

Abstract
Zinc is essential for normal brain development. Gestational severe zinc deficiency can lead to overt fetal brain malformations. Although not teratogenic, suboptimal zinc nutrition during gestation can have long-term effects on the offspring's nervous system. This paper will review current knowledge on the role of zinc in modulating neurogenesis and neuronal apoptosis, as well as the proposed underlying mechanisms. A decrease in neuronal zinc causes cell cycle arrest, which in part involves a deregulation of select signals (ERK1/2, p53, NF-κB). Zinc deficiency also induces apoptotic neuronal death through the intrinsic (mitochondrial) pathway, which can be triggered by the activation of the zinc-regulated enzyme caspase-3, and as a consequence of abnormal regulation of pro-survival signals (ERK1/2, NF-κB). Alterations in the finely-tuned processes of neurogenesis, neuronal migration, differentiation and apoptosis, that involve the developmental shaping of the nervous system, could have a long-term impact on brain health. Zinc deficiency during gestation, even at the marginal levels observed in human populations, could increase the risk for behavioral/neurological disorders in infancy, adolescence and adulthood.
Keywords: Zinc, neuron, proliferation, apoptosis, caspase, NF-κB, ERK, zinc deficiency, p53, Bad
1. Introduction
Zinc is one of the most abundant essential elements in the human body. It is present in the structure of many proteins, like enzymes and transcription factors and modulates the function of other proteins, including receptors, enzymes and proteins involved in cell signalling. Zinc is a key micronutrient in the physiology of the nervous system, participating in the modulation of synaptic transmission [1, 2], regulation of neuronal cell oxidant production [3, 4], regulation of signalling cascades [5-9]; neural cell proliferation [10-13], cell survival [9-12, 14] and neuronal differentiation [15, 16]. The physiological functions of zinc in the nervous system depend on a fine balance of intra and extracellular zinc concentrations [2, 17]. Elevations in neuronal zinc levels that are associated with select brain injury conditions lead to apoptotic cell death [18, 19]. On the other hand, severe gestational zinc deficiency is characterized by fetal malformations including agenesis and dysmorphogenesis of the brain, spinal cord, eye and olfactory tract [20, 21]. A marginal zinc nutrition during gestation and early postnatal development is not teratogenic but affects neurotransmission and cell signalling [5, 22]. Furthermore, early postnatal zinc deficiency affects the number and differentiation of basket and stellate [15], Purkinje [16], and cerebellum granule neurons [23].
The adverse effects of zinc deficiency on brain development could constitute a risk for the onset of neurological disorders during infancy, adolescence and adulthood [24]. In this review we will addess current knowledge on the impact of zinc deficiency on two processes that are central to normal brain development: neurogenesis and neuronal apoptosis. The proposed mechanisms (summarized in Fig.1) will be discussed.
Figure 1. Mechanisms involved in the impaired neurogenesis and triggering of apoptotic neuronal death ehrn neuronal zinc decreases.
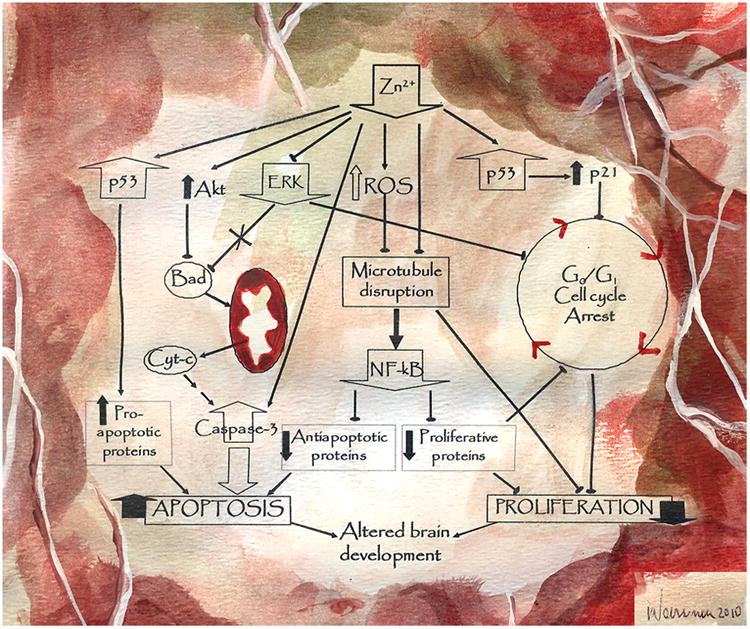
A decreased zinc availability can affect cell proliferation and neuronal survival by affecting multiple signaling pathways. Apoptosis can occur as a consequence of: a) a direct activation of caspase-3, b) the inhibition of ERK1/2, and the consequent translocation of Bad to the mitochondria and the occurrence of MOMP, c) p53-mediated increased expression of pro-apoptotic genes, and d) a microtubule-mediated inhibition of the pro-survival signal NF-κB. As a consequence of zinc deficiency, a decreased cell proliferation occurs due to an arrest of the cell cycle in G0/G1 phase. The potential mechanisms involved include: a) an upregulation of p53 that leads to an increased expression of p21, inhibitor of cdks, b) ERK1/2 inhibition and a consequent decreased expression of cyclins D and E, c) alterations in microtubule assembly that can affect the formation of the mitotic spindle, and d) NF-κB inhibition.
Illustrated by the artist Andres Waissman (http://www.andreswaissman.com)
2. Zinc and neuronal apoptosis
Neuronal apoptosis occurs as a physiological and critical event in the normal development of the nervous system. However, it also occurs as a consequence of brain injury and disease. Extracellular and intracellular stimuli such as trophic factors, oxidative stress, neurotoxins and DNA damage act upon neurons leading to life or apoptotic death decisions. A finely tuned regulation of the cell cycle is essential for cell proliferation and differentiation, but it can also participate in the triggering of neuronal death. In this regards, the re-entry of postmitotic neurons in the cell cycle induces cell death instead of proliferation [25][26].
The mechanisms of apoptosis are specific for particular cell types and also depend on the apoptotic signal that initiates the cascade. Zinc depletion induces apoptosis in different cells and tissues (reviewed in [27-29]). In neuronal cells, both low and high zinc concentrations can trigger apoptosis. The accumulation of chelatable zinc in neurons can induce neuronal death after ischemia, epileptic seizures or brain injury by trauma [18, 19]. Cho et al. [30] proposed that endogenous neuronal zinc can physiologically regulate apoptosis. This was based on the finding that the subcutaneous injection of the chelator N,N,N′,N′-tetra-kis(2-pyridylmethyl)ethylenediamine (TPEN) into 7-day-old rats decreased the number of apoptotic neurons in the brain as compared to controls. However, these results as well as others that use TPEN to decrease cellular zinc should be carefully interpreted. TPEN will chelate zinc pools based on its relatively very high affinity for zinc compared to physiological cell zinc ligands. Thus, TPEN-chelatable zinc will not necessarily reflect the zinc pools that are physiologically involved in the regulation of neuronal function and survival.
