observed robust major histocompatibility complex (MHC) class I–restricted, LCMV NP118–specific CTL responses in H2O2-LCMV vaccinated mice (Fig. 2d), indicating that viral infection was not required for the development of cytolytic activity in vivo.
To determine protective efficacy, we challenged H2O2-LCMV vaccinated mice with 2 × 106 PFU of LCMV clone 13. This viral variant of LCMV causes chronic infection in naïve mice that lasts for several weeks or months and provides a rigorous test for CD8+ T cell–mediated protection20 (Fig. 2e). As expected, mice that had previously cleared acute LCMV-Armstrong infection were protected from chronic LCMV clone 13 infection. Mice that were vaccinated once with H2O2-inactivated LCMV at either 7 d or 28 d before challenge were also protected against chronic infection (Fig. 2e). Similar to LCMV-Armstrong immune mice and mice vaccinated with recombinant Listeria monocytogenes expressing LCMV NP in previous studies23, H2O2-LCMV–vaccinated mice had reduced virus titers by >99% or had fully cleared LCMV within 7 d after challenge, whereas unvaccinated mice remained viremic for at least 35 d (Fig. 2e). In vivo depletion of CD8+ T cells before LCMV clone 13 challenge completely abrogated protection against chronic infection, indicating that vaccine-induced immunity was mediated by CD8+ T cells (data not shown). Together, these results demonstrate that multifunctional CD8+ T cells can be elicited with a H2O2-inactivated vaccine and provide protective immunity against chronic viral infection.
H2O2-based vaccines elicit neutralizing antibody responses
Neutralizing antibody responses are the hallmark of protective immunity for most licensed vaccines24,25, and the H2O2 vaccine platform is well suited for inducing neutralizing antibody responses because it does not seem to damage native viral epitopes to the extent observed with other methods of inactivation (Fig. 1b,c). However, the only method to formally determine immunogenicity is through in vivo vaccination studies using appropriate model systems in which neutralizing antibody responses have an important role in protection. The live, attenuated smallpox vaccine (consisting of VV) is often described as the ‘gold standard’ in vaccine efficacy, having achieved total eradication of its target virus, variola. Prior studies have demonstrated that VV-induced neutralizing antibodies are both necessary and sufficient for protection against lethal orthopoxvirus (for example, monkeypox) infection26. To determine how H2O2-based vaccines might compare in this model system, we measured neutralizing antibody responses against VV in mice vaccinated subcutaneously with either live VV, live modified vaccinia Ankara (MVA, which is replication deficient in humans and mice), or purified VV inactivated with either H2O2, formaldehyde or ultraviolet (UV) irradiation. At 28 d after booster vaccination, we determined antibody responses to VV. H2O2-VV immunization induced significantly higher virus-specific neutralizing antibody responses than vaccines produced by other inactivation approaches, and the response most closely mimicked the immunity induced by live viral infection (Fig. 3a). In addition to the subcutaneous administration experiment, we infected another group of mice with 1 × 106 PFU of VV by the intraperitoneal route, and they generated half-maximal plaque reduction neutralization (NT50) responses with a geometric mean value of 5,858, but this was not significantly different from the antibody responses elicited by H2O2-VV immunization (NT50 = 5,528, P = 0.99; data not shown). To determine whether H2O2-VV induced protective immunity, we challenged mice immunized with H2O2-VV with a lethal dose of VV, and 100% of the vaccinated animals survived (Fig. 3b). These studies indicate that H2O2-inactivated VV not only stimulates high titers of neutralizing antibodies (NT50 = 5,528), but also protects against a rigorous model of encephalitic disease.
Figure 3.