In neuronal cells, human neuroblastoma IMR-32 cells and primary rat cortical neurons, zinc deficiency induced apoptotic cell death via the intrinsic pathway [10]. In this pathway [31], mitochondrial components (cytochrome c) present in the space between the inner and outer membranes are released to the cytosol as a consequence of mitochondrial outer membrane permeabilization (MOMP). Members of the Bcl-2 family of proteins regulate MOMP, with select protein components (Bcl2, Bcl-xL) that prevent MOMP; and pro-apoptotic proteins (Bad, Bax) that act promoting MOMP. In the cytosol, cytochrome c assembles with the proteins Apaf-1 and caspase-9 forming the apoptosome, with a consequent activation of caspase-9. Caspase-9 activates the executioner caspase-3, which will subsequently cleave several proteins initiating a sequence of catabolic events. In IMR-32 cells and cortical neurons the occurrence of apoptosis via the intrinsic pathway was evidenced by an increase of the pro-apoptotic protein Bad in the mitochondrial fraction of zinc-deficient cells, the associated release of cytochrome c to the cytosol, caspase-3 activation, and subsequent DNA fragmentation [10] (Fig.1).
Although the mechanisms underlying the induction of apoptosis by zinc deprivation are still unclear, the activation of p53, of certain members of the caspase family of cystein-proteases, and oxidative stress, have been proposed to play a role.
3. Mechanisms of zinc-deficiency-induced neuronal apoptosis
3.a. Zinc deficiency and the tumor suppressor gene product p53
The tumor suppressor gene product p53 participates in cellular stress responses by regulating cell cycle arrest, apoptosis, senescence, DNA repair and genetic stability [32]. Lane [33] called p53 “the guardian of the genome” because it causes cell cycle arrest in response to DNA damage, allowing DNA repair before the progression of the cell cycle occurs [34]. Depending on the cell type, p53 can also trigger apoptosis as a consequence of DNA damage [35].
During apoptosis, p53 acts as a transcription factor increasing the expression levels of pro-apoptotic proteins such as PUMA (p53-up-regulated modulator of apoptosis) and NOXA [36]. Cytoplasmic p53 also interacts with the anti-apoptotic members Bcl-xL and Bcl-2, leading to mitochondrial membrane permeabilization [37]. In human hepatoblastoma HepG2 cells, zinc depletion increases p53 mRNA and nuclear p53 protein levels, and zinc repletion results in a normal abundance of the transcript [38]. Zinc plays a structural role in maintaining p53 stability and its DNA binding capacity [39, 40]. In cultured human aortic endothelial cells, p53 and downstream targets, as well as events resulting from p53 increase (Bax, Bcl-2 and caspase-3 activation) are responsive to low or elevated cellular zinc levels [41].
We studied the role of zinc in the modulation of neuronal apoptosis, by investigating the effects of incubating human neuroblastoma IMR-32 cells and primary cultures of rat cortical neurons in zinc-depleted media. Zinc depleted media was obtained by incubating the serum in the presence of the chelator diethylenetriamine pentaacetic acid (DTPA), the subsequent removal of DTPA by extensive dialysis, and final adjustment of zinc, iron, copper and protein concentrations of the 10% (v/v) serum-containing media [42]. An increase in p53 protein levels was observed after 6 to 24 h of culturing cells in zinc deficient medium. Zinc deficiency induced IMR-32 cell cycle arrest at the G1 phase, and apoptotic death in both IMR-32 cells and primary cortical neurons [10] . Accordingly, Ra et al. [43] demonstrated that p53 is essential for TPEN-induced neuronal apoptosis showing the involvement of p53-dependent induction of PUMA/NOXA, and caspase-11-mediated activation of caspase-3. The genetic depletion of p53 blocks the effects of TPEN on neuronal apoptosis [43]. TPEN-induced apoptotic death in neuronal precursor cells is also associated with the expression of the p53-dependent pro-apoptotic signal Rb-1 [11]. On the contrary, while long-term exposure of a rat glioma cells to zinc-depleted media induces oxidative DNA damage and p53 expression; the binding of p53 to DNA decreases, potentially affecting the expression of p53-regulated genes [3]. Thus, p53 activation can have a central role in zinc deficiency- triggered apoptotic neuronal death. Its role in the associated cell cycle arrest will be subsequently discussed (
3.b. Zinc deficiency and caspase activation
While zinc depletion induces apoptosis in different cell types, zinc supplementation inhibits internucleosomal DNA fragmentation and other characteristic morphological features of apoptosis in vivo and in vitro [44-46]. Although there are many potential targets for the antiapoptotic effects of zinc, the apoptotic nucleases emerge as central antiapoptotic targets. Among these nucleases, the caspases (cysteine-aspartic acid proteases) family of proteases, specially caspase-3, are important in determining brain cell number [47]. Caspase-3 is also recognized to participate in other essential processes in neurodevelopment, including neuronal differentiation and dendrite pruning (reviewed in [48]).
Caspase-3 activation involves the proteolityc cleavage of a 32kDa cytosolic precursor, with the generation of two peptides that dimerize to generate the active enzyme. Perry et al. [49] demonstrated that zinc inhibits caspase-3 activation at micromolar concentrations; while millimolar concentrations of zinc inhibit caspase-6/Mch 2α, a caspase involved in the cleavage of lamin A and in the activation of caspase-3 [50]. Furthermore, zinc inhibits the processing of the proenzyme into the caspase-3 active form [51]. Zinc supplementation of neuroblastoma cells suppresses butyrate/staurosporine-induced activation of caspase-3 [52]. TPEN-induced zinc depletion in HeLa cells induces apoptosis through the activation of caspases-3, -8 and -9 [53]. In this cellular model, zinc supplementation inhibited caspase-3 activation but not caspase-8 and -9 cleavages, suggesting that zinc protects cells from apoptosis by maintaining caspase-3 inactive [53]. In the human breast cancer cell lines MCF-7 and MDA-MB468, zinc depletion caused apoptosis through the activation of the mitochondrial death pathway [54]. Supporting a role of caspase-3 in zinc deficiency-associated apoptosis, MCF-7 cells lacking the expression of caspase-3, were less sensitive to zinc depletion [54]
In IMR-32 cells, the increase in caspase-3 activation was significant after 24 h of incubation in zinc deficient media, remaining high up to 48 h, when neuronal apoptosis was in an advanced stage [10]. In IMR-32 cells and cortical neurons, caspase-3 activity was normalized by supplementing the media with zinc, which was associated with the inhibition of apoptotic death. On the other hand, the antioxidant lipoic acid which prevents oxidant accumulation in zinc deficient neurons [4], did not decrease caspase-3 activation and apoptosis in IMR-32 cells and rat cortical neurons [10]. The above findings stress the relevance of caspase-3 activation as a key factor in the triggering of apoptosis when neuronal zinc concentrations decrease.