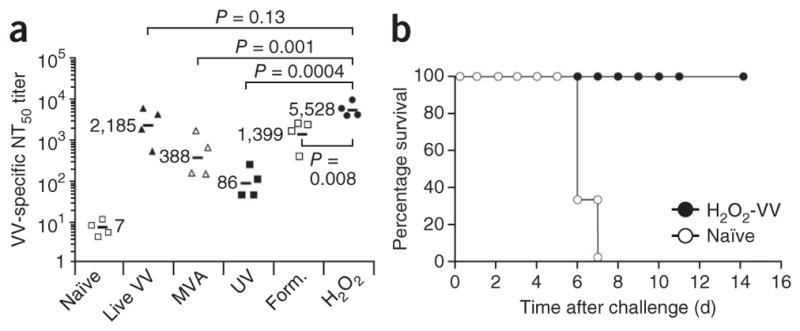
Induction of protective orthopoxvirus-specific neutralizing antibody responses following H2O2-VV immunization. (a) BALB/c mice (n = 4 per group) were vaccinated with live VV, MVA or purified VV inactivated with ultraviolet light (UV), 1% formaldehyde (Form.) for 2 h or 1% H2O2 for 2 h. Each group of mice was boosted 28 d later with the same vaccine preparation, and neutralizing antibody titers (NT50) were determined 28 d after secondary immunization. The black bars indicate group geometric mean NT50 titers with statistical comparisons determined using analysis of variance (ANOVA). (b) Naïve mice or mice that received primary H2O2-VV immunization 28 d previously (n = 6–8 mice per group) were challenged intranasally with 10 LD50 of VV.
Similar to the protective role of antibody against orthopoxvirus infection26, the key role of humoral immunity in controlling flavivirus infection is also well documented27,28. Although there is no WNV vaccine licensed for human use, there is an effective formaldehyde-inactivated horse vaccine, the Ft. Dodge Innovator29. For comparison, we immunized mice with H2O2-WNV or the Ft. Dodge Innovator and measured virus-specific antibody titers. Both groups mounted similar WNV-specific antibody responses after primary vaccination (Fig. 4a). After booster vaccination, H2O2-WNV–immunized mice showed antibody responses that were nearly tenfold higher than those induced in mice given the horse vaccine (Fig. 4a). Antiviral antibody titers peaked within 28 d after booster vaccination and then stabilized with no evidence of decline during the ensuing 6 months of observation (Fig. 4a). For a point of reference, we compared neutralizing anti-body levels in H2O2-WNV–immunized mice with those observed in human subjects who recovered from WNV infection (Fig. 4b). Within 2 months after booster vaccination (that is, the plateau phase of the response, Fig. 4a), H2O2-WNV–vaccinated mice developed neutralizing titers of 14,400 ± 2,442 by NT50. In comparison, human subjects who contracted WNV naturally had a geometric mean NT50 = 1,400 ± 1,115 when measured at ~1 year after infection. Although it is unknown what level of immunity is required for protection in humans, those who survive WNV infection are believed to be immune for life, and H2O2-WNV vaccination of mice elicits antibody responses that seem to meet or exceed the levels of immunity that develop after natural infection in humans.
Figure 4.
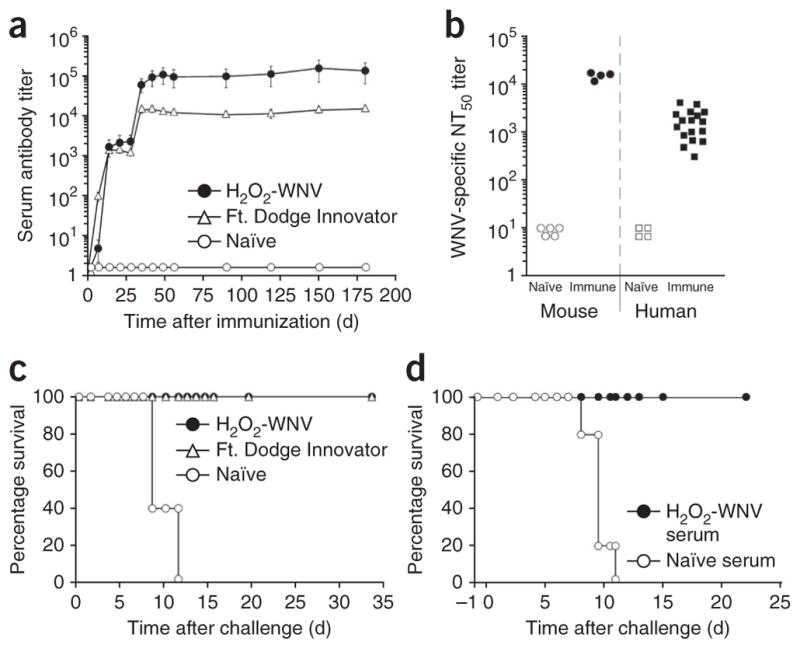
Vaccination with H2O2-WNV induces strong neutralizing antibody responses and protects against lethal WNV infection. (a) BALB/c mice (n = 4 mice per group) were vaccinated with H2O2-WNV or the Ft. Dodge WNV Innovator horse vaccine and boosted with the same vaccine at 28 d after primary vaccination. WNV-specific IgG responses ± the s.e.m. were determined by ELISA. (b) Virus-specific neutralizing antibody levels were determined for mice vaccinated with H2O2-WNV (2 months after booster vaccination) and compared to the levels observed after natural WNV infection in human subjects (~1 year after exposure). (c) Naïve mice and mice vaccinated with H2O2-WNV or Ft. Dodge WNV Innovator (75 d after booster vaccination) were challenged with 20 LD50 of WNV-NY99 (n = 4 mice per group). (d) Naïve mice received immune serum from H2O2-WNV-vaccinated mice or naïve serum from unvaccinated mice 1 d before challenge with 20 LD50 of WNV-NY99 (n = 3–5 mice per group).
To determine whether an H2O2-based vaccine can protect against lethal flavivirus infection, we challenged H2O2-WNV–immunized mice and age-matched naïve controls with 200 PFU of WNV-NY99, representing 20 times the half-maximal lethal dose (20 LD50) of WNV-NY99 by intraperitoneal injection (Fig. 4c). Similarly to C57BL/6 mice, naïve BALB/c mice showed weight loss starting at day 6 after WNV infection, with a mean survival time of 9.9 ± 1.6 d. All H2O2-WNV–vaccinated mice survived virulent WNV-NY99 challenge with no signs of disease or weight loss (Fig. 4c and data not shown). Unlike C57BL/6 mice, which lose consistent susceptibility to peripheral WNV challenge after ~12 weeks of age30, unvaccinated BALB/c mice remain susceptible to lethal WNV infection and thus provide an excellent model for determining long-term, vaccine-mediated immunity. Indeed, in this strain of mice we observed full protective immunity against WNV-NY99 challenge at >280 d after vaccination (data not shown), indicating the induction of long-term protective immunity by the H2O2-based vaccine platform. Both neutralizing antibody and CD8+ T cells are important for protection against WNV infection30. Therefore, to determine whether vaccine-induced neutralizing antibodies are sufficient to provide protection in this model, we transferred 300 μl of immune serum from H2O2-WNV–vaccinated mice (60–90 d after vaccination) into naïve recipient mice and challenged them with a lethal dose of WNV-NY99 (Fig. 4d). Serum was transferred by intraperitoneal injection, and for this reason the mice were challenged by the subcutaneous route 1 d later with 1,000 PFU (20 LD50 by subcutaneous injection). Mice that received the same amount of naïve serum before WNV challenge died or required euthanasia, whereas 100% of the mice that received WNV-specific immune serum from H2O2-WNV–vaccinated mice were protected and showed no signs of disease or weight loss (data not shown). Together, these results demonstrate that full protective immunity against a lethal WNV infection can be attained by vaccination with a H2O2-inactivated vaccine and that the protective immunity achieved by this approach can be mediated by neutralizing antibodies.
DISCUSSION
In these studies, we demonstrate that H2O2 is effective at inactivating viruses for vaccine production. Exposure to H2O2 resulted in minimal damage to viral epitopes and provided improved antigenicity and immunogenicity when compared to other standard approaches used for virus inactivation. Immunization with H2O2-inactivated viruses resulted in effective CD8+ T cell responses with enhanced antiviral function as well as strong neutralizing antibody responses that protected against lethal VV or WNV infection. Together, these results indicate that the use of H2O2 represents a new approach to preparing inactivated antiviral vaccines with improved potency and protective efficacy.