3.c. Zinc deficiency and apoptosis: neuronal oxidative stress and the regulation of redox sensitive signals
Zinc deficiency can cause oxidative stress as evidenced by its association to increased levels of cell oxidants [3, 4, 9, 55], altered activity and concentration of antioxidant enzymes [56, 57], decreased glutathione levels [58, 59], and tissue oxidative damage, including increased lipid, protein and DNA oxidation [3, 60, 61]. An adequate zinc status can be key to the regulation of the oxidant/antioxidant neuronal balance. In human neuroblastoma IMR-32 cells, zinc deficiency causes an increase in cell oxidants levels, particularly of H2O2 [9], a decrease in glutathione concentrations [4], a key antioxidant defense in neurons, and an increased activity of CuZn superoxide dismutase, and of activity and expression of Mn superoxide dismutase [62]. Culturing IMR-32 cells in the presence of the H2O2–metabolizing enzyme catalase, or the antioxidant α-lipoic acid, prevented the increase in H2O2 [4, 9]. Catalase also inhibited the associated activation of the redox-sensitive Mitogen Activated Protein Kinases (MAPKs) p38 and JNK in zinc deficient neuronal cells [9]. However, caspase-3 activation and DNA fragmentation were not prevented by catalase [9]. Furthermore, the treatment with α-lipoic acid did not inhibit the activation of caspase-3 in IMR-32 cells and in cortical neurons [10]. Thus, the above results suggest that oxidative stress is not the triggering event in zinc-deficiency-induced neuronal apoptosis and that JNK and p38 are not relevant apoptosis-triggering cascades. However, although an initial increase in neuronal oxidants may not trigger the initial apoptotic events in zinc deficient neurons, the permeabilization of the mitochondrial membrane would ultimately result in the uncoupling of the respiratory chain and in an increased production of reactive oxygen species. However, this still represent an advanced stage in the apoptotic process rather than a triggering factor. In section 5, we will discuss the potential involvement of the redox-sensitive transcription factor NF-κB in zinc deficiency-associated neuronal apoptosis.
4. Zinc deficiency and cell cycle progression
Zinc is also actively involved in cell proliferation and differentiation in different tissues. Dwarfism is a characteristic symptom of zinc deficiency and is associated to a decreased activity of the epiphyseal growth plate [63]. The epiphyseal growth plate is responsible for longitudinal bone growth because of proliferation and differentiation of chondrocytes [64], a process that is stimulated by zinc [65]. In vivo, zinc deficiency causes a significant decrease of chicken chondrocyte proliferation [66], that is not affected by local growth factors such as FGF-2, PTHrP and IGF-1. Zinc deficiency also affects the immune system and a decreased cell proliferation could in part explain some of these effects. In mice, zinc deficiency causes depletion of precursor lymphoid cells in the lymphoid organs causing lymphopenia and thymus atrophy [67]. Although the involved mechanisms are not clear, zinc deficiency caused a decreased T cell deoxythymidine kinase gene expression and cell cycle arrest at the G1 phase [68]. Protein kinase C could also be involved in zinc-deficiency-mediated lymphocyte-impaired responses to mitogen stimulation [69].
Although neurogenesis is essential during normal brain development, it is also relevant in the adult brain [11]. Adult rats fed a severely zinc-restricted diet for three weeks showed a significant decrease in neural precursor cells in the subgranular zone and granular cell layer of the dentate gyrus [11]. In cultured human Ntera-2 neuronal precursor cells depleted of zinc by treatment with TPEN, a significant decrease in cell proliferation was observed with an increase in nuclear p53 [11]. A role of p53, through the upregulation of the gene reprimo, was proposed to be involved in Ntera-2 cell cycle arrest as a consequence of zinc depletion. A significant reduction in proliferating cells and immature neurons in the dentate gyrus of the hippocampus was observed in zinc-deficient mice [12]. Decreased neurogenesis was also found in the dentate gyrus of rats fed zinc deficient diets for 6 weeks [13]. Although the above results constitute an important starting evidence, and supports a role of zinc in neurogenesis, it should be stressed that the conditions of zinc deficiency used are extremely severe [11-13]. Furthermore, some studies did not include a restrict-fed group, necessary to control for the confounding factor of the anorexia associated with severe zinc deficiency [12, 13].
Zinc deficiency causes a decreased proliferation of human neuroblastoma IMR-32 cells, which is due to an arrest of the cell cycle in the G1 to phase S transition [10]. Cell proliferation is induced by the activation of cyclin-dependent kinases (cdks) by the different cyclins, while the progression of the cell cycle is inhibited by cdks inhibitors. p21, a member of the Cip/Kip family of cdks inhibitors, is regulated by various mechanism including transcriptional regulation, epigenetic silencing, mRNA stability and ubiquitin-dependent and –independent degradation [70]. p21 can also be regulated by posttranslational modifications such as phosphorylation [71]. Transcriptional modulation of p21 involves p53-dependent and –independent mechanisms. Two p53-binding sites exist in the promoter of p21 and one of them is involved in the response of p53 to DNA damage [72]. LaBaer et al., [73] demonstrated that p21 can also positively modulate cell cycle progression by stabilizing the cyclinD1/cdk4/cdk6 complexes. Wong et al., [70], found that zinc depletion in HepG2 cells down regulates p21 mRNA and protein expression due to a decreased p21 promoter accessibility. In the zinc deficient HepG2 cells, the arrest at the G1/S phase of the cell cycle seems unrelated to a decrease in p21 protein levels. On the contrary, in zinc-deficient IMR-32 neuroblastoma cells the inhibition of the G1 to phase S transition occurs concomitantly with the upregulation of p53 and an increase of the downstream target protein p21 [10].
Other mechanism that could underly the decreased cell proliferation observed in a zinc deficient state is the requirement of a functional tubulin network for the proper assembly of the mitotic spindle during mitosis. In support of this, in IMR-32 neuronal cells as well as in gestation day 19 fetal brain, zinc deficiency causes an impaired assembly of microtubules [8, 74].