The ability of H2O2-inactivated LCMV vaccination to induce antiviral CD8+ T cell responses was particularly noteworthy. Within 8 d after vaccination, the frequency of LCMV-specific CD8+ T cells reached levels similar to those observed following infection with recombinant VV expressing LCMV NP21 or GP22. It is unclear whether MHC class I presentation is due to truncated or residual translation of viral proteins in the absence of productive replication or whether cross-presentation of virus particles is the underlying mechanism. Further mechanistic analysis is underway, but the results presented here indicate that the H2O2 platform induces cytolytic, multifunctional T cells (Fig. 2) and is similar to live recombinant vaccines at inducing protective antigen-specific CD8+ T cell responses.
The majority of licensed vaccines provide protective immunity via the induction of neutralizing antibodies24. Thus, any new approach to vaccine development will require the capacity to induce strong and effective antiviral antibody responses. To assess retained viral antigenicity, we examined antibody binding after 2–16 h exposure to varying concentrations of H2O2, formaldehyde or BPL. Inactivated hepatitis vaccines are produced using 0.025% formaldehyde at 37 °C for 15 d (ref. 15), and complete inactivation of Japanese encephalitis virus requires ~0.02% formaldehyde at 37 °C for several weeks17. We chose 0.1% formaldehyde because it is within the same general range of concentration used for these other vaccines (albeit under less harsh conditions than 37 °C for 2 or more weeks), and it matched the routine time and concentration of 0.1% BPL used for comparison. Moreover, 0.01% H2O2, BPL and formaldehyde were insufficient to provide complete virus inactivation within 16 h at 20–24 °C (data not shown) and were not examined further. To assess immunogenicity and protective efficacy, we compared H2O2-VV to the live, attenuated smallpox vaccine (that is, VV infection) and found that H2O2-inactivated VV immunization induced neutralizing antibodies that were comparable to those elicited by the live vaccine and protected mice against lethal VV infection. To further evaluate the ability of H2O2 to function as a vaccine platform, we examined immunity against WNV. Our results indicate that H2O2-WNV vaccination induces virus-specific neutralizing antibody responses that equal or exceed the levels observed following natural WNV infection and that full protective immunity against lethal challenge can be elicited by direct vaccination or by adoptive transfer of immune serum from H2O2-WNV–vaccinated mice. Induction of vaccine-mediated antiviral antibody responses that exceed those observed after live viral infection is not unprecedented. For example, virus-like particle-based human papillomavirus (HPV) vaccines induce HPV-specific antibody responses that are more than tenfold higher than those observed after natural HPV infection31. Our data showing H2O2-WNV vaccine–mediated protection, together with the studies performed with VV (Fig. 3), support the potential of H2O2-inactivated vaccines for inducing protective immunity against a variety of viral pathogens in which humoral immunity is involved in host defense.
For many years, the standard approach to preparing inactivated vaccines has been to inactivate the pathogens with formaldehyde or BPL3,7. In particular, the utility of formaldehyde as the standard mode of pathogen inactivation for vaccine development has been questioned3. Other inactivation approaches such as psoralens, ethylenimine and nonionic detergents have also been tested for development of various vaccines, but these are believed to have disadvantages similar to formaldehyde or BPL in terms of chemical modification of immunogenic proteins3,7. In contrast, we have found that H2O2 represents a new and versatile platform for the development of inactivated vaccines. In the studies presented here, we used the H2O2 vaccine platform to demonstrate protective immunity against chronic viral infection (LCMV clone 13), and lethal viral challenge in two independent model systems, VV and WNV. The current commercial use of H2O2 for sterilizing manufacturing equipment and the availability of assay kits for determining H2O2 identity and potency (for example, the PeroXOquant kit from Thermo Scientific) add to the feasibility of using H2O2 for vaccine production. Moreover, the ability of H2O2 to inactivate a wide spectrum of pathogens, including viruses32, bacteria32, parasites33 and potentially even bacterial spores34, may allow expansion into new areas of vaccine research, fulfilling previously unmet needs for combating a number of human pathogens.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Mice
Female BALB/c mice were bred at Oregon Health & Science University (OHSU) or purchased from The Jackson Laboratory (Bar Harbor, ME). Female BALB/c severe combined immunodeficiency and C57BL/6 Rag2−/− mice were purchased from The Jackson Laboratory. Mice were used at 6–12 weeks of age. During lethal challenge studies, mice that became moribund or that experienced greater than 30% weight loss were euthanized. The OHSU Institutional Animal Care and Use Committee approved all animal use protocols.