5. Other signaling cascades involved in zinc deficiency-induced impaired neurogenesis and neuronal apoptosis
5.a. Transcription factor NF-κB
Initially described in the immune system, transcription factor NF-κB was subsequently found in multiple tissues, and its role in the nervous system is a current subject of active research. The active NF-κB is constituted of protein dimers that belong to the Rel/NF-κB family of proteins, including c-Rel, RelB, p65 (RelA), p50/p105 and p52/p100 [5]. NF-κB dimers are localized in the cytosol in an inactive form, given their binding to the inhibitory IκB proteins. In the canonical pathway, activation involves the phosphorylation of two conserved serine groups in the IκB peptide by the specific IκB kinase complex (IKK) [33], which signals the protein for polyubiquitination and degradation by the 26S proteasome. IκB degradation unmasks a nuclear localization signal in the NF-κB dimer which drives its translocation through nuclear pores into the nucleus. NF-κB is involved in the development of the nervous system, regulating neuronal proliferation, migration, differentiation, survival and cell interactions [75-78]. The NF-κB subunits are selectively expressed in the subventricular zone and in the rostral migratory stream during early postnatal development and also in the adult brain. This suggests their involvement in regulating the generation and migration of new neurons [79]. In NF-κB p-50 deficient adult mice, the rate of neural precursor proliferation was not affected [80]. However, the p50 deficient animals showed a significant decrease in the number of hippocampus newborn neurons, and defects in short-term special memory performance [80]. NF-κB also participates in the regulation of synaptic plasticity, which in turn impacts learning and memory. NF-κB regulates the transcription of multiple genes, which includes those codifying for proteins that modulate cell proliferation (e.g. cyclin D1, c-Myc, and IL-1), and survival (Bcl2, Bcl-xL, c-IAP1, XIAP).
Zinc deficiency affects the NF-κB signaling cascade in neuronal cells in culture and in the developing nervous system. In IMR-32 cells, a decreased zinc availability causes the activation of the initial events in the pathway (IκBα phosphorylation and degradation), which occurs as a consequence of zinc deficiency-induced increase in oxidant production [4]. However, the active NF-κB is not efficiently translocated into the nucleus secondary to zinc deficiency-induced alterations in the normal assembly of the microtubule network [6, 8]. Functional microtubules are required for the axonal retrograde transport and nuclear translocation of NF-κB in neurons [6, 81]. In zinc deficient neurons, a decreased nuclear content of the NF-κB components Rel A and p50 and of NF-κB-DNA binding is observed [6, 8]. As a consequence, a decreased expression of NF-κB-dependent genes occurs, including those involved in the regulation of the NF-κB pathway [6], modulators of the cell cycle progression [10] and of cell survival [6, 10]. In this regard, zinc deficiency leads to decreased neuronal levels of the NF-κB-regulated antiapoptotic proteins c-IAP-1, which inhibits caspases; XIAP, which interacts with critical partners in the apoptotic/antiapoptotic cascade (caspase-3, caspase-9, DIABLO/Smac); Bcl-xL and Bcl-2 that act in the intrinsic apoptotic pathway, inhibiting the nuclear pore opening. Furthermore, low levels of the NF-κB-regulated cyclins D and E, could in part contribute to the cell cycle arrest and decreased proliferation occurring as a consequence of zinc deficiency. Although the impaired activity of NF-κB would not be an initial triggering factor in zinc deficiency-induced neuronal apoptosis and cell cycle arrest, it could contribute to the irreversibility of both processes.
5.b. The serine-threonine kinase (Akt) and the extracellular-signal-regulated kinase (ERK)
Akt plays a key role in cell survival. Activated by phosphorylation, Akt acts preventing apoptosis induced by different external stimuli and in different cell types [82]. An important event in the intrinsic apoptotic pathway is the translocation of the proapoptotic protein Bad from the cytosol to the mitochondrial membrane, where it initiates steps leading to MOMP. When Bad is phosphorylated at Ser112, Ser136 or Ser155, it is retained in the cytosol through its binding to the 14-3-3 protein. Unphosphorylated Bad forms a complex with Bcl-xL and inhibits its anti-apoptotic function [83] [84]. Bad phosphorylation is catalyzed by ERK [85] and protein kinase A [83] that phosphorylate Ser-112, by Akt that phosphorylates Ser-136 [86] and protein kinase A also phophorylates Ser-155 [87]. In zinc deficient neuronal cells we observed high levels of Akt phosphorylation [10], that could be triggered by the associated increase in cellular oxidants [9]. In this regard, Akt activation protects NIH3T3 against apoptosis induced by H2O2 [88]. In hepatoma cells, H2O2 increases Akt phosphorylation promoting cell proliferation, while antioxidants induces apoptosis [89]. Although Akt is in general an antiapoptotic signal, high cellular zinc concentrations activates Akt and induces apoptosis in immortalized embryonic hippocampal cells via the non-conventional activation of GSK-3β and tau phosphorylation [90]
In zinc- deficient IMR-32 cells and cortical neurons we observed differential patterns of activation of the pro-survival kinases ERK and Akt [10]. While Akt was activated, ERK was inhibited; nevertheless, the resultant was the translocation of Bad to the mitochondria and the triggering of apoptosis in the zinc-deficient cells. This is explained by previous findings [91] showing that the phosphorylation of Bad at its three serine residues is required to inhibit Bad mitochondrial translocation and the prevention of apoptosis. The above results stress an important role of ERK inactivation in zinc deficiency-induced neuronal apoptosis.
The Raf/MEK/ERK1/2 pathway regulates the cell cycle in response to different stimuli including nutrients, mitogens, cytokines and growth factors [92]. This pathway could be involved in the arrest of the cell cycle at the G0/G1 phase in zinc deficient IMR-32 neuroblastoma cells [10]. A low zinc availability induced a decreased ERK phosphorylation both in human IMR-32 cells and in cortical neurons [10]. ERK phosphorylation is also decreased in the brain of gestation day 19 fetuses from dams fed marginal zinc diets throughout gestation, compared to controls [5]. ERK is central to multiple pathways given that it phosphorylates and activates several targets in the cytosol and in the nucleus. Among these targets, several of them promote cell growth including RSK, c-Myc and ELK. In the nervous system, ERK participates in the modulation of brain development and function [93], being involved in the promotion of neurogenesis [94]. For example, ERK2 deletion in mice causes decreased cortical neurogenesis and impaired neuronal differentiation [95]. Supporting the finding in zinc deficient neuroblastoma cells [96], the activation of ERK during the cell G1 phase is necessary for the subsequent entry into S phase [97].
Summary
The development of the nervous system is a tightly regulated process that includes neurogenesis and removal of redundant neurons by apoptotic cell death. Zinc deficiency affects neurogenesis in animal models, causes the arrest in the G1/S phase of the cell cycle in human IMR-32 neuroblastoma cells, and induces apoptosis in animal and cell models. These alterations can occur as a consequence of the deregulation of select signalling cascades and cellular events that are summarized in Fig.1. Zinc deficiency-induced deregulation of neuronal progenitor proliferation and increased neuronal apoptosis during early development could irreversibly affect the nervous system and lead to impaired behavior and locomotor performance, learning and memory deficits, and increased risk for the development of neurological disorders later in life.
Acknowledgments
Supported by grants from the US National Institutes of Health (grant # HD 01743) and the University of California, Davis, and the University of Buenos Aires and CONICET, Argentina. We are honoured and deeply thankful to the artist Andrés Waissman for his illustration of Figure 1.
Abbreviations
- Akt
serine-threonine kinase
- Cdks
cyclin-dependent kinases
- DTPA
diethylenetriamine pentaacetic acid
- ERK
extracellular-signal-regulated kinase
- MAPK
mitogen activated kinases
- MOMP
mitochondrial outer membrane permeabilization
- NF-κB
nuclear factor-kappa B
- TPEN
N,N,N′,N′-tetra-kis(2-pyridylmethyl)ethylenediamine
References
- 1.Bitanihirwe BK, Cunningham MG. Zinc: the brain's dark horse. Synapse. 2009;63:1029–1049. doi: 10.1002/syn.20683. [DOI] [PubMed] [Google Scholar]
- 2.Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci. 2005;6:449–462. doi: 10.1038/nrn1671. [DOI] [PubMed] [Google Scholar]
- 3.Ho E, Ames BN. Low intracellular zinc induces oxidative DNA damage, disrupts p53, NFkappa B, and AP1 DNA binding, and affects DNA repair in a rat glioma cell line. Proc Natl Acad Sci U S A. 2002;99:16770–16775. doi: 10.1073/pnas.222679399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mackenzie GG, Zago MP, Erlejman AG, Aimo L, Keen CL, Oteiza PI. alpha-Lipoic acid and N-acetyl cysteine prevent zinc deficiency-induced activation of NF-kappaB and AP-1 transcription factors in human neuroblastoma IMR-32 cells. Free Radic Res. 2006;40:75–84. doi: 10.1080/10715760500312305. [DOI] [PubMed] [Google Scholar]
- 5.Aimo L, Mackenzie GG, Keenan A, Oteiza PI. Gestational zinc deficiency affects the regulation of transcription factors AP-1, NF-kB and NFAT in fetal brain. J Nutr Biochem. 2010 doi: 10.1016/j.jnutbio.2009.09.003. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mackenzie GG, Keen CL, Oteiza PI. Microtubules are required for NF-kappaB nuclear translocation in neuroblastoma IMR-32 cells: modulation by zinc. J Neurochem. 2006;99:402–415. doi: 10.1111/j.1471-4159.2006.04005.x. [DOI] [PubMed] [Google Scholar]
- 7.Mackenzie GG, Oteiza PI. Zinc and the cytoskeleton in the neuronal modulation of transcription factor NFAT. J Cell Physiol. 2007;210:246–256. doi: 10.1002/jcp.20861. [DOI] [PubMed] [Google Scholar]
- 8.Mackenzie GG, Zago MP, Keen CL, Oteiza PI. Low intracellular zinc impairs the translocation of activated NF-kappa B to the nuclei in human neuroblastoma IMR-32 cells. J Biol Chem. 2002;277:34610–34617. doi: 10.1074/jbc.M203616200. [DOI] [PubMed] [Google Scholar]
- 9.Zago MP, Mackenzie GG, Adamo AM, Keen CL, Oteiza PI. Differential Modulation of MAP Kinases by Zinc Deficiency in IMR-32 Cells: Role of H(2)O(2) Antioxid Redox Signal. 2005;7:1773–1782. doi: 10.1089/ars.2005.7.1773. [DOI] [PubMed] [Google Scholar]
- 10.Adamo AM, Zago MP, Mackenzie GG, Keen CL, Keenan A, Oteiza PI. The role of zinc in the modulation of neuronal proliferation and apoptosis. Neurotox Res. 2010;17:1–14. doi: 10.1007/s12640-009-9067-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corniola RS, Tassabehji NM, Hare J, Sharma G, Levenson CW. Zinc deficiency impairs neuronal precursor cell proliferation and induces apoptosis via p53-mediated mechanisms. Brain Res. 2008;1237:52–61. doi: 10.1016/j.brainres.2008.08.040. [DOI] [PubMed] [Google Scholar]
- 12.Gao HL, Zheng W, Xin N, Chi ZH, Wang ZY, Chen J. Zinc deficiency reduces neurogenesis accompanied by neuronal apoptosis through caspase-dependent and -independent signaling pathways. Neurotox Res. 2009;16:416–425. doi: 10.1007/s12640-009-9072-7. [DOI] [PubMed] [Google Scholar]
- 13.Suh SW, Won SJ, Hamby AM, Yoo BH, Fan Y, Sheline CT, Tamano H, Takeda A, Liu J. Decreased brain zinc availability reduces hippocampal neurogenesis in mice and rats. J Cereb Blood Flow Metab. 2009;29:1579–1588. doi: 10.1038/jcbfm.2009.80. [DOI] [PubMed] [Google Scholar]
- 14.Cho E, Hwang JJ, Han SH, Chung SJ, Koh JY, Lee JY. Endogenous zinc mediates apoptotic programmed cell death in the developing brain. Neurotox Res. 2010;17:156–166. doi: 10.1007/s12640-009-9085-2. [DOI] [PubMed] [Google Scholar]
- 15.Dvergsten CL, Johnson LA, Sandstead HH. Alterations in the postnatal development of the cerebellar cortex due to zinc deficiency. III. Impaired dendritic differentiation of basket and stellate cells. Brain Res. 1984;318:21–26. doi: 10.1016/0165-3806(84)90058-0. [DOI] [PubMed] [Google Scholar]
- 16.Dvergsten CL, Fosmire GJ, Ollerich DA, Sandstead HH. Alterations in the postnatal development of the cerebellar cortex due to zinc deficiency. II. Impaired maturation of Purkinje cells. Brain Res. 1984;318:11–20. doi: 10.1016/0165-3806(84)90057-9. [DOI] [PubMed] [Google Scholar]
- 17.Colvin RA, Fontaine CP, Laskowski M, Thomas D. Zn2+ transporters and Zn2+ homeostasis in neurons. Eur J Pharmacol. 2003;479:171–185. doi: 10.1016/j.ejphar.2003.08.067. [DOI] [PubMed] [Google Scholar]
- 18.Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996;272:1013–1016. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- 19.Choi DW, Koh JY. Zinc and brain injury. Annu Rev Neurosci. 1998;21:347–375. doi: 10.1146/annurev.neuro.21.1.347. [DOI] [PubMed] [Google Scholar]
- 20.Keen CL, Taubeneck MW, Daston GP, Gershwin ME, Ansari A, Rogers J. Primary and secondary Zn deficiency as factors contributing to abnormal central nervous system development. Dev Brain Dysfunct. 1995;8:79–89. [Google Scholar]
- 21.Rogers JM, Oteiza PI, Keen CL. En: (Mal)Nutrition and infant brain. New York: Wiley-Liss; 1990. Zinc and manganese deficiencies in prenatal and neonatal development with special reference to the central nervous system. [Google Scholar]
- 22.Chowanadisai W, Kelleher SL, Lonnerdal B. Maternal zinc deficiency reduces NMDA receptor expression in neonatal rat brain, which persists into early adulthood. J Neurochem. 2005;94:510–519. doi: 10.1111/j.1471-4159.2005.03246.x. [DOI] [PubMed] [Google Scholar]