Human subjects
Human subjects who had experienced symptoms of WNV infection (n = 21, 71% female, 29% male, average age: 38 years (range, 19–55 years)) were recruited ~1 year after a WNV outbreak in Colorado35. Subjects (n = 4) from Oregon with no history of flavivirus infection were included as negative controls. Each subject provided written informed consent before participating in the study, and the OHSU Institutional Review Board approved all human studies.
Flavivirus studies
YFV-17D was grown on Vero cells, and for antigenicity studies purified virus was exposed to dilutions of H2O2 (Fisher Scientific), formaldehyde (Fisher Scientific) or BPL (Sigma-Aldrich) at 20–24 °C, diluted to 1 μg ml−1 and used to coat ELISA plates. YFV-immune mice were infected intraperitoneally with 1 × 106 PFU of YFV-17D, and immune serum was obtained at 220 d after infection. WNV-NY99 was grown on Vero cells and purified by ultracentrifugation using 36% (w/v) sucrose cushions in PBS (pH 7.4) at 110,000g for 2.5 h at 4 °C. Pellets were resuspended in PBS, pH 7.4 and inactivated with 3% H2O2 for 2 h at 20–24 °C, with inactivation confirmed by plaque assay on Vero cells before final formulation and immunization. Mice were immunized subcutaneously with 10-μg doses of H2O2-WNV formulated with 50 μg of aluminum hydroxide (alum, Sigma-Aldrich) and 5 μg of monophosphoryl lipid A (MPL, List Biological Laboratories, Campbell, CA) or with 100 μl (that is, one-tenth the horse dose) of Ft. Dodge Innovator vaccine (Wyeth, Fort Dodge, IA). Immunity following vaccination was assessed by ELISA using WNV-NY99 lysate or by neutralizing assays similar to previous studies36. The NT50 was defined as the reciprocal serum dilution that reduced the number of initial WNV-NY99 PFU (70–100 PFU per well) by 50%. For WNV challenge experiments, the 50% lethal dose (LD50) value was determined using the method of Reed and Muench37 by either subcutaneous or intraperitoneal routes, yielding different LD50 values of ~50 PFU and ~5 PFU, respectively.
LCMV studies
LCMV-Armstrong (Arm-53b) was grown on BHK-21 cells and purified by ultracentrifugation on a 25% glycerol cushion in TNE (10 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, pH 8) at 80,000g for 3 h at 4 °C. Pellets were resuspended in PBS pH 7.4 and inactivated with 3% H2O2 for 2 h at 20–24 °C. Inactivation was confirmed by Vero cell plaque assay, co-culture on BHK cells and injection into BALB/c severe combined immunodeficient or C57BL/6 Rag2−/− mice. Subcutaneous vaccination was performed with 50 μg H2O2-LCMV formulated with 5 μg of MPL. Intracellular cytokine staining has been described previously38. Briefly, single-cell splenocyte suspensions were stimulated for 6 h at 37 °C, 6% CO2 with 1 × 10−7 M NP118–126 peptide (Sigma-Aldrich) followed by staining for CD8a (eBioscience) overnight at 4 °C. Cells were fixed, permeabilized and stained for IFN-γ (Invitrogen), TNF-α (eBioscience) and IL-2 (BD Biosciences, San Jose, CA). In parallel, unstimulated virus-specific T cells were stained for CD8, CD11a (BioLegend) and H-2Ld-restricted NP118 tetramers (National Institutes of Health Tetramer Core Facility) overnight at 4 °C followed by being fixed, permeabilized and stained intracellularly for granzyme B (BD Biosciences). Nonviable cells were excluded using a live cell gate based on the viability stain, Aqua (LIVE/DEAD Fixable Dead Cell Stain, Invitrogen), followed by an optimized lymphocyte gate. In vivo CTL assays were performed as previously described39. Splenocytes from naïve BALB/c mice were labeled with 1 μM (CFSElo) or 10 μM (CFSEhi) CFSE for 5 min at 20–24 °C, washed and incubated with or without 1 × 10−7 M NP118 peptide at 37 °C for 1 h. CFSE-labeled cell suspensions were washed, mixed at a 1:1 ratio (5 × 106 of each population) and injected intravenously into recipient mice. After 15 h, mice were euthanized, and peptide-specific depletion of target cells was analyzed by flow cytometry. Percentage in vivo lysis was calculated from the ratio of NP118-coated (CFSElo) to uncoated (CFSEhi) target cells after normalizing to the ratio of cell populations before injection.