- 23.Dvergsten CL, Fosmire GJ, Ollerich DA, Sandstead HH. Alterations in the postnatal development of the cerebellar cortex due to zinc deficiency. I. Impaired acquisition of granule cells. Brain Res. 1983;271:217–226. doi: 10.1016/0006-8993(83)90284-6. [DOI] [PubMed] [Google Scholar]
- 24.DiGirolamo AM, Ramirez-Zea M. Role of zinc in maternal and child mental health. Am J Clin Nutr. 2009;89:940S–945S. doi: 10.3945/ajcn.2008.26692C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frade JM. Unscheduled re-entry into the cell cycle induced by NGF precedes cell death in nascent retinal neurones. J Cell Sci. 2000;113(Pt 7):1139–1148. doi: 10.1242/jcs.113.7.1139. [DOI] [PubMed] [Google Scholar]
- 26.Persengiev SP, Li J, Poulin ML, Kilpatrick DL. E2F2 converts reversibly differentiated PC12 cells to an irreversible, neurotrophin-dependent state. Oncogene. 2001;20:5124–5131. doi: 10.1038/sj.onc.1204663. [DOI] [PubMed] [Google Scholar]
- 27.Clegg MS, Hanna LA, Niles BJ, Momma TY, Keen CL. Zinc deficiency-induced cell death. IUBMB Life. 2005;57:661–669. doi: 10.1080/15216540500264554. [DOI] [PubMed] [Google Scholar]
- 28.Fraker PJ. Roles for cell death in zinc deficiency. J Nutr. 2005;135:359–362. doi: 10.1093/jn/135.3.359. [DOI] [PubMed] [Google Scholar]
- 29.Truong-Tran AQ, Grosser D, Ruffin RE, Murgia C, Zalewski PD. Apoptosis in the normal and inflamed airway epithelium: role of zinc in epithelial protection and procaspase-3 regulation. Biochem Pharmacol. 2003;66:1459–1468. doi: 10.1016/s0006-2952(03)00498-2. [DOI] [PubMed] [Google Scholar]
- 30.Cho E, Hwang JJ, Han SH, Chung SJ, Koh JY, Lee JY. Endogenous Zinc Mediates Apoptotic Programmed Cell Death in the Developing Brain. Neurotox Res. 2009 doi: 10.1007/s12640-009-9085-2. [DOI] [PubMed] [Google Scholar]
- 31.Galluzzi L, Blomgren K, Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci. 2009;10:481–494. doi: 10.1038/nrn2665. [DOI] [PubMed] [Google Scholar]
- 32.Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochim Biophys Acta. 2009;1787:414–420. doi: 10.1016/j.bbabio.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 34.Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–6311. [PubMed] [Google Scholar]
- 35.Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature. 1993;362:847–849. doi: 10.1038/362847a0. [DOI] [PubMed] [Google Scholar]
- 36.Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 37.Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309:1732–1735. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- 38.Reaves SK, Fanzo JC, Arima K, Wu JY, Wang YR, Lei KY. Expression of the p53 tumor suppressor gene is up-regulated by depletion of intracellular zinc in HepG2 cells. J Nutr. 2000;130:1688–1694. doi: 10.1093/jn/130.7.1688. [DOI] [PubMed] [Google Scholar]
- 39.Hainaut P, Milner J. A structural role for metal ions in the “wild-type” conformation of the tumor suppressor protein p53. Cancer Res. 1993;53:1739–1742. [PubMed] [Google Scholar]
- 40.Rainwater R, Parks D, Anderson ME, Tegtmeyer P, Mann K. Role of cysteine residues in regulation of p53 function. Mol Cell Biol. 1995;15:3892–3903. doi: 10.1128/mcb.15.7.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fanzo JC, Reaves SK, Cui L, Zhu L, Lei KY. p53 protein and p21 mRNA levels and caspase-3 activity are altered by zinc status in aortic endothelial cells. Am J Physiol Cell Physiol. 2002;283:C631–638. doi: 10.1152/ajpcell.00248.2001. [DOI] [PubMed] [Google Scholar]
- 42.Niles BJ, Clegg MS, Hanna LA, Chou SS, Momma TY, Hong H, Keen CL. Zinc deficiency-induced iron accumulation, a consequence of alterations in iron regulatory protein-binding activity, iron transporters, and iron storage proteins. J Biol Chem. 2008;283:5168–5177. doi: 10.1074/jbc.M709043200. [DOI] [PubMed] [Google Scholar]
- 43.Ra H, Kim HL, Lee HW, Kim YH. Essential role of p53 in TPEN-induced neuronal apoptosis. FEBS Lett. 2009;583:1516–1520. doi: 10.1016/j.febslet.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 44.Zalewski PD, Forbes IJ, Giannakis C. Physiological role for zinc in prevention of apoptosis (gene-directed death) Biochem Int. 1991;24:1093–1101. [PubMed] [Google Scholar]
- 45.Cohen GM, Sun XM, Snowden RT, Dinsdale D, Skilleter DN. Key morphological features of apoptosis may occur in the absence of internucleosomal DNA fragmentation. Biochem J. 1992;286(Pt 2):331–334. doi: 10.1042/bj2860331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lazebnik YA, Cole S, Cooke CA, Nelson WG, Earnshaw WC. Nuclear events of apoptosis in vitro in cell-free mitotic extracts: a model system for analysis of the active phase of apoptosis. J Cell Biol. 1993;123:7–22. doi: 10.1083/jcb.123.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 48.D'Amelio M, Cavallucci V, Cecconi F. Neuronal caspase-3 signaling: not only cell death. Cell Death Differ. 2009 doi: 10.1038/cdd.2009.180. [DOI] [PubMed] [Google Scholar]
- 49.Perry DK, Smyth MJ, Stennicke HR, Salvesen GS, Duriez P, Poirier GG, Hannun YA. Zinc is a potent inhibitor of the apoptotic protease, caspase-3. A novel target for zinc in the inhibition of apoptosis. J Biol Chem. 1997;272:18530–18533. doi: 10.1074/jbc.272.30.18530. [DOI] [PubMed] [Google Scholar]
- 50.Takahashi A, Alnemri ES, Lazebnik YA, Fernandes-Alnemri T, Litwack G, Moir RD, Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC. Cleavage of lamin A by Mch2 alpha but not CPP32: multiple interleukin 1 beta-converting enzyme-related proteases with distinct substrate recognition properties are active in apoptosis. Proc Natl Acad Sci U S A. 1996;93:8395–8400. doi: 10.1073/pnas.93.16.8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aiuchi T, Mihara S, Nakaya M, Masuda Y, Nakajo S, Nakaya K. Zinc ions prevent processing of caspase-3 during apoptosis induced by geranylgeraniol in HL-60 cells. J Biochem. 1998;124:300–303. doi: 10.1093/oxfordjournals.jbchem.a022111. [DOI] [PubMed] [Google Scholar]
- 52.Ho LH, Ratnaike RN, Zalewski PD. Involvement of intracellular labile zinc in suppression of DEVD-caspase activity in human neuroblastoma cells. Biochem Biophys Res Commun. 2000;268:148–154. doi: 10.1006/bbrc.2000.2090. [DOI] [PubMed] [Google Scholar]
- 53.Chimienti F, Seve M, Richard S, Mathieu J, Favier A. Role of cellular zinc in programmed cell death: temporal relationship between zinc depletion, activation of caspases, and cleavage of Sp family transcription factors. Biochem Pharmacol. 2001;62:51–62. doi: 10.1016/s0006-2952(01)00624-4. [DOI] [PubMed] [Google Scholar]
- 54.Hashemi M, Ghavami S, Eshraghi M, Booy EP, Los M. Cytotoxic effects of intra and extracellular zinc chelation on human breast cancer cells. Eur J Pharmacol. 2007;557:9–19. doi: 10.1016/j.ejphar.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 55.Oteiza PI, Clegg MS, Zago MP, Keen CL. Zinc deficiency induces oxidative stress and AP-1 activation in 3T3 cells. Free Radic Biol Med. 2000;28:1091–1099. doi: 10.1016/s0891-5849(00)00200-8. [DOI] [PubMed] [Google Scholar]
- 56.Oteiza PL, Olin KL, Fraga CG, Keen CL. Oxidant defense systems in testes from zinc-deficient rats. Proc Soc Exp Biol Med. 1996;213:85–91. doi: 10.3181/00379727-213-44040. [DOI] [PubMed] [Google Scholar]
- 57.Virgili F, Canali R, Figus E, Vignolini F, Nobili F, Mengheri E. Intestinal damage induced by zinc deficiency is associated with enhanced CuZn superoxide dismutase activity in rats: effect of dexamethasone or thyroxine treatment. Free Radic Biol Med. 1999;26:1194–1201. doi: 10.1016/s0891-5849(98)00307-4. [DOI] [PubMed] [Google Scholar]
- 58.Bagchi D, Vuchetich PJ, Bagchi M, Tran MX, Krohn RL, Ray SD, Stohs SJ. Protective effects of zinc salts on TPA-induced hepatic and brain lipid peroxidation, glutathione depletion, DNA damage and peritoneal macrophage activation in mice. Gen Pharmacol. 1998;30:43–50. doi: 10.1016/s0306-3623(97)00072-4. [DOI] [PubMed] [Google Scholar]
- 59.Kraus A, Roth HP, Kirchgessner M. Supplementation with vitamin C, vitamin E or beta-carotene influences osmotic fragility and oxidative damage of erythrocytes of zinc-deficient rats. J Nutr. 1997;127:1290–1296. doi: 10.1093/jn/127.7.1290. [DOI] [PubMed] [Google Scholar]
- 60.Oteiza PI, Olin KL, Fraga CG, Keen CL. Zinc deficiency causes oxidative damage to proteins, lipids and DNA in rat testes. J Nutr. 1995;125:823–829. doi: 10.1093/jn/125.4.823. [DOI] [PubMed] [Google Scholar]
- 61.Olin KL, Shigenaga MK, Ames BN, Golub MS, Gershwin ME, Hendrickx AG, Keen CL. Maternal dietary zinc influences DNA strand break and 8-hydroxy-2′-deoxyguanosine levels in infant rhesus monkey liver. Proc Soc Exp Biol Med. 1993;203:461–466. doi: 10.3181/00379727-203-43623. [DOI] [PubMed] [Google Scholar]
- 62.Mackenzie GG, Zago MP, Aimo L, Oteiza PI. Zinc deficiency in neuronal biology. IUBMB Life. 2007;59:299–307. doi: 10.1080/15216540701225966. [DOI] [PubMed] [Google Scholar]
- 63.Rossi L, Migliaccio S, Corsi A, Marzia M, Bianco P, Teti A, Gambelli L, Cianfarani S, Paoletti F, Branca F. Reduced growth and skeletal changes in zinc-deficient growing rats are due to impaired growth plate activity and inanition. J Nutr. 2001;131:1142–1146. doi: 10.1093/jn/131.4.1142. [DOI] [PubMed] [Google Scholar]
- 64.Hunziker EB. Mechanism of longitudinal bone growth and its regulation by growth plate chondrocytes. Microsc Res Tech. 1994;28:505–519. doi: 10.1002/jemt.1070280606. [DOI] [PubMed] [Google Scholar]
- 65.Rodriguez JP, Rosselot G. Effects of zinc on cell proliferation and proteoglycan characteristics of epiphyseal chondrocytes. J Cell Biochem. 2001;82:501–511. doi: 10.1002/jcb.1178. [DOI] [PubMed] [Google Scholar]
- 66.Wang X, Fosmire GJ, Gay CV, Leach RM., Jr Short-term zinc deficiency inhibits chondrocyte proliferation and induces cell apoptosis in the epiphyseal growth plate of young chickens. J Nutr. 2002;132:665–673. doi: 10.1093/jn/132.4.665. [DOI] [PubMed] [Google Scholar]
- 67.Fraker PJ, King LE, Laakko T, Vollmer TL. The dynamic link between the integrity of the immune system and zinc status. J Nutr. 2000;130:1399S–1406S. doi: 10.1093/jn/130.5.1399S. [DOI] [PubMed] [Google Scholar]
- 68.Prasad AS, Beck FW, Endre L, Handschu W, Kukuruga M, Kumar G. Zinc deficiency affects cell cycle and deoxythymidine kinase gene expression in HUT-78 cells. J Lab Clin Med. 1996;128:51–60. doi: 10.1016/s0022-2143(96)90113-4. [DOI] [PubMed] [Google Scholar]
- 69.Klecha AJ, Salgueiro J, Wald M, Boccio J, Zubillaga M, Leonardi NM, Gorelik G, Cremaschi GA. In vivo iron and zinc deficiency diminished T- and B-selective mitogen stimulation of murine lymphoid cells through protein kinase C-mediated mechanism. Biol Trace Elem Res. 2005;104:173–183. doi: 10.1385/BTER:104:2:173. [DOI] [PubMed] [Google Scholar]
- 70.Wong SH, Zhao Y, Schoene NW, Han CT, Shih RS, Lei KY. Zinc deficiency depresses p21 gene expression: inhibition of cell cycle progression is independent of the decrease in p21 protein level in HepG2 cells. Am J Physiol Cell Physiol. 2007;292:C2175–2184. doi: 10.1152/ajpcell.00256.2006. [DOI] [PubMed] [Google Scholar]
- 71.Li Y, Dowbenko D, Lasky LA. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J Biol Chem. 2002;277:11352–11361. doi: 10.1074/jbc.M109062200. [DOI] [PubMed] [Google Scholar]
- 72.el-Deiry WS, Tokino T, Waldman T, Oliner JD, Velculescu VE, Burrell M, Hill DE, Healy E, Rees JL, Hamilton SR, et al. Topological control of p21WAF1/CIP1 expression in normal and neoplastic tissues. Cancer Res. 1995;55:2910–2919. [PubMed] [Google Scholar]