Orthopoxvirus studies
VV (Western Reserve strain) and monkeypox virus (Zaire strain) were grown on BSC-40 cells and purified by ultracentrifugation as previously described40. Virus titers were determined by plaque assay on Vero cells. MVA was grown on BHK-21 cells. For inactivated VV immunization, purified VV was inactivated with 1% formaldehyde or 1% H2O2 in 10 mM Tris-HCl pH 7.4 for 2 h at 20–24 °C or inactivated with 10 J of UV irradiation and tested for residual live virus on Vero cells. VV inactivation for 2 h with 0.1% or 0.01% formaldehyde was suboptimal, and breakthrough of infectious virus precluded further analysis (data not shown). Mice were vaccinated subcutaneously with live VV (1 × 106 PFU), MVA (1.5 × 107 PFU), or 10 μg of inactivated VV formulated with 50 μg of alum plus 5 μg of MPL. Neutralizing antibody responses were determined as described36. The LD50 (5 × 105 PFU) was determined by intranasal infection of groups of naïve mice with serial dilutions of virus.
Statistical analyses
Statistical comparisons were performed by one-way ANOVA with post hoc testing using StatPlus;mac (AnalystSoft Inc.) for Microsoft Excel. P values of ≤ 0.05 were considered statistically significant.
Acknowledgments
We thank E. Poore and M. Dubois for the growth of YFV and E. Hammarlund for the growth of monkeypox virus. This work was supported by US National Institutes of Health grants R56 AI076506 (to M.K.S.), UO1 AI082196 (to M.K.S.) and R43 AI079898 (to I.J.A. and M.K.S.) and Oregon National Primate Research Center grant 8P51 OD011092-53 (to M.K.S.).
Footnotes
AUTHOR CONTRIBUTIONS
M.K.S. conceived of the project. H.-P.R. performed the in vivo experiments, data analysis and figure preparation for the LCMV studies. I.J.A. performed the experiments, data analysis and figure preparation for the VV, YFV and WNV studies. I.J.A., H.-P.R. and M.K.S. wrote the manuscript, discussed the results and reviewed the manuscript before submission.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
References
- 1.Plotkin SL, Plotkin SA. A short history of vaccination. In: Plotkin SA, Orenstein WA, Offit PA, editors. Vaccines. Saunders/Elsevier; Philadelphia: 2008. pp. 1–16. [Google Scholar]
- 2.Logrippo GA, Hartman FW. Antigenicity of β-propiolactone–inactivated virus vaccines. J Immunol. 1955;75:123–128. [PubMed] [Google Scholar]
- 3.Brown F. Review of accidents caused by incomplete inactivation of viruses. Dev Biol Stand. 1993;81:103–107. [PubMed] [Google Scholar]
- 4.Kim HW, et al. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am J Epidemiol. 1969;89:422–434. doi: 10.1093/oxfordjournals.aje.a120955. [DOI] [PubMed] [Google Scholar]
- 5.Delgado MF, et al. Lack of antibody affinity maturation due to poor Toll-like receptor stimulation leads to enhanced respiratory syncytial virus disease. Nat Med. 2009;15:34–41. doi: 10.1038/nm.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Annunziato D, et al. Atypical measles syndrome: pathologic and serologic findings. Pediatrics. 1982;70:203–209. [PubMed] [Google Scholar]
- 7.Stauffer F, El-Bacha T, Da Poian AT. Advances in the development of inactivated virus vaccines. Recent Pat Antiinfect Drug Disc. 2006;1:291–296. doi: 10.2174/157489106778777673. [DOI] [PubMed] [Google Scholar]