- 73.LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- 74.Oteiza PI, Cuellar S, Lonnerdal B, Hurley LS, Keen CL. Influence of maternal dietary zinc intake on in vitro tubulin polymerization in fetal rat brain. Teratology. 1990;41:97–104. doi: 10.1002/tera.1420410110. [DOI] [PubMed] [Google Scholar]
- 75.Albensi BC, Mattson MP. Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse. 2000;35:151–159. doi: 10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 76.Mattson MP, Culmsee C, Yu Z, Camandola S. Roles of nuclear factor kappaB in neuronal survival and plasticity. J Neurochem. 2000;74:443–456. doi: 10.1046/j.1471-4159.2000.740443.x. [DOI] [PubMed] [Google Scholar]
- 77.Mattson MP. NF-kappaB in the survival and plasticity of neurons. Neurochem Res. 2005;30:883–893. doi: 10.1007/s11064-005-6961-x. [DOI] [PubMed] [Google Scholar]
- 78.Mattson MP, Meffert MK. Roles for NF-kappaB in nerve cell survival, plasticity, and disease. Cell Death Differ. 2006;13:852–860. doi: 10.1038/sj.cdd.4401837. [DOI] [PubMed] [Google Scholar]
- 79.Denis-Donini S, Caprini A, Frassoni C, Grilli M. Members of the NF-kappaB family expressed in zones of active neurogenesis in the postnatal and adult mouse brain. Brain Res Dev Brain Res. 2005;154:81–89. doi: 10.1016/j.devbrainres.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 80.Denis-Donini S, Dellarole A, Crociara P, Francese MT, Bortolotto V, Quadrato G, Canonico PL, Orsetti M, Ghi P, Memo M, Bonini SA, Ferrari-Toninelli G, Grilli M. Impaired adult neurogenesis associated with short-term memory defects in NF-kappaB p50-deficient mice. J Neurosci. 2008;28:3911–3919. doi: 10.1523/JNEUROSCI.0148-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hanz S, Fainzilber M. Integration of retrograde axonal and nuclear transport mechanisms in neurons: implications for therapeutics. Neuroscientist. 2004;10:404–408. doi: 10.1177/1073858404267884. [DOI] [PubMed] [Google Scholar]
- 82.Mathieu AL, Gonin S, Leverrier Y, Blanquier B, Thomas J, Dantin C, Martin G, Baverel G, Marvel J. Activation of the phosphatidylinositol 3-kinase/Akt pathway protects against interleukin-3 starvation but not DNA damage-induced apoptosis. J Biol Chem. 2001;276:10935–10942. doi: 10.1074/jbc.M007147200. [DOI] [PubMed] [Google Scholar]
- 83.Yang X, Liu L, Sternberg D, Tang L, Galinsky I, DeAngelo D, Stone R. The FLT3 Internal tandem duplication mutation prevents apoptosis in interleukin-3-deprived BaF3 cells due to protein kinase A and ribosomal S6 kinase 1-mediated BAD phosphorylation at serine 112. Cancer Res. 2005;65:7338–7347. doi: 10.1158/0008-5472.CAN-04-2263. [DOI] [PubMed] [Google Scholar]
- 84.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 85.Zhu Y, Yang GY, Ahlemeyer B, Pang L, Che XM, Culmsee C, Klumpp S, Krieglstein J. Transforming growth factor-beta 1 increases bad phosphorylation and protects neurons against damage. J Neurosci. 2002;22:3898–3909. doi: 10.1523/JNEUROSCI.22-10-03898.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Blume-Jensen P, Janknecht R, Hunter T. The kit receptor promotes cell survival via activation of PI 3-kinase and subsequent Akt-mediated phosphorylation of Bad on Ser136. Curr Biol. 1998;8:779–782. doi: 10.1016/s0960-9822(98)70302-1. [DOI] [PubMed] [Google Scholar]
- 87.Virdee K, Parone PA, Tolkovsky AM. Phosphorylation of the proapoptotic protein BAD on serine 155, a novel site, contributes to cell survival. Curr Biol. 2000;10:1151–1154. doi: 10.1016/s0960-9822(00)00702-8. [DOI] [PubMed] [Google Scholar]
- 88.Wang X, McCullough KD, Franke TF, Holbrook NJ. Epidermal growth factor receptor-dependent Akt activation by oxidative stress enhances cell survival. J Biol Chem. 2000;275:14624–14631. doi: 10.1074/jbc.275.19.14624. [DOI] [PubMed] [Google Scholar]
- 89.Dong-Yun S, Yu-Ru D, Shan-Lin L, Ya-Dong Z, Lian W. Redox stress regulates cell proliferation and apoptosis of human hepatoma through Akt protein phosphorylation. FEBS Lett. 2003;542:60–64. doi: 10.1016/s0014-5793(03)00338-7. [DOI] [PubMed] [Google Scholar]
- 90.Min YK, Lee JE, Chung KC. Zinc induces cell death in immortalized embryonic hippocampal cells via activation of Akt-GSK-3beta signaling. Exp Cell Res. 2007;313:312–321. doi: 10.1016/j.yexcr.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 91.Wang XT, Pei DS, Xu J, Guan QH, Sun YF, Liu XM, Zhang GY. Opposing effects of Bad phosphorylation at two distinct sites by Akt1 and JNK1/2 on ischemic brain injury. Cell Signal. 2007;19:1844–1856. doi: 10.1016/j.cellsig.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 92.Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18:189–218. doi: 10.1038/sj.leu.2403241. [DOI] [PubMed] [Google Scholar]
- 93.Samuels IS, Saitta SC, Landreth GE. MAP'ing CNS development and cognition: an ERKsome process. Neuron. 2009;61:160–167. doi: 10.1016/j.neuron.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Miller FD, Gauthier AS. Timing is everything: making neurons versus glia in the developing cortex. Neuron. 2007;54:357–369. doi: 10.1016/j.neuron.2007.04.019. [DOI] [PubMed] [Google Scholar]
- 95.Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, Saitta SC, Landreth GE. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci. 2008;28:6983–6995. doi: 10.1523/JNEUROSCI.0679-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Adamo AM, Zago MP, Mackenzie GG, Aimo L, Keen CL, Keenan A, Oteiza PI. The Role of Zinc in the Modulation of Neuronal Proliferation and Apoptosis. Neurotox Res. 2009 doi: 10.1007/s12640-009-9067-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yamamoto T, Ebisuya M, Ashida F, Okamoto K, Yonehara S, Nishida E. Continuous ERK activation downregulates antiproliferative genes throughout G1 phase to allow cell-cycle progression. Curr Biol. 2006;16:1171–1182. doi: 10.1016/j.cub.2006.04.044. [DOI] [PubMed] [Google Scholar]