- 8.Swanson MC, et al. IgE and IgG antibodies to β-propiolactone and human serum albumin associated with urticarial reactions to rabies vaccine. J Infect Dis. 1987;155:909–913. doi: 10.1093/infdis/155.5.909. [DOI] [PubMed] [Google Scholar]
- 9.Monath TP, et al. An inactivated cell-culture vaccine against yellow fever. N Engl J Med. 2011;364:1326–1333. doi: 10.1056/NEJMoa1009303. [DOI] [PubMed] [Google Scholar]
- 10.Guenter TE. Hydrogen peroxide. In: McKetta JJ, editor. Encyclopedia of Chemical Processing and Design. Vol. 27. Marcel Dekker; New York: 1988. pp. 27–43. [Google Scholar]
- 11.Sykes G. Disinfection and Sterilization. Ch 2. Lippincott; Philadelphia: 1965. The theory and mode of action of disinfection. [Google Scholar]
- 12.Valko M, et al. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Siber GR, et al. Safety and immunogenicity of hydrogen peroxide-inactivated pertussis toxoid in 18-month-old children. Vaccine. 1991;9:735–740. doi: 10.1016/0264-410x(91)90289-i. [DOI] [PubMed] [Google Scholar]
- 14.Termini J. Hydroperoxide-induced DNA damage and mutations. Mutat Res. 2000;450:107–124. doi: 10.1016/s0027-5107(00)00019-1. [DOI] [PubMed] [Google Scholar]
- 15.Fiore AE, Finestone SM, Bell BP. Hepatitis A vaccine. In: Plotkin SA, Orenstein WA, Offit PA, editors. Vaccines. Saunders/Elsevier; Philadelphia: 2008. pp. 177–203. [Google Scholar]
- 16.Plotkin SA, Vidor E. Poliovirus vaccine—inactivated. In: Plotkin SA, Orenstein WA, Offit PA, editors. Vaccines. Saunders/Elsevier; Phildelphia: 2008. pp. 605–630. [Google Scholar]
- 17.Srivastava AK, et al. A purified inactivated Japanese encephalitis virus vaccine made in Vero cells. Vaccine. 2001;19:4557–4565. doi: 10.1016/s0264-410x(01)00208-0. [DOI] [PubMed] [Google Scholar]
- 18.Monath TP, et al. Inactivated yellow fever 17D vaccine: development and nonclinical safety, immunogenicity and protective activity. Vaccine. 2010;28:3827–3840. doi: 10.1016/j.vaccine.2010.03.023. [DOI] [PubMed] [Google Scholar]
- 19.Robinson HL, Amara RR. T cell vaccines for microbial infections. Nat Med. 2005;11:S25–S32. doi: 10.1038/nm1212. [DOI] [PubMed] [Google Scholar]
- 20.Lau LL, Jamieson BD, Somasundaram T, Ahmed R. Cytotoxic T-cell memory without antigen. Nature. 1994;369:648–652. doi: 10.1038/369648a0. [DOI] [PubMed] [Google Scholar]
- 21.Hassett DE, Slifka MK, Zhang J, Whitton JL. Direct ex vivo kinetic and phenotypic analyses of CD8+ T cell responses induced by DNA immunization. J Virol. 2000;74:8286–8291. doi: 10.1128/jvi.74.18.8286-8291.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwarz K, et al. Efficient homologous prime-boost strategies for T cell vaccination based on virus-like particles. Eur J Immunol. 2005;35:816–821. doi: 10.1002/eji.200425755. [DOI] [PubMed] [Google Scholar]
- 23.Slifka MK, et al. Antiviral cytotoxic T-cell memory by vaccination with recombinant Listeria monocytogenes. J Virol. 1996;70:2902–2910. doi: 10.1128/jvi.70.5.2902-2910.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plotkin SA. Correlates of protection induced by vaccination. Clin Vaccine Immunol. 2010;17:1055–1065. doi: 10.1128/CVI.00131-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amanna IJ, Messaoudi I, Slifka MK. Protective immunity following vaccination: how is it defined? Hum Vaccin. 2008;4:316–319. doi: 10.4161/hv.4.4.5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edghill-Smith Y, et al. Smallpox vaccine–induced antibodies are necessary and sufficient for protection against monkeypox virus. Nat Med. 2005;11:740–747. doi: 10.1038/nm1261. [DOI] [PubMed] [Google Scholar]
- 27.Eckels KH, Putnak R. Formalin-inactivated whole virus and recombinant subunit flavivirus vaccines. Adv Virus Res. 2003;61:395–418. doi: 10.1016/s0065-3527(03)61010-9. [DOI] [PubMed] [Google Scholar]
- 28.Amanna IJ, Slifka MK. Wanted, dead or alive: new viral vaccines. Antiviral Res. 2009;84:119–130. doi: 10.1016/j.antiviral.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ng T, et al. Equine vaccine for West Nile virus. Dev Biol (Basel) 2003;114:221–227. [PubMed] [Google Scholar]
- 30.Shrestha B, Ng T, Chu HJ, Noll M, Diamond MS. The relative contribution of antibody and CD8+ T cells to vaccine immunity against West Nile encephalitis virus. Vaccine. 2008;26:2020–2033. doi: 10.1016/j.vaccine.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giannini SL, et al. Enhanced humoral and memory B cellular immunity using HPV16/18 L1 VLP vaccine formulated with the MPL/aluminium salt combination (AS04) compared to aluminium salt only. Vaccine. 2006;24:5937–5949. doi: 10.1016/j.vaccine.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 32.Neighbor NK, et al. The effect of microaerosolized hydrogen peroxide on bacterial and viral poultry pathogens. Poult Sci. 1994;73:1511–1516. doi: 10.3382/ps.0731511. [DOI] [PubMed] [Google Scholar]
- 33.Channon JY, Blackwell JM. A study of the sensitivity of Leishmania donovani promastigotes and amastigotes to hydrogen peroxide. I Differences in sensitivity correlate with parasite-mediated removal of hydrogen peroxide. Parasitology. 1985;91:197–206. doi: 10.1017/s0031182000057309. [DOI] [PubMed] [Google Scholar]
- 34.DeQueiroz GA, Day DF. Disinfection of Bacillus subtilis spore-contaminated surface materials with a sodium hypochlorite and a hydrogen peroxide-based sanitizer. Lett Appl Microbiol. 2008;46:176–180. doi: 10.1111/j.1472-765X.2007.02283.x. [DOI] [PubMed] [Google Scholar]
- 35.Gujral IB, Zielinski-Gutierrez EC, LeBailly A, Nasci R. Behavioral risks for West Nile virus disease, northern Colorado, 2003. Emerg Infect Dis. 2007;13:419–425. doi: 10.3201/eid1303.060941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hammarlund E, et al. Duration of antiviral immunity after smallpox vaccination. Nat Med. 2003;9:1131–1137. doi: 10.1038/nm917. [DOI] [PubMed] [Google Scholar]
- 37.Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. Am J Hyg. 1938;27:493–497. [Google Scholar]
- 38.Raué HP, Slifka MK. Pivotal Advance: CTLA-4+ T cells exhibit normal antiviral functions during acute viral infection. J Leukoc Biol. 2007;81:1165–1175. doi: 10.1189/jlb.0806535. [DOI] [PubMed] [Google Scholar]
- 39.Barber DL, Wherry EJ, Ahmed R. Cutting edge: rapid in vivo killing by memory CD8 T cells. J Immunol. 2003;171:27–31. doi: 10.4049/jimmunol.171.1.27. [DOI] [PubMed] [Google Scholar]
- 40.Hammarlund E, et al. Monkeypox virus evades antiviral CD4+ and CD8+ T cell responses by suppressing cognate T cell activation. Proc Natl Acad Sci USA. 2008;105:14567–14572. doi: 10.1073/pnas.0800589105. [DOI] [PMC free article] [PubMed] [Google Scholar